
Drug Information
General Information of This Drug
| Drug ID | DRG00005 | |||||
|---|---|---|---|---|---|---|
| Drug Name | Primaquine | |||||
| Synonyms |
PRIMAQUINE; 90-34-6; Primachin; Neo-Quipenyl; 8-(4-Amino-1-methylbutylamino)-6-methoxyquinoline; Primaquin; Primachinum; Primaquinum; Primaquina; Kanaprim; Primachina; (+/-)-Primaquine; 1,4-Pentanediamine, N4-(6-methoxy-8-quinolinyl)-; Maliride; SN 13,272; 6-Methoxy-8-(4-amino-1-methylbutylamino)quinoline; Primaquinum [INN-Latin]; Primaquina [INN-Spanish]; 4-N-(6-methoxyquinolin-8-yl)pentane-1,4-diamine; N-(5-aminopentan-2-yl)-6-methoxyquinolin-8-amine; NSC 27296; CCRIS 4109; CHEBI:8405; S. N. 13272; HSDB 6516; 8-((4-Amino-1-methylbutyl)amino)-6-methoxyquinoline; EINECS 201-987-2; UNII-MVR3634GX1; NSC-27296; Primaquine (INN); WR 2975; MVR3634GX1; N4-(6-Methoxy-8-quinolinyl)-1,4-pentanediamine; Quinoline, 8-(4-amino-1-methylbutylamino)-6-methoxy; WR-2975; SN-13272; DTXSID8023509; N-(6-methoxyquinolin-8-yl)pentane-1,4-diamine; (+)-Primaquine; (-)-Primaquine; NSC27296; N(4)-(6-methoxyquinolin-8-yl)pentane-1,4-diamine; 1,4-Pentanediamine, N(4)-(6-methoxy-8-quinolinyl)-; 8-[(4-Amino-1-methylbutyl)amino]-6-methoxyquinoline; Quinoline, 8-(4-amino-1-methylbutylamino)-6-methoxy-; N-[(S)-4-Amino-1-methylbutyl]-6-methoxy-8-quinolinamine; Quinoline, 8-((4-amino-1-methylbutyl)amino)-6-methoxy-; Primachina [DCIT]; dl-Primaquine; PRIMAQUINE [INN]; Primaquinum (INN-Latin); Primaquina (INN-Spanish); Primaquine [INN:BAN]; BRN 0019337; Maliride; NSC 27296; Neo-Quipenyl; Primachin; N4-(6-methoxy-8-quinolyl)pentane-1,4-diamine; 6-Methoxy-8-[(4-amino-1-methylbutyl)amino]quinoline; N~4~-[6-(methyloxy)quinolin-8-yl]pentane-1,4-diamine; CHEMBL506; MLS001334045; Kanaprim (TN); n4-[6-methoxy-8-quinolinyl]-1,4-pentanediamine; 4-22-00-05817 (Beilstein Handbook Reference); NCGC00178754-06; N~4~-(6-methoxyquinolin-8-yl)pentane-1,4-diamine; SMR000875314; 6-Methoxy-8-((4-amino-1-methylbutyl)amino)quinoline; Quinoline, 8-[(4-amino-1-methylbutyl)amino]-6-methoxy-; (RS)-primaquine; TG1-296; TG1-297; Spectrum_000830; PRIMAQUINE [MI]; Prestwick0_000476; Prestwick1_000476; Prestwick2_000476; Prestwick3_000476; Spectrum2_000887; Spectrum3_000552; Spectrum4_000484; Spectrum5_001363; PRIMAQUINE [VANDF]; Epitope ID:131792; PRIMAQUINE [WHO-DD]; Oprea1_546209; SCHEMBL22207; BSPBio_000612; BSPBio_002223; KBioGR_000967; KBioSS_001310; 4-amino-1-methylbutyl(6-methoxy-8-quinolyl)amine; DivK1c_000806; SPBio_000674; SPBio_002551; (+)-N4-(6-Methoxy-8-quinolinyl)-1,4-pentanediamine; (-)-N4-(6-Methoxy-8-quinolinyl)-1,4-pentanediamine; BPBio1_000674; cid_359247; DTXCID903509; GTPL9952; BDBM71542; KBio1_000806; KBio2_001310; KBio2_003878; KBio2_006446; KBio3_001723; P01BA03; NINDS_000806; HMS2090J17; BCP29271; BBL011330; HY-12651A; MFCD00598906; STL146416; 1, N4-(6-methoxy-8-quinolinyl)-; AKOS005721199; DB01087; WLN: T66 BNJ HO1 JMY1&3Z; IDI1_000806; SMP1_000263; NCGC00178754-01; NCGC00178754-02; NCGC00178754-03; AC-23007; AS-30679; NCI60_001035; NCI60_005887; SBI-0051491.P003; AB00053529; CS-0013754; NS00000469; C07627; D08420; EN300-144245; AB00053529-11; AB00053529_12; AB00053529_14; N-(6-Methoxy-8-quinolinyl)-1,4-pentanediamine; A843518; N4-(6-methoxyquinolin-8-yl)pentane-1,4-diamine; Q419834; BRD-A55913614-316-06-2; SR-05000001864-11; N4-(6-methoxy-8-quinolyl)pentane-1,4-diamine;Primaquine; N4-(6-Methoxyquinolin-8-yl)pentane-1,4-diamine (2 H3PO4); N4-(6-methoxy-8-quinolinyl)pentane-1,4-diamine;phosphoric acid; N4-(6-methoxyquinolin-8-yl)pentane-1,4-diamine;phosphoric acid; (4-amino-1-methyl-butyl)-(6-methoxy-8-quinolyl)amine;phosphoric acid
Click to Show/Hide
|
|||||
| Target(s) | Plasmodium Deoxyribonucleic acid (Malaria DNA) | Target Info | ||||
| Structure |
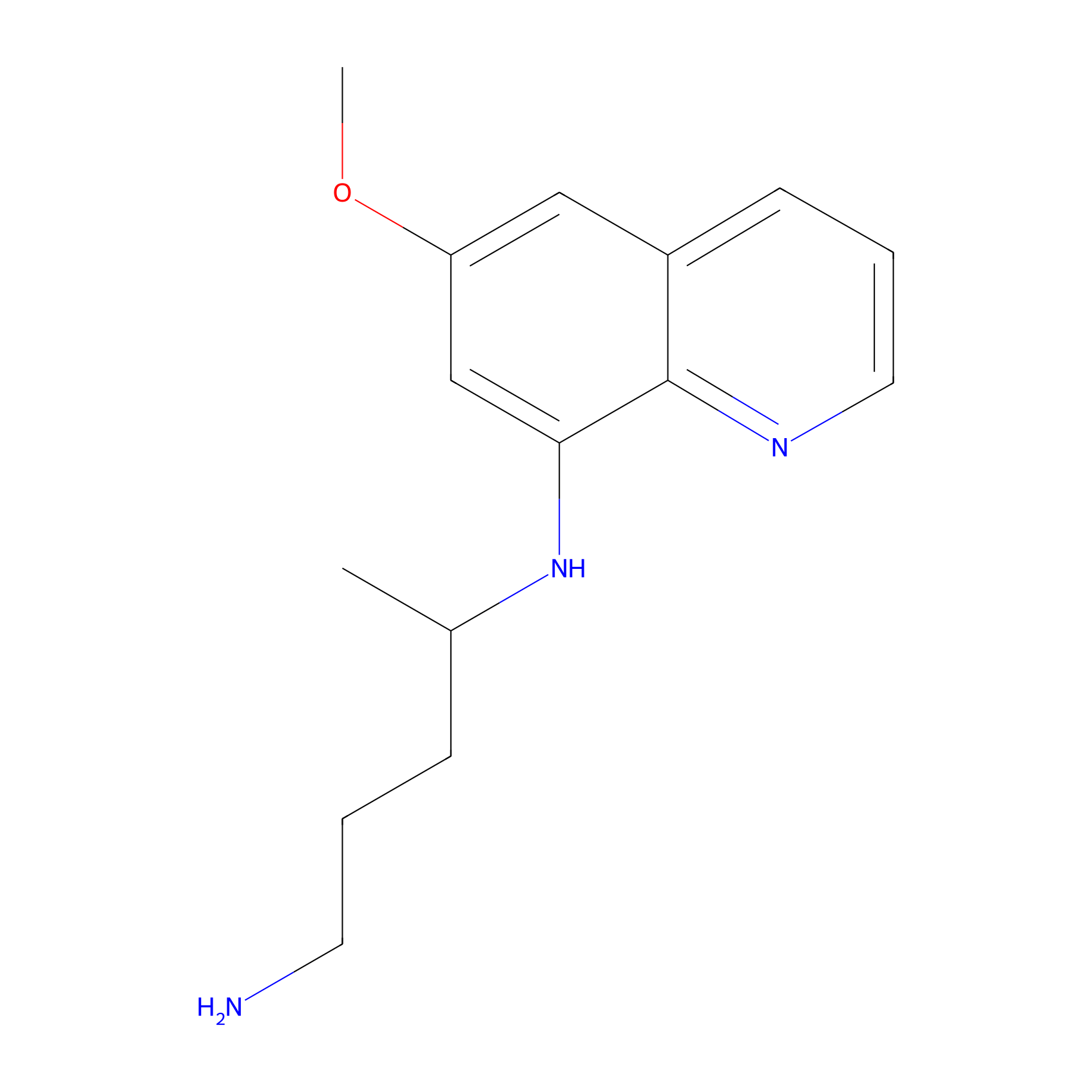
|
|||||
| Formula |
C15H21N3O
|
|||||
| #Ro5 Violations (Lipinski): 0 | Molecular Weight (mw) | 259.35 | ||||
| Lipid-water partition coefficient (xlogp) | 2.2 | |||||
| Hydrogen Bond Donor Count (hbonddonor) | 2 | |||||
| Hydrogen Bond Acceptor Count (hbondacc) | 4 | |||||
| Rotatable Bond Count (rotbonds) | 6 | |||||
| PubChem CID | ||||||
| Canonical smiles |
CC(CCCN)NC1=C2C(=CC(=C1)OC)C=CC=N2
|
|||||
| InChI |
InChI=1S/C15H21N3O/c1-11(5-3-7-16)18-14-10-13(19-2)9-12-6-4-8-17-15(12)14/h4,6,8-11,18H,3,5,7,16H2,1-2H3
|
|||||
| InChIKey |
INDBQLZJXZLFIT-UHFFFAOYSA-N
|
|||||
| IUPAC Name |
4-N-(6-methoxyquinolin-8-yl)pentane-1,4-diamine
|
|||||
The activity data of This Drug
| Standard Type | Value | Administration times | Cell line | Cell line ID | Ref. | |
|---|---|---|---|---|---|---|
| Half Maximal Inhibitory Concentration (IC50) | 2.07 µM | 18-24 h | Plasmodium falciparum strain 3D7 | 5833 | [1] | |
| Half Maximal Cytotoxicity Concentration (CC50) | 120.03 uM | N.A. | Hep-G2 cell | CVCL_0027 | [2] | |
| Half Maximal Inhibitory Concentration (IC50) | 6.9 uM | N.A. | MCF-7 cell | CVCL_0031 | [3] | |
| Half Maximal Inhibitory Concentration (IC50) | 69.7 uM | N.A. | HT29 cell | CVCL_A8EZ | [3] | |
| Half Maximal Inhibitory Concentration (IC50) | ≥100 uM | N.A. | SW620 cell | CVCL_0547 | [4] | |
Each Peptide-drug Conjugate Related to This Drug
Full Information of The Activity Data of The PDC(s) Related to This Drug
PDIP-DBCO-SS-PQ [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 4.25 µM | |||
| Evaluation Method | Flow cytometry assay | ||||
| Administration Time | 18-24 h | ||||
| MOA of PDC |
As a proof of concept, we aimed to produce first-generation PDCs by conjugating the antimalarial drug primaquine (PQ) onto PDIP. Although PQ is one of the few drugs without clinically relevant resistance, it does not have widespread use because it causes hemolysis in individuals who are deficient in glucose-6-phosphate dehydrogenase (G6PD), a genetic trait common in malaria-endemic areas. Furthermore, PQ is metabolized into carboxyprimaquine in the body, which does not have any activity against the parasite. The proposed PDC approach provides the potential to deliver PQ directly to the parasite, which could prevent its interaction with healthy tissues and slow the conversion of PQ into inactive byproducts. Further, the combination of the peptide and drug, each with distinct antiplasmodial mechanisms of action, provides the potential to avoid the formation of drug-resistant parasites. Herein, we report the design, synthesis, and biological evaluation of a library of PDIP-PQ conjugates. Various design elements of the PDCs were probed to investigate their effect on biological activity, including: (i) the location of the PDIP conjugation site, (ii) the hydrophilicity of the linker between the peptide and drug, (iii) the spacing between the peptide and drug, and (iv) whether the linker can be cleaved to release the drug cargo under conditions which mimic the intracellular environment of infected RBCs. This work demonstrates that conjugation within the flexible interhelix spacer of PDIP and incorporation of traceless cleavable linkersbearing either a disulfide or trioxolane moietyare important for maintaining the low micromolar potency of the PQ drug cargo against P. falciparum.
Click to Show/Hide
|
||||
| Description |
The six PDIP-PQ conjugates were analyzed for their ability to inhibit the in vitro growth of P. falciparum asexual blood stage parasites (strain 3D7) in RBCs and were compared to the activity of the parent drug and peptide. We were encouraged to discover that most of the PDIP-PQ PDCs retained antiplasmodial activity similar to PDIP, with IC50 values in the low micromolar range. Notably, the various design elements probed provided valuable information regarding which PDC characteristics can be modified to improve activity.
Click to Show/Hide
|
||||
| In Vivo Model | Plasmodium falciparum 3D7. | ||||
| Half life period | 4.55 h | ||||
PDIP-DBCO-triox-PQ [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 4.68 µM | |||
| Evaluation Method | Flow cytometry assay | ||||
| Administration Time | 18-24 h | ||||
| MOA of PDC |
As a proof of concept, we aimed to produce first-generation PDCs by conjugating the antimalarial drug primaquine (PQ) onto PDIP. Although PQ is one of the few drugs without clinically relevant resistance, it does not have widespread use because it causes hemolysis in individuals who are deficient in glucose-6-phosphate dehydrogenase (G6PD), a genetic trait common in malaria-endemic areas. Furthermore, PQ is metabolized into carboxyprimaquine in the body, which does not have any activity against the parasite. The proposed PDC approach provides the potential to deliver PQ directly to the parasite, which could prevent its interaction with healthy tissues and slow the conversion of PQ into inactive byproducts. Further, the combination of the peptide and drug, each with distinct antiplasmodial mechanisms of action, provides the potential to avoid the formation of drug-resistant parasites. Herein, we report the design, synthesis, and biological evaluation of a library of PDIP-PQ conjugates. Various design elements of the PDCs were probed to investigate their effect on biological activity, including: (i) the location of the PDIP conjugation site, (ii) the hydrophilicity of the linker between the peptide and drug, (iii) the spacing between the peptide and drug, and (iv) whether the linker can be cleaved to release the drug cargo under conditions which mimic the intracellular environment of infected RBCs. This work demonstrates that conjugation within the flexible interhelix spacer of PDIP and incorporation of traceless cleavable linkersbearing either a disulfide or trioxolane moietyare important for maintaining the low micromolar potency of the PQ drug cargo against P. falciparum.
Click to Show/Hide
|
||||
| Description |
The six PDIP-PQ conjugates were analyzed for their ability to inhibit the in vitro growth of P. falciparum asexual blood stage parasites (strain 3D7) in RBCs and were compared to the activity of the parent drug and peptide. We were encouraged to discover that most of the PDIP-PQ PDCs retained antiplasmodial activity similar to PDIP, with IC50 values in the low micromolar range. Notably, the various design elements probed provided valuable information regarding which PDC characteristics can be modified to improve activity.
Click to Show/Hide
|
||||
| In Vivo Model | Plasmodium falciparum 3D7. | ||||
| Half life period | 3.99 h | ||||
PDIP-PEG3-PQ [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 8.81 µM | |||
| Evaluation Method | Flow cytometry assay | ||||
| Administration Time | 18-24 h | ||||
| MOA of PDC |
As a proof of concept, we aimed to produce first-generation PDCs by conjugating the antimalarial drug primaquine (PQ) onto PDIP. Although PQ is one of the few drugs without clinically relevant resistance, it does not have widespread use because it causes hemolysis in individuals who are deficient in glucose-6-phosphate dehydrogenase (G6PD), a genetic trait common in malaria-endemic areas. Furthermore, PQ is metabolized into carboxyprimaquine in the body, which does not have any activity against the parasite. The proposed PDC approach provides the potential to deliver PQ directly to the parasite, which could prevent its interaction with healthy tissues and slow the conversion of PQ into inactive byproducts. Further, the combination of the peptide and drug, each with distinct antiplasmodial mechanisms of action, provides the potential to avoid the formation of drug-resistant parasites. Herein, we report the design, synthesis, and biological evaluation of a library of PDIP-PQ conjugates. Various design elements of the PDCs were probed to investigate their effect on biological activity, including: (i) the location of the PDIP conjugation site, (ii) the hydrophilicity of the linker between the peptide and drug, (iii) the spacing between the peptide and drug, and (iv) whether the linker can be cleaved to release the drug cargo under conditions which mimic the intracellular environment of infected RBCs. This work demonstrates that conjugation within the flexible interhelix spacer of PDIP and incorporation of traceless cleavable linkersbearing either a disulfide or trioxolane moietyare important for maintaining the low micromolar potency of the PQ drug cargo against P. falciparum.
Click to Show/Hide
|
||||
| Description |
The six PDIP-PQ conjugates were analyzed for their ability to inhibit the in vitro growth of P. falciparum asexual blood stage parasites (strain 3D7) in RBCs and were compared to the activity of the parent drug and peptide. We were encouraged to discover that most of the PDIP-PQ PDCs retained antiplasmodial activity similar to PDIP, with IC50 values in the low micromolar range. Notably, the various design elements probed provided valuable information regarding which PDC characteristics can be modified to improve activity.
Click to Show/Hide
|
||||
| In Vivo Model | Plasmodium falciparum 3D7. | ||||
PDIP-PEG1-PQ [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 11.3 µM | |||
| Evaluation Method | Flow cytometry assay | ||||
| Administration Time | 18-24 h | ||||
| MOA of PDC |
As a proof of concept, we aimed to produce first-generation PDCs by conjugating the antimalarial drug primaquine (PQ) onto PDIP. Although PQ is one of the few drugs without clinically relevant resistance, it does not have widespread use because it causes hemolysis in individuals who are deficient in glucose-6-phosphate dehydrogenase (G6PD), a genetic trait common in malaria-endemic areas. Furthermore, PQ is metabolized into carboxyprimaquine in the body, which does not have any activity against the parasite. The proposed PDC approach provides the potential to deliver PQ directly to the parasite, which could prevent its interaction with healthy tissues and slow the conversion of PQ into inactive byproducts. Further, the combination of the peptide and drug, each with distinct antiplasmodial mechanisms of action, provides the potential to avoid the formation of drug-resistant parasites. Herein, we report the design, synthesis, and biological evaluation of a library of PDIP-PQ conjugates. Various design elements of the PDCs were probed to investigate their effect on biological activity, including: (i) the location of the PDIP conjugation site, (ii) the hydrophilicity of the linker between the peptide and drug, (iii) the spacing between the peptide and drug, and (iv) whether the linker can be cleaved to release the drug cargo under conditions which mimic the intracellular environment of infected RBCs. This work demonstrates that conjugation within the flexible interhelix spacer of PDIP and incorporation of traceless cleavable linkersbearing either a disulfide or trioxolane moietyare important for maintaining the low micromolar potency of the PQ drug cargo against P. falciparum.
Click to Show/Hide
|
||||
| Description |
The six PDIP-PQ conjugates were analyzed for their ability to inhibit the in vitro growth of P. falciparum asexual blood stage parasites (strain 3D7) in RBCs and were compared to the activity of the parent drug and peptide. We were encouraged to discover that most of the PDIP-PQ PDCs retained antiplasmodial activity similar to PDIP, with IC50 values in the low micromolar range. Notably, the various design elements probed provided valuable information regarding which PDC characteristics can be modified to improve activity.
Click to Show/Hide
|
||||
| In Vivo Model | Plasmodium falciparum 3D7. | ||||
PDIP-alk-PQ [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 12.9 µM | |||
| Evaluation Method | Flow cytometry assay | ||||
| Administration Time | 18-24 h | ||||
| MOA of PDC |
As a proof of concept, we aimed to produce first-generation PDCs by conjugating the antimalarial drug primaquine (PQ) onto PDIP. Although PQ is one of the few drugs without clinically relevant resistance, it does not have widespread use because it causes hemolysis in individuals who are deficient in glucose-6-phosphate dehydrogenase (G6PD), a genetic trait common in malaria-endemic areas. Furthermore, PQ is metabolized into carboxyprimaquine in the body, which does not have any activity against the parasite. The proposed PDC approach provides the potential to deliver PQ directly to the parasite, which could prevent its interaction with healthy tissues and slow the conversion of PQ into inactive byproducts. Further, the combination of the peptide and drug, each with distinct antiplasmodial mechanisms of action, provides the potential to avoid the formation of drug-resistant parasites. Herein, we report the design, synthesis, and biological evaluation of a library of PDIP-PQ conjugates. Various design elements of the PDCs were probed to investigate their effect on biological activity, including: (i) the location of the PDIP conjugation site, (ii) the hydrophilicity of the linker between the peptide and drug, (iii) the spacing between the peptide and drug, and (iv) whether the linker can be cleaved to release the drug cargo under conditions which mimic the intracellular environment of infected RBCs. This work demonstrates that conjugation within the flexible interhelix spacer of PDIP and incorporation of traceless cleavable linkersbearing either a disulfide or trioxolane moietyare important for maintaining the low micromolar potency of the PQ drug cargo against P. falciparum.
Click to Show/Hide
|
||||
| Description |
The six PDIP-PQ conjugates were analyzed for their ability to inhibit the in vitro growth of P. falciparum asexual blood stage parasites (strain 3D7) in RBCs and were compared to the activity of the parent drug and peptide. We were encouraged to discover that most of the PDIP-PQ PDCs retained antiplasmodial activity similar to PDIP, with IC50 values in the low micromolar range. Notably, the various design elements probed provided valuable information regarding which PDC characteristics can be modified to improve activity.
Click to Show/Hide
|
||||
| In Vivo Model | Plasmodium falciparum 3D7. | ||||
[GG]PDIP-alk-PQ [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 22.5 µM | |||
| Evaluation Method | Flow cytometry assay | ||||
| Administration Time | 18-24 h | ||||
| MOA of PDC |
As a proof of concept, we aimed to produce first-generation PDCs by conjugating the antimalarial drug primaquine (PQ) onto PDIP. Although PQ is one of the few drugs without clinically relevant resistance, it does not have widespread use because it causes hemolysis in individuals who are deficient in glucose-6-phosphate dehydrogenase (G6PD), a genetic trait common in malaria-endemic areas. Furthermore, PQ is metabolized into carboxyprimaquine in the body, which does not have any activity against the parasite. The proposed PDC approach provides the potential to deliver PQ directly to the parasite, which could prevent its interaction with healthy tissues and slow the conversion of PQ into inactive byproducts. Further, the combination of the peptide and drug, each with distinct antiplasmodial mechanisms of action, provides the potential to avoid the formation of drug-resistant parasites. Herein, we report the design, synthesis, and biological evaluation of a library of PDIP-PQ conjugates. Various design elements of the PDCs were probed to investigate their effect on biological activity, including: (i) the location of the PDIP conjugation site, (ii) the hydrophilicity of the linker between the peptide and drug, (iii) the spacing between the peptide and drug, and (iv) whether the linker can be cleaved to release the drug cargo under conditions which mimic the intracellular environment of infected RBCs. This work demonstrates that conjugation within the flexible interhelix spacer of PDIP and incorporation of traceless cleavable linkersbearing either a disulfide or trioxolane moietyare important for maintaining the low micromolar potency of the PQ drug cargo against P. falciparum.
Click to Show/Hide
|
||||
| Description |
The six PDIP-PQ conjugates were analyzed for their ability to inhibit the in vitro growth of P. falciparum asexual blood stage parasites (strain 3D7) in RBCs and were compared to the activity of the parent drug and peptide. We were encouraged to discover that most of the PDIP-PQ PDCs retained antiplasmodial activity similar to PDIP, with IC50 values in the low micromolar range. Notably, the various design elements probed provided valuable information regarding which PDC characteristics can be modified to improve activity.
Click to Show/Hide
|
||||
| In Vivo Model | Plasmodium falciparum 3D7. | ||||
References