
Linker Information
General Information of This Linker
| Linker ID |
LIN00031
|
|||||
|---|---|---|---|---|---|---|
| Linker Name |
4-yl 6-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)hexanoate
|
|||||
| Structure |
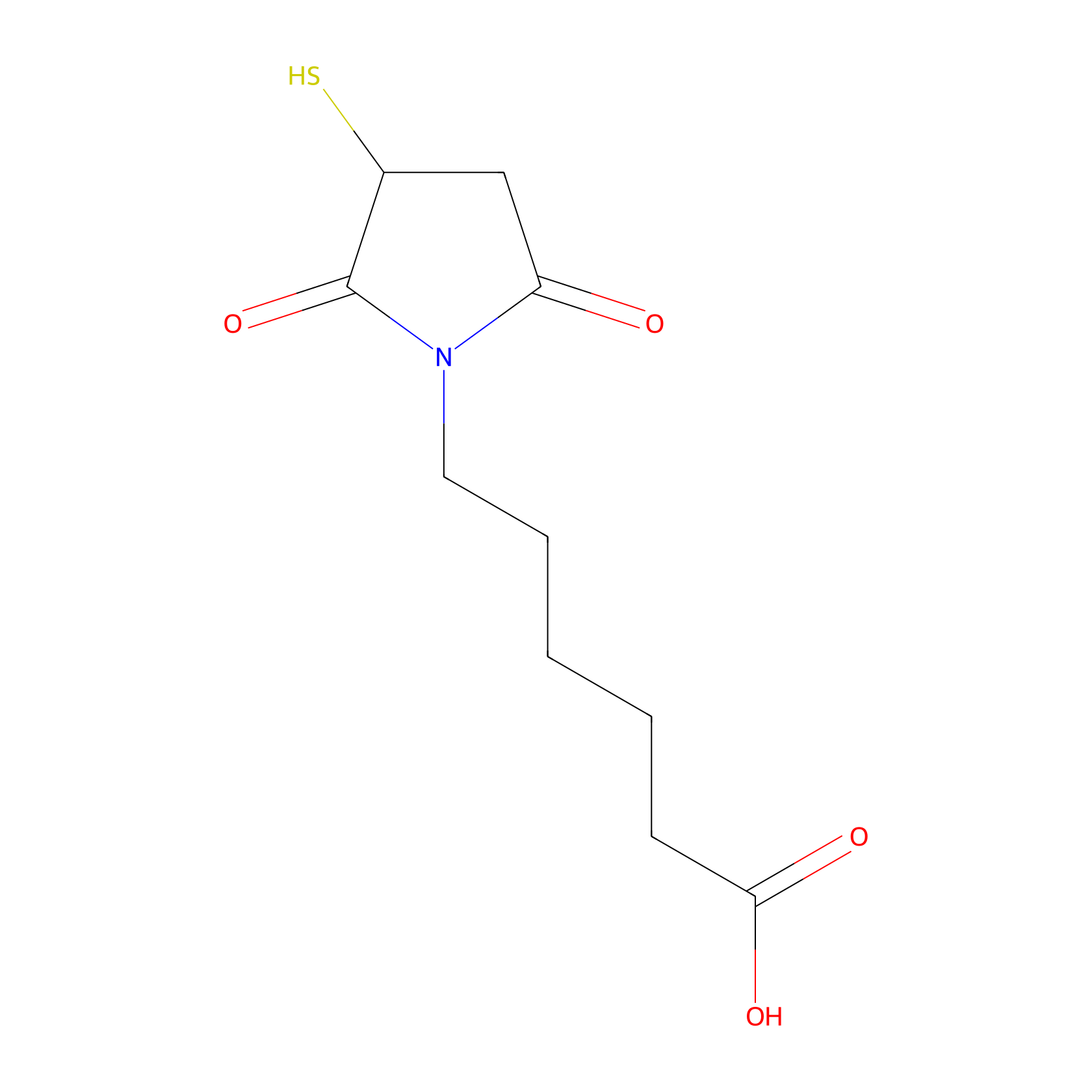
|
|||||
| Formula |
C10H15NO4S
|
|||||
| #Ro5 Violations (Lipinski): 0 | Molecular Weight (mw) | 245.3 | ||||
| Lipid-water partition coefficient (xlogp) | 0.1 | |||||
| Hydrogen Bond Donor Count (hbonddonor) | 2 | |||||
| Hydrogen Bond Acceptor Count (hbondacc) | 5 | |||||
| Rotatable Bond Count (rotbonds) | 6 | |||||
| PubChem CID | ||||||
| Canonical smiles |
C1C(C(=O)N(C1=O)CCCCCC(=O)O)S
|
|||||
| InChI |
InChI=1S/C10H15NO4S/c12-8-6-7(16)10(15)11(8)5-3-1-2-4-9(13)14/h7,16H,1-6H2,(H,13,14)
|
|||||
| InChIKey |
WCUXBNLBBGRJCW-UHFFFAOYSA-N
|
|||||
| IUPAC Name |
6-(2,5-dioxo-3-sulfanylpyrrolidin-1-yl)hexanoic acid
|
|||||
Each Peptide-drug Conjugate Related to This Linker
Full Information of The Activity Data of The PDC(s) Related to This Linker
FA-P7-PTX [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Hepatoma | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
0.00%
|
|||
| Administration Time | 4 days | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
To study the anticancer activity of FA-P7-PTX in vivo, we performed tumor-bearing mice model with H22cells by administering once every two days peritumoral injection of FA-P7-PTX (12 μmol/kg), PTX (12 μmol/kg, as the positive control), or 0.9% saline as the negative control for 2 weeks. Compared with control group, the tumor volumes of the FA-P7-PTX group were dramatically reduced by 69% with no significant variation in mouse body weight. Meanwhile FA-P7-PTX exhibited stronger inhibitory effects on tumor volume compared with PTX (69% versus 49%). The result confirmed that FA-P7-PTX possessed higher potency in slowing the growth of solid tumors.
Click to Show/Hide
|
||||
| In Vivo Model | Tumor-bearing mice model with H22 cells. | ||||
| In Vitro Model | Hepatoma | H22 cell | CVCL_H613 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Hepatoma | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
16.70%
|
|||
| Administration Time | 6 days | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
To study the anticancer activity of FA-P7-PTX in vivo, we performed tumor-bearing mice model with H22cells by administering once every two days peritumoral injection of FA-P7-PTX (12 μmol/kg), PTX (12 μmol/kg, as the positive control), or 0.9% saline as the negative control for 2 weeks. Compared with control group, the tumor volumes of the FA-P7-PTX group were dramatically reduced by 69% with no significant variation in mouse body weight. Meanwhile FA-P7-PTX exhibited stronger inhibitory effects on tumor volume compared with PTX (69% versus 49%). The result confirmed that FA-P7-PTX possessed higher potency in slowing the growth of solid tumors.
Click to Show/Hide
|
||||
| In Vivo Model | Tumor-bearing mice model with H22 cells. | ||||
| In Vitro Model | Hepatoma | H22 cell | CVCL_H613 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Hepatoma | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
38.90%
|
|||
| Administration Time | 8 days | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
To study the anticancer activity of FA-P7-PTX in vivo, we performed tumor-bearing mice model with H22cells by administering once every two days peritumoral injection of FA-P7-PTX (12 μmol/kg), PTX (12 μmol/kg, as the positive control), or 0.9% saline as the negative control for 2 weeks. Compared with control group, the tumor volumes of the FA-P7-PTX group were dramatically reduced by 69% with no significant variation in mouse body weight. Meanwhile FA-P7-PTX exhibited stronger inhibitory effects on tumor volume compared with PTX (69% versus 49%). The result confirmed that FA-P7-PTX possessed higher potency in slowing the growth of solid tumors.
Click to Show/Hide
|
||||
| In Vivo Model | Tumor-bearing mice model with H22 cells. | ||||
| In Vitro Model | Hepatoma | H22 cell | CVCL_H613 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Hepatoma | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
50.00%
|
|||
| Administration Time | 10 days | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
To study the anticancer activity of FA-P7-PTX in vivo, we performed tumor-bearing mice model with H22cells by administering once every two days peritumoral injection of FA-P7-PTX (12 μmol/kg), PTX (12 μmol/kg, as the positive control), or 0.9% saline as the negative control for 2 weeks. Compared with control group, the tumor volumes of the FA-P7-PTX group were dramatically reduced by 69% with no significant variation in mouse body weight. Meanwhile FA-P7-PTX exhibited stronger inhibitory effects on tumor volume compared with PTX (69% versus 49%). The result confirmed that FA-P7-PTX possessed higher potency in slowing the growth of solid tumors.
Click to Show/Hide
|
||||
| In Vivo Model | Tumor-bearing mice model with H22 cells. | ||||
| In Vitro Model | Hepatoma | H22 cell | CVCL_H613 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Hepatoma | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
63.00%
|
|||
| Administration Time | 12 days | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
To study the anticancer activity of FA-P7-PTX in vivo, we performed tumor-bearing mice model with H22cells by administering once every two days peritumoral injection of FA-P7-PTX (12 μmol/kg), PTX (12 μmol/kg, as the positive control), or 0.9% saline as the negative control for 2 weeks. Compared with control group, the tumor volumes of the FA-P7-PTX group were dramatically reduced by 69% with no significant variation in mouse body weight. Meanwhile FA-P7-PTX exhibited stronger inhibitory effects on tumor volume compared with PTX (69% versus 49%). The result confirmed that FA-P7-PTX possessed higher potency in slowing the growth of solid tumors.
Click to Show/Hide
|
||||
| In Vivo Model | Tumor-bearing mice model with H22 cells. | ||||
| In Vitro Model | Hepatoma | H22 cell | CVCL_H613 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Hepatoma | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
65.20%
|
|||
| Administration Time | 16 days | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
To study the anticancer activity of FA-P7-PTX in vivo, we performed tumor-bearing mice model with H22cells by administering once every two days peritumoral injection of FA-P7-PTX (12 μmol/kg), PTX (12 μmol/kg, as the positive control), or 0.9% saline as the negative control for 2 weeks. Compared with control group, the tumor volumes of the FA-P7-PTX group were dramatically reduced by 69% with no significant variation in mouse body weight. Meanwhile FA-P7-PTX exhibited stronger inhibitory effects on tumor volume compared with PTX (69% versus 49%). The result confirmed that FA-P7-PTX possessed higher potency in slowing the growth of solid tumors.
Click to Show/Hide
|
||||
| In Vivo Model | Tumor-bearing mice model with H22 cells. | ||||
| In Vitro Model | Hepatoma | H22 cell | CVCL_H613 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Hepatoma | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
66.90%
|
|||
| Administration Time | 14 days | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
To study the anticancer activity of FA-P7-PTX in vivo, we performed tumor-bearing mice model with H22cells by administering once every two days peritumoral injection of FA-P7-PTX (12 μmol/kg), PTX (12 μmol/kg, as the positive control), or 0.9% saline as the negative control for 2 weeks. Compared with control group, the tumor volumes of the FA-P7-PTX group were dramatically reduced by 69% with no significant variation in mouse body weight. Meanwhile FA-P7-PTX exhibited stronger inhibitory effects on tumor volume compared with PTX (69% versus 49%). The result confirmed that FA-P7-PTX possessed higher potency in slowing the growth of solid tumors.
Click to Show/Hide
|
||||
| In Vivo Model | Tumor-bearing mice model with H22 cells. | ||||
| In Vitro Model | Hepatoma | H22 cell | CVCL_H613 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Hepatoma | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
67.30%
|
|||
| Administration Time | 18 days | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
To study the anticancer activity of FA-P7-PTX in vivo, we performed tumor-bearing mice model with H22cells by administering once every two days peritumoral injection of FA-P7-PTX (12 μmol/kg), PTX (12 μmol/kg, as the positive control), or 0.9% saline as the negative control for 2 weeks. Compared with control group, the tumor volumes of the FA-P7-PTX group were dramatically reduced by 69% with no significant variation in mouse body weight. Meanwhile FA-P7-PTX exhibited stronger inhibitory effects on tumor volume compared with PTX (69% versus 49%). The result confirmed that FA-P7-PTX possessed higher potency in slowing the growth of solid tumors.
Click to Show/Hide
|
||||
| In Vivo Model | Tumor-bearing mice model with H22 cells. | ||||
| In Vitro Model | Hepatoma | H22 cell | CVCL_H613 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Hepatoma | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
69.10%
|
|||
| Administration Time | 20 days | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
To study the anticancer activity of FA-P7-PTX in vivo, we performed tumor-bearing mice model with H22cells by administering once every two days peritumoral injection of FA-P7-PTX (12 μmol/kg), PTX (12 μmol/kg, as the positive control), or 0.9% saline as the negative control for 2 weeks. Compared with control group, the tumor volumes of the FA-P7-PTX group were dramatically reduced by 69% with no significant variation in mouse body weight. Meanwhile FA-P7-PTX exhibited stronger inhibitory effects on tumor volume compared with PTX (69% versus 49%). The result confirmed that FA-P7-PTX possessed higher potency in slowing the growth of solid tumors.
Click to Show/Hide
|
||||
| In Vivo Model | Tumor-bearing mice model with H22 cells. | ||||
| In Vitro Model | Hepatoma | H22 cell | CVCL_H613 | ||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Invasive breast carcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
1.39 ± 0.12 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Ovarian endometrioid adenocarcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
1.42 ± 0.08 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Invasive ductal carcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
2.92 ± 0.2 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive ductal carcinoma | MCF7/PTX cell | CVCL_C5RS | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Chronic myeloid leukemia | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
3.85 ± 0.9 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Chronic myeloid leukemia | K562 cell | CVCL_0004 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Ovarian serous cystadenocarcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
5.49 ± 0.36 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [1] | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
45.21 ± 1.79 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cells | Homo sapiens | ||
Peptide 18-4 analog doxorubicin conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
75%
|
|||
| Administration Time | 30 days | ||||
| Administration Dosage | 2.5 mg DOX equivalent/kg | ||||
| MOA of PDC |
Keratin 1 (K1) is a novel receptor, present on the surface of cancer cells (breast and neuroblastoma) and cells that have undergone oxidative stress, that is being used for targeted drug delivery. We showed that K1 is present on the surface of MCF-7 breast cancer cells, and a comparison of the total K1 levels in cell lysates using Western blot showed that cancer cells (MCF-7 and MDA-MB-435) have a much higher expression of K1 compared to non-cancerous breast tissue derived epithelial (MCF-10A) cells. We engineered peptides, such as linear 18-4 and cyclic analogues, for specific uptake by breast cancer cells (MCF-7 and MDA-MB-231) via cell surface K1 mediated endocytosis. Further, K1 targeting linear peptide 18-4 was used to synthesize four peptide-doxorubicin conjugates with different linker chemistries, such as ester, amide, succinimidyl thioether, and hydrazone. We showed specific uptake of the targeted PDCs via receptor mediated endocytosis in MCF-7 and MDA-MB-435-MDR cancer cells. The PDCs with K1 targeting peptide 18-4 were more cytotoxic to TNBC cells (IC50 1.2-4.7 μM) compared to non-cancerous human mammary epithelial MCF-10A cells (IC50 15.1-38.6 μM), while free drug (doxorubicin) was equally cytotoxic to both cancer and non-cancerous cells (IC50 0.24-1.5 μM). To explore the in vivo efficacy and evaluate the potential of K1 targeting PDC for TNBC treatment, we report here the antitumor activity of one of these peptide-doxorubicin conjugates, where the peptide (18-4) is conjugated to Dox via an acid-sensitive N-acyl hydrazone linker in a mouse model for TNBC. TNBC MDA-MB-231 cells were subcutaneously injected into female NOD/SCID mice to generate TNBC cell-derived xenograft models. Mice treated with the conjugate showed better efficacy, pharmacokinetics, and safety profile compared to the Dox treated mice, supporting the future clinical development of K1 targeted PDCs for treatment of TNBC.
Click to Show/Hide
|
||||
| Description |
After the tumor xenografts reached a volume of around 100150 mm3, mice were randomized into three groups (n = 7), namely, saline (negative control), free doxorubicin (positive control), and hydrazone PDC. A low dose of 2.5 mg/kg Dox or 2.5 mg/kg Dox equivalent for PDC was chosen to study the antitumor efficacy in vivo. Mice were intravenously administered treatment by tail vein every seventh day for six doses. Compared to the saline group, the PDC reduced tumor growth significantly (3.8 times) on day 35 after treatment, whereas the reduction of tumor growth after free Dox treatment was 2.5 times, suggesting the PDC, at the same equivalent dose, was more potent than the free Dox. In addition, the mice treated with PDC remained in overall good health condition, as evidenced by the general appearance, behavior, diet consumption, and body weight. On day 32 during the treatment period, there were no significant differences observed between the PDC and saline groups in the average body weight (p > 0.05). However, the mice treated with Dox showed significant body weight loss (reduced by 11.2%) compared to the PDC group. Twenty-four hours after the last treatment with PDC or free Dox, mice were euthanized. Mice treated with saline were euthanized on day 32 because of the tumor size per IACUC policy and the conditions for euthanasia. Tumor and other major tissues were collected and weighted for further analysis. The mice with PDC treatment exhibited a greater reduction (three times reduction compared to saline) of tumor weight compared to that of free Dox treated (two times reduction compared to saline).
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.2 µM
|
|||
| Evaluation Method | MTT assay | ||||
| Description |
The results showed that the cytotoxicity of both conjugates 1 and 2 (IC50 = 1.3 and 2.2 uM, respectively), as well as the free Dox (IC50 = 1.5 uM) on MDA-MB-231 breast cancer cell line, were in the low micromolar range (Figure 4). For the breast cancer cell line MDA-MB-468, the free Dox (IC50 = 0.35 uM) was slightly more toxic compared to conjugates 1 (4.7 uM) and 2 (1.2 uM). For the non-cancerous cell line MCF 10A, the free Dox was highly toxic (IC50 = 0.24 uM) whereas conjugates 1 and 2 displayed much-reduced toxicity (IC50 = 38.6 and 15.1 uM, respectively).
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-468 cell | CVCL_0419 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.2 µM
|
|||
| Evaluation Method | MTT assay | ||||
| Description |
The results showed that the cytotoxicity of both conjugates 1 and 2 (IC50 = 1.3 and 2.2 uM, respectively), as well as the free Dox (IC50 = 1.5 uM) on MDA-MB-231 breast cancer cell line, were in the low micromolar range (Figure 4). For the breast cancer cell line MDA-MB-468, the free Dox (IC50 = 0.35 uM) was slightly more toxic compared to conjugates 1 (4.7 uM) and 2 (1.2 uM). For the non-cancerous cell line MCF 10A, the free Dox was highly toxic (IC50 = 0.24 uM) whereas conjugates 1 and 2 displayed much-reduced toxicity (IC50 = 38.6 and 15.1 uM, respectively).
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
15.1 µM
|
|||
| Evaluation Method | MTT assay | ||||
| Description |
The results showed that the cytotoxicity of both conjugates 1 and 2 (IC50 = 1.3 and 2.2 uM, respectively), as well as the free Dox (IC50 = 1.5 uM) on MDA-MB-231 breast cancer cell line, were in the low micromolar range (Figure 4). For the breast cancer cell line MDA-MB-468, the free Dox (IC50 = 0.35 uM) was slightly more toxic compared to conjugates 1 (4.7 uM) and 2 (1.2 uM). For the non-cancerous cell line MCF 10A, the free Dox was highly toxic (IC50 = 0.24 uM) whereas conjugates 1 and 2 displayed much-reduced toxicity (IC50 = 38.6 and 15.1 uM, respectively).
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
FA-P3-PTX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Invasive breast carcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
1.79 ± 0.09 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Ovarian endometrioid adenocarcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
1.95 ± 0.20 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Invasive ductal carcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
4.54 ± 0.71 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive ductal carcinoma | MCF7/PTX cell | CVCL_C5RS | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Chronic myeloid leukemia | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
5.38 ± 0.25 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Chronic myeloid leukemia | K562 cell | CVCL_0004 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Ovarian serous cystadenocarcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
5.92 ± 0.84 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [1] | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
48.55 ± 2.94 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cells | Homo sapiens | ||
Lytic peptides 6 - Paclitaxel conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Invasive breast carcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
1.98 ± 0.14 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Ovarian endometrioid adenocarcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
2.79 ± 0.17 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Chronic myeloid leukemia | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
3.82 ± 0.29 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Chronic myeloid leukemia | K562 cell | CVCL_0004 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Invasive ductal carcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
5.53 ± 0.76 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive ductal carcinoma | MCF7/PTX cell | CVCL_C5RS | ||
| Experiment 5 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Ovarian serous cystadenocarcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
6.61 ± 0.94 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [1] | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
37.22 ± 2.36 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cells | Homo sapiens | ||
Lytic peptides 5 - Paclitaxel conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Invasive breast carcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
2.15 ± 0.18 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Ovarian endometrioid adenocarcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
2.69 ± 0.19 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Chronic myeloid leukemia | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
5.90 ± 0.92 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Chronic myeloid leukemia | K562 cell | CVCL_0004 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Invasive ductal carcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
6.11 ± 0.61 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive ductal carcinoma | MCF7/PTX cell | CVCL_C5RS | ||
| Experiment 5 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Ovarian serous cystadenocarcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
7.17 ± 0.77 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [1] | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
31.60 ± 1.88 μM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We have previously reported that structural optimized lytic peptides I-3 and I-7 can be used as cell-disrupting peptides and molecular carriers. Meanwhile, PTX, a firstline antitumor drug, its poor aqueous solubility (no more than 0.004mg/mL) and acquired drug resistant need to be addressed urgently. In this work, we choose the 16-site cysteine-substituted I-3 and I-7 (namely P3 and P7, respectively) served as peptide backbone and we designed a novel folate targeting peptide-PTX conjugates to achieve selective tumor delivery, enhance cellular uptake, make FA-P3/P7-PTX conjugates water-soluble and overcome drug resistance. The conjugates were evaluated for the antiproliferative activity in different cancer cell lines, the inhibitory rate of tubulin polymerization, hemolytic toxicity and water solubility. Furthermore, we assessed the conjugates for their cellular uptake, Membrane permeability, pro-apoptosis, alternation of mitochondrial membrane potential, rat plasma stability and cell apoptosis pathway in PTX resistant MCF-7/PTX cells. Finally, we researched the most optimized conjugate in vivo antitumor efficacy compared with free PTX.
Click to Show/Hide
|
||||
| Description |
The anticancer activities of the conjugates were evaluated using various cancer cells (MCF-7, MCF-7/PTX, K562, A2780 and SKOV3). The IC50 values are listed in Table 3, and PTX was used for comparison. All the conjugates exhibited improved cytotoxic effects on various cancer cells. According to the results, all the conjugates showed significantly stronger antiproliferative activity than former lytic peptides (P3 and P7), and FA-P3-PTX and FA-P7-PTX showed more excellent antiproliferative activity than P3-PTX and P7-PTX in FA-overexpressing cancer cells MCF-7 (1.79 μM versus 2.15 μM; 1.39 μM versus 1.98 μM), MCF-7/PTX (4.54 μM versus 6.11 μM; 2.92 μM versus 5.53 μM), A2780 (1.95 μM versus 2.69 μM; 1.42 μM versus 2.79 μM), respectively. Thus, the conjugate FA-P3-PTX and FA-P7-PTX exhibited great antiproliferative activity on folate receptors overexpressing cancer cells, and almost equal potency to both drug resistant and -sensitive cells. Meanwhile, the conjugates showed weak toxicity to the normal cell lines HUVEC. To assess the safety profile of the designed conjugates, we examined their hemolytic activity using RBCs. As depicted in Fig. 1, all the tested peptides exhibited modest hemolytic activity.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cells | Homo sapiens | ||
I-6 (NPNWGRSWYNQRFKGC=(-S-Maleimide-CPT)GC=(-S-Maleimide-CPT)GRIKPRKGYTR) [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
6.72 ± 1.25 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
10.91 ± 1.23 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
20.35 ± 1.21 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
25.16 ± 2.12 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
28.24 ± 1.36 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
SMAC-P1 CPT conjugates 9 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
12.58 ± 2.45 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
24.15 ± 3.76 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
31.35 ± 3.17 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
I-3 (NPNWGRSWYNQRFKGC=(-S-Maleimide-CPT)GRIKPRKGYTR) [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
15.32 ± 2.25 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
23.09 ± 1.77 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
24.39 ± 1.45 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
25.45 ± 0.64 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
35.20 ± 2.64 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
SMAC-P1 CPT conjugates 6 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
15.68 ± 1.14 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
20.14 ± 2.12 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
28.21 ± 1.28 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
SMAC-P1 CPT conjugates 3 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
18.34 ± 1.42 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
25.31 ± 1.48 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
30.58 ± 2.76 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
CPT-Cyclo-GCGPep Conjugate 3 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell viability |
70.00%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 10 μM | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell viability |
90.00%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 2.5 μM | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Gastric tubular adenocarcinoma | ||||
| Efficacy Data | Cell viability |
90.00%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 10 μM | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell viability |
98.00%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 0.156 μM | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell viability |
99.00%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 0.625 μM | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Gastric tubular adenocarcinoma | ||||
| Efficacy Data | Cell viability |
100.00%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 0.156 μM | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Gastric tubular adenocarcinoma | ||||
| Efficacy Data | Cell viability |
100.00%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 0.625 μM | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Gastric tubular adenocarcinoma | ||||
| Efficacy Data | Cell viability |
100.00%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 2.5 μM | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
References