
Linker Information
General Information of This Linker
| Linker ID |
LIN00012
|
|||||
|---|---|---|---|---|---|---|
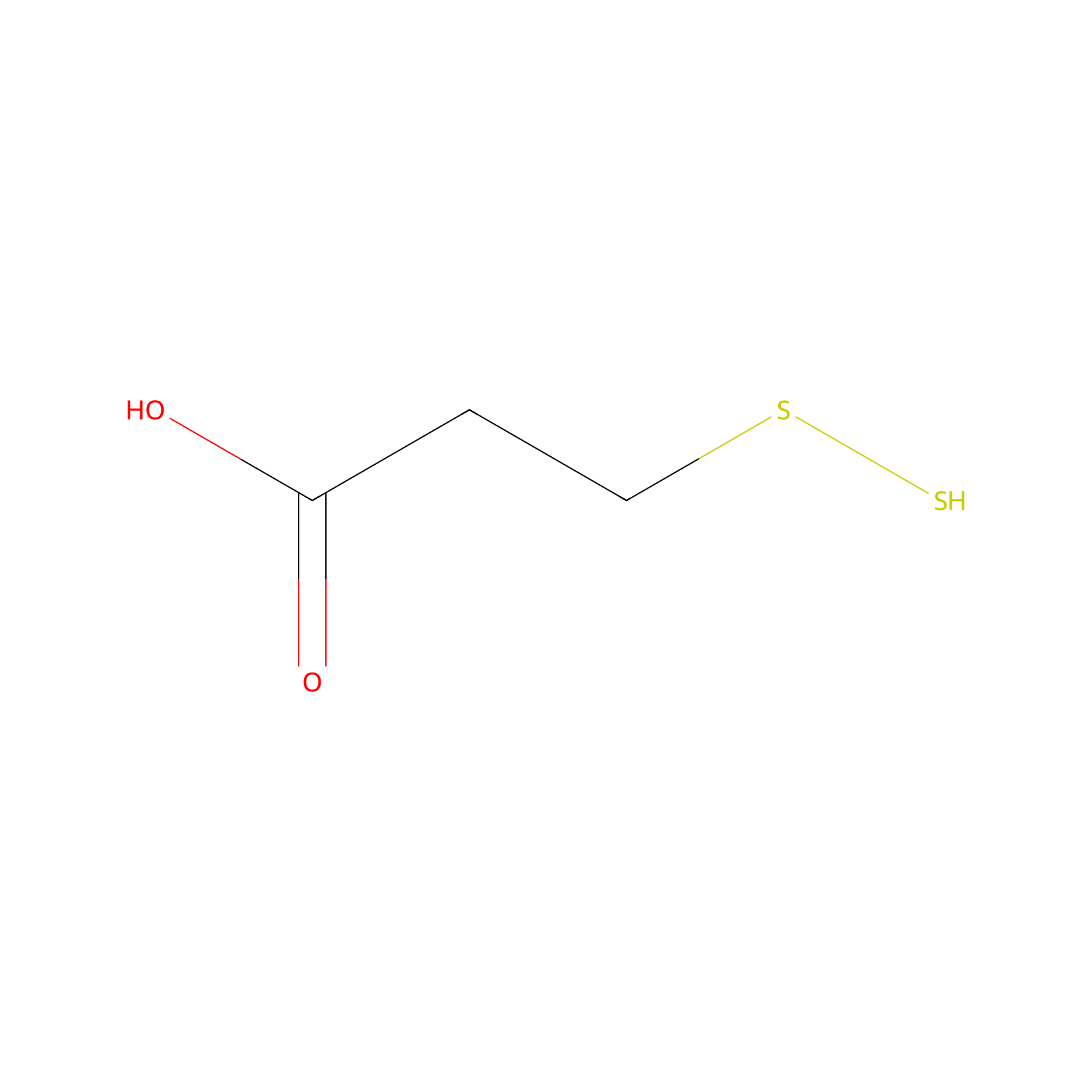
| Linker Name |
3-Disulfanylpropanoic Acid
|
|||||
| Linker Type |
GSH concentration-sensitive linkers
|
|||||
| Structure |
|
|||||
| Formula |
C3H6O2S2
|
|||||
| #Ro5 Violations (Lipinski): 0 | Molecular Weight (mw) | 138.21 | ||||
| Lipid-water partition coefficient (xlogp) | 0.5 | |||||
| Hydrogen Bond Donor Count (hbonddonor) | 2 | |||||
| Hydrogen Bond Acceptor Count (hbondacc) | 4 | |||||
| Rotatable Bond Count (rotbonds) | 3 | |||||
| PubChem CID | ||||||
| Canonical smiles |
C(CSS)C(=O)O
|
|||||
| InChI |
InChI=1S/C3H6O2S2/c4-3(5)1-2-7-6/h6H,1-2H2,(H,4,5)
|
|||||
| InChIKey |
CXXDUKKBFUFJDF-UHFFFAOYSA-N
|
|||||
| IUPAC Name |
3-(disulfanyl)propanoic acid
|
|||||
Each Peptide-drug Conjugate Related to This Linker
Full Information of The Activity Data of The PDC(s) Related to This Linker
LLC2B-SS-DM1 [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
58.76%
|
|||
| Administration Time | 14 d | ||||
| Administration Dosage | 0.5 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
58.77%
|
|||
| Administration Time | 6 d | ||||
| Administration Dosage | 0.5 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
60.43%
|
|||
| Administration Time | 16 d | ||||
| Administration Dosage | 0.5 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
61.33%
|
|||
| Administration Time | 10 d | ||||
| Administration Dosage | 0.5 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
63.36%
|
|||
| Administration Time | Once a week for 3 weeks | ||||
| Administration Dosage | 2.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
64.57%
|
|||
| Administration Time | 8 d | ||||
| Administration Dosage | 0.5 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
65.31%
|
|||
| Administration Time | Once a week for 3 weeks | ||||
| Administration Dosage | 0.5 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
66.73%
|
|||
| Administration Time | 2 d | ||||
| Administration Dosage | 0.5 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
67.10%
|
|||
| Administration Time | 2 d | ||||
| Administration Dosage | 2.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
67.68%
|
|||
| Administration Time | Once a week for 3 weeks | ||||
| Administration Dosage | 1.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
68.27%
|
|||
| Administration Time | 12 d | ||||
| Administration Dosage | 0.5 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
69.20%
|
|||
| Administration Time | 20 d | ||||
| Administration Dosage | 0.5 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
69.39%
|
|||
| Administration Time | 10 d | ||||
| Administration Dosage | 1.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
70.04%
|
|||
| Administration Time | 8 d | ||||
| Administration Dosage | 2.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
70.26%
|
|||
| Administration Time | 6 d | ||||
| Administration Dosage | 2.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
71.16%
|
|||
| Administration Time | 6 d | ||||
| Administration Dosage | 1.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 17 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
72.14%
|
|||
| Administration Time | 16 d | ||||
| Administration Dosage | 1.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 18 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
72.20%
|
|||
| Administration Time | 8 d | ||||
| Administration Dosage | 1.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 19 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
72.45%
|
|||
| Administration Time | 10 d | ||||
| Administration Dosage | 2.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 20 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
72.90%
|
|||
| Administration Time | 2 d | ||||
| Administration Dosage | 1.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 21 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
72.91%
|
|||
| Administration Time | 16 d | ||||
| Administration Dosage | 2.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 22 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
72.98%
|
|||
| Administration Time | 12 d | ||||
| Administration Dosage | 2.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 23 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
73.90%
|
|||
| Administration Time | 20 d | ||||
| Administration Dosage | 2.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 24 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
75.40%
|
|||
| Administration Time | 14 d | ||||
| Administration Dosage | 1.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 25 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
75.49%
|
|||
| Administration Time | 20 d | ||||
| Administration Dosage | 1.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 26 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
77.25%
|
|||
| Administration Time | 12 d | ||||
| Administration Dosage | 1.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 27 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
78.30%
|
|||
| Administration Time | 14 d | ||||
| Administration Dosage | 2.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Completely suppress concentration |
0.1 μg DM1 equiv./mL
|
|||
| Administration Time | 12 h | ||||
| Evaluation Method | Clone formation experiment | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
The results show that DM1 could completely suppress the clone formation of MCF-7 cells at 0.001 μg/mL, while LLC2B-SS-DM1 could completely suppress the clone formation of MCF-7 cells at 0.1 μg DM1 equiv./mL. A concentration of 1 μg DM1 equiv./mL was required for LLC2B-Mal-DM1 to achieve the same suppression effect.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
DOX-ApoA-1 mimic peptide [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
83%
|
|||
| Administration Time | 15 days | ||||
| MOA of PDC |
Herein, we report a dual-functional sHDL (DP-sHDL) that is co-assembled from DOX-ApoA1 mimetic peptide conjugate, PSB-603 (an A2BR inhibitor), phospholipid and cholesterol oleate (CO) through a microfluidic-based methodology. We expect that DP-sHDL could accumulate in tumor, where they can target cancerous cells, CAF and tumor-associated macrophages (TAMs). The DOX would induce the ICD of cancer cells and promote DC maturation and T lymphocyte infiltration and activity. PSB-603 is expected to block the A2BR, which would down-regulate CD73 and thus ADO, leading to a decrease in the intratumoral densities of several immunosuppressive cells. By simultaneously activating anti-tumor immunity and relieving immunosuppressive microenvironment, DP-sHDL may inhibit TNBC tumor growth and prolong the survival of animals, which could be further improved when used in combination with an immune checkpoint inhibitor.
Click to Show/Hide
|
||||
| Description |
Given the potent effects of DP-sHDL in boosting anti-tumor immunity and relieving immunosuppressive microenvironment, its efficacy in treating TNBC model mice was evaluated. Through a 3-dose regime, DP-sHDL inhibited the growth of tumors by 84% and significantly reduced tumor burden. In contrast, DOX was less effective and caused a progressive decline in body weight. Further biochemical assays revealed that the levels of AST, CREA, CK and LDH in DOX-treated mice were significantly higher than those receiving PBS. Accordingly, inflammatory cell infiltration, cell necrosis and local swelling were observed in the heart, liver and kidneys, respectively, in the DOX-injection group. With the same regime, no visible damage was monitored in the major organs in DP-sHDL group. Based on these findings, the long-term efficacy of D-sHDL, P-sHDL and DP-sHDL were further evaluated. DP-sHDL retarded the tumor growth, and prolonged the median survival time (MST) from 18 days (PBS-treated mice) to 35 days, without body weight loss. These results proved that DP-sHDL could reduce the toxicity of DOX and effectively inhibit tumor growth.
Click to Show/Hide
|
||||
| In Vivo Model | Female BALB/c mice murine 4T1 breast cancer cells xenograft tumor models. | ||||
| In Vitro Model | Breast cancer | Murine 4T1 breast cancer cell | Homo sapiens | ||
I-5 (NPNWGRSWYNQRFKGC=(-SS-COO-CPT)GC=(-SS-COO-CPT)GRIKPRKGYTR) [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.49 ± 0.58 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4.12 ± 0.87 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
7.37 ± 1.18 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
11.51 ± 1.37 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
18.25 ± 1.76 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
SMAC-P1 CPT conjugates 5 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.78 ± 0.35 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
5.81 ± 1.08 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
6.53 ± 1.02 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
16.24 ± 1.04 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
22.12 ± 1.83 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
SMAC-P1 CPT conjugates 8 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.30 ± 0.48 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4.73 ± 1.33 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
8.01 ± 1.34 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
15.11 ± 1.80 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
28.36 ± 2.12 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
SMAC-P1 CPT conjugates 2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.59 ± 0.40 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
5.93 ± 0.46 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
8.24 ± 0.57 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
10.64 ± 1.32 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
15.13 ± 1.24 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
I-2 (NPNWGRSWYNQRFKGC=(-SS-COO-CPT)GRIKPRKGYTR) [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
5.76 ± 0.79 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
6.25 ± 0.96 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
6.42 ± 1.21 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
10.93 ± 1.14 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
20.41 ± 1.04 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
References