
Drug Information
General Information of This Drug
| Drug ID | DRG00009 | |||||
|---|---|---|---|---|---|---|
| Drug Name | Camptothecin | |||||
| Synonyms |
camptothecin; 7689-03-4; Camptothecine; (S)-(+)-Camptothecin; Campathecin; (+)-Camptothecine; d-Camptothecin; (+)-Camptothecin; 20(S)-Camptothecine; 21,22-Secocamptothecin-21-oic acid lactone; NSC94600; Camptothecine (S,+); CHEMBL65; (S)-4-ethyl-4-hydroxy-1H-Pyrano[3',4':6,7]indolizino[1,2-b]quinoline-3,14(4H,12H)-dione; NSC-94600; (4S)-4-ethyl-4-hydroxy-1H-pyrano[3',4':6,7]indolizino[1,2-b]quinoline-3,14(4H,12H)-dione; MLS000766223; XT3Z54Z28A; CHEBI:27656; MFCD00081076; (19S)-19-ethyl-19-hydroxy-17-oxa-3,13-diazapentacyclo[11.8.0.02,11.04,9.015,20]henicosa-1(21),2,4,6,8,10,15(20)-heptaene-14,18-dione; NSC 100880; (19S)-19-ethyl-19-hydroxy-17-oxa-3,13-diazapentacyclo[11.8.0.0^{2,11}.0^{4,9}.0^{15,20}]henicosa-1(21),2,4(9),5,7,10,15(20)-heptaene-14,18-dione; (S)-Camptothecin; 1H-Pyrano(3',4':6,7)indolizino(1,2-b)quinoline-3,14(4H,12H)-dione, 4-ethyl-4-hydroxy-, (4S)-; 1H-Pyrano(3',4':6,7)indolizino(1,2-b)quinoline-3,14(4H,12H)-dione, 4-ethyl-4-hydroxy-, (S)-; 1H-Pyrano[3',4':6,7]indolizino[1,2-b]quinoline-3,14(4H,12H)-dione, 4-ethyl-4-hydroxy-, (4S)-; 20(S)-Camptothecin; 4-ETHYL-4-HYDROXY-1,12-DIHYDRO-4H-2-OXA-6,12A-DIAZA-DIBENZO[B,H]FLUORENE-3,13-DIONE; SR-01000075798; SR-01000597379; d-camptothecine; (s)-camptothecine; Camptothecin,(S); (4S)-4-ETHYL-4-HYDROXY-1H-PYRANO(3',4':6,7)INDOLIZINO(1,2-B)QUINOLINE-3,14(4H,12H)-DIONE; (S)-4-ethyl-4-hydroxy-1H-Pyrano(3',4':6,7)indolizino(1,2-b)quinoline-3,14(4H,12H)-dione; (S)-4-Ethyl-4-hydroxy-1H-pyrano[3',4':6,7]indolizino[1,2-b]quinoline-3,14-(4H,12H)-dione; 1H-Pyrano[3',4':6,7]indolizino[1,2-b]quinoline-3,14(4H,12H)-dione, 4-ethyl-4-hydroxy-, (S)-; Prestwick_102; (+)-Camptothecin;; Camptothecine (8CI); Spectrum_000299; Tocris-1100; SpecPlus_000712; Prestwick0_000200; Prestwick1_000200; Prestwick2_000200; Prestwick3_000200; Spectrum2_000903; Spectrum3_001203; Spectrum4_000738; Spectrum5_001126; CAMPTOTHECIN [MI]; Lopac-C-9911; SCHEMBL6038; UNII-XT3Z54Z28A; Lopac0_000341; BSPBio_000159; BSPBio_002586; KBioGR_001036; KBioSS_000779; KBioSS_002283; cid_24360; CAMPTOTHECIN [WHO-DD]; DivK1c_000826; DivK1c_006808; SPECTRUM1502232; SPBio_000746; SPBio_002080; BPBio1_000175; CCRIS 8162; DTXSID0030956; HMS502J08; KBio1_000826; KBio1_001752; KBio2_000779; KBio2_003347; KBio2_005915; KBio3_002086; 4-Ethyl-4-hydroxy-1H-pyrano-[3',4':6,7]indolizino[1,2-b]quinoline-3,14(4H,12H)-dione; NINDS_000826; Bio1_000400; Bio1_000889; Bio1_001378; GLXC-10346; HMS1568H21; HMS1921N08; HMS2089F08; HMS2095H21; HMS3261E03; HMS3414J17; HMS3654D13; HMS3678J15; HMS3712H21; BCP02857; Tox21_500341; AC-202; BBL033963; BDBM50008923; CCG-40255; GR-301; NSC 94600; s1288; STK801886; AKOS004119861; CS-1049; DB04690; KS-5235; LP00341; SDCCGMLS-0066688.P001; SDCCGSBI-0050329.P003; BRN 0631069; CAS-2114454; IDI1_000826; NCGC00015290-01; NCGC00016994-01; NCGC00016994-02; NCGC00016994-03; NCGC00016994-04; NCGC00016994-05; NCGC00016994-06; NCGC00016994-07; NCGC00016994-08; NCGC00016994-09; NCGC00016994-10; NCGC00016994-11; NCGC00016994-12; NCGC00016994-16; NCGC00016994-29; NCGC00024997-01; NCGC00024997-02; NCGC00024997-03; NCGC00024997-04; NCGC00024997-05; NCGC00024997-06; NCGC00178592-01; NCGC00178592-02; NCGC00261026-01; 1ST40312; HY-16560; NCI60_042105; SMR000445686; SY010324; AI3-62475; EU-0100341; NS00011856; SW196414-3; C 9911; C01897; M01564; AB00052452-08; AB00052452-09; AB00052452_10; EN300-1725804; (S)-(+)-Camptothecin, >=90% (HPLC), powder; A838882; Q419964; Q-200785; SR-01000075798-1; SR-01000075798-4; SR-01000597379-1; SR-01000597379-3; BRD-K37890730-001-09-4; BRD-K37890730-001-10-2; Z1741982070; (S)-4-ethyl-4-hydroxy-1,12-dihydro-4H-2-oxa-6,12a-diaza-dibenzo[b,h]florene-3,13-dione; (S)-4-ethyl-4-hydroxy-1,12-dihydro-4H-2-oxa-6,12a-diaza-dibenzo[b,h]fluorene-3,13-dione; 4-Ethyl-4-hydroxy-1H-pyrano-[3,4:6,7]indolizino[1,2-b]quinoline-3,14(4H,12H)-dione; 4-Ethyl-4-hydroxy-1H-pyrano-[3[,4[:6,7]indolizino[1,2-b]quinoline-3,14(4H,12H)-dione; (19S)-19-ethyl-19-hydroxy-17-oxa-3,13-diazapentacyclo[11.8.0.0^{2,11}.0^{4,9}.0^{15,20}]henicosa-1(21),2(11),3,5,7,9,15(20)-heptaene-14,18-dione; (S)-4-Ethyl-4-hydroxy-1H-pyrano-[3',4':6,7]indolizino[1,2-b]quinoline-3,14(4H,12H)-dione;(S)-(+)-Camptothecin; (S)-4-Ethyl-4-hydroxy-1H-pyrano[3 inverted exclamation mark ,4 inverted exclamation mark :6,7]indolizino[1,2-b]quinoline-3,14-(4H,12H)-dione; 1H-Pyrano[3',7]indolizino[1,2-b]quinoline-3,14(4H,12H)-dione, 4-ethyl-4-hydroxy-, (S)-; 4(S)-Ethyl-4-hydroxy-1H-pyrano-[3',4':6,7]indolizino[1,2-b]quinoline-3,14 (4H,12H)-dione; 4-ethyl-4-hydroxy-(4S)-3,4,12,14-tetrahydro-1H-pyrano[3'',4'':6,7]indolizino[1,2-b]quinoline-3,14-dione; 4-Ethyl-4-hydroxy-1,12-dihydro-4H-2-oxa-6,12a-diaza-dibenzo[b,h]fluorene-3,13-dione (camptothecin or CPT); 4-Ethyl-4-hydroxy-1,12-dihydro-4H-2-oxa-6,12a-diaza-dibenzo[b,h]fluorene-3,13-dione (Camptothecin); 4-Ethyl-4-hydroxy-1,12-dihydro-4H-2-oxa-6,12a-diaza-dibenzo[b,h]fluorene-3,13-dione (CPT, Camptothecin)
Click to Show/Hide
|
|||||
| Target(s) | DNA topoisomerase 1 (TOP1) | Target Info | ||||
| Structure |
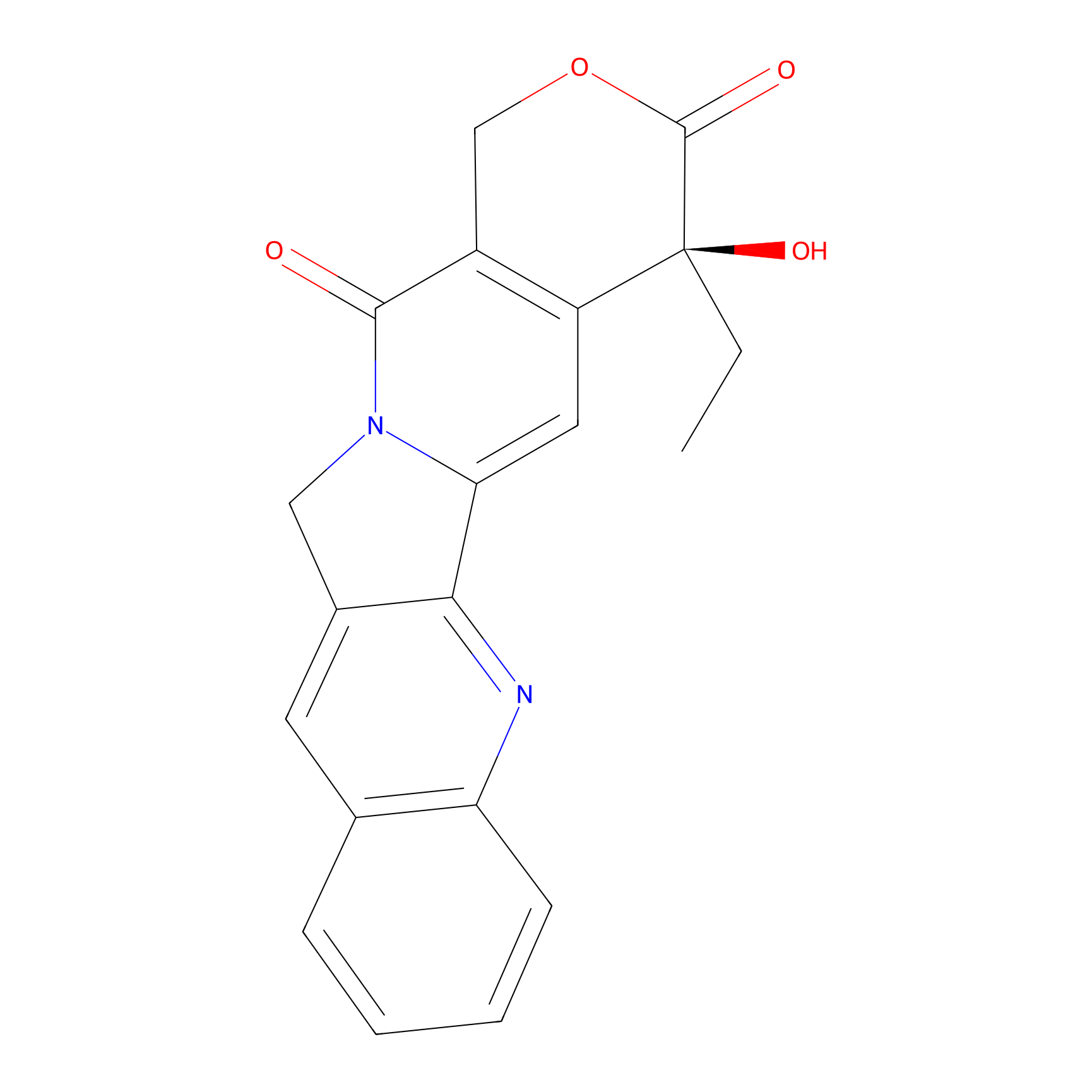
|
|||||
| Formula |
C20H16N2O4
|
|||||
| #Ro5 Violations (Lipinski): 0 | Molecular Weight (mw) | 348.4 | ||||
| Lipid-water partition coefficient (xlogp) | 1 | |||||
| Hydrogen Bond Donor Count (hbonddonor) | 1 | |||||
| Hydrogen Bond Acceptor Count (hbondacc) | 5 | |||||
| Rotatable Bond Count (rotbonds) | 1 | |||||
| PubChem CID | ||||||
| Canonical smiles |
CCC1(C2=C(COC1=O)C(=O)N3CC4=CC5=CC=CC=C5N=C4C3=C2)O
|
|||||
| InChI |
InChI=1S/C20H16N2O4/c1-2-20(25)14-8-16-17-12(7-11-5-3-4-6-15(11)21-17)9-22(16)18(23)13(14)10-26-19(20)24/h3-8,25H,2,9-10H2,1H3/t20-/m0/s1
|
|||||
| InChIKey |
VSJKWCGYPAHWDS-FQEVSTJZSA-N
|
|||||
| IUPAC Name |
(19S)-19-ethyl-19-hydroxy-17-oxa-3,13-diazapentacyclo[11.8.0.02,11.04,9.015,20]henicosa-1(21),2,4,6,8,10,15(20)-heptaene-14,18-dione
|
|||||
The activity data of This Drug
| Standard Type | Value | Administration times | Administration dosage | Cell line | Cell line ID | Ref. |
|---|---|---|---|---|---|---|
| Apoptosis rate | 55.80% | 24 h | 2 μM | SK-BR-3 cell | CVCL_0033 | [1] |
| Cell survival rate | 46% | 72 h | 10 μM | B16-F10 cell | CVCL_0159 | [2] |
| Cell survival rate | 67% | 72 h | 5 μM | B16-F10 cell | CVCL_0159 | [2] |
| Cell survival rate | 71% | 72 h | 2.5 μM | B16-F10 cell | CVCL_0159 | [2] |
| Cell survival rate | 82% | 72 h | 1.25 μM | B16-F10 cell | CVCL_0159 | [2] |
| Cell survival rate | 95% | 72 h | 0.625 μM | B16-F10 cell | CVCL_0159 | [2] |
| Tumor Growth Inhibition value (TGI) | 39% | Injected via tail vein every three days | 8 μmol/kg | SK-BR-3 cell | CVCL_0033 | [1] |
| Half Maximal Inhibitory Concentration (IC50) | 0.21±0.03 µM | 48 h | N.A. | HCT 116 cell | CVCL_0291 | [3] |
| Half Maximal Inhibitory Concentration (IC50) | 0.21±0.04 µM | 48 h | N.A. | SK-BR-3 cell | CVCL_0033 | [4] |
| Half Maximal Inhibitory Concentration (IC50) | 0.26±0.06 µM | 48 h | N.A. | SK-BR-3 cell | CVCL_0033 | [1] |
| Half Maximal Inhibitory Concentration (IC50) | 0.38±0.11 µM | 48 h | N.A. | SK-BR-3 cell | CVCL_0033 | [5] |
| Half Maximal Inhibitory Concentration (IC50) | 0.53±0.21 µM | 48 h | N.A. | MDA-MB-231 cell | CVCL_0062 | [5] |
| Half Maximal Inhibitory Concentration (IC50) | 0.65±0.03 µM | 48 h | N.A. | SGC-7901 cell | CVCL_0520 | [3] |
| Half Maximal Inhibitory Concentration (IC50) | 0.68±0.04 µM | 48 h | N.A. | MCF-7 cell | CVCL_0031 | [3] |
| Half Maximal Inhibitory Concentration (IC50) | 1.06±0.42 µM | 48 h | N.A. | MDA-MB-231 cell | CVCL_0062 | [4] |
| Half Maximal Inhibitory Concentration (IC50) | 1.12±0.11 µM | 48 h | N.A. | NCM460 cell | CVCL_0460 | [3] |
| Half Maximal Inhibitory Concentration (IC50) | 2.04±0.20 µM | 48 h | N.A. | GES1 cell | CVCL_EQ22 | [3] |
| Half Maximal Inhibitory Concentration (IC50) | 2.07±0.67 µM | 48 h | N.A. | NCI-N87 cell | CVCL_1603 | [5] |
| Half Maximal Inhibitory Concentration (IC50) | 2.07±0.67 µM | 48 h | N.A. | NCI-N87 cell | CVCL_1603 | [5] |
| Half Maximal Inhibitory Concentration (IC50) | 2.10±1.34 µM | 48 h | N.A. | NCI-N87 cell | CVCL_1603 | [1] |
| Half Maximal Inhibitory Concentration (IC50) | 2.10±1.34 µM | 48 h | N.A. | NCI-N87 cell | CVCL_1603 | [1] |
| Half Maximal Inhibitory Concentration (IC50) | 2.32±0.24 µM | 48 h | N.A. | NCI-N87 cell | CVCL_1603 | [4] |
| Half Maximal Inhibitory Concentration (IC50) | 2.32±0.24 µM | 48 h | N.A. | NCI-N87 cell | CVCL_1603 | [4] |
| Half Maximal Inhibitory Concentration (IC50) | 2.52±0.22 µM | 48 h | N.A. | LO #2 cell | CVCL_C7SD | [3] |
| Half Maximal Inhibitory Concentration (IC50) | 2.52±0.22 µM | 48 h | N.A. | LO #2 cell | CVCL_C7SD | [3] |
| Half Maximal Inhibitory Concentration (IC50) | 2.58±0.77 µM | 48 h | N.A. | MCF-10A cell | CVCL_0598 | [4] |
| Half Maximal Inhibitory Concentration (IC50) | 3.02±0.49 µM | 48 h | N.A. | MCF-10A cell | CVCL_0598 | [5] |
| Half Maximal Inhibitory Concentration (IC50) | 3.05±1.24 µM | 48 h | N.A. | MCF-10A cell | CVCL_0598 | [1] |
| Half Maximal Inhibitory Concentration (IC50) | 3.17±1.15 µM | 48 h | N.A. | MDA-MB-231 cell | CVCL_0062 | [1] |
| Half Maximal Inhibitory Concentration (IC50) | 3.82±0.91 µM | 48 h | N.A. | SK-OV-3 cell | CVCL_0532 | [4] |
| Half Maximal Inhibitory Concentration (IC50) | 3.82±0.91 µM | 48 h | N.A. | SK-OV-3 cell | CVCL_0532 | [4] |
| Half Maximal Inhibitory Concentration (IC50) | 4.07±1.82 µM | 48 h | N.A. | SK-OV-3 cell | CVCL_0532 | [5] |
| Half Maximal Inhibitory Concentration (IC50) | 4.07±1.82 µM | 48 h | N.A. | SK-OV-3 cell | CVCL_0532 | [5] |
| Half Maximal Inhibitory Concentration (IC50) | 5.21±0.31 µM | 48 h | N.A. | HCT-116/CPT cell | CVCL_0291 | [3] |
| Half Maximal Inhibitory Concentration (IC50) | 15.8±0.7 µM | 48 h | N.A. | MCF7/C4 cell | CVCL_GX99 | [3] |
| Half Maximal Inhibitory Concentration (IC50) | 18.1±1.1 µM | 48 h | N.A. | SGC-7901/CPT cell | CVCL_0520 | [3] |
| Half Maximal Cytotoxicity Concentration (CC50) | 3 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [6] |
| Half Maximal Cytotoxicity Concentration (CC50) | 4 nM | N.A. | N.A. | MT4 cell | CVCL_2632 | [6] |
| Half Maximal Cytotoxicity Concentration (CC50) | 5 nM | N.A. | N.A. | WIL2-NS cell | CVCL_2761 | [7] |
| Half Maximal Cytotoxicity Concentration (CC50) | 6 nM | N.A. | N.A. | KB cell | CVCL_0372 | [8] |
| Half Maximal Cytotoxicity Concentration (CC50) | 10 nM | N.A. | N.A. | MT4 cell | CVCL_2632 | [8] |
| Half Maximal Cytotoxicity Concentration (CC50) | 15 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [7] |
| Half Maximal Cytotoxicity Concentration (CC50) | 20 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [7] |
| Half Maximal Cytotoxicity Concentration (CC50) | 50 nM | N.A. | N.A. | SK-MES-1 cell | CVCL_0630 | [6] |
| Half Maximal Cytotoxicity Concentration (CC50) | 80 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [6] |
| Half Maximal Cytotoxicity Concentration (CC50) | 200 nM | N.A. | N.A. | MRC5 cell | CVCL_0440 | [7] |
| Half Maximal Effective Concentration (EC50) | 10 ng/mL | N.A. | N.A. | KB cell | CVCL_0372 | [9] |
| Half Maximal Effective Concentration (EC50) | 0.15 nM | N.A. | N.A. | SW620 cell | CVCL_0547 | [10] |
| Half Maximal Effective Dosage (ED50) | 8.6 pmol/ml | N.A. | N.A. | P388 cell | CVCL_7222 | [11] |
| Half Maximal Effective Dosage (ED50) | 3.2 nM | N.A. | N.A. | A2780-1A9 cell | CVCL_H619 | [12] |
| Half Maximal Effective Dosage (ED50) | 3.2 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [12] |
| Half Maximal Effective Dosage (ED50) | 20.9 nM | N.A. | N.A. | KB cell | CVCL_0372 | [12] |
| Half Maximal Effective Dosage (ED50) | 57 nM | N.A. | N.A. | Col2 cell | CVCL_D645 | [13] |
| Half Maximal Effective Dosage (ED50) | >10 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [14] |
| Half Maximal Growth Inhibition (GI50) | 23 pM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [15] |
| Half Maximal Growth Inhibition (GI50) | 2 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [16] |
| Half Maximal Growth Inhibition (GI50) | 7 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [17] |
| Half Maximal Growth Inhibition (GI50) | 8 nM | N.A. | N.A. | LoVo cell | CVCL_0399 | [18] |
| Half Maximal Growth Inhibition (GI50) | 10 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [19] |
| Half Maximal Growth Inhibition (GI50) | 10 nM | N.A. | N.A. | HOP-62 cell | CVCL_1285 | [20] |
| Half Maximal Growth Inhibition (GI50) | <10 nM | N.A. | N.A. | UACC-62 cell | CVCL_1780 | [21] |
| Half Maximal Growth Inhibition (GI50) | 10 nM | N.A. | N.A. | UACC-62 cell | CVCL_1780 | [19] |
| Half Maximal Growth Inhibition (GI50) | 10 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [22] |
| Half Maximal Growth Inhibition (GI50) | 10 nM | N.A. | N.A. | SF539 cell | CVCL_1691 | [23] |
| Half Maximal Growth Inhibition (GI50) | 13 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [19] |
| Half Maximal Growth Inhibition (GI50) | 19 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [24] |
| Half Maximal Growth Inhibition (GI50) | 20 nM | N.A. | N.A. | SN12C cell | CVCL_1705 | [19] |
| Half Maximal Growth Inhibition (GI50) | 30 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [25] |
| Half Maximal Growth Inhibition (GI50) | 40 nM | N.A. | N.A. | MDA-MB-435 cell | CVCL_0417 | [26] |
| Half Maximal Growth Inhibition (GI50) | 50 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [27] |
| Half Maximal Growth Inhibition (GI50) | 60 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [28] |
| Half Maximal Growth Inhibition (GI50) | 80 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [29] |
| Half Maximal Growth Inhibition (GI50) | 83 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [30] |
| Half Maximal Growth Inhibition (GI50) | 90 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [31] |
| Half Maximal Growth Inhibition (GI50) | 99 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [32] |
| Half Maximal Growth Inhibition (GI50) | 170 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [30] |
| Half Maximal Growth Inhibition (GI50) | 210 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [32] |
| Half Maximal Growth Inhibition (GI50) | 210 nM | N.A. | N.A. | SW1573 cell | CVCL_1720 | [33] |
| Half Maximal Growth Inhibition (GI50) | 220 nM | N.A. | N.A. | OVCAR-3 cell | CVCL_0465 | [21] |
| Half Maximal Growth Inhibition (GI50) | 230 nM | N.A. | N.A. | HBL-100 cell | CVCL_4362 | [33] |
| Half Maximal Growth Inhibition (GI50) | 280 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [34] |
| Half Maximal Growth Inhibition (GI50) | 460 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [33] |
| Half Maximal Growth Inhibition (GI50) | 480 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [18] |
| Half Maximal Growth Inhibition (GI50) | 550 nM | N.A. | N.A. | SiHa cell | CVCL_0032 | [35] |
| Half Maximal Growth Inhibition (GI50) | 570 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [28] |
| Half Maximal Growth Inhibition (GI50) | 600 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [36] |
| Half Maximal Growth Inhibition (GI50) | >1000 nM | N.A. | N.A. | CEM/C2 cell | CVCL_3497 | [16] |
| Half Maximal Growth Inhibition (GI50) | 1.3 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [18] |
| Half Maximal Growth Inhibition (GI50) | 1.7 uM | N.A. | N.A. | WiDr cell | CVCL_2760 | [33] |
| Half Maximal Growth Inhibition (GI50) | 2.8 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [29] |
| Half Maximal Growth Inhibition (GI50) | 6 uM | N.A. | N.A. | SAS cell | CVCL_1675 | [31] |
| Half Maximal Growth Inhibition (GI50) | 6 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [18] |
| Half Maximal Growth Inhibition (GI50) | >287 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [30] |
| Half Maximal Growth Inhibition (GI50) | >287 uM | N.A. | N.A. | U2OS cell | CVCL_0042 | [30] |
| Half Maximal Inhibitory Concentration (IC50) | 0.19 ug/mL | N.A. | N.A. | HeLa cell | CVCL_0030 | [37] |
| Half Maximal Inhibitory Concentration (IC50) | 1.54 ug/mL | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [38] |
| Half Maximal Inhibitory Concentration (IC50) | 2.05 ug/mL | N.A. | N.A. | RPMI-8226 cell | CVCL_7353 | [39] |
| Half Maximal Inhibitory Concentration (IC50) | 2.72 ug/mL | N.A. | N.A. | Bel-7402 cell | CVCL_5492 | [40] |
| Half Maximal Inhibitory Concentration (IC50) | 4.63 ng/mL | N.A. | N.A. | QG-56 cell | CVCL_6943 | [41] |
| Half Maximal Inhibitory Concentration (IC50) | 4.74 ng/mL | N.A. | N.A. | P388 cell | CVCL_7222 | [41] |
| Half Maximal Inhibitory Concentration (IC50) | 5 ng/mL | N.A. | N.A. | P388 cell | CVCL_7222 | [42] |
| Half Maximal Inhibitory Concentration (IC50) | <5 ng/mL | N.A. | N.A. | KB cell | CVCL_0372 | [40] |
| Half Maximal Inhibitory Concentration (IC50) | 10 ng/mL | N.A. | N.A. | KB cell | CVCL_0372 | [43] |
| Half Maximal Inhibitory Concentration (IC50) | 14 mM | N.A. | N.A. | DLD-1 cell | CVCL_0248 | [44] |
| Half Maximal Inhibitory Concentration (IC50) | 20 ng/mL | N.A. | N.A. | Vero cell | CVCL_0059 | [45] |
| Half Maximal Inhibitory Concentration (IC50) | 40 ng/mL | N.A. | N.A. | KB cell | CVCL_0372 | [43] |
| Half Maximal Inhibitory Concentration (IC50) | 54 ng/mL | N.A. | N.A. | Vero cell | CVCL_0059 | [46] |
| Half Maximal Inhibitory Concentration (IC50) | 70 ng/mL | N.A. | N.A. | A2780 cell | CVCL_0134 | [47] |
| Half Maximal Inhibitory Concentration (IC50) | 70 ng/mL | N.A. | N.A. | WiDr cell | CVCL_2760 | [37] |
| Half Maximal Inhibitory Concentration (IC50) | 72 ng/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [38] |
| Half Maximal Inhibitory Concentration (IC50) | 90 ng/mL | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [43] |
| Half Maximal Inhibitory Concentration (IC50) | 0.32 nM | N.A. | N.A. | Huh-7 cell | CVCL_0336 | [48] |
| Half Maximal Inhibitory Concentration (IC50) | 0.32 nM | N.A. | N.A. | LN-229 cell | CVCL_0393 | [48] |
| Half Maximal Inhibitory Concentration (IC50) | 1 nM | N.A. | N.A. | NCI-H69 cell | CVCL_1579 | [49] |
| Half Maximal Inhibitory Concentration (IC50) | 1 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [50] |
| Half Maximal Inhibitory Concentration (IC50) | 1.4 nM | N.A. | N.A. | Jurkat cell | CVCL_0065 | [51] |
| Half Maximal Inhibitory Concentration (IC50) | 1.6 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [52] |
| Half Maximal Inhibitory Concentration (IC50) | <1.8 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [53] |
| Half Maximal Inhibitory Concentration (IC50) | 2 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [54] |
| Half Maximal Inhibitory Concentration (IC50) | 2 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [55] |
| Half Maximal Inhibitory Concentration (IC50) | 2 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [56] |
| Half Maximal Inhibitory Concentration (IC50) | 3 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [57] |
| Half Maximal Inhibitory Concentration (IC50) | 3 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [58] |
| Half Maximal Inhibitory Concentration (IC50) | 3 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [59] |
| Half Maximal Inhibitory Concentration (IC50) | 3 nM | N.A. | N.A. | KB cell | CVCL_0372 | [60] |
| Half Maximal Inhibitory Concentration (IC50) | 3 nM | N.A. | N.A. | SMMC-7721 cell | CVCL_0534 | [61] |
| Half Maximal Inhibitory Concentration (IC50) | 3 nM | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [55] |
| Half Maximal Inhibitory Concentration (IC50) | 3.1 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [62] |
| Half Maximal Inhibitory Concentration (IC50) | 4 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [9] |
| Half Maximal Inhibitory Concentration (IC50) | 4 nM | N.A. | N.A. | MT4 cell | CVCL_2632 | [63] |
| Half Maximal Inhibitory Concentration (IC50) | 4 nM | N.A. | N.A. | P388 cell | CVCL_7222 | [64] |
| Half Maximal Inhibitory Concentration (IC50) | 5 nM | N.A. | N.A. | RPMI-8402 cell | CVCL_1667 | [65] |
| Half Maximal Inhibitory Concentration (IC50) | 5.6 nM | N.A. | N.A. | Jurkat cell | CVCL_0065 | [66] |
| Half Maximal Inhibitory Concentration (IC50) | 6 nM | N.A. | N.A. | RPMI-8402 cell | CVCL_1667 | [67] |
| Half Maximal Inhibitory Concentration (IC50) | 6 nM | N.A. | N.A. | RPMI-8226 cell | CVCL_7353 | [68] |
| Half Maximal Inhibitory Concentration (IC50) | 6 nM | N.A. | N.A. | T24 cell | CVCL_0554 | [69] |
| Half Maximal Inhibitory Concentration (IC50) | 6 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [55] |
| Half Maximal Inhibitory Concentration (IC50) | 6.3 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [70] |
| Half Maximal Inhibitory Concentration (IC50) | 7 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [71] |
| Half Maximal Inhibitory Concentration (IC50) | 7 nM | N.A. | N.A. | P388 cell | CVCL_7222 | [72] |
| Half Maximal Inhibitory Concentration (IC50) | 8 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [73] |
| Half Maximal Inhibitory Concentration (IC50) | 8.63 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [74] |
| Half Maximal Inhibitory Concentration (IC50) | 9 nM | N.A. | N.A. | KB cell | CVCL_0372 | [75] |
| Half Maximal Inhibitory Concentration (IC50) | 9 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [76] |
| Half Maximal Inhibitory Concentration (IC50) | 10 nM | N.A. | N.A. | 833K cell | CVCL_2292 | [77] |
| Half Maximal Inhibitory Concentration (IC50) | <10 nM | N.A. | N.A. | MCF-12A cell | CVCL_3744 | [78] |
| Half Maximal Inhibitory Concentration (IC50) | 10 nM | N.A. | N.A. | SK-MES-1 cell | CVCL_0630 | [58] |
| Half Maximal Inhibitory Concentration (IC50) | 10 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [60] |
| Half Maximal Inhibitory Concentration (IC50) | <10 nM | N.A. | N.A. | ZR-75-1 cell | CVCL_0588 | [78] |
| Half Maximal Inhibitory Concentration (IC50) | <10 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [78] |
| Half Maximal Inhibitory Concentration (IC50) | <10 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [78] |
| Half Maximal Inhibitory Concentration (IC50) | <10 nM | N.A. | N.A. | SK-BR-3 cell | CVCL_0033 | [78] |
| Half Maximal Inhibitory Concentration (IC50) | 11.5 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [79] |
| Half Maximal Inhibitory Concentration (IC50) | 12 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [49] |
| Half Maximal Inhibitory Concentration (IC50) | 13 nM | N.A. | N.A. | P388 cell | CVCL_7222 | [80] |
| Half Maximal Inhibitory Concentration (IC50) | 13 nM | N.A. | N.A. | A-375 cell | CVCL_0132 | [81] |
| Half Maximal Inhibitory Concentration (IC50) | 13 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [82] |
| Half Maximal Inhibitory Concentration (IC50) | 14 nM | N.A. | N.A. | P388 cell | CVCL_7222 | [68] |
| Half Maximal Inhibitory Concentration (IC50) | 14 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [83] |
| Half Maximal Inhibitory Concentration (IC50) | 15 nM | N.A. | N.A. | L1210 cell | CVCL_0382 | [84] |
| Half Maximal Inhibitory Concentration (IC50) | 15 nM | N.A. | N.A. | U-937 cell | CVCL_0007 | [85] |
| Half Maximal Inhibitory Concentration (IC50) | 15.8 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [86] |
| Half Maximal Inhibitory Concentration (IC50) | 16 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [87] |
| Half Maximal Inhibitory Concentration (IC50) | 17 nM | N.A. | N.A. | MKN45 cell | CVCL_0434 | [83] |
| Half Maximal Inhibitory Concentration (IC50) | 18 nM | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [88] |
| Half Maximal Inhibitory Concentration (IC50) | 18 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [89] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [90] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [83] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | KB cell | CVCL_0372 | [91] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | AGS cell | CVCL_0139 | [92] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [93] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [94] |
| Half Maximal Inhibitory Concentration (IC50) | 24 nM | N.A. | N.A. | A-427 cell | CVCL_1055 | [62] |
| Half Maximal Inhibitory Concentration (IC50) | 26 nM | N.A. | N.A. | Jurkat cell | CVCL_0065 | [73] |
| Half Maximal Inhibitory Concentration (IC50) | 28.4 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [95] |
| Half Maximal Inhibitory Concentration (IC50) | 28.7 nM | N.A. | N.A. | KB cell | CVCL_0372 | [96] |
| Half Maximal Inhibitory Concentration (IC50) | 28.7 nM | N.A. | N.A. | SW626 cell | CVCL_1725 | [96] |
| Half Maximal Inhibitory Concentration (IC50) | 29 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [97] |
| Half Maximal Inhibitory Concentration (IC50) | 30 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [98] |
| Half Maximal Inhibitory Concentration (IC50) | 30 nM | N.A. | N.A. | L1210 cell | CVCL_0382 | [99] |
| Half Maximal Inhibitory Concentration (IC50) | 30 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [100] |
| Half Maximal Inhibitory Concentration (IC50) | 30 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [101] |
| Half Maximal Inhibitory Concentration (IC50) | 30 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [101] |
| Half Maximal Inhibitory Concentration (IC50) | 30 nM | N.A. | N.A. | KB cell | CVCL_0372 | [102] |
| Half Maximal Inhibitory Concentration (IC50) | 30 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [101] |
| Half Maximal Inhibitory Concentration (IC50) | 30 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [101] |
| Half Maximal Inhibitory Concentration (IC50) | 32 nM | N.A. | N.A. | P388 cell | CVCL_7222 | [103] |
| Half Maximal Inhibitory Concentration (IC50) | 32 nM | N.A. | N.A. | U-937 cell | CVCL_0007 | [85] |
| Half Maximal Inhibitory Concentration (IC50) | 34 nM | N.A. | N.A. | CT26 cell | CVCL_7254 | [88] |
| Half Maximal Inhibitory Concentration (IC50) | 35 nM | N.A. | N.A. | KB cell | CVCL_0372 | [102] |
| Half Maximal Inhibitory Concentration (IC50) | 37 nM | N.A. | N.A. | KB cell | CVCL_0372 | [97] |
| Half Maximal Inhibitory Concentration (IC50) | 40 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [92] |
| Half Maximal Inhibitory Concentration (IC50) | 44 nM | N.A. | N.A. | HCC70 cell | CVCL_1270 | [104] |
| Half Maximal Inhibitory Concentration (IC50) | 45 nM | N.A. | N.A. | Col2 cell | CVCL_D645 | [89] |
| Half Maximal Inhibitory Concentration (IC50) | 50 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [104] |
| Half Maximal Inhibitory Concentration (IC50) | 56 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [105] |
| Half Maximal Inhibitory Concentration (IC50) | 57 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [62] |
| Half Maximal Inhibitory Concentration (IC50) | 58 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [106] |
| Half Maximal Inhibitory Concentration (IC50) | 60 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [107] |
| Half Maximal Inhibitory Concentration (IC50) | 60 nM | N.A. | N.A. | Huh-7 cell | CVCL_0336 | [78] |
| Half Maximal Inhibitory Concentration (IC50) | 65 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [108] |
| Half Maximal Inhibitory Concentration (IC50) | 67 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [109] |
| Half Maximal Inhibitory Concentration (IC50) | 67 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [110] |
| Half Maximal Inhibitory Concentration (IC50) | 69 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [111] |
| Half Maximal Inhibitory Concentration (IC50) | 70 nM | N.A. | N.A. | T-47D cell | CVCL_0553 | [112] |
| Half Maximal Inhibitory Concentration (IC50) | 70 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [113] |
| Half Maximal Inhibitory Concentration (IC50) | 70 nM | N.A. | N.A. | KB cell | CVCL_0372 | [102] |
| Half Maximal Inhibitory Concentration (IC50) | 70 nM | N.A. | N.A. | SF268 cell | CVCL_1689 | [114] |
| Half Maximal Inhibitory Concentration (IC50) | 71 nM | N.A. | N.A. | THP-1 cell | CVCL_0006 | [73] |
| Half Maximal Inhibitory Concentration (IC50) | 80 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [115] |
| Half Maximal Inhibitory Concentration (IC50) | 80 nM | N.A. | N.A. | T-47D cell | CVCL_0553 | [116] |
| Half Maximal Inhibitory Concentration (IC50) | 80 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [109] |
| Half Maximal Inhibitory Concentration (IC50) | 80 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [117] |
| Half Maximal Inhibitory Concentration (IC50) | 86.1 nM | N.A. | N.A. | LNCaP cell | CVCL_0395 | [96] |
| Half Maximal Inhibitory Concentration (IC50) | 87.5 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [118] |
| Half Maximal Inhibitory Concentration (IC50) | 88 nM | N.A. | N.A. | T24 cell | CVCL_0554 | [109] |
| Half Maximal Inhibitory Concentration (IC50) | 89 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [106] |
| Half Maximal Inhibitory Concentration (IC50) | 90 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [119] |
| Half Maximal Inhibitory Concentration (IC50) | 90 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [120] |
| Half Maximal Inhibitory Concentration (IC50) | 90 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [60] |
| Half Maximal Inhibitory Concentration (IC50) | 91 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [121] |
| Half Maximal Inhibitory Concentration (IC50) | 98 nM | N.A. | N.A. | SNU-638 cell | CVCL_0102 | [122] |
| Half Maximal Inhibitory Concentration (IC50) | 100 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [123] |
| Half Maximal Inhibitory Concentration (IC50) | 100 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [123] |
| Half Maximal Inhibitory Concentration (IC50) | <100 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [124] |
| Half Maximal Inhibitory Concentration (IC50) | <100 nM | N.A. | N.A. | Huh-7 cell | CVCL_0336 | [124] |
| Half Maximal Inhibitory Concentration (IC50) | <100 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [124] |
| Half Maximal Inhibitory Concentration (IC50) | 110 nM | N.A. | N.A. | Jurkat cell | CVCL_0065 | [125] |
| Half Maximal Inhibitory Concentration (IC50) | 120 nM | N.A. | N.A. | LX-1 cell | CVCL_5791 | [9] |
| Half Maximal Inhibitory Concentration (IC50) | 120 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [126] |
| Half Maximal Inhibitory Concentration (IC50) | 120 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [127] |
| Half Maximal Inhibitory Concentration (IC50) | 130 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [60] |
| Half Maximal Inhibitory Concentration (IC50) | 130 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [93] |
| Half Maximal Inhibitory Concentration (IC50) | 130 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [128] |
| Half Maximal Inhibitory Concentration (IC50) | 130 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [129] |
| Half Maximal Inhibitory Concentration (IC50) | 140 nM | N.A. | N.A. | A-172 cell | CVCL_0131 | [130] |
| Half Maximal Inhibitory Concentration (IC50) | 160 nM | N.A. | N.A. | SW948 cell | CVCL_0632 | [128] |
| Half Maximal Inhibitory Concentration (IC50) | 166 nM | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [121] |
| Half Maximal Inhibitory Concentration (IC50) | 170 nM | N.A. | N.A. | MDA-MB-361 cell | CVCL_0620 | [78] |
| Half Maximal Inhibitory Concentration (IC50) | 180 nM | N.A. | N.A. | PANC-1 cell | CVCL_0480 | [131] |
| Half Maximal Inhibitory Concentration (IC50) | 180 nM | N.A. | N.A. | MIA PaCa-2 cell | CVCL_0428 | [132] |
| Half Maximal Inhibitory Concentration (IC50) | 180 nM | N.A. | N.A. | L1210 cell | CVCL_0382 | [130] |
| Half Maximal Inhibitory Concentration (IC50) | 180 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [133] |
| Half Maximal Inhibitory Concentration (IC50) | 180 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [90] |
| Half Maximal Inhibitory Concentration (IC50) | 200 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [60] |
| Half Maximal Inhibitory Concentration (IC50) | 200 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [134] |
| Half Maximal Inhibitory Concentration (IC50) | 210 nM | N.A. | N.A. | BGC-823 cell | CVCL_3360 | [135] |
| Half Maximal Inhibitory Concentration (IC50) | 210 nM | N.A. | N.A. | SW480 cell | CVCL_0546 | [136] |
| Half Maximal Inhibitory Concentration (IC50) | 210 nM | N.A. | N.A. | DLD-1 cell | CVCL_0248 | [137] |
| Half Maximal Inhibitory Concentration (IC50) | 240 nM | N.A. | N.A. | BxPC-3 cell | CVCL_0186 | [60] |
| Half Maximal Inhibitory Concentration (IC50) | 260 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [138] |
| Half Maximal Inhibitory Concentration (IC50) | 260 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [126] |
| Half Maximal Inhibitory Concentration (IC50) | 260 nM | N.A. | N.A. | Bel-7402 cell | CVCL_5492 | [138] |
| Half Maximal Inhibitory Concentration (IC50) | 260 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [49] |
| Half Maximal Inhibitory Concentration (IC50) | 260 nM | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [139] |
| Half Maximal Inhibitory Concentration (IC50) | 280 nM | N.A. | N.A. | SNU-638 cell | CVCL_0102 | [140] |
| Half Maximal Inhibitory Concentration (IC50) | 290 nM | N.A. | N.A. | SW480 cell | CVCL_0546 | [141] |
| Half Maximal Inhibitory Concentration (IC50) | 291 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [69] |
| Half Maximal Inhibitory Concentration (IC50) | 300 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [142] |
| Half Maximal Inhibitory Concentration (IC50) | 304 nM | N.A. | N.A. | Renca cell | CVCL_2174 | [88] |
| Half Maximal Inhibitory Concentration (IC50) | 307 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [74] |
| Half Maximal Inhibitory Concentration (IC50) | 320 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [143] |
| Half Maximal Inhibitory Concentration (IC50) | 330 nM | N.A. | N.A. | L1210 cell | CVCL_0382 | [144] |
| Half Maximal Inhibitory Concentration (IC50) | 340 nM | N.A. | N.A. | CNE cell | CVCL_6888 | [145] |
| Half Maximal Inhibitory Concentration (IC50) | 370 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [146] |
| Half Maximal Inhibitory Concentration (IC50) | 400 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [147] |
| Half Maximal Inhibitory Concentration (IC50) | 400 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [94] |
| Half Maximal Inhibitory Concentration (IC50) | 400 nM | N.A. | N.A. | MIA PaCa-2 cell | CVCL_0428 | [148] |
| Half Maximal Inhibitory Concentration (IC50) | 400 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [149] |
| Half Maximal Inhibitory Concentration (IC50) | 400 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [150] |
| Half Maximal Inhibitory Concentration (IC50) | 420 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [125] |
| Half Maximal Inhibitory Concentration (IC50) | 430 nM | N.A. | N.A. | IMR-90 cell | CVCL_0347 | [114] |
| Half Maximal Inhibitory Concentration (IC50) | 450 nM | N.A. | N.A. | SMMC-7721 cell | CVCL_0534 | [141] |
| Half Maximal Inhibitory Concentration (IC50) | 490 nM | N.A. | N.A. | T-47D cell | CVCL_0553 | [93] |
| Half Maximal Inhibitory Concentration (IC50) | 500 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [151] |
| Half Maximal Inhibitory Concentration (IC50) | 500 nM | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [152] |
| Half Maximal Inhibitory Concentration (IC50) | 520 nM | N.A. | N.A. | CA46 cell | CVCL_1101 | [153] |
| Half Maximal Inhibitory Concentration (IC50) | 540 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [154] |
| Half Maximal Inhibitory Concentration (IC50) | 580 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [110] |
| Half Maximal Inhibitory Concentration (IC50) | 590 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [155] |
| Half Maximal Inhibitory Concentration (IC50) | 600 nM | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [119] |
| Half Maximal Inhibitory Concentration (IC50) | 600 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [73] |
| Half Maximal Inhibitory Concentration (IC50) | 610 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [155] |
| Half Maximal Inhibitory Concentration (IC50) | 630 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [107] |
| Half Maximal Inhibitory Concentration (IC50) | 660 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [107] |
| Half Maximal Inhibitory Concentration (IC50) | 680 nM | N.A. | N.A. | U-937 cell | CVCL_0007 | [85] |
| Half Maximal Inhibitory Concentration (IC50) | 700 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [156] |
| Half Maximal Inhibitory Concentration (IC50) | 711 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [94] |
| Half Maximal Inhibitory Concentration (IC50) | 760 nM | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [107] |
| Half Maximal Inhibitory Concentration (IC50) | 800 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [150] |
| Half Maximal Inhibitory Concentration (IC50) | 800 nM | N.A. | N.A. | NCI-H1299 cell | CVCL_0060 | [156] |
| Half Maximal Inhibitory Concentration (IC50) | 800 nM | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [155] |
| Half Maximal Inhibitory Concentration (IC50) | 800 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [157] |
| Half Maximal Inhibitory Concentration (IC50) | 850 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [108] |
| Half Maximal Inhibitory Concentration (IC50) | 860 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [108] |
| Half Maximal Inhibitory Concentration (IC50) | 890 nM | N.A. | N.A. | MRC5 cell | CVCL_0440 | [100] |
| Half Maximal Inhibitory Concentration (IC50) | 940 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [158] |
| Half Maximal Inhibitory Concentration (IC50) | 950 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [159] |
| Half Maximal Inhibitory Concentration (IC50) | 980 nM | N.A. | N.A. | CNE-2 cell | CVCL_6889 | [145] |
| Half Maximal Inhibitory Concentration (IC50) | 1000 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [110] |
| Half Maximal Inhibitory Concentration (IC50) | >1000 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [49] |
| Half Maximal Inhibitory Concentration (IC50) | <1000 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [160] |
| Half Maximal Inhibitory Concentration (IC50) | 1000 nM | N.A. | N.A. | Raji cell | CVCL_0511 | [161] |
| Half Maximal Inhibitory Concentration (IC50) | <1000 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [162] |
| Half Maximal Inhibitory Concentration (IC50) | 1000 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [82] |
| Half Maximal Inhibitory Concentration (IC50) | 1.01 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [163] |
| Half Maximal Inhibitory Concentration (IC50) | 1.04 uM | N.A. | N.A. | KB cell | CVCL_0372 | [102] |
| Half Maximal Inhibitory Concentration (IC50) | 1.07 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [110] |
| Half Maximal Inhibitory Concentration (IC50) | 1.08 uM | N.A. | N.A. | SMMC-7721 cell | CVCL_0534 | [145] |
| Half Maximal Inhibitory Concentration (IC50) | 1.08 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [164] |
| Half Maximal Inhibitory Concentration (IC50) | 1.1 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [92] |
| Half Maximal Inhibitory Concentration (IC50) | 1.1 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [165] |
| Half Maximal Inhibitory Concentration (IC50) | 1.18 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [163] |
| Half Maximal Inhibitory Concentration (IC50) | 1.22 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [166] |
| Half Maximal Inhibitory Concentration (IC50) | 1.22 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [166] |
| Half Maximal Inhibitory Concentration (IC50) | 1.37 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [112] |
| Half Maximal Inhibitory Concentration (IC50) | 1.4 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [167] |
| Half Maximal Inhibitory Concentration (IC50) | 1.4144 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [168] |
| Half Maximal Inhibitory Concentration (IC50) | 1.46 uM | N.A. | N.A. | DU145 cell | CVCL_0105 | [169] |
| Half Maximal Inhibitory Concentration (IC50) | 1.59 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [170] |
| Half Maximal Inhibitory Concentration (IC50) | 1.6 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [153] |
| Half Maximal Inhibitory Concentration (IC50) | 1.61 uM | N.A. | N.A. | HEK293 cell | CVCL_0045 | [171] |
| Half Maximal Inhibitory Concentration (IC50) | 1.62 uM | N.A. | N.A. | T-47D cell | CVCL_0553 | [170] |
| Half Maximal Inhibitory Concentration (IC50) | 1.7 uM | N.A. | N.A. | MX1 cell | CVCL_4774 | [172] |
| Half Maximal Inhibitory Concentration (IC50) | 1.8 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [173] |
| Half Maximal Inhibitory Concentration (IC50) | 1.84 uM | N.A. | N.A. | T-47D cell | CVCL_0553 | [174] |
| Half Maximal Inhibitory Concentration (IC50) | 1.88 uM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [171] |
| Half Maximal Inhibitory Concentration (IC50) | 1.98 uM | N.A. | N.A. | U-937 cell | CVCL_0007 | [73] |
| Half Maximal Inhibitory Concentration (IC50) | 2 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [142] |
| Half Maximal Inhibitory Concentration (IC50) | 2.21 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [175] |
| Half Maximal Inhibitory Concentration (IC50) | 2.3 uM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [165] |
| Half Maximal Inhibitory Concentration (IC50) | 2.31 uM | N.A. | N.A. | T-47D cell | CVCL_0553 | [176] |
| Half Maximal Inhibitory Concentration (IC50) | 2.5 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [120] |
| Half Maximal Inhibitory Concentration (IC50) | 2.53 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [167] |
| Half Maximal Inhibitory Concentration (IC50) | 2.544 uM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [121] |
| Half Maximal Inhibitory Concentration (IC50) | 2.7 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [173] |
| Half Maximal Inhibitory Concentration (IC50) | 3.04 uM | N.A. | N.A. | HEp-2 cell | CVCL_1906 | [177] |
| Half Maximal Inhibitory Concentration (IC50) | 3.09 uM | N.A. | N.A. | T24 cell | CVCL_0554 | [178] |
| Half Maximal Inhibitory Concentration (IC50) | 3.1 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [179] |
| Half Maximal Inhibitory Concentration (IC50) | 3.1 uM | N.A. | N.A. | LoVo cell | CVCL_0399 | [180] |
| Half Maximal Inhibitory Concentration (IC50) | 3.2 uM | N.A. | N.A. | DU145 cell | CVCL_0105 | [165] |
| Half Maximal Inhibitory Concentration (IC50) | 3.57 uM | N.A. | N.A. | DU145 cell | CVCL_0105 | [108] |
| Half Maximal Inhibitory Concentration (IC50) | 3.6 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [181] |
| Half Maximal Inhibitory Concentration (IC50) | 3.8 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [182] |
| Half Maximal Inhibitory Concentration (IC50) | 3.97 uM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [164] |
| Half Maximal Inhibitory Concentration (IC50) | 4.16 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [82] |
| Half Maximal Inhibitory Concentration (IC50) | 4.2 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [165] |
| Half Maximal Inhibitory Concentration (IC50) | 4.79 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [183] |
| Half Maximal Inhibitory Concentration (IC50) | 5.5 uM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [184] |
| Half Maximal Inhibitory Concentration (IC50) | 5.5 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [185] |
| Half Maximal Inhibitory Concentration (IC50) | 5.93 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [177] |
| Half Maximal Inhibitory Concentration (IC50) | 6.08 uM | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [170] |
| Half Maximal Inhibitory Concentration (IC50) | 6.4 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [186] |
| Half Maximal Inhibitory Concentration (IC50) | 6.96 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [187] |
| Half Maximal Inhibitory Concentration (IC50) | 7.42 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [169] |
| Half Maximal Inhibitory Concentration (IC50) | 7.94 uM | N.A. | N.A. | HEK293 cell | CVCL_0045 | [188] |
| Half Maximal Inhibitory Concentration (IC50) | 8.81 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [133] |
| Half Maximal Inhibitory Concentration (IC50) | 8.87 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [189] |
| Half Maximal Inhibitory Concentration (IC50) | 9.17 uM | N.A. | N.A. | T-47D cell | CVCL_0553 | [188] |
| Half Maximal Inhibitory Concentration (IC50) | 9.29 uM | N.A. | N.A. | DU145 cell | CVCL_0105 | [188] |
| Half Maximal Inhibitory Concentration (IC50) | 9.55 uM | N.A. | N.A. | Caco-2 cell | CVCL_0025 | [190] |
| Half Maximal Inhibitory Concentration (IC50) | 9.92 uM | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [188] |
| Half Maximal Inhibitory Concentration (IC50) | 10 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [142] |
| Half Maximal Inhibitory Concentration (IC50) | 10.1 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [191] |
| Half Maximal Inhibitory Concentration (IC50) | 11.17 uM | N.A. | N.A. | T-47D cell | CVCL_0553 | [192] |
| Half Maximal Inhibitory Concentration (IC50) | 12.5 uM | N.A. | N.A. | Bel-7402 cell | CVCL_5492 | [193] |
| Half Maximal Inhibitory Concentration (IC50) | 13.62 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [189] |
| Half Maximal Inhibitory Concentration (IC50) | 13.64 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [191] |
| Half Maximal Inhibitory Concentration (IC50) | 14 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [153] |
| Half Maximal Inhibitory Concentration (IC50) | 14.2 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [165] |
| Half Maximal Inhibitory Concentration (IC50) | 14.76 uM | N.A. | N.A. | MDA-MB-468 cell | CVCL_0419 | [194] |
| Half Maximal Inhibitory Concentration (IC50) | 18.24 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [175] |
| Half Maximal Inhibitory Concentration (IC50) | 23.76 uM | N.A. | N.A. | AGS cell | CVCL_0139 | [195] |
| Half Maximal Inhibitory Concentration (IC50) | >40 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [94] |
| Half Maximal Inhibitory Concentration (IC50) | >40 uM | N.A. | N.A. | BJ cell | CVCL_E483 | [94] |
| Half Maximal Inhibitory Concentration (IC50) | >40 uM | N.A. | N.A. | TERT-RPE1 cell | CVCL_4388 | [94] |
| Half Maximal Inhibitory Concentration (IC50) | 51.5 uM | N.A. | N.A. | Jurkat cell | CVCL_0065 | [196] |
| Half Maximal Inhibitory Concentration (IC50) | 60.01 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [197] |
| Half Maximal Inhibitory Concentration (IC50) | 130 uM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [198] |
| Half Maximal Lethal Concentration (IC50) | 31.62 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [199] |
| Half Maximal Lethal Concentration (IC50) | >100 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [200] |
| Half Maximal Lethal Concentration (IC50) | >100 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [199] |
| Tumor Growth Inhibition value (TGI) | 160 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [200] |
| Tumor Growth Inhibition value (TGI) | 31.62 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [199] |
| Tumor Growth Inhibition value (TGI) | 50.12 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [199] |
Each Peptide-drug Conjugate Related to This Drug
Full Information of The Activity Data of The PDC(s) Related to This Drug
PDC-Z8 [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) | 60% (Day 14) | |||
| Administration Time | Injected via tail vein every t hree days | ||||
| Administration Dosage | 8 µmol/kg | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To study the in vivo anti-tumor activity of Z8, SK-BR-3 tumor bearing nude mice were administered once every three days. As shown in Fig. 11A-11D, CPT and Z8 demonstrated significant inhibitory effects on SK-BR-3 tumor growth, with the Z8 group exhibiting more pronounced inhibition compared to CPT. Notably, the average tumor volume and weight in the Z8 group were the smallest among all groups, including the control and CPT groups. Moreover, to further study the toxicity of CPT and Z8, serums of mice were collected for blood biochemical analysis. As shown in Fig. 11E-H, treatment with both CPT and Z8 resulted in increased levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST), but the effect of Z8 group was relatively low, indicating that Z8 was safer than CPT in vivo. Furthermore, in CPT group, urea nitrogen levels increased significantly, whereas no significant change was observed in the Z8 group, indicating potential renal function impairment following CPT treatment, whereas Z8 showed better safety in this regard. Additionally, the levels of alkaline phosphatase (ALP), total protein (TP), albumin (ALB), globulin (GLOB) and creatinine (CREA) did not significantly differ between the control group and the treatment groups. Finally, compared to the control group, histological examination using H&E staining (Hematoxylin and Eosin staining) of the major organs in the treatment group did not reveal obvious cell necrosis and inflammatory cell infiltration, indicating that Z8 would not bring additional toxicity.
Click to Show/Hide
|
||||
| In Vivo Model | SK-BR-3 tumor-bearing nude mice xenograft model. | ||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.04 ± 0.24 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.91 ± 0.71 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 13.49 ± 3.59 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 26.34 ± 1.08 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Apoptosis rate | 27.10% | |||
| Evaluation Method | Flow cytometry assay | ||||
| Administration Time | 24 h | ||||
| Administration Dosage | 1 µM | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
The proapoptotic activity of Z8 was studied by flow cytometry combined with Annexin V-FITC/PI double staining. As shown in Fig. 4, after treatment with 1 uM Z8, the apoptosis rate of SK-BR-3 cells was approximately 27.1 % which increased to 41.1 % following treatment with 2 uM Z8 (4.4 % in the blank control group and 55.8 % in CPT group. These findings suggest that Z8 can significantly induce apoptosis in a dose-dependent manner.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Apoptosis rate | 41.10% | |||
| Evaluation Method | Flow cytometry assay | ||||
| Administration Time | 24 h | ||||
| Administration Dosage | 2 µM | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
The proapoptotic activity of Z8 was studied by flow cytometry combined with Annexin V-FITC/PI double staining. As shown in Fig. 4, after treatment with 1 uM Z8, the apoptosis rate of SK-BR-3 cells was approximately 27.1 % which increased to 41.1 % following treatment with 2 uM Z8 (4.4 % in the blank control group and 55.8 % in CPT group. These findings suggest that Z8 can significantly induce apoptosis in a dose-dependent manner.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
diCPTiRGD [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [201] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Tumer volume | 0 mm3 | |||
| Administration Time | 30 days | ||||
| MOA of PDC |
In this context, we developed a drug-bearing supramolecular hydrogel system to intratumourally (i.t.) deliver CDNs against malignant tumours to achieve cancer chemoimmunotherapy. Our strategy was to chemically conjugate the hydrophilic pep-tide moiety iRGD (a tumour-penetrating peptide that can bind to neuropilin-1 (NRP-1) and trigger tumour tissue penetration) to the hydrophobic anticancer drug CPT to form a self-assembling and self-formulating peptide-drug conjugate (diCPT-iRGD). In aqueous solution, the designed drug amphiphile spontaneously assembles into supramolecular nanotubes (NTs). The negatively charged STING agonist (c-di-AMP (CDA)) can condense on the surface of these positively charged NTs through electrostatic complexations. After injection into the tumour site, the CDA-NT solution can immediately form a hydrogel, functioning as a local reservoir for extended localized release of CDA and CPT to awaken both the innate and adaptive immune systems.
Click to Show/Hide
|
||||
| Description |
When using the diCPT-iRGD NT hydrogel, the CDA-NT treatment led to substantial tumour regression (Fig. 3c-e) and demonstrated a 100% survival rate in mice (Fig. 3f).
|
||||
| In Vivo Model | GL-261 brain cancer C57BL/6 mice. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [201] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Survival rate | 100.00% | |||
| Administration Time | 30 days | ||||
| MOA of PDC |
In this context, we developed a drug-bearing supramolecular hydrogel system to intratumourally (i.t.) deliver CDNs against malignant tumours to achieve cancer chemoimmunotherapy. Our strategy was to chemically conjugate the hydrophilic pep-tide moiety iRGD (a tumour-penetrating peptide that can bind to neuropilin-1 (NRP-1) and trigger tumour tissue penetration) to the hydrophobic anticancer drug CPT to form a self-assembling and self-formulating peptide-drug conjugate (diCPT-iRGD). In aqueous solution, the designed drug amphiphile spontaneously assembles into supramolecular nanotubes (NTs). The negatively charged STING agonist (c-di-AMP (CDA)) can condense on the surface of these positively charged NTs through electrostatic complexations. After injection into the tumour site, the CDA-NT solution can immediately form a hydrogel, functioning as a local reservoir for extended localized release of CDA and CPT to awaken both the innate and adaptive immune systems.
Click to Show/Hide
|
||||
| Description |
When using the diCPT-iRGD NT hydrogel, the CDA-NT treatment led to substantial tumour regression (Fig. 3c-e) and demonstrated a 100% survival rate in mice (Fig. 3f).
|
||||
| In Vivo Model | GL-261 brain cancer C57BL/6 mice. | ||||
JF-10-71 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [202], [203] | ||||
| Indication | Neuroblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 1363 nM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 3 days | ||||
| MOA of PDC |
In a previous study, we used a potent somatostatin analog (SSA) with high affinity for SSTR2 for attachment to an antisense peptide nucleic acid (PNA) directed against the n-myc oncogene and showed that PNA-SSA conjugates produced receptor-specific inhibition of cell growth. Cleavable-linker chemistry was then developed that allowed this approach to be extended to well-known cytotoxic compounds such as camptothecin and combretastatin. In the present report two CPT-SSA conjugates, JF-10-71 and JF-10-81, displaying differing stabilities were chosen to potentially treat SSTR2-positive rat pancreatic CA20948 tumors in Lewis rats and SCLC NCI-H69 tumors in athymic nude mice.
Click to Show/Hide
|
||||
| Description |
Previously, a series of CPT-SSA conjugates were tested for their stability in phosphate buffer/rat plasma and in vitro inhibitory activity in tumor cell growth (data not shown). Two of these conjugates, JF-10-71 and JF-10-81, displaying higher and lower stability, respectively, were chosen for further experiments in serial tumor cell lines. Also, free CPT and SSA itself were tested as controls. The results (Table 1) show that the IC50 values increased compared with CPT to JF-10-81 and further to JF-10-71 with the exception of the CA20948 cells. CPT and both conjugates were particularly effective in SSTR2-overexpressing IMR32 cells displaying IC50 values 3.1, 64.13, and 282.50 nM, respectively. SSTR2-overexpressing CA20948 cells were poorly responsive to CPT itself (IC50, 3077 nM); however, its somatostatin conjugates were actually more potent (IC50: JF-10-81, 1790 nM; JF-10-71, 1363 nM). SSA itself exhibited little effect on growth of all tested cell lines even at doses up to 10-5 M.
Click to Show/Hide
|
||||
JF-10-81 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [202], [203] | ||||
| Indication | Neuroblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 1790 nM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 3 days | ||||
| MOA of PDC |
In a previous study, we used a potent somatostatin analog (SSA) with high affinity for SSTR2 for attachment to an antisense peptide nucleic acid (PNA) directed against the n-myc oncogene and showed that PNA-SSA conjugates produced receptor-specific inhibition of cell growth. Cleavable-linker chemistry was then developed that allowed this approach to be extended to well-known cytotoxic compounds such as camptothecin and combretastatin. In the present report two CPT-SSA conjugates, JF-10-71 and JF-10-81, displaying differing stabilities were chosen to potentially treat SSTR2-positive rat pancreatic CA20948 tumors in Lewis rats and SCLC NCI-H69 tumors in athymic nude mice.
Click to Show/Hide
|
||||
| Description |
Previously, a series of CPT-SSA conjugates were tested for their stability in phosphate buffer/rat plasma and in vitro inhibitory activity in tumor cell growth (data not shown). Two of these conjugates, JF-10-71 and JF-10-81, displaying higher and lower stability, respectively, were chosen for further experiments in serial tumor cell lines. Also, free CPT and SSA itself were tested as controls. The results (Table 1) show that the IC50 values increased compared with CPT to JF-10-81 and further to JF-10-71 with the exception of the CA20948 cells. CPT and both conjugates were particularly effective in SSTR2-overexpressing IMR32 cells displaying IC50 values 3.1, 64.13, and 282.50 nM, respectively. SSTR2-overexpressing CA20948 cells were poorly responsive to CPT itself (IC50, 3077 nM); however, its somatostatin conjugates were actually more potent (IC50: JF-10-81, 1790 nM; JF-10-71, 1363 nM). SSA itself exhibited little effect on growth of all tested cell lines even at doses up to 10-5 M.
Click to Show/Hide
|
||||
Kb-CC07 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.11 ± 0.01 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT 116 cell | CVCL_0291 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.23 ± 0.01 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT-116/CPT cell | CVCL_0291 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.33 ± 0.02 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric carcinoma | SGC-7901 cell | CVCL_0520 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.42 ± 0.01 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.80 ± 0.08 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Human papillomavirus-related cervical adenocarcinoma | SGC-7901/CPT cell | CVCL_0520 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.01 ± 0.05 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma of no special type | MCF7/C4 cell | CVCL_GX99 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 13.7 ± 1.1 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | NCM460 cell | CVCL_0460 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Liver cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 15.5 ± 1.0 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | LO #2 cell | CVCL_C7SD | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 17.3 ± 0.8 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | GES1 cell | CVCL_EQ22 | ||
I-4 (NPNWGRSWYNQRFKGC=(-SS-O-COO-CPT)GC=(-SS-O-COO-CPT)GRIKPRKGYTR) [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.12 ± 0.07 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.27 ± 0.38 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.78 ± 0.47 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 7.72 ± 0.92 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 17.14 ± 2.42 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
Kb-CC04 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.14 ± 0.02 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT 116 cell | CVCL_0291 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.32 ± 0.02 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT-116/CPT cell | CVCL_0291 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.38 ± 0.01 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric carcinoma | SGC-7901 cell | CVCL_0520 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.43 ± 0.02 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.00 ± 0.08 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Human papillomavirus-related cervical adenocarcinoma | SGC-7901/CPT cell | CVCL_0520 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.05 ± 0.03 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma of no special type | MCF7/C4 cell | CVCL_GX99 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 13.4 ± 0.4 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | NCM460 cell | CVCL_0460 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 15.5 ± 0.6 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | GES1 cell | CVCL_EQ22 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Liver cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 16.4 ± 0.2 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | LO #2 cell | CVCL_C7SD | ||
Kb-CC08 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.14 ± 0.02 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT 116 cell | CVCL_0291 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.35 ± 0.02 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT-116/CPT cell | CVCL_0291 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.41 ± 0.02 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.47 ± 0.01 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric carcinoma | SGC-7901 cell | CVCL_0520 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.03 ± 0.06 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma of no special type | MCF7/C4 cell | CVCL_GX99 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.06 ± 0.07 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Human papillomavirus-related cervical adenocarcinoma | SGC-7901/CPT cell | CVCL_0520 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 14.8 ± 0.9 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | NCM460 cell | CVCL_0460 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 15.2 ± 0.7 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | GES1 cell | CVCL_EQ22 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Liver cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 16.1 ± 0.5 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | LO #2 cell | CVCL_C7SD | ||
Kb-CC03 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.15 ± 0.03 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT 116 cell | CVCL_0291 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.35 ± 0.03 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT-116/CPT cell | CVCL_0291 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.42 ± 0.01 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.46 ± 0.02 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric carcinoma | SGC-7901 cell | CVCL_0520 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.03 ± 0.03 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma of no special type | MCF7/C4 cell | CVCL_GX99 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.19 ± 0.06 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Human papillomavirus-related cervical adenocarcinoma | SGC-7901/CPT cell | CVCL_0520 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 13.3 ± 1.0 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | NCM460 cell | CVCL_0460 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 14.9 ± 0.9 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | GES1 cell | CVCL_EQ22 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Liver cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 17.3 ± 0.8 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | LO #2 cell | CVCL_C7SD | ||
Kb-CC02 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.17 ± 0.02 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT 116 cell | CVCL_0291 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.47 ± 0.01 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT-116/CPT cell | CVCL_0291 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.50 ± 0.01 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.51 ± 0.03 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric carcinoma | SGC-7901 cell | CVCL_0520 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.23 ± 0.10 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Human papillomavirus-related cervical adenocarcinoma | SGC-7901/CPT cell | CVCL_0520 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.26 ± 0.11 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma of no special type | MCF7/C4 cell | CVCL_GX99 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 14.4 ± 0.5 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | GES1 cell | CVCL_EQ22 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 15.4 ± 0.2 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | NCM460 cell | CVCL_0460 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Liver cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 15.4 ± 0.5 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | LO #2 cell | CVCL_C7SD | ||
Kb-CC01 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.18 ± 0.01 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT 116 cell | CVCL_0291 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.45 ± 0.02 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT-116/CPT cell | CVCL_0291 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.57 ± 0.02 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric carcinoma | SGC-7901 cell | CVCL_0520 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.59 ± 0.02 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.34 ± 0.12 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Human papillomavirus-related cervical adenocarcinoma | SGC-7901/CPT cell | CVCL_0520 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.48 ± 0.18 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma of no special type | MCF7/C4 cell | CVCL_GX99 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 14.0 ± 0.5 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | NCM460 cell | CVCL_0460 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Liver cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 15.0 ± 0.8 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | LO #2 cell | CVCL_C7SD | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 15.5 ± 0.5 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | GES1 cell | CVCL_EQ22 | ||
Kb-CC06 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.18 ± 0.02 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT 116 cell | CVCL_0291 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.41 ± 0.02 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric carcinoma | SGC-7901 cell | CVCL_0520 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.44 ± 0.01 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT-116/CPT cell | CVCL_0291 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.51 ± 0.04 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.02 ± 0.11 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Human papillomavirus-related cervical adenocarcinoma | SGC-7901/CPT cell | CVCL_0520 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.20 ± 0.13 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma of no special type | MCF7/C4 cell | CVCL_GX99 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 15.1 ± 0.4 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | NCM460 cell | CVCL_0460 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 16.0 ± 0.8 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | GES1 cell | CVCL_EQ22 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Liver cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 16.3 ± 0.4 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | LO #2 cell | CVCL_C7SD | ||
Kb-CC05 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.19 ± 0.02 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT 116 cell | CVCL_0291 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.46 ± 0.02 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT-116/CPT cell | CVCL_0291 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.51 ± 0.02 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.54 ± 0.03 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric carcinoma | SGC-7901 cell | CVCL_0520 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.18 ± 0.10 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma of no special type | MCF7/C4 cell | CVCL_GX99 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.20 ± 0.10 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Human papillomavirus-related cervical adenocarcinoma | SGC-7901/CPT cell | CVCL_0520 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Liver cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 15.6 ± 0.2 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | LO #2 cell | CVCL_C7SD | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 16.0 ± 0.5 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | GES1 cell | CVCL_EQ22 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 16.2 ± 1.0 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Cationic anticancer peptides (CAPs) refer to peptide-based molecules possessing a net positive charge, allowing them to preferentially combined with cancer cells which exhibit an overabundance of anionic components on their surface. Cationic peptides additionally induce cell membrane destabilization through carpet-like, toroidal pore or sinking raft et al. mechanism, or penetrate cell membrane and act on cellular sub-structures. Besides, cell-penetrating peptides (CPPs) represent a special subset of CAPs, renowned for their rapid and low-toxic cellular translocation abilities, making them promising vehicles for drug delivery. Therefore, CAPs are emerging as a class of potential anticancer agents and gaining widespread attention by researchers. As reported, the construction of bioactive peptides with cytotoxic payloads via a chemical linker provides a kind of prodrugs known as peptide-drug conjugates (PDCs), which represents an efficient approach to address the limitations of small molecules by leveraging the properties of peptides. In previous study, we have discovered the CAP of KM8-Aib (sequence: Lys-Leu-Leu-Lys-Lys-Asn-Leu-Lys-Aib-Leu-Ala-Ala-Leu-Ala-Lys-Lys-Ile-Leu-NH2), which derived from venom peptide Mastoparan, exhibiting favorable antiproliferative effect against several cancer cells but less toxic to benign cells. Herein, we attempted to integrate CPT with KM8-Aib, obtaining a series of peptide-CPT conjugates, to improve the solubility, selectivity of CPT, and achieve enhanced therapeutic outcomes. Based on preliminary in vitro assays, the PDC Kb-CC07 demonstrated exceptional selective cytotoxicity against various cancer cell lines, including drug-resistant variants, surpassing that of other conjugates. The solubility of Kb-CC07 was also significantly increased, which is ˜ 100 fold higher than that of CPT. In addition, we evaluated the in vivo antitumor efficacy of Kb-CC07 via nude-xenograft model. It is showed that mice treated with Kb-CC07 had substantially lower tumor volumes and minimum organ toxicities than CPT-treated group. As such, Kb-CC07 is suggested to be a promising anticancer agent and warrant thorough exploration.
Click to Show/Hide
|
||||
| Description |
MTT assay was preformed to measure the in vitro cytotoxicities of the compounds against cancer cells (HCT-116, SGC-7901, MCF-7, HCT-116/CPT, SGC-7901/CPT and MCF-7/C4) and benign cells (NCM460, GES-1 and LO2). As present in Table 1, all the peptides (Kb-C01, 0207) exhibited certain selective anticancer activities, which are comparable to their parent counterpart KM8-Aib. The target PDCs (Kb-CC01, 0207) were further obtained by coupling the peptides with CPT, displaying markedly improved antiproliferative activities and selectivities against cancer cell lines over CPT. Studies have revealed that cancer cell membranes are negatively charged due to the presence of substantial amounts of acid phospholipids. Therefore, positively charged CAPs are capable of binding to cancer cell surface through electrostatic interactions, making them specific to cancer cells. In this project, since the PDCs were composed of CAP KM8-Aib and CPT, they potentially inherited the antineoplastic activities of KM8-Aib against cancer cells, as well as low toxicities to non-malignant cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | NCM460 cell | CVCL_0460 | ||
PDC-Z11 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.41 ± 0.13 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 3.93 ± 1.41 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 10.25 ± 1.34 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 12.93 ± 1.15 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
SMAC-P1 CPT conjugates 4 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.53 ± 0.17 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.58 ± 0.41 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 2.50 ± 0.78 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 17.76 ± 1.35 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 18.44 ± 2.09 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
SMAC-P1 CPT conjugates 7 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.58 ± 0.21 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 3.86 ± 1.84 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 4.73 ± 1.55 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 6.17 ± 1.21 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 15.15 ± 1.44 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
SMAC-P1 CPT conjugates 1 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.85 ± 0.23 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 3.04 ± 0.75 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 4.03 ± 0.25 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 6.43 ± 1.20 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 18.23 ± 2.06 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
PDC-Z4 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.37 ± 0.42 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 3.45 ± 1.09 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 13.91 ± 2.61 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 28.25 ± 3.12 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
PDC-Z5 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.44 ± 0.63 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 7.83 ± 1.77 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 17.84 ± 3.14 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 20.36 ± 1.82 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
I-1 (NPNWGRSWYNQRFKGC=(-SS-O-COO-CPT)GRIKPRKGYTR) [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.47 ± 0.54 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 3.29 ± 0.67 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 4.29 ± 1.11 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 7.60 ± 1.23 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 23.90 ± 1.58 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
I-5 (NPNWGRSWYNQRFKGC=(-SS-COO-CPT)GC=(-SS-COO-CPT)GRIKPRKGYTR) [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.49 ± 0.58 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 4.12 ± 0.87 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 7.37 ± 1.18 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 11.51 ± 1.37 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 18.25 ± 1.76 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
PDC-Z12 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.76 ± 0.33 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 6.12 ± 0.45 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 13.21 ± 1.15 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 15.25 ± 2.08 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
SMAC-P1 CPT conjugates 5 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.78 ± 0.35 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 5.81 ± 1.08 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 6.53 ± 1.02 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 16.24 ± 1.04 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 22.12 ± 1.83 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
SMAC-P1 CPT conjugates 8 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 2.30 ± 0.48 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 4.73 ± 1.33 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 8.01 ± 1.34 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 15.11 ± 1.80 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 28.36 ± 2.12 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
PDC-Z2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 2.37 ± 0.32 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 7.25 ± 1.75 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 19.10 ± 2.94 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 24.45 ± 2.04 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
SMAC-P1 CPT conjugates 2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 2.59 ± 0.40 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 5.93 ± 0.46 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 8.24 ± 0.57 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 10.64 ± 1.32 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 15.13 ± 1.24 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
PDC-Z3 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 3.47 ± 0.67 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 5.51 ± 1.74 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 30.12 ± 1.38 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 32.44 ± 5.49 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
PDC-Z10 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 3.53 ± 0.53 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 17.25 ± 2.31 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 22.44 ± 2.21 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 25.12 ± 2.04 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
PDC-Z1 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 4.96 ± 0.98 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 17.94 ± 2.78 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 27.24 ± 1.73 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 38.72 ± 4.62 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
I-2 (NPNWGRSWYNQRFKGC=(-SS-COO-CPT)GRIKPRKGYTR) [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 5.76 ± 0.79 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 6.25 ± 0.96 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 6.42 ± 1.21 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 10.93 ± 1.14 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 20.41 ± 1.04 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
PDC-Z7 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 5.96 ± 2.41 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 10.23 ± 1.65 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 15.44 ± 2.41 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 33.12 ± 3.84 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
I-6 (NPNWGRSWYNQRFKGC=(-S-Maleimide-CPT)GC=(-S-Maleimide-CPT)GRIKPRKGYTR) [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 6.72 ± 1.25 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 10.91 ± 1.23 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 20.35 ± 1.21 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 25.16 ± 2.12 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 28.24 ± 1.36 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
PDC-Z6 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 11.02 ± 1.78 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
PDC-Z9 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 11.63 ± 2.35 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
SMAC-P1 CPT conjugates 9 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 12.58 ± 2.45 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 24.15 ± 3.76 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 31.35 ± 3.17 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
I-3 (NPNWGRSWYNQRFKGC=(-S-Maleimide-CPT)GRIKPRKGYTR) [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 15.32 ± 2.25 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 23.09 ± 1.77 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 24.39 ± 1.45 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 25.45 ± 0.64 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 35.20 ± 2.64 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
SMAC-P1 CPT conjugates 6 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 15.68 ± 1.14 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 20.14 ± 2.12 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 28.21 ± 1.28 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
SMAC-P1 CPT conjugates 3 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 18.34 ± 1.42 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 25.31 ± 1.48 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 30.58 ± 2.76 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
CPT-Cyclo-GCGPep Conjugate 1 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [204] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell viability | 33.00% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 10 μM | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [204] | ||||
| Indication | Gastric tubular adenocarcinoma | ||||
| Efficacy Data | Cell viability | 44.00% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 10 μM | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [204] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell viability | 53.00% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 2.5 μM | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [204] | ||||
| Indication | Gastric tubular adenocarcinoma | ||||
| Efficacy Data | Cell viability | 53.00% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 2.5 μM | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [204] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell viability | 61.00% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 0.625 μM | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [204] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell viability | 76.00% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 0.156 μM | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [204] | ||||
| Indication | Gastric tubular adenocarcinoma | ||||
| Efficacy Data | Cell viability | 82.00% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 0.625 μM | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [204] | ||||
| Indication | Gastric tubular adenocarcinoma | ||||
| Efficacy Data | Cell viability | 85.00% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 0.156 μM | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
PDC-CPT1 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [205] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 38% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 5 µM | ||||
| Description |
Antiproliferative results showed that PTX1 inhibited cell proliferation by 18.7%. The anti-proliferative activity of CPT1 was diminished by 1.9-fold as compared to CPT whereas the activity of CPT2 was comparable to CPT, since CPT2 reduced the cell viability to 61%.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
CPT-Cyclo-GCGPep Conjugate 2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [204] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell viability | 45.00% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 10 μM | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [204] | ||||
| Indication | Gastric tubular adenocarcinoma | ||||
| Efficacy Data | Cell viability | 45.00% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 10 μM | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [204] | ||||
| Indication | Gastric tubular adenocarcinoma | ||||
| Efficacy Data | Cell viability | 62.00% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 2.5 μM | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [204] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell viability | 69.00% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 2.5 μM | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [204] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell viability | 78.00% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 0.625 μM | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [204] | ||||
| Indication | Gastric tubular adenocarcinoma | ||||
| Efficacy Data | Cell viability | 80.00% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 0.625 μM | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [204] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell viability | 85.00% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 0.156 μM | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [204] | ||||
| Indication | Gastric tubular adenocarcinoma | ||||
| Efficacy Data | Cell viability | 100.00% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 0.156 μM | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
CPT-Cyclo-GCGPep Conjugate 3 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [204] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell viability | 70.00% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 10 μM | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [204] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell viability | 90.00% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 2.5 μM | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [204] | ||||
| Indication | Gastric tubular adenocarcinoma | ||||
| Efficacy Data | Cell viability | 90.00% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 10 μM | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [204] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell viability | 98.00% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 0.156 μM | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [204] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell viability | 99.00% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 0.625 μM | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [204] | ||||
| Indication | Gastric tubular adenocarcinoma | ||||
| Efficacy Data | Cell viability | 100.00% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 0.156 μM | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [204] | ||||
| Indication | Gastric tubular adenocarcinoma | ||||
| Efficacy Data | Cell viability | 100.00% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 0.625 μM | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [204] | ||||
| Indication | Gastric tubular adenocarcinoma | ||||
| Efficacy Data | Cell viability | 100.00% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 2.5 μM | ||||
| MOA of PDC |
In this research, based on the binding mode between Trastuzumab and HER2 protein, we first discovered a peptide sequence Leadpep that plays a vital role when antibody binds with HER2 protein. In silico mutations were conducted on Leadpep to screen out HER2-targeted peptides. The binding affinity of Cyclo-GCGPep1 increased when cyclized (Kd = 2.555 10-6M). It was used for constructing PDCs with the cytotoxin Camptothecin. Among them, Conjugate 1 showed best antiproliferative activities. Further research demonstrated that Conjugate 1 has a similar mechanism to Camptothecin. It is worth noting that Conjugate 1 showed a good specific delivery capacity and better penetration than Camptothecin. It could be a new therapeutic tool for HER2-positive cancer.
Click to Show/Hide
|
||||
| Description |
Antiproliferative activity of conjugates were evaluated using HER2-positive SK-BR-3 and NCI-N87 cells. As shown in Fig. 5A, Conjugate 1 reduced the cell viability by 23, 38, 47 and 66% at 0.156, 0.625, 2.5 and 10 μM. Its cytotoxicity is significantly more than Conjugate 2 at 0.625 μM and 10 μM. It showed comparable cytotoxicity with CPT at 10 μM. And Conjugate 3 showed little cytotoxicity at any concentrations other than 10 μM, which may be related to the structure of Conjugate 3 where CPT is conjugated on the peptide by non-breakable bond preventing CPT from acting as an anticancer agent. Cyclo-GCGPep1 exhibited no significant cytotoxicity at any concentrations indicating that peptide part of PDCs is inactive for inducing cancer cell death. Similar results can be seen on NCI-N87 cells. Conjugate 1 reduced significantly more cell viability than Conjugate 2 at 0.156 μM and 2.5 μM. And Conjugate 1 exhibited comparable antiproliferation activity as CPT at 2.5 μM and 10 μM. The cytotoxicity of Conjugate 3 is still weak at any concentrations. Cyclo-GCGPep1 is non-toxic to NCI-N87 cells as well. These results told us antiproliferative activity of conjugates is closely related to the ways drugs are conjugated. The cleavable disulfide bonds in Conjugate 1 and 2 could be broken and then Camptothecin derivatives can be released in cells, which is much related to anti-proliferative activity. In general, Conjugate 1 showed best antiproliferative activity and it was selected for further research.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
PDC-CPT2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [205] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 80% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 5 µM | ||||
| Description |
Antiproliferative results showed that PTX1 inhibited cell proliferation by 18.7%. The anti-proliferative activity of CPT1 was diminished by 1.9-fold as compared to CPT whereas the activity of CPT2 was comparable to CPT, since CPT2 reduced the cell viability to 61%.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
CPT-AAM-1 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Cell survival rate | 43% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 10 µM | ||||
| MOA of PDC |
In this study, we developed novel melittin analogs with pH-responsive cell-penetrating and membrane-lytic activities by replacing arginines and lysines with histidines. Importantly, we found that the conjugation of cargoes to the N-terminus of melittin analogs decreased their cell-penetrating and membrane-lytic activity compared to the C-terminus, implying that the C-terminus of analogs is more suitable for cargo conjugation. After the attachment of CPT, CPT-AAM-1 and CPT-AAM-2 displayed obvious pH-responsive antitumor activity. CPT-AAM-1 and CPT-AAM-2 destroyed tumor cells through the release of CPT and membrane disruption. Compared with CPT-AAM-2, CPT-AAM-1 showed stronger antitumor activity under acidic conditions. Notably, CPT-AAM-1 significantly inhibited the tumor growth in vivo compared with AAM, AAM-1, and CPT. In addition, CPT-AAM-1 showed relatively low toxicity compared with melittin and CPT. Taken together, our results demonstrate that CPT-AAM-1, with efficient pH-responsive cell-penetrating and membrane-lytic activities, possesses significant therapeutic potential for tumor therapy. This study provides a novel strategy for the development of PDCs based on pH-responsive AMPs for oncology therapeutics.
Click to Show/Hide
|
||||
| Description |
Subsequently, the cytotoxicity of these conjugates against B16-F10 cells was determined. As shown in Fig. 4B, CPT-AAM-1 and CPT-AAM-2 displayed significant pH-dependent antitumor activity after 72 h of treatment, whereas the cytotoxicity of CPT showed no obvious difference at pH 7.4 and 5.5. Notably, CPT-AAM-1 and CPT-AAM-2 exhibited greater cytotoxicity than free CPT under acidic conditions, particularly at high concentrations. Compared to CPT-AAM-2, CPT-AAM-1 displayed strong antitumor activity, suggesting that AAM-1 could deliver more CPT molecules into cells. In addition, this result further demonstrates that the C-terminus of AAM is more suitable for drug conjugation than the N-terminus.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Cell survival rate | 60% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 5 µM | ||||
| MOA of PDC |
In this study, we developed novel melittin analogs with pH-responsive cell-penetrating and membrane-lytic activities by replacing arginines and lysines with histidines. Importantly, we found that the conjugation of cargoes to the N-terminus of melittin analogs decreased their cell-penetrating and membrane-lytic activity compared to the C-terminus, implying that the C-terminus of analogs is more suitable for cargo conjugation. After the attachment of CPT, CPT-AAM-1 and CPT-AAM-2 displayed obvious pH-responsive antitumor activity. CPT-AAM-1 and CPT-AAM-2 destroyed tumor cells through the release of CPT and membrane disruption. Compared with CPT-AAM-2, CPT-AAM-1 showed stronger antitumor activity under acidic conditions. Notably, CPT-AAM-1 significantly inhibited the tumor growth in vivo compared with AAM, AAM-1, and CPT. In addition, CPT-AAM-1 showed relatively low toxicity compared with melittin and CPT. Taken together, our results demonstrate that CPT-AAM-1, with efficient pH-responsive cell-penetrating and membrane-lytic activities, possesses significant therapeutic potential for tumor therapy. This study provides a novel strategy for the development of PDCs based on pH-responsive AMPs for oncology therapeutics.
Click to Show/Hide
|
||||
| Description |
Subsequently, the cytotoxicity of these conjugates against B16-F10 cells was determined. As shown in Fig. 4B, CPT-AAM-1 and CPT-AAM-2 displayed significant pH-dependent antitumor activity after 72 h of treatment, whereas the cytotoxicity of CPT showed no obvious difference at pH 7.4 and 5.5. Notably, CPT-AAM-1 and CPT-AAM-2 exhibited greater cytotoxicity than free CPT under acidic conditions, particularly at high concentrations. Compared to CPT-AAM-2, CPT-AAM-1 displayed strong antitumor activity, suggesting that AAM-1 could deliver more CPT molecules into cells. In addition, this result further demonstrates that the C-terminus of AAM is more suitable for drug conjugation than the N-terminus.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Cell survival rate | 62% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 2.5 µM | ||||
| MOA of PDC |
In this study, we developed novel melittin analogs with pH-responsive cell-penetrating and membrane-lytic activities by replacing arginines and lysines with histidines. Importantly, we found that the conjugation of cargoes to the N-terminus of melittin analogs decreased their cell-penetrating and membrane-lytic activity compared to the C-terminus, implying that the C-terminus of analogs is more suitable for cargo conjugation. After the attachment of CPT, CPT-AAM-1 and CPT-AAM-2 displayed obvious pH-responsive antitumor activity. CPT-AAM-1 and CPT-AAM-2 destroyed tumor cells through the release of CPT and membrane disruption. Compared with CPT-AAM-2, CPT-AAM-1 showed stronger antitumor activity under acidic conditions. Notably, CPT-AAM-1 significantly inhibited the tumor growth in vivo compared with AAM, AAM-1, and CPT. In addition, CPT-AAM-1 showed relatively low toxicity compared with melittin and CPT. Taken together, our results demonstrate that CPT-AAM-1, with efficient pH-responsive cell-penetrating and membrane-lytic activities, possesses significant therapeutic potential for tumor therapy. This study provides a novel strategy for the development of PDCs based on pH-responsive AMPs for oncology therapeutics.
Click to Show/Hide
|
||||
| Description |
Subsequently, the cytotoxicity of these conjugates against B16-F10 cells was determined. As shown in Fig. 4B, CPT-AAM-1 and CPT-AAM-2 displayed significant pH-dependent antitumor activity after 72 h of treatment, whereas the cytotoxicity of CPT showed no obvious difference at pH 7.4 and 5.5. Notably, CPT-AAM-1 and CPT-AAM-2 exhibited greater cytotoxicity than free CPT under acidic conditions, particularly at high concentrations. Compared to CPT-AAM-2, CPT-AAM-1 displayed strong antitumor activity, suggesting that AAM-1 could deliver more CPT molecules into cells. In addition, this result further demonstrates that the C-terminus of AAM is more suitable for drug conjugation than the N-terminus.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Cell survival rate | 72% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 1.25 µM | ||||
| MOA of PDC |
In this study, we developed novel melittin analogs with pH-responsive cell-penetrating and membrane-lytic activities by replacing arginines and lysines with histidines. Importantly, we found that the conjugation of cargoes to the N-terminus of melittin analogs decreased their cell-penetrating and membrane-lytic activity compared to the C-terminus, implying that the C-terminus of analogs is more suitable for cargo conjugation. After the attachment of CPT, CPT-AAM-1 and CPT-AAM-2 displayed obvious pH-responsive antitumor activity. CPT-AAM-1 and CPT-AAM-2 destroyed tumor cells through the release of CPT and membrane disruption. Compared with CPT-AAM-2, CPT-AAM-1 showed stronger antitumor activity under acidic conditions. Notably, CPT-AAM-1 significantly inhibited the tumor growth in vivo compared with AAM, AAM-1, and CPT. In addition, CPT-AAM-1 showed relatively low toxicity compared with melittin and CPT. Taken together, our results demonstrate that CPT-AAM-1, with efficient pH-responsive cell-penetrating and membrane-lytic activities, possesses significant therapeutic potential for tumor therapy. This study provides a novel strategy for the development of PDCs based on pH-responsive AMPs for oncology therapeutics.
Click to Show/Hide
|
||||
| Description |
Subsequently, the cytotoxicity of these conjugates against B16-F10 cells was determined. As shown in Fig. 4B, CPT-AAM-1 and CPT-AAM-2 displayed significant pH-dependent antitumor activity after 72 h of treatment, whereas the cytotoxicity of CPT showed no obvious difference at pH 7.4 and 5.5. Notably, CPT-AAM-1 and CPT-AAM-2 exhibited greater cytotoxicity than free CPT under acidic conditions, particularly at high concentrations. Compared to CPT-AAM-2, CPT-AAM-1 displayed strong antitumor activity, suggesting that AAM-1 could deliver more CPT molecules into cells. In addition, this result further demonstrates that the C-terminus of AAM is more suitable for drug conjugation than the N-terminus.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Cell survival rate | 88% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 0.625 µM | ||||
| MOA of PDC |
In this study, we developed novel melittin analogs with pH-responsive cell-penetrating and membrane-lytic activities by replacing arginines and lysines with histidines. Importantly, we found that the conjugation of cargoes to the N-terminus of melittin analogs decreased their cell-penetrating and membrane-lytic activity compared to the C-terminus, implying that the C-terminus of analogs is more suitable for cargo conjugation. After the attachment of CPT, CPT-AAM-1 and CPT-AAM-2 displayed obvious pH-responsive antitumor activity. CPT-AAM-1 and CPT-AAM-2 destroyed tumor cells through the release of CPT and membrane disruption. Compared with CPT-AAM-2, CPT-AAM-1 showed stronger antitumor activity under acidic conditions. Notably, CPT-AAM-1 significantly inhibited the tumor growth in vivo compared with AAM, AAM-1, and CPT. In addition, CPT-AAM-1 showed relatively low toxicity compared with melittin and CPT. Taken together, our results demonstrate that CPT-AAM-1, with efficient pH-responsive cell-penetrating and membrane-lytic activities, possesses significant therapeutic potential for tumor therapy. This study provides a novel strategy for the development of PDCs based on pH-responsive AMPs for oncology therapeutics.
Click to Show/Hide
|
||||
| Description |
Subsequently, the cytotoxicity of these conjugates against B16-F10 cells was determined. As shown in Fig. 4B, CPT-AAM-1 and CPT-AAM-2 displayed significant pH-dependent antitumor activity after 72 h of treatment, whereas the cytotoxicity of CPT showed no obvious difference at pH 7.4 and 5.5. Notably, CPT-AAM-1 and CPT-AAM-2 exhibited greater cytotoxicity than free CPT under acidic conditions, particularly at high concentrations. Compared to CPT-AAM-2, CPT-AAM-1 displayed strong antitumor activity, suggesting that AAM-1 could deliver more CPT molecules into cells. In addition, this result further demonstrates that the C-terminus of AAM is more suitable for drug conjugation than the N-terminus.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
CPT-AAM-2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Cell survival rate | 55% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 10 µM | ||||
| MOA of PDC |
In this study, we developed novel melittin analogs with pH-responsive cell-penetrating and membrane-lytic activities by replacing arginines and lysines with histidines. Importantly, we found that the conjugation of cargoes to the N-terminus of melittin analogs decreased their cell-penetrating and membrane-lytic activity compared to the C-terminus, implying that the C-terminus of analogs is more suitable for cargo conjugation. After the attachment of CPT, CPT-AAM-1 and CPT-AAM-2 displayed obvious pH-responsive antitumor activity. CPT-AAM-1 and CPT-AAM-2 destroyed tumor cells through the release of CPT and membrane disruption. Compared with CPT-AAM-2, CPT-AAM-1 showed stronger antitumor activity under acidic conditions. Notably, CPT-AAM-1 significantly inhibited the tumor growth in vivo compared with AAM, AAM-1, and CPT. In addition, CPT-AAM-1 showed relatively low toxicity compared with melittin and CPT. Taken together, our results demonstrate that CPT-AAM-1, with efficient pH-responsive cell-penetrating and membrane-lytic activities, possesses significant therapeutic potential for tumor therapy. This study provides a novel strategy for the development of PDCs based on pH-responsive AMPs for oncology therapeutics.
Click to Show/Hide
|
||||
| Description |
Subsequently, the cytotoxicity of these conjugates against B16-F10 cells was determined. As shown in Fig. 4B, CPT-AAM-1 and CPT-AAM-2 displayed significant pH-dependent antitumor activity after 72 h of treatment, whereas the cytotoxicity of CPT showed no obvious difference at pH 7.4 and 5.5. Notably, CPT-AAM-1 and CPT-AAM-2 exhibited greater cytotoxicity than free CPT under acidic conditions, particularly at high concentrations. Compared to CPT-AAM-2, CPT-AAM-1 displayed strong antitumor activity, suggesting that AAM-1 could deliver more CPT molecules into cells. In addition, this result further demonstrates that the C-terminus of AAM is more suitable for drug conjugation than the N-terminus.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Cell survival rate | 57% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 5 µM | ||||
| MOA of PDC |
In this study, we developed novel melittin analogs with pH-responsive cell-penetrating and membrane-lytic activities by replacing arginines and lysines with histidines. Importantly, we found that the conjugation of cargoes to the N-terminus of melittin analogs decreased their cell-penetrating and membrane-lytic activity compared to the C-terminus, implying that the C-terminus of analogs is more suitable for cargo conjugation. After the attachment of CPT, CPT-AAM-1 and CPT-AAM-2 displayed obvious pH-responsive antitumor activity. CPT-AAM-1 and CPT-AAM-2 destroyed tumor cells through the release of CPT and membrane disruption. Compared with CPT-AAM-2, CPT-AAM-1 showed stronger antitumor activity under acidic conditions. Notably, CPT-AAM-1 significantly inhibited the tumor growth in vivo compared with AAM, AAM-1, and CPT. In addition, CPT-AAM-1 showed relatively low toxicity compared with melittin and CPT. Taken together, our results demonstrate that CPT-AAM-1, with efficient pH-responsive cell-penetrating and membrane-lytic activities, possesses significant therapeutic potential for tumor therapy. This study provides a novel strategy for the development of PDCs based on pH-responsive AMPs for oncology therapeutics.
Click to Show/Hide
|
||||
| Description |
Subsequently, the cytotoxicity of these conjugates against B16-F10 cells was determined. As shown in Fig. 4B, CPT-AAM-1 and CPT-AAM-2 displayed significant pH-dependent antitumor activity after 72 h of treatment, whereas the cytotoxicity of CPT showed no obvious difference at pH 7.4 and 5.5. Notably, CPT-AAM-1 and CPT-AAM-2 exhibited greater cytotoxicity than free CPT under acidic conditions, particularly at high concentrations. Compared to CPT-AAM-2, CPT-AAM-1 displayed strong antitumor activity, suggesting that AAM-1 could deliver more CPT molecules into cells. In addition, this result further demonstrates that the C-terminus of AAM is more suitable for drug conjugation than the N-terminus.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Cell survival rate | 74% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 2.5 µM | ||||
| MOA of PDC |
In this study, we developed novel melittin analogs with pH-responsive cell-penetrating and membrane-lytic activities by replacing arginines and lysines with histidines. Importantly, we found that the conjugation of cargoes to the N-terminus of melittin analogs decreased their cell-penetrating and membrane-lytic activity compared to the C-terminus, implying that the C-terminus of analogs is more suitable for cargo conjugation. After the attachment of CPT, CPT-AAM-1 and CPT-AAM-2 displayed obvious pH-responsive antitumor activity. CPT-AAM-1 and CPT-AAM-2 destroyed tumor cells through the release of CPT and membrane disruption. Compared with CPT-AAM-2, CPT-AAM-1 showed stronger antitumor activity under acidic conditions. Notably, CPT-AAM-1 significantly inhibited the tumor growth in vivo compared with AAM, AAM-1, and CPT. In addition, CPT-AAM-1 showed relatively low toxicity compared with melittin and CPT. Taken together, our results demonstrate that CPT-AAM-1, with efficient pH-responsive cell-penetrating and membrane-lytic activities, possesses significant therapeutic potential for tumor therapy. This study provides a novel strategy for the development of PDCs based on pH-responsive AMPs for oncology therapeutics.
Click to Show/Hide
|
||||
| Description |
Subsequently, the cytotoxicity of these conjugates against B16-F10 cells was determined. As shown in Fig. 4B, CPT-AAM-1 and CPT-AAM-2 displayed significant pH-dependent antitumor activity after 72 h of treatment, whereas the cytotoxicity of CPT showed no obvious difference at pH 7.4 and 5.5. Notably, CPT-AAM-1 and CPT-AAM-2 exhibited greater cytotoxicity than free CPT under acidic conditions, particularly at high concentrations. Compared to CPT-AAM-2, CPT-AAM-1 displayed strong antitumor activity, suggesting that AAM-1 could deliver more CPT molecules into cells. In addition, this result further demonstrates that the C-terminus of AAM is more suitable for drug conjugation than the N-terminus.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Cell survival rate | 86% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 1.25 µM | ||||
| MOA of PDC |
In this study, we developed novel melittin analogs with pH-responsive cell-penetrating and membrane-lytic activities by replacing arginines and lysines with histidines. Importantly, we found that the conjugation of cargoes to the N-terminus of melittin analogs decreased their cell-penetrating and membrane-lytic activity compared to the C-terminus, implying that the C-terminus of analogs is more suitable for cargo conjugation. After the attachment of CPT, CPT-AAM-1 and CPT-AAM-2 displayed obvious pH-responsive antitumor activity. CPT-AAM-1 and CPT-AAM-2 destroyed tumor cells through the release of CPT and membrane disruption. Compared with CPT-AAM-2, CPT-AAM-1 showed stronger antitumor activity under acidic conditions. Notably, CPT-AAM-1 significantly inhibited the tumor growth in vivo compared with AAM, AAM-1, and CPT. In addition, CPT-AAM-1 showed relatively low toxicity compared with melittin and CPT. Taken together, our results demonstrate that CPT-AAM-1, with efficient pH-responsive cell-penetrating and membrane-lytic activities, possesses significant therapeutic potential for tumor therapy. This study provides a novel strategy for the development of PDCs based on pH-responsive AMPs for oncology therapeutics.
Click to Show/Hide
|
||||
| Description |
Subsequently, the cytotoxicity of these conjugates against B16-F10 cells was determined. As shown in Fig. 4B, CPT-AAM-1 and CPT-AAM-2 displayed significant pH-dependent antitumor activity after 72 h of treatment, whereas the cytotoxicity of CPT showed no obvious difference at pH 7.4 and 5.5. Notably, CPT-AAM-1 and CPT-AAM-2 exhibited greater cytotoxicity than free CPT under acidic conditions, particularly at high concentrations. Compared to CPT-AAM-2, CPT-AAM-1 displayed strong antitumor activity, suggesting that AAM-1 could deliver more CPT molecules into cells. In addition, this result further demonstrates that the C-terminus of AAM is more suitable for drug conjugation than the N-terminus.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Cell survival rate | 94% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 0.625 µM | ||||
| MOA of PDC |
In this study, we developed novel melittin analogs with pH-responsive cell-penetrating and membrane-lytic activities by replacing arginines and lysines with histidines. Importantly, we found that the conjugation of cargoes to the N-terminus of melittin analogs decreased their cell-penetrating and membrane-lytic activity compared to the C-terminus, implying that the C-terminus of analogs is more suitable for cargo conjugation. After the attachment of CPT, CPT-AAM-1 and CPT-AAM-2 displayed obvious pH-responsive antitumor activity. CPT-AAM-1 and CPT-AAM-2 destroyed tumor cells through the release of CPT and membrane disruption. Compared with CPT-AAM-2, CPT-AAM-1 showed stronger antitumor activity under acidic conditions. Notably, CPT-AAM-1 significantly inhibited the tumor growth in vivo compared with AAM, AAM-1, and CPT. In addition, CPT-AAM-1 showed relatively low toxicity compared with melittin and CPT. Taken together, our results demonstrate that CPT-AAM-1, with efficient pH-responsive cell-penetrating and membrane-lytic activities, possesses significant therapeutic potential for tumor therapy. This study provides a novel strategy for the development of PDCs based on pH-responsive AMPs for oncology therapeutics.
Click to Show/Hide
|
||||
| Description |
Subsequently, the cytotoxicity of these conjugates against B16-F10 cells was determined. As shown in Fig. 4B, CPT-AAM-1 and CPT-AAM-2 displayed significant pH-dependent antitumor activity after 72 h of treatment, whereas the cytotoxicity of CPT showed no obvious difference at pH 7.4 and 5.5. Notably, CPT-AAM-1 and CPT-AAM-2 exhibited greater cytotoxicity than free CPT under acidic conditions, particularly at high concentrations. Compared to CPT-AAM-2, CPT-AAM-1 displayed strong antitumor activity, suggesting that AAM-1 could deliver more CPT molecules into cells. In addition, this result further demonstrates that the C-terminus of AAM is more suitable for drug conjugation than the N-terminus.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
References