
Drug Information
General Information of This Drug
| Drug ID | DRG00024 | |||||
|---|---|---|---|---|---|---|
| Drug Name | Thapsigargin | |||||
| Synonyms |
thapsigargin; 67526-95-8; MFCD00083511; Z96BQ26RZD; CHEMBL96926; CHEBI:9516; (-)-Thapsigargin; (3S,3aR,4S,6S,6aR,7S,8S,9bS)-6-(acetyloxy)-4-(butanoyloxy)-3,3a-dihydroxy-3,6,9-trimethyl-8-{[(2Z)-2-methylbut-2-enoyl]oxy}-2-oxo-2,3,3a,4,5,6,6a,7,8,9b-decahydroazuleno[4,5-b]furan-7-yl octanoate; TG1; SR-01000076181; UNII-Z96BQ26RZD; OCTANOIC ACID [3S-[3ALPHA, 3ABETA, 4ALPHA, 6BETA, 6ABETA, 7BETA, 8ALPHA(Z), 9BALPHA]]-6-(ACETYLOXY)-2,3,-3A,4,5,6,6A,7,8,9B-DECAHYDRO-3,3A-DIHYDROXY-3,6,9-TRIMETHYL-8-[(2-METHYL-1-OXO-2-BUTENYL)OX Y]-2-OXO-4-(1-OXOBUTOXY)-AZULENO[4,5-B]FURAN-7-YL ESTER; -7-yl ester; C34H50O12; Azuleno[4,5-b]furan, octanoic acid deriv.; THAPSIGARGIN [MI]; n-7-yl ester (9CI); Lopac0_001262; SCHEMBL82423; BSPBio_001501; MLS006010944; GTPL5351; CHEBI:93212; IXFPJGBNCFXKPI-FSIHEZPISA-N; HMS1361L03; HMS1791L03; HMS1989L03; HMS3263N06; HMS3402L03; Tox21_501262; BDBM50035612; HB1118; s7895; AKOS024456410; AT23806; CCG-205336; LMPR0103410001; LP01262; SDCCGSBI-0051229.P002; IDI1_033971; NCGC00016060-08; NCGC00162381-05; NCGC00162381-06; NCGC00261947-01; AC-32567; AS-56389; HY-13433; Octanoic acid, 6-(acetyloxy)-2,3,3a,4,5,6,6a,7,8,9b-decahydro-3,3a-dihydroxy-3,6,9-trimethyl-8-((2-methyl-1-oxo-2-butenyl)oxy)-2-oxo-4-(1-oxobutoxy)azuleno(4,5-b)furan-7-yl ester, (3S-(3alpha,3abeta,4alpha,6beta,6abeta,7beta,8alpha(Z),9balpha))-; SMR001456557; Thapsigargin, >=98% (HPLC), solid film; CS-0006886; EU-0101262; C09561; T 9033; alpha,6beta,6abeta,7beta,8alpha(Z),9balpha]]-; Q3981006; SR-01000076181-1; SR-01000076181-5; BRD-K69023402-001-02-5; Y]-2-OXO-4-(1-OXOBUTOXY)-AZULENO[4,5-B]FURAN-7-YL ESTER; OCTANOIC ACID [3S-[3ALPHA, 3ABETA, 4ALPHA, 6BETA, 6ABETA, 7BETA, 8ALPHA(Z),; (3S,3aR,4S,6S,6AR,7S,8S,9bS)-6-(Acetyloxy)-2,3,3a,4,5,6,6a,7,8,9b- decahydro-3,3a-dihydroxy-3,6,9-trimethyl-8-[[(2Z)-2-methyl-1-oxo-2-butenyl]oxy]-2-oxo-4-(1-oxobutoxy)azuleno[4,5-b]furan-7-yl octanoate; (3S,3aR,4S,6S,6aR,7S,8S,9bS)-6-(acetyloxy)-2,3,3a,4,5,6,6a,7,8,9b-decahydro-3,3a-dihydroxy-3,6,9-trimethyl-8-[[(2Z)-2-methyl-1-oxo-2-buten-1-yl]oxy]-2-oxo-4-(1-oxobutoxy)azuleno[4,5-b]f uran-7-yl ester; (3S,3aR,4S,6S,6aR,7S,8S,9bS)-6-(acetyloxy)-4-(butanoyloxy)-3,3a-dihydroxy-3,6,9-trimethyl-8-{[(2Z)-2-methylbut-2-enoyl]oxy}-2-oxo-2H,3H,3aH,4H,5H,6H,6aH,7H,8H,9bH-azuleno[4,5-b]furan-7-yl octanoate; (3S,3aR,4S,6S,6aR,7S,8S,9bS)-6-Acetoxy-4-(butyryloxy)-3,3a-dihydroxy-3,6,9-trimethyl-8-(((Z)-2-methylbut-2-enoyl)oxy)-2-oxo-2,3,3a,4,5,6,6a,7,8,9b-decahydroazuleno[4,5-b]furan-7-yl octanoate; 123269-03-4; 9BALPHA]]-6-(ACETYLOXY)-2,3,-3A,4,5,6,6A,7,8,9B-DECAHYDRO-3,3A-DIHYDROXY-3,6,9-TRIMETHYL-8-[(2-METHYL-1-OXO-2-BUTENYL)OX; octanoic acid {3S-[3alpha,3abeta,4alpha,6beta,6abeta,7beta,8alpha(Z),9balpha]}-6-(acetoxy)-2,3,3a,4,5,6,6a,7,8,9b-decahydro-3,3a-dihydroxy-3,6,9-trimethyl-8-[(2-methyl-1-oxo-2-butenyl)oxy]-2-oxo-4-(1-oxobutoxy)-azuleno[4,5-b]furan-7-yl ester; OCTANOIC ACID, (3S,3AR,4S,6S,6AR,7S,8S,9BS)-6-(ACETYLOXY)-2,3,3A,4,5,6,6A,7,8,9B-DECAHYDRO-3,3A-DIHYDROXY-3,6,9-TRIMETHYL-8-(((2Z)-2-METHYL-1-OXO-2-BUTEN-1-YL)OXY)-2-OXO-4-(1-OXOBUTOXY)AZULENO(4,5-B)FURAN-7-YL ESTER; octanoic acid, (3S,3aR,4S,6S,6aR,7S,8S,9bS)-6-(acetyloxy)-2,3,3a,4,5,6,6a,7,8,9b-decahydro-3,3a-dihydroxy-3,6,9-trimethyl-8-[[(2Z)-2-methyl-1-oxo-2-butenyl]oxy]-2-oxo-4-(-oxobutoxy)azuleno[4,5-b]furan; Octanoic acid, (3S,3aR,4S,6S,6aR,7S,8S,9bS)-6-(acetyloxy)-2,3,3a,4,5,6,6a,7,8,9b-decahydro-3,3a-dihydroxy-3,6,9-trimethyl-8-[[(2Z)-2-methyl-1-oxo-2-butenyl]oxy]-2-oxo-4-(1-oxobutoxy)azuleno[4,5-b]fura; Octanoic acid, 6-(acetyloxy)-2,3,3a,4,5,6,6a,7,8,9b-decahydro-3,3a-dihydroxy-3,6,9-trimethyl-8-[(2-methyl-1-oxo-2-butenyl)oxy]-2-oxo-4-(1-oxobutoxy)azuleno[4,5-b]furan-7-yl ester, [3S-[3alpha,3abeta,4
Click to Show/Hide
|
|||||
| Target(s) | Calcium-transporting ATPase type 2C member 1 (ATP2C1) | Target Info | ||||
| Structure |
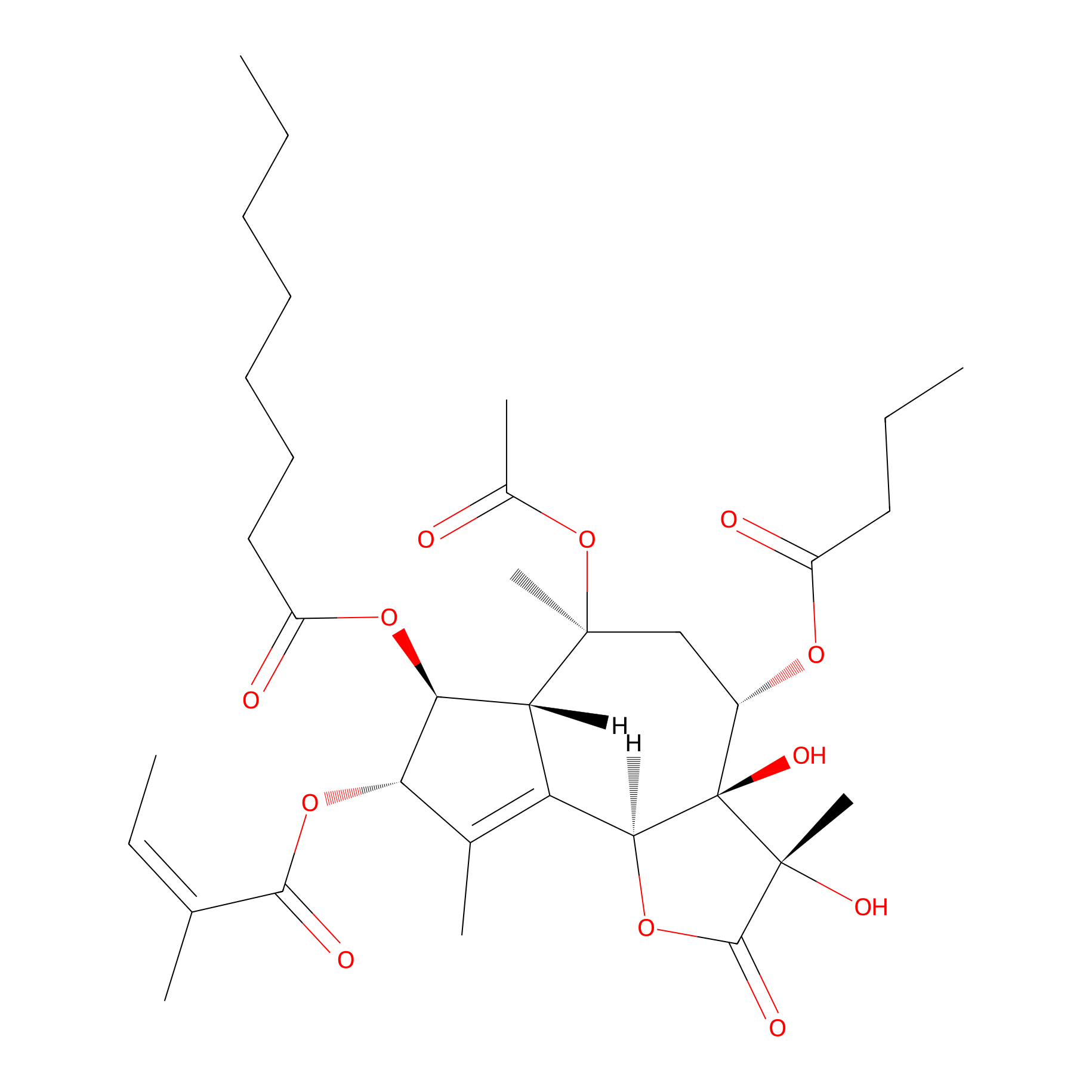
|
|||||
| Formula |
C34H50O12
|
|||||
| #Ro5 Violations (Lipinski): 3 | Molecular Weight (mw) | 650.8 | ||||
| Lipid-water partition coefficient (xlogp) | 3.6 | |||||
| Hydrogen Bond Donor Count (hbonddonor) | 2 | |||||
| Hydrogen Bond Acceptor Count (hbondacc) | 12 | |||||
| Rotatable Bond Count (rotbonds) | 17 | |||||
| PubChem CID | ||||||
| Canonical smiles |
CCCCCCCC(=O)OC1C2C(=C(C1OC(=O)C(=CC)C)C)C3C(C(CC2(C)OC(=O)C)OC(=O)CCC)(C(C(=O)O3)(C)O)O
|
|||||
| InChI |
InChI=1S/C34H50O12/c1-9-12-13-14-15-17-24(37)43-28-26-25(20(5)27(28)44-30(38)19(4)11-3)29-34(41,33(8,40)31(39)45-29)22(42-23(36)16-10-2)18-32(26,7)46-21(6)35/h11,22,26-29,40-41H,9-10,12-18H2,1-8H3/b19-11-/t22-,26+,27-,28-,29-,32-,33+,34+/m0/s1
|
|||||
| InChIKey |
IXFPJGBNCFXKPI-FSIHEZPISA-N
|
|||||
| IUPAC Name |
[(3S,3aR,4S,6S,6aR,7S,8S,9bS)-6-acetyloxy-4-butanoyloxy-3,3a-dihydroxy-3,6,9-trimethyl-8-[(Z)-2-methylbut-2-enoyl]oxy-2-oxo-4,5,6a,7,8,9b-hexahydroazuleno[4,5-b]furan-7-yl] octanoate
|
|||||
The activity data of This Drug
| Standard Type | Value | Disease Model | Cell line | Cell line ID | Ref. | |
|---|---|---|---|---|---|---|
| Half Maximal Inhibitory Concentration (IC50) | 170 nM | Chronic myeloid leukemia | K562 cell | CVCL_0004 | [1] | |
| Half Maximal Inhibitory Concentration (IC50) | 270 nM | T acute lymphoblastic leukemia | CCRF-CEM cell | CVCL_0207 | [1] | |
| Half Maximal Inhibitory Concentration (IC50) | 1.4 uM | Thymoma | EL4 cell | CVCL_0255 | [2] | |
| Half Maximal Inhibitory Concentration (IC50) | 2.7 uM | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | [2] | |
| Half Maximal Lethal dose (LD50) | 10 nM | Prostate carcinoma | LNCaP cell | CVCL_0395 | [3] | |
Each Peptide-drug Conjugate Related to This Drug
Full Information of The Activity Data of The PDC(s) Related to This Drug
HK2-TG prodrug [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Peak serum prodrug concentration | ˜36 µM | |||
| Administration Dosage | 3.67 µmoles/kg/dose | ||||
| MOA of PDC |
There are at least three additional cancer-associated extracellular proteases as potential candidates for the activation of 12-ADT-based prodrugs. These are Human Glandular Kallikrein 2 (HK2 also known as KLK2), Fibroblast Activation Protein (FAP), and Prostate Specific Membrane Antigen (PSMA also known as FOLH1). HK2 is a trypsin-like protease, uniquely secreted into the extracellular fluid at high enzymatically active levels only by normal and malignant prostate epithelial cells. Like PSA, once in the blood, its enzymatic activity is inhibited by serum protease inhibitors, making it an alternative candidate for prostate-targeted prodrug activation. Another alternative candidate is the serine protease, FAP. This is based upon the studies of W. Nathaniel Brennen, initiated while a graduate student with Sam Denmeade, and then, as a post-doctoral fellow with John Isaacs. Subsequently, Dr. Brennen continued this collaboration when he became an independent faculty investigator at Hopkins. His studies focused on the tumor-promoting activity of the influx within sites of metastatic prostate cancer of tumor-infiltrating host-derived fibroblasts that have a highly increased plasma membrane expression of FAP. FAP is a type II integral membrane serine prolyl protease of the dipeptidyl peptidase IV family, which is characterized by a unique post-prolyl cleavage specificity. Based on its restricted expression and unique substrate requirements, FAP is an ideal potential candidate for prodrug activation. PSMA is highly expressed on the extracellular plasma membranes of prostate cancer cells and, as originally discovered by Warren D. W. Heston, has folate hydrolase enzymatic activity. PSMA expression is also upregulated after ADT in resistant metastatic prostate cancer.
Click to Show/Hide
|
||||
| Description |
For the HK2-TG prodrug, the maximally tolerated multiday intravenous dose of prodrug is 6 mg/kg (3.67 umoles/kg/dose), which produced a peak serum concentration of ~36 uM and has a half-life of ~40 min. In addition, over a 24 h period, <0.5% of free L12ADT analog is observed in plasma, while, within the cancer, the level of 12ADT-released toxin at this MTD is ~1 uM. The prodrug demonstrated a significant antitumor effect in vivo while being administered, but prolonged intravenous administration is not possible due to local toxicity to tail veins.
Click to Show/Hide
|
||||
| In Vivo Model | LNCaP human prostate cancer cells xenografts in intact male immune-deficient mice. | ||||
| In Vitro Model | Prostate carcinoma | LNCaP cell | CVCL_0395 | ||
| Half life period | ˜ 40 min | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Maximum tolerated dose (MTD) | 6 mg/kg | |||
| Administration Dosage | 3.67 µmoles/kg/dose | ||||
| MOA of PDC |
There are at least three additional cancer-associated extracellular proteases as potential candidates for the activation of 12-ADT-based prodrugs. These are Human Glandular Kallikrein 2 (HK2 also known as KLK2), Fibroblast Activation Protein (FAP), and Prostate Specific Membrane Antigen (PSMA also known as FOLH1). HK2 is a trypsin-like protease, uniquely secreted into the extracellular fluid at high enzymatically active levels only by normal and malignant prostate epithelial cells. Like PSA, once in the blood, its enzymatic activity is inhibited by serum protease inhibitors, making it an alternative candidate for prostate-targeted prodrug activation. Another alternative candidate is the serine protease, FAP. This is based upon the studies of W. Nathaniel Brennen, initiated while a graduate student with Sam Denmeade, and then, as a post-doctoral fellow with John Isaacs. Subsequently, Dr. Brennen continued this collaboration when he became an independent faculty investigator at Hopkins. His studies focused on the tumor-promoting activity of the influx within sites of metastatic prostate cancer of tumor-infiltrating host-derived fibroblasts that have a highly increased plasma membrane expression of FAP. FAP is a type II integral membrane serine prolyl protease of the dipeptidyl peptidase IV family, which is characterized by a unique post-prolyl cleavage specificity. Based on its restricted expression and unique substrate requirements, FAP is an ideal potential candidate for prodrug activation. PSMA is highly expressed on the extracellular plasma membranes of prostate cancer cells and, as originally discovered by Warren D. W. Heston, has folate hydrolase enzymatic activity. PSMA expression is also upregulated after ADT in resistant metastatic prostate cancer.
Click to Show/Hide
|
||||
| Description |
For the HK2-TG prodrug, the maximally tolerated multiday intravenous dose of prodrug is 6 mg/kg (3.67 umoles/kg/dose), which produced a peak serum concentration of ~36 uM and has a half-life of ~40 min. In addition, over a 24 h period, <0.5% of free L12ADT analog is observed in plasma, while, within the cancer, the level of 12ADT-released toxin at this MTD is ~1 uM. The prodrug demonstrated a significant antitumor effect in vivo while being administered, but prolonged intravenous administration is not possible due to local toxicity to tail veins.
Click to Show/Hide
|
||||
| In Vivo Model | LNCaP human prostate cancer cells xenografts in intact male immune-deficient mice. | ||||
| In Vitro Model | Prostate carcinoma | LNCaP cell | CVCL_0395 | ||
| Half life period | ˜ 40 min | ||||
FAP-TG prodrug [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Peak serum prodrug concentration | ˜35-40 µM | |||
| Administration Dosage | 4 µmoles/kg/dose | ||||
| MOA of PDC |
There are at least three additional cancer-associated extracellular proteases as potential candidates for the activation of 12-ADT-based prodrugs. These are Human Glandular Kallikrein 2 (HK2 also known as KLK2), Fibroblast Activation Protein (FAP), and Prostate Specific Membrane Antigen (PSMA also known as FOLH1). HK2 is a trypsin-like protease, uniquely secreted into the extracellular fluid at high enzymatically active levels only by normal and malignant prostate epithelial cells. Like PSA, once in the blood, its enzymatic activity is inhibited by serum protease inhibitors, making it an alternative candidate for prostate-targeted prodrug activation. Another alternative candidate is the serine protease, FAP. This is based upon the studies of W. Nathaniel Brennen, initiated while a graduate student with Sam Denmeade, and then, as a post-doctoral fellow with John Isaacs. Subsequently, Dr. Brennen continued this collaboration when he became an independent faculty investigator at Hopkins. His studies focused on the tumor-promoting activity of the influx within sites of metastatic prostate cancer of tumor-infiltrating host-derived fibroblasts that have a highly increased plasma membrane expression of FAP. FAP is a type II integral membrane serine prolyl protease of the dipeptidyl peptidase IV family, which is characterized by a unique post-prolyl cleavage specificity. Based on its restricted expression and unique substrate requirements, FAP is an ideal potential candidate for prodrug activation. PSMA is highly expressed on the extracellular plasma membranes of prostate cancer cells and, as originally discovered by Warren D. W. Heston, has folate hydrolase enzymatic activity. PSMA expression is also upregulated after ADT in resistant metastatic prostate cancer.
Click to Show/Hide
|
||||
| Description |
For the FAP-TG prodrug, the maximally tolerated multiday intravenous dose of prodrug is 6.8 mg/kg (4 umoles/kg/dose), which produced peak serum concentration of ~35-40 uM, and has a half-life of 4-6 h. In addition, over a 24 h period, <1% of free L12ADT analog is observed in plasma while, within the cancer, the level of S12ADT-released toxin at this MTD is 3-5 uM. The prodrug demonstrated a significant antitumor effect in vivo. The antitumor effect is comparable to that observed with a maximally tolerated multiday intravenous dose of docetaxel, but results in significantly less toxicity.
Click to Show/Hide
|
||||
| In Vivo Model | LNCaP human prostate cancer cells xenografts in intact male immune-deficient mice. | ||||
| In Vitro Model | Prostate carcinoma | LNCaP cell | CVCL_0395 | ||
| Half life period | 46 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Maximum tolerated dose (MTD) | 6.8 mg/kg | |||
| Administration Dosage | 4 µmoles/kg/dose | ||||
| MOA of PDC |
There are at least three additional cancer-associated extracellular proteases as potential candidates for the activation of 12-ADT-based prodrugs. These are Human Glandular Kallikrein 2 (HK2 also known as KLK2), Fibroblast Activation Protein (FAP), and Prostate Specific Membrane Antigen (PSMA also known as FOLH1). HK2 is a trypsin-like protease, uniquely secreted into the extracellular fluid at high enzymatically active levels only by normal and malignant prostate epithelial cells. Like PSA, once in the blood, its enzymatic activity is inhibited by serum protease inhibitors, making it an alternative candidate for prostate-targeted prodrug activation. Another alternative candidate is the serine protease, FAP. This is based upon the studies of W. Nathaniel Brennen, initiated while a graduate student with Sam Denmeade, and then, as a post-doctoral fellow with John Isaacs. Subsequently, Dr. Brennen continued this collaboration when he became an independent faculty investigator at Hopkins. His studies focused on the tumor-promoting activity of the influx within sites of metastatic prostate cancer of tumor-infiltrating host-derived fibroblasts that have a highly increased plasma membrane expression of FAP. FAP is a type II integral membrane serine prolyl protease of the dipeptidyl peptidase IV family, which is characterized by a unique post-prolyl cleavage specificity. Based on its restricted expression and unique substrate requirements, FAP is an ideal potential candidate for prodrug activation. PSMA is highly expressed on the extracellular plasma membranes of prostate cancer cells and, as originally discovered by Warren D. W. Heston, has folate hydrolase enzymatic activity. PSMA expression is also upregulated after ADT in resistant metastatic prostate cancer.
Click to Show/Hide
|
||||
| Description |
For the FAP-TG prodrug, the maximally tolerated multiday intravenous dose of prodrug is 6.8 mg/kg (4 umoles/kg/dose), which produced peak serum concentration of ~35-40 uM, and has a half-life of 4-6 h. In addition, over a 24 h period, <1% of free L12ADT analog is observed in plasma while, within the cancer, the level of S12ADT-released toxin at this MTD is 3-5 uM. The prodrug demonstrated a significant antitumor effect in vivo. The antitumor effect is comparable to that observed with a maximally tolerated multiday intravenous dose of docetaxel, but results in significantly less toxicity.
Click to Show/Hide
|
||||
| In Vivo Model | LNCaP human prostate cancer cells xenografts in intact male immune-deficient mice. | ||||
| In Vitro Model | Prostate carcinoma | LNCaP cell | CVCL_0395 | ||
| Half life period | 46 h | ||||
PSMA-TG prodrug [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Peak serum prodrug concentration | ˜800 µM | |||
| Administration Dosage | 40 µmole/kg/dose | ||||
| MOA of PDC |
There are at least three additional cancer-associated extracellular proteases as potential candidates for the activation of 12-ADT-based prodrugs. These are Human Glandular Kallikrein 2 (HK2 also known as KLK2), Fibroblast Activation Protein (FAP), and Prostate Specific Membrane Antigen (PSMA also known as FOLH1). HK2 is a trypsin-like protease, uniquely secreted into the extracellular fluid at high enzymatically active levels only by normal and malignant prostate epithelial cells. Like PSA, once in the blood, its enzymatic activity is inhibited by serum protease inhibitors, making it an alternative candidate for prostate-targeted prodrug activation. Another alternative candidate is the serine protease, FAP. This is based upon the studies of W. Nathaniel Brennen, initiated while a graduate student with Sam Denmeade, and then, as a post-doctoral fellow with John Isaacs. Subsequently, Dr. Brennen continued this collaboration when he became an independent faculty investigator at Hopkins. His studies focused on the tumor-promoting activity of the influx within sites of metastatic prostate cancer of tumor-infiltrating host-derived fibroblasts that have a highly increased plasma membrane expression of FAP. FAP is a type II integral membrane serine prolyl protease of the dipeptidyl peptidase IV family, which is characterized by a unique post-prolyl cleavage specificity. Based on its restricted expression and unique substrate requirements, FAP is an ideal potential candidate for prodrug activation. PSMA is highly expressed on the extracellular plasma membranes of prostate cancer cells and, as originally discovered by Warren D. W. Heston, has folate hydrolase enzymatic activity. PSMA expression is also upregulated after ADT in resistant metastatic prostate cancer.
Click to Show/Hide
|
||||
| Description |
For the PSMA-TG prodrug, the maximally tolerated multiday intravenous dose of prodrug is 56 mg/kg (i.e., 40 umole/kg/dose), which produced a peak serum concentration of ~800 uM and had a half-life of 4.9 h. In addition, over a 24 h period, <1% of free 12ADTAsp analog is observed in plasma while, within the cancer, the level of released toxin at this MTD is >8 uM. The prodrug demonstrated a significant antitumor effect in vivo. The antitumor effect is comparable to that observed with docetaxel, but results in significantly less toxicity.
Click to Show/Hide
|
||||
| In Vivo Model | LNCaP human prostate cancer cells xenografts in intact male immune-deficient mice. | ||||
| In Vitro Model | Prostate carcinoma | LNCaP cell | CVCL_0395 | ||
| Half life period | 4.9 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Maximum tolerated dose (MTD) | 56 mg/kg | |||
| Administration Dosage | 40 µmole/kg/dose | ||||
| MOA of PDC |
There are at least three additional cancer-associated extracellular proteases as potential candidates for the activation of 12-ADT-based prodrugs. These are Human Glandular Kallikrein 2 (HK2 also known as KLK2), Fibroblast Activation Protein (FAP), and Prostate Specific Membrane Antigen (PSMA also known as FOLH1). HK2 is a trypsin-like protease, uniquely secreted into the extracellular fluid at high enzymatically active levels only by normal and malignant prostate epithelial cells. Like PSA, once in the blood, its enzymatic activity is inhibited by serum protease inhibitors, making it an alternative candidate for prostate-targeted prodrug activation. Another alternative candidate is the serine protease, FAP. This is based upon the studies of W. Nathaniel Brennen, initiated while a graduate student with Sam Denmeade, and then, as a post-doctoral fellow with John Isaacs. Subsequently, Dr. Brennen continued this collaboration when he became an independent faculty investigator at Hopkins. His studies focused on the tumor-promoting activity of the influx within sites of metastatic prostate cancer of tumor-infiltrating host-derived fibroblasts that have a highly increased plasma membrane expression of FAP. FAP is a type II integral membrane serine prolyl protease of the dipeptidyl peptidase IV family, which is characterized by a unique post-prolyl cleavage specificity. Based on its restricted expression and unique substrate requirements, FAP is an ideal potential candidate for prodrug activation. PSMA is highly expressed on the extracellular plasma membranes of prostate cancer cells and, as originally discovered by Warren D. W. Heston, has folate hydrolase enzymatic activity. PSMA expression is also upregulated after ADT in resistant metastatic prostate cancer.
Click to Show/Hide
|
||||
| Description |
For the PSMA-TG prodrug, the maximally tolerated multiday intravenous dose of prodrug is 56 mg/kg (i.e., 40 umole/kg/dose), which produced a peak serum concentration of ~800 uM and had a half-life of 4.9 h. In addition, over a 24 h period, <1% of free 12ADTAsp analog is observed in plasma while, within the cancer, the level of released toxin at this MTD is >8 uM. The prodrug demonstrated a significant antitumor effect in vivo. The antitumor effect is comparable to that observed with docetaxel, but results in significantly less toxicity.
Click to Show/Hide
|
||||
| In Vivo Model | LNCaP human prostate cancer cells xenografts in intact male immune-deficient mice. | ||||
| In Vitro Model | Prostate carcinoma | LNCaP cell | CVCL_0395 | ||
| Half life period | 4.9 h | ||||
L-Leu-12ADT [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Peak serum prodrug concentration | 15.4 ± 1.1 µM | |||
| Administration Dosage | 4.4 µmoles/kg/dose | ||||
| MOA of PDC |
This approach was initially based upon the discovery by Hans Lilja at the University of Lund, Sweden, that Prostate-Specific Antigen (PSA), besides being a serum marker for prostate cancer detection, is a unique prostate differentiation restricted chymotrypsin-like serine protease whose enzymatic activity, once secreted by normal or malignant prostate epithelial cells into the serum, is inhibited by serum inhibitors. Based upon these results, a collaboration was established between the Lilja laboratory at Lund and the Isaacs laboratory at Hopkins, with Sam Denmeade leading the Hopkins effort to identify specific peptide substrates whose efficient hydrolysis is restricted to PSA. This led to the identification of His-Ser-Ser-Lys-Leu-Gln-x-Leu (i.e., a 7AA peptide where -x- is the PSA cleavage site) as the lead peptide for such PSA-activated prodrug development. As an in vitro proof of principle for such a PSA-activated prodrug approach, we collaborated with Andrew V. Schally, Department of Medicine, Tulane University School of Medicine and Veterans Affairs Medical Center, New Orleans, Louisiana to couple the primary amine of doxorubicin via a peptide bond to the carboxylic acid group of the 7AA lead peptide. These studies documented that such a doxorubicin-peptide prodrug selectively kills cells that express enzymatically active PSA.
Click to Show/Hide
|
||||
| Description |
The maximum tolerated multiday intravenous dose (MTD) of this PSA-TG prodrug is 7 mg/kg (4.4 umoles/kg/dose), which results in a peak serum prodrug concentration of 15.4 ± 1.1 uM and an elimination half-life of ~2.8 h. After 24 h, less than 0.5% of free L12ADT is observed in the plasma. At this MTD, intratumoral concentration of the PSA-TG prodrug is 640 ± 80 nM (i.e., 8.5 times the LD50 for LNCaP cells in vitro) and 170 ± 58 nM for the liberated L12ADT (i.e., 13 times the LD50 for L12ADT in vitro).
Click to Show/Hide
|
||||
| In Vivo Model | LNCaP human prostate cancer cells xenografts in intact male immune-deficient mice. | ||||
| In Vitro Model | Prostate carcinoma | LNCaP cell | CVCL_0395 | ||
| Half life period | ˜ 2.8 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Maximum tolerated dose (MTD) | 7 mg/kg | |||
| Administration Dosage | 4.4 µmoles/kg/dose | ||||
| MOA of PDC |
This approach was initially based upon the discovery by Hans Lilja at the University of Lund, Sweden, that Prostate-Specific Antigen (PSA), besides being a serum marker for prostate cancer detection, is a unique prostate differentiation restricted chymotrypsin-like serine protease whose enzymatic activity, once secreted by normal or malignant prostate epithelial cells into the serum, is inhibited by serum inhibitors. Based upon these results, a collaboration was established between the Lilja laboratory at Lund and the Isaacs laboratory at Hopkins, with Sam Denmeade leading the Hopkins effort to identify specific peptide substrates whose efficient hydrolysis is restricted to PSA. This led to the identification of His-Ser-Ser-Lys-Leu-Gln-x-Leu (i.e., a 7AA peptide where -x- is the PSA cleavage site) as the lead peptide for such PSA-activated prodrug development. As an in vitro proof of principle for such a PSA-activated prodrug approach, we collaborated with Andrew V. Schally, Department of Medicine, Tulane University School of Medicine and Veterans Affairs Medical Center, New Orleans, Louisiana to couple the primary amine of doxorubicin via a peptide bond to the carboxylic acid group of the 7AA lead peptide. These studies documented that such a doxorubicin-peptide prodrug selectively kills cells that express enzymatically active PSA.
Click to Show/Hide
|
||||
| Description |
The maximum tolerated multiday intravenous dose (MTD) of this PSA-TG prodrug is 7 mg/kg (4.4 umoles/kg/dose), which results in a peak serum prodrug concentration of 15.4 ± 1.1 uM and an elimination half-life of ~2.8 h. After 24 h, less than 0.5% of free L12ADT is observed in the plasma. At this MTD, intratumoral concentration of the PSA-TG prodrug is 640 ± 80 nM (i.e., 8.5 times the LD50 for LNCaP cells in vitro) and 170 ± 58 nM for the liberated L12ADT (i.e., 13 times the LD50 for L12ADT in vitro).
Click to Show/Hide
|
||||
| In Vivo Model | LNCaP human prostate cancer cells xenografts in intact male immune-deficient mice. | ||||
| In Vitro Model | Prostate carcinoma | LNCaP cell | CVCL_0395 | ||
| Half life period | ˜ 2.8 h | ||||
References