
Peptide Information
General Information of This Peptide
| Peptide ID |
PEP00083
|
|||||
|---|---|---|---|---|---|---|
| Peptide Name |
LRRY-fQWAV-β-Ala-H-Aib-Nle
|
|||||
| Structure |
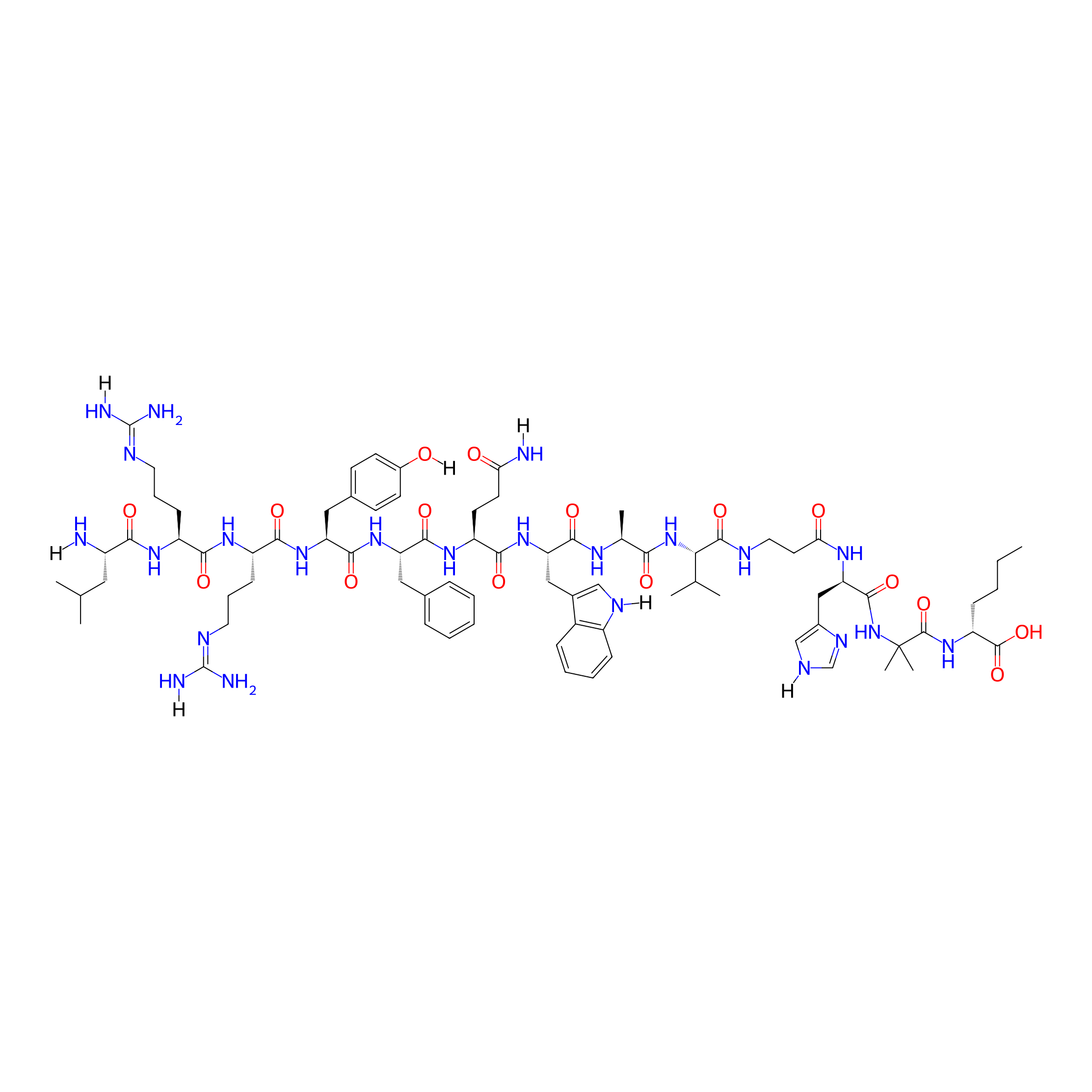
|
|||||
| Sequence |
LRRY-fQWAVAH-Aib-Nle
|
|||||
| Peptide Type |
Linear
|
|||||
| Receptor Name |
Gastrin-releasing peptide receptor (GRPR)
|
Receptor Info | ||||
| PDC Transmembrane Types | Cell targeting peptides (CTPs) | |||||
| Formula |
C79H117N23O16
|
|||||
| Isosmiles |
[H]NC(=O)CC[C@H](NC(=O)[C@H](Cc1ccccc1)NC(=O)[C@H](Cc1ccc(O[H])cc1)NC(=O)[C@H](CCC/N=C(\N)N[H])NC(=O)[C@H](CCC/N=C(\N)N[H])NC(=O)[C@H](CC(C)C)N[H])C(=O)N[C@@H](Cc1cn([H])c2ccccc12)C(=O)N[C@@H](C)C(=O)N[C@H](C(=O)NCCC(=O)N[C@H](Cc1cn([H])cn1)C(=O)NC(C)(C)C(=O)N[C@H](CCCC)C(=O)O)C(C)C
|
|||||
| InChI |
InChI=1S/C79H117N23O16/c1-9-10-21-57(75(116)117)100-76(118)79(7,8)102-73(114)61(39-49-41-86-42-91-49)93-63(105)31-34-87-74(115)64(44(4)5)101-65(106)45(6)92-70(111)60(38-48-40-90-53-22-15-14-20-51(48)53)99-69(110)56(29-30-62(81)104)96-71(112)58(36-46-18-12-11-13-19-46)98-72(113)59(37-47-25-27-50(103)28-26-47)97-68(109)55(24-17-33-89-78(84)85)95-67(108)54(23-16-32-88-77(82)83)94-66(107)52(80)35-43(2)3/h11-15,18-20,22,25-28,40-45,52,54-61,64,90,103H,9-10,16-17,21,23-24,29-39,80H2,1-8H3,(H2,81,104)(H,86,91)(H,87,115)(H,92,111)(H,93,105)(H,94,107)(H,95,108)(H,96,112)(H,97,109)(H,98,113)(H,99,110)(H,100,118)(H,101,106)(H,102,114)(H,116,117)(H4,82,83,88)(H4,84,85,89)/t45-,52-,54-,55-,56-,57+,58-,59-,60-,61+,64-/m0/s1
|
|||||
| InChIKey |
RBAHKJYIHKUSOL-VNRUPWOQSA-N
|
|||||
| Pharmaceutical Properties |
Molecule Weight
|
1644.95
|
Polar area
|
649.11
|
||
|
Complexity
|
1643.904865
|
xlogp Value
|
-2.1869
|
|||
|
Heavy Count
|
118
|
Rot Bonds
|
53
|
|||
|
Hbond acc
|
19
|
Hbond Donor
|
22
|
|||
Each Peptide-drug Conjugate Related to This Peptide
Full Information of The Activity Data of The PDC(s) Related to This Peptide
PDC-L3 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Uptake Concentration (UC50) |
24.59 µM
|
|||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
We quantified the ability of the produced PDCs to promote the internalisation of daunorubicin after binding to GRP-R by flow cytometry. Each bioconjugate was incubated for 1.5 h in four concentrations (25 uM, 12.5 uM, 6.25 uM and 3.125 uM), with the three cell lines that we have also used for the evaluation of the cytostatic effect. For better comparison, the uptake was described as the necessary concentration to internalise 50% of the compound (UC50). The uptake reflects the GRP-R protein expression: the lower UC50 values, hence the highest uptakes, are noticed in the human breast cancer cell line MDA-MB-453, whereas they are comparable in the two other cell lines. As far as the internalisation of the individual conjugates is concerned, L5 and L6 are the most promising ones in these three cell lines. Notably, both hold the LRRY spacer and the Sta13. L1 and L2 have satisfactory UC50s, too. On the other hand, majority of the conjugates with the GFLG spacer are poorly internalised.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-453 cell | CVCL_0418 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Uptake Concentration (UC50) | > 25 µM | |||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
We quantified the ability of the produced PDCs to promote the internalisation of daunorubicin after binding to GRP-R by flow cytometry. Each bioconjugate was incubated for 1.5 h in four concentrations (25 uM, 12.5 uM, 6.25 uM and 3.125 uM), with the three cell lines that we have also used for the evaluation of the cytostatic effect. For better comparison, the uptake was described as the necessary concentration to internalise 50% of the compound (UC50). The uptake reflects the GRP-R protein expression: the lower UC50 values, hence the highest uptakes, are noticed in the human breast cancer cell line MDA-MB-453, whereas they are comparable in the two other cell lines. As far as the internalisation of the individual conjugates is concerned, L5 and L6 are the most promising ones in these three cell lines. Notably, both hold the LRRY spacer and the Sta13. L1 and L2 have satisfactory UC50s, too. On the other hand, majority of the conjugates with the GFLG spacer are poorly internalised.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Uptake Concentration (UC50) | > 25 µM | |||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
We quantified the ability of the produced PDCs to promote the internalisation of daunorubicin after binding to GRP-R by flow cytometry. Each bioconjugate was incubated for 1.5 h in four concentrations (25 uM, 12.5 uM, 6.25 uM and 3.125 uM), with the three cell lines that we have also used for the evaluation of the cytostatic effect. For better comparison, the uptake was described as the necessary concentration to internalise 50% of the compound (UC50). The uptake reflects the GRP-R protein expression: the lower UC50 values, hence the highest uptakes, are noticed in the human breast cancer cell line MDA-MB-453, whereas they are comparable in the two other cell lines. As far as the internalisation of the individual conjugates is concerned, L5 and L6 are the most promising ones in these three cell lines. Notably, both hold the LRRY spacer and the Sta13. L1 and L2 have satisfactory UC50s, too. On the other hand, majority of the conjugates with the GFLG spacer are poorly internalised.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
18.96 ± 3.23 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
The cytostatic effect of the conjugates and the free peptides was evaluated on the three mentioned human cancer cell lines expressing GRP-R. The free Dau was used as a positive control for comparison purposes; it displays an IC50 in the high nanomolar range. Overall, the conjugates containing the LRRY spacer have a higher cytostatic effect than the ones with the GFLG spacer. L1, which has the original BBN (7-14) sequence, and L5, bearing a D-Phe in position 6 and the Sta13-Leu14 bond at the C-terminus, shows the best activity in all three cell lines, with IC50 values in the low micromolar range. The Gly11/-Ala11 and Leu13-Met14/Sta13-Nle14 substitutions, which led to a new BBN (6-14) peptide sequence, held by the conjugate L6, affect the activity only slightly. Contrarily, the two free peptides, bearing the sequences of L5 and L6, do not show any activity on any of the cell lines. The lack of toxicity of the conjugates on healthy cells was checked on MRC-5 human fibroblasts, proving that they are non-toxic on non-cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 25 µM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
The cytostatic effect of the conjugates and the free peptides was evaluated on the three mentioned human cancer cell lines expressing GRP-R. The free Dau was used as a positive control for comparison purposes; it displays an IC50 in the high nanomolar range. Overall, the conjugates containing the LRRY spacer have a higher cytostatic effect than the ones with the GFLG spacer. L1, which has the original BBN (7-14) sequence, and L5, bearing a D-Phe in position 6 and the Sta13-Leu14 bond at the C-terminus, shows the best activity in all three cell lines, with IC50 values in the low micromolar range. The Gly11/-Ala11 and Leu13-Met14/Sta13-Nle14 substitutions, which led to a new BBN (6-14) peptide sequence, held by the conjugate L6, affect the activity only slightly. Contrarily, the two free peptides, bearing the sequences of L5 and L6, do not show any activity on any of the cell lines. The lack of toxicity of the conjugates on healthy cells was checked on MRC-5 human fibroblasts, proving that they are non-toxic on non-cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-453 cell | CVCL_0418 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 25 µM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
The cytostatic effect of the conjugates and the free peptides was evaluated on the three mentioned human cancer cell lines expressing GRP-R. The free Dau was used as a positive control for comparison purposes; it displays an IC50 in the high nanomolar range. Overall, the conjugates containing the LRRY spacer have a higher cytostatic effect than the ones with the GFLG spacer. L1, which has the original BBN (7-14) sequence, and L5, bearing a D-Phe in position 6 and the Sta13-Leu14 bond at the C-terminus, shows the best activity in all three cell lines, with IC50 values in the low micromolar range. The Gly11/-Ala11 and Leu13-Met14/Sta13-Nle14 substitutions, which led to a new BBN (6-14) peptide sequence, held by the conjugate L6, affect the activity only slightly. Contrarily, the two free peptides, bearing the sequences of L5 and L6, do not show any activity on any of the cell lines. The lack of toxicity of the conjugates on healthy cells was checked on MRC-5 human fibroblasts, proving that they are non-toxic on non-cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
References