
Drug Information
General Information of This Drug
| Drug ID | DRG00019 | |||||
|---|---|---|---|---|---|---|
| Drug Name | Methotrexate | |||||
| Synonyms |
methotrexate; 59-05-2; Rheumatrex; Amethopterin; Methylaminopterin; Metatrexan; Hdmtx; Abitrexate; Trexall; Mexate; Ledertrexate; Methylaminopterinum; (S)-2-(4-(((2,4-Diaminopteridin-6-yl)methyl)(methyl)amino)benzamido)pentanedioic acid; Methotrexatum; Antifolan; MTX; 4-Amino-10-methylfolic acid; Amethopterine; Metotrexato; Emtexate; Maxtrex; Rasuvo; Methotrexate hydrate; A-Methopterin; L-Amethopterin; A-Methpterin; Amethopterin L-; Folex-Pfs; Methotrexat-Ebewe; NSC-740; Methotrexate, L-; N-Bismethylpteroylglutamic acid; Farmitrexat; Medsatrexate; Methoblastin; Methotextrate; Methotrexat; Metotressato; Brimexate; Emthexat; Emthexate; Fauldexato; Lantarel; Lumexon; Metrotex; Novatrex; Otrexup; Tremetex; Trexeron; Trixilem; Texate; Metex; Mexate-Aq; alpha-Methopterin; CL-14377; Folex; NCI-C04671; JYLAMVO; Methotrexate Lpf; CCRIS 1109; Nordimet; Xatmep; EMT 25,299; 133073-73-1; 4-Aminomethylpteroylglutamic acid; HSDB 3123; NSC 740; CL 14377; UNII-YL5FZ2Y5U1; Methotrexatum [INN-Latin]; Metotrexato [INN-Spanish]; EINECS 200-413-8; YL5FZ2Y5U1; R 9985; REDITREX; NSC740; DTXSID4020822; CHEBI:44185; AI3-25299; TCMDC-125858; X 133; 4-Amino-N(sup 10)-methylpteroylglutamic acid; ADX-2191; MPI-2505; WR-19039; CHEMBL34259; DTXCID80822; (2S)-2-[[4-[(2,4-diaminopteridin-6-yl)methyl-methylamino]benzoyl]amino]pentanedioic acid; R-9985; Kyselina 4-amino-N(sup 10)-methylpteroylglutamova; N-(p-(((2,4-Diamino-6-pteridyl)methyl)methylamino)benzoyl)glutamic acid; L-(+)-N-(p-(((2,4-Diamino-6-pteridinyl)methyl)methylamino)benzoyl)glutamic acid; N-(4-(((2,4-Diamino-6-pteridinyl)methyl)methylamino)benzoyl)-L-glutamicacid; N-[4-[[(2,4-diamino-6-pteridinyl)methyl]methylamino]benzoyl]-L-glutamic acid; N-(p-(((2,4-Diamino-6-pteridinyl)methyl)methylamino)benzoyl)-L-(+)-glutamic acid; N-[(4-{[(2,4-diaminopteridin-6-yl)methyl](methyl)amino}phenyl)carbonyl]-L-glutamic acid; Methotrexate [USAN:USP:INN:BAN:JAN]; NCGC00025060-04; 4-amino-N(10)-methylpteroylglutamic acid; MFCD00150847; Kyselina N-(p-((2,4-diamino-6-pteridinylmethyl)methylamino)benzoyl)-L-glutamova; METHOTREXATE (IARC); METHOTREXATE [IARC]; Methotrexatum (INN-Latin); Metotrexato (INN-Spanish); (2S)-2-[(4-{[(2,4-diaminopteridin-6-yl)methyl](methyl)amino}phenyl)formamido]pentanedioic acid; Glutamic acid, N-(p-(((2,4-diamino-6-pteridinyl)methyl)methylamino)benzoyl)-, L-; METHOTREXATE (MART.); METHOTREXATE [MART.]; METHOTREXATE (USP-RS); METHOTREXATE [USP-RS]; Metotressato [DCIT]; Methotrexate, d-; METHOTREXATE (EP MONOGRAPH); METHOTREXATE (USP IMPURITY); METHOTREXATE [EP MONOGRAPH]; METHOTREXATE [USP IMPURITY]; METHOTREXATE (USP MONOGRAPH); METHOTREXATE [USP MONOGRAPH]; MLS001401431; [3H]methotrexate; Methotrexate (hydrate); Methotrexate (USAN:USP:INN:BAN:JAN); SMR000112001; [3H]-methotrexate; folic acid antagonist; 4-Amino-10-methylfolic acid hydrate; SR-01000075682; SMR000449324; Methotrexate (1.0mg/mL in Methanol with 0.1N NaOH); TCMDC-125488; Metolate; Antifolan hydrate; Intradose-MTX; MTX hydrate; 1dhi; 1dhj; 2drc; 4ocx; (2S)-2-[[4-[(2,4-diaminopteridin-6-yl)methyl-methyl-amino]benzoyl]amino]pentanedioic acid; CAS-59-05-2; Prestwick_322; Otrexup (TN); Xatmep (TN); L-METHOTREXATE; OTREXUP PFS; Kyselina 4-amino-N(sup 10)-methylpteroylglutamova [Czech]; Methylaminopterin; MTX; DL-Amethopterin hydrate; Spectrum_001836; Tocris-1230; 4kn0; Methylaminopterin hydrate; Prestwick0_000135; Prestwick1_000135; Prestwick2_000135; Spectrum2_001077; Spectrum3_000497; Spectrum4_000616; Spectrum5_000958; Abitrexate (Methotrexate); METHOTREXATE [MI]; METHOTREXATE [INN]; METHOTREXATE [JAN]; METHOTREXATE [HSDB]; METHOTREXATE [USAN]; NCIMech_000767; SCHEMBL3711; Kyselina N-(p-((2,4-diamino-6-pteridinylmethyl)methylamino)benzoyl)-L-glutamova [Czech]; METHOTREXATE [VANDF]; BIDD:PXR0175; Lopac0_000020; KBioGR_001172; KBioSS_002341; Methotrexate Methylaminopterin; MLS000049968; MLS002154208; DivK1c_000114; METHOTREXATE [WHO-DD]; METHOTREXATE [WHO-IP]; SPBio_001094; SPBio_002149; Amethopterin (hydrate); CL14377 (hydrate); WR19039 (hydrate); AMY235; cid_126941; cid_165528; GTPL4674; GTPL4815; SCHEMBL12421860; SCHEMBL23111732; BDBM18050; BDBM66082; HMS500F16; KBio1_000114; KBio2_002338; KBio2_004906; KBio2_007474; KBio3_001493; Methotrexate (JP17/USP/INN); L01BA01; L04AX03; g301; NINDS_000114; Bio1_000486; Bio1_000975; Bio1_001464; HMS1568K12; HMS2095K12; HMS2233O18; HMS3260C21; HMS3414L09; HMS3678L07; HMS3712K12; METHOTREXATE [ORANGE BOOK]; APC-2002; BCP13701; GLUTAMIC ACID, N-(P-(((2,4-DIAMINO-6-PTERIDINYL)METHYL)METHYLAMINO)BENZOYL)-, L-(+)-; MPI-5004; Tox21_110944; Tox21_300269; Tox21_500020; CCG-35800; EMT-25299; METHOTREXATUM [WHO-IP LATIN]; MFCD00064370; s1210; STL535338; (4-(((2,4-diaminopteridin-6-yl)methyl)(methyl)amino)benzoyl)-L-glutamic acid; AKOS016340329; Tox21_110944_1; CL14377; CS-1732; DB00563; KS-5093; LP00020; SDCCGSBI-0050009.P003; IDI1_000114; SMP2_000020; (methyl)amino)benzamido)pentanedioic acid; NCGC00025060-01; NCGC00025060-02; NCGC00025060-03; NCGC00025060-05; NCGC00025060-06; NCGC00025060-07; NCGC00025060-08; NCGC00025060-09; NCGC00025060-10; NCGC00025060-11; NCGC00025060-12; NCGC00025060-13; NCGC00025060-15; NCGC00025060-16; NCGC00254216-01; NCGC00260705-01; HY-14519; EU-0100020; FT-0601523; FT-0630651; G-301; NS00098764; SW198601-3; Methotrexate 1.0 mg/ml in Dimethyl Sulfoxide; C01937; D00142; EN300-119523; DL-4-Amino-N10-methylpteroylglutamic acid hydrate; Q422232; SR-01000597411; W-60383; (S)-2-(4-(((2,4-diaminopteridin-6-yl)methyl); Q-201366; SR-01000075682-1; SR-01000075682-2; SR-01000075682-6; SR-01000597411-1; W-105347; BRD-K59456551-001-09-3; BRD-K59456551-001-11-9; WLN: T66 BN DN GN JNJ CZ EZ H1N1&R DVMYVQ2VQ; Z1521553982; 4-AMINO-4-DEOXY-N(SUP 10)-METHYLPTEROYLGLUTAMATE; Methotrexate, European Pharmacopoeia (EP) Reference Standard; Methotrexate, United States Pharmacopeia (USP) Reference Standard; Glutamic acid,4-diamino-6-pteridinyl)methyl] methylamino]benzoyl]-, L-(+)-; L-Glutamic acid,4-diamino-6-pteridinyl)methyl]- methylamino]benzoyl]-; (S)-2-(4-(((2,4-Diaminopteridin-6-yl)methyl)-(methyl)amino)benzamido)pentanedioic acid; GLUTAMIC ACID, N-(P-(((2,4-DIAMINO-6-PTERIDINYL)METHYL)METHYLAMINO)BENZOYL)-,L; L-Glutamic acid,N-[4-[[(2,4-diamino-6-pteridinyl)methyl]methylamino]benzoyl]-,hydrate(9ci); Methotrexate for peak identification, European Pharmacopoeia (EP) Reference Standard; Methotrexate for system suitability, European Pharmacopoeia (EP) Reference Standard; Methotrexate, Pharmaceutical Secondary Standard; Certified Reference Material; N-(4-{[(2,4-diaminopteridin-6-yl)methyl](methyl)amino}benzoyl)-L-glutamic acid; N-[4-[[(2,4-Diamino-6-pteridinyl)methyl] methylamino]benzoyl]-L-glutamic acid; (2S)-2-((4-(((2,4-DIAMINOPTERIDIN-6-YL)METHYL)(METHYL)AMINO)BENZOYL)AMINO)PENTANEDIOIC ACID; (2S)-2-[[[4-[(2,4-diamino-6-pteridinyl)methyl-methylamino]phenyl]-oxomethyl]amino]pentanedioic acid;hydrate; (2S)-2-[[4-[(2,4-diaminopteridin-6-yl)methyl-methyl-amino]benzoyl]amino]glutaric acid;hydrate; (2S)-2-[[4-[[2,4-bis(azanyl)pteridin-6-yl]methyl-methyl-amino]phenyl]carbonylamino]pentanedioic acid;hydrate; 102613-64-9
Click to Show/Hide
|
|||||
| Target(s) | Reduced folate transporter (SLC19A1) | Target Info | ||||
| Structure |
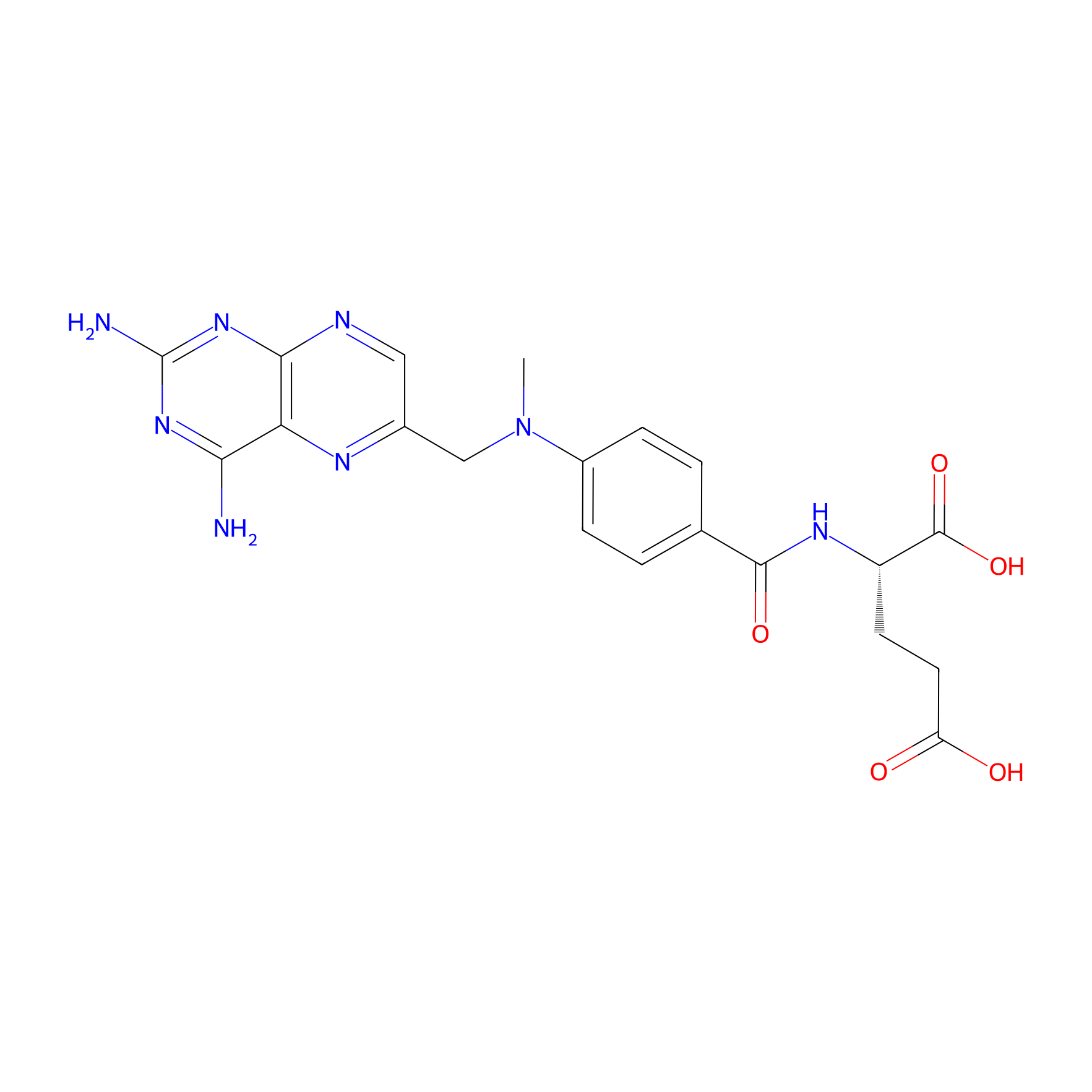
|
|||||
| Formula |
C20H22N8O5
|
|||||
| #Ro5 Violations (Lipinski): 1 | Molecular Weight (mw) | 454.4 | ||||
| Lipid-water partition coefficient (xlogp) | -1.8 | |||||
| Hydrogen Bond Donor Count (hbonddonor) | 5 | |||||
| Hydrogen Bond Acceptor Count (hbondacc) | 12 | |||||
| Rotatable Bond Count (rotbonds) | 9 | |||||
| PubChem CID | ||||||
| Canonical smiles |
CN(CC1=CN=C2C(=N1)C(=NC(=N2)N)N)C3=CC=C(C=C3)C(=O)NC(CCC(=O)O)C(=O)O
|
|||||
| InChI |
InChI=1S/C20H22N8O5/c1-28(9-11-8-23-17-15(24-11)16(21)26-20(22)27-17)12-4-2-10(3-5-12)18(31)25-13(19(32)33)6-7-14(29)30/h2-5,8,13H,6-7,9H2,1H3,(H,25,31)(H,29,30)(H,32,33)(H4,21,22,23,26,27)/t13-/m0/s1
|
|||||
| InChIKey |
FBOZXECLQNJBKD-ZDUSSCGKSA-N
|
|||||
| IUPAC Name |
(2S)-2-[[4-[(2,4-diaminopteridin-6-yl)methyl-methylamino]benzoyl]amino]pentanedioic acid
|
|||||
The activity data of This Drug
| Standard Type | Value | Administration times | Cell line | Cell line ID | Ref. | |
|---|---|---|---|---|---|---|
| Half Maximal Inhibitory Concentration (IC50) | 3.29-0.44 µM | 72 h | HeLa cell | CVCL_0030 | [1] | |
| Half Maximal Inhibitory Concentration (IC50) | 10 µM | 1 h | Human coronary artery endothelial cell | N.A. | [2] | |
| Half Maximal Inhibitory Concentration (IC50) | 40 µM | 72 h | MDA-MB-231 cell | CVCL_0062 | [1] | |
| Half Maximal Inhibitory Concentration (IC50) | 500 µM | 1 h | Molt-3 T cell | CVCL_0624 | [2] | |
| Half Maximal Effective Concentration (EC50) | 8.2 nM | N.A. | CCRF-CEM cell | CVCL_0207 | [3] | |
| Half Maximal Effective Concentration (EC50) | 11.3 nM | N.A. | FaDu cell | CVCL_1218 | [4] | |
| Half Maximal Effective Concentration (EC50) | 12 nM | N.A. | CCRF-CEM cell | CVCL_0207 | [5] | |
| Half Maximal Effective Concentration (EC50) | 14 nM | N.A. | CCRF-CEM cell | CVCL_0207 | [6] | |
| Half Maximal Effective Concentration (EC50) | 14.4 nM | N.A. | CCRF-CEM cell | CVCL_0207 | [4] | |
| Half Maximal Effective Concentration (EC50) | 14.5 nM | N.A. | CCRF-CEM cell | CVCL_0207 | [7] | |
| Half Maximal Effective Concentration (EC50) | 15 nM | N.A. | CCRF-CEM cell | CVCL_0207 | [8] | |
| Half Maximal Effective Concentration (EC50) | 15.5 nM | N.A. | CCRF-CEM cell | CVCL_0207 | [9] | |
| Half Maximal Effective Concentration (EC50) | 17 nM | N.A. | FaDu cell | CVCL_1218 | [6] | |
| Half Maximal Effective Concentration (EC50) | 17.5 nM | N.A. | CCRF-CEM cell | CVCL_0207 | [10] | |
| Half Maximal Effective Concentration (EC50) | 18 nM | N.A. | CCRF-CEM cell | CVCL_0207 | [6] | |
| Half Maximal Effective Concentration (EC50) | 18 nM | N.A. | R30dm-CCRF-CEM cell | CVCL_V325 | [11] | |
| Half Maximal Effective Concentration (EC50) | 19 nM | N.A. | FaDu cell | CVCL_1218 | [11] | |
| Half Maximal Effective Concentration (EC50) | 31 nM | N.A. | FaDu cell | CVCL_1218 | [12] | |
| Half Maximal Effective Concentration (EC50) | 567 nM | N.A. | CCRF-CEM cell | CVCL_0207 | [13] | |
| Half Maximal Effective Concentration (EC50) | 595 nM | N.A. | CCRF-CEM cell | CVCL_0207 | [12] | |
| Half Maximal Effective Concentration (EC50) | 615 nM | N.A. | CCRF-CEM cell | CVCL_0207 | [7] | |
| Half Maximal Effective Concentration (EC50) | 620 nM | N.A. | CCRF-CEM cell | CVCL_0207 | [14] | |
| Half Maximal Effective Concentration (EC50) | 680 nM | N.A. | MV4-11 cell | CVCL_0064 | [10] | |
| Half Maximal Effective Concentration (EC50) | 720 nM | N.A. | R1 cell | CVCL_2167 | [11] | |
| Half Maximal Effective Concentration (EC50) | 900 nM | N.A. | MCF-7 cell | CVCL_0031 | [15] | |
| Half Maximal Effective Concentration (EC50) | 1.05 uM | N.A. | R1 cell | CVCL_2167 | [16] | |
| Half Maximal Effective Concentration (EC50) | 1.1 uM | N.A. | Caco-2 cell | CVCL_0025 | [15] | |
| Half Maximal Effective Concentration (EC50) | 1.5 uM | N.A. | CCRF-CEM cell | CVCL_0207 | [7] | |
| Half Maximal Effective Concentration (EC50) | 2.03 uM | N.A. | CCRF-CEM cell | CVCL_0207 | [9] | |
| Half Maximal Effective Concentration (EC50) | 2.63 uM | N.A. | R2-CCRF-CEM cell | CVCL_S650 | [17] | |
| Half Maximal Effective Concentration (EC50) | 2.76 uM | N.A. | CCRF-CEM cell | CVCL_0207 | [11] | |
| Half Maximal Effective Concentration (EC50) | 3.1 uM | N.A. | CCRF-CEM cell | CVCL_0207 | [12] | |
| Half Maximal Effective Dosage (ED50) | 2.4 nM | N.A. | HEp-2 cell | CVCL_1906 | [18] | |
| Half Maximal Effective Dosage (ED50) | 14.5 nM | N.A. | CCRF-CEM cell | CVCL_0207 | [19] | |
| Half Maximal Effective Dosage (ED50) | 15.5 nM | N.A. | CCRF-CEM cell | CVCL_0207 | [20] | |
| Half Maximal Effective Dosage (ED50) | 20 nM | N.A. | W256 cell | CVCL_3537 | [21] | |
| Half Maximal Effective Dosage (ED50) | <50 nM | N.A. | HL-60 cell | CVCL_0002 | [22] | |
| Half Maximal Effective Dosage (ED50) | 655 nM | N.A. | CCRF-CEM cell | CVCL_0207 | [20] | |
| Half Maximal Growth Inhibition (GI50) | 2.512 nM | N.A. | OVCAR-4 cell | CVCL_1627 | [23] | |
| Half Maximal Growth Inhibition (GI50) | 10 nM | N.A. | HCT 116 cell | CVCL_0291 | [24] | |
| Half Maximal Growth Inhibition (GI50) | 24 nM | N.A. | MCF-7 cell | CVCL_0031 | [25] | |
| Half Maximal Growth Inhibition (GI50) | 40 nM | N.A. | HT29 cell | CVCL_A8EZ | [26] | |
| Half Maximal Growth Inhibition (GI50) | 70 nM | N.A. | HT29 cell | CVCL_A8EZ | [24] | |
| Half Maximal Growth Inhibition (GI50) | 10.25 uM | N.A. | NCI-H226 cell | CVCL_1544 | [27] | |
| Half Maximal Growth Inhibition (GI50) | >100 uM | N.A. | HeLa cell | CVCL_0030 | [28] | |
| Half Maximal Growth Inhibition (GI50) | >100 uM | N.A. | HT29 cell | CVCL_A8EZ | [28] | |
| Half Maximal Infective Dose (ID50) | 3 ng/mL | N.A. | CCRF-CEM cell | CVCL_0207 | [29] | |
| Half Maximal Infective Dose (ID50) | 6 nmol/L | N.A. | CCRF-CEM cell | CVCL_0207 | [30] | |
| Half Maximal Infective Dose (ID50) | 8 mM | N.A. | TA3 cell | CVCL_4315 | [31] | |
| Half Maximal Infective Dose (ID50) | 3 nM | N.A. | CCRF-CEM cell | CVCL_0207 | [32] | |
| Half Maximal Infective Dose (ID50) | 10 nM | N.A. | L1210 cell | CVCL_0382 | [32] | |
| Half Maximal Infective Dose (ID50) | 20 nM | N.A. | Raji cell | CVCL_0511 | [33] | |
| Half Maximal Infective Dose (ID50) | 30 nM | N.A. | H9 cell | CVCL_1240 | [33] | |
| Half Maximal Inhibitory Concentration (IC50) | 0.23 ug/mL | N.A. | HT29 cell | CVCL_A8EZ | [34] | |
| Half Maximal Inhibitory Concentration (IC50) | 0.29 ug/mL | N.A. | NCI-H23 cell | CVCL_1547 | [35] | |
| Half Maximal Inhibitory Concentration (IC50) | 0.32 ug/mL | N.A. | Caco-2 cell | CVCL_0025 | [34] | |
| Half Maximal Inhibitory Concentration (IC50) | 2 ng/mL | N.A. | P388 cell | CVCL_7222 | [36] | |
| Half Maximal Inhibitory Concentration (IC50) | 4 ng/mL | N.A. | CCRF-CEM cell | CVCL_0207 | [37] | |
| Half Maximal Inhibitory Concentration (IC50) | 4.5 ug/mL | N.A. | Caki-1 cell | CVCL_0234 | [38] | |
| Half Maximal Inhibitory Concentration (IC50) | 10 ng/mL | N.A. | HT29 cell | CVCL_A8EZ | [36] | |
| Half Maximal Inhibitory Concentration (IC50) | 25 ug/mL | N.A. | Caki-1 cell | CVCL_0234 | [38] | |
| Half Maximal Inhibitory Concentration (IC50) | 27 ng/mL | N.A. | PC-3 cell | CVCL_0035 | [35] | |
| Half Maximal Inhibitory Concentration (IC50) | 35 ng/mL | N.A. | A-549 cell | CVCL_0023 | [39] | |
| Half Maximal Inhibitory Concentration (IC50) | 47.82 ug/mL | N.A. | HT29 cell | CVCL_A8EZ | [40] | |
| Half Maximal Inhibitory Concentration (IC50) | >60 ug/mL | N.A. | ZR-75-1 cell | CVCL_0588 | [35] | |
| Half Maximal Inhibitory Concentration (IC50) | 108 ug/mL | N.A. | M21 cell | CVCL_D031 | [38] | |
| Half Maximal Inhibitory Concentration (IC50) | 109 ug/mL | N.A. | M21 cell | CVCL_D031 | [38] | |
| Half Maximal Inhibitory Concentration (IC50) | 115 ug/mL | N.A. | M21 cell | CVCL_D031 | [38] | |
| Half Maximal Inhibitory Concentration (IC50) | 117 ug/mL | N.A. | M21 cell | CVCL_D031 | [38] | |
| Half Maximal Inhibitory Concentration (IC50) | 119 ug/mL | N.A. | M21 cell | CVCL_D031 | [38] | |
| Half Maximal Inhibitory Concentration (IC50) | 121 ug/mL | N.A. | M21 cell | CVCL_D031 | [38] | |
| Half Maximal Inhibitory Concentration (IC50) | 1 nM | N.A. | PC-3 cell | CVCL_0035 | [41] | |
| Half Maximal Inhibitory Concentration (IC50) | 2 nM | N.A. | PC-3 cell | CVCL_0035 | [42] | |
| Half Maximal Inhibitory Concentration (IC50) | 2.1 nM | N.A. | L1210 cell | CVCL_0382 | [43] | |
| Half Maximal Inhibitory Concentration (IC50) | 2.7 nM | N.A. | L1210 cell | CVCL_0382 | [44] | |
| Half Maximal Inhibitory Concentration (IC50) | 3.28 nM | N.A. | L1210 cell | CVCL_0382 | [45] | |
| Half Maximal Inhibitory Concentration (IC50) | 3.4 nM | N.A. | L1210 cell | CVCL_0382 | [46] | |
| Half Maximal Inhibitory Concentration (IC50) | 3.9 nM | N.A. | L1210 cell | CVCL_0382 | [47] | |
| Half Maximal Inhibitory Concentration (IC50) | 4.4 nM | N.A. | L1210 cell | CVCL_0382 | [48] | |
| Half Maximal Inhibitory Concentration (IC50) | 4.6 nM | N.A. | L1210 cell | CVCL_0382 | [49] | |
| Half Maximal Inhibitory Concentration (IC50) | 5 nM | N.A. | L1210 cell | CVCL_0382 | [50] | |
| Half Maximal Inhibitory Concentration (IC50) | 5 nM | N.A. | A-549 cell | CVCL_0023 | [41] | |
| Half Maximal Inhibitory Concentration (IC50) | 5.4 nM | N.A. | CCRF S-180 cell | CVCL_2874 | [51] | |
| Half Maximal Inhibitory Concentration (IC50) | 5.8 nM | N.A. | SCC-7 cell | CVCL_V412 | [52] | |
| Half Maximal Inhibitory Concentration (IC50) | 6 nM | N.A. | KB cell | CVCL_0372 | [53] | |
| Half Maximal Inhibitory Concentration (IC50) | 6.92 nM | N.A. | HL-60 cell | CVCL_0002 | [54] | |
| Half Maximal Inhibitory Concentration (IC50) | 7.5 nM | N.A. | SCC-25 cell | CVCL_1682 | [55] | |
| Half Maximal Inhibitory Concentration (IC50) | 8.1 nM | N.A. | HL-60 cell | CVCL_0002 | [56] | |
| Half Maximal Inhibitory Concentration (IC50) | 8.8 nM | N.A. | 143B cell | CVCL_2270 | [57] | |
| Half Maximal Inhibitory Concentration (IC50) | 8.9 nM | N.A. | HL-60 cell | CVCL_0002 | [58] | |
| Half Maximal Inhibitory Concentration (IC50) | 9 nM | N.A. | L1210 cell | CVCL_0382 | [56] | |
| Half Maximal Inhibitory Concentration (IC50) | 9.2 nM | N.A. | Vero cell | CVCL_0059 | [57] | |
| Half Maximal Inhibitory Concentration (IC50) | 9.5 nM | N.A. | NCI-H460 cell | CVCL_0459 | [57] | |
| Half Maximal Inhibitory Concentration (IC50) | 10 nM | N.A. | H35 cell | CVCL_0285 | [59] | |
| Half Maximal Inhibitory Concentration (IC50) | 10 nM | N.A. | L1210 cell | CVCL_0382 | [60] | |
| Half Maximal Inhibitory Concentration (IC50) | 10 nM | N.A. | KB cell | CVCL_0372 | [61] | |
| Half Maximal Inhibitory Concentration (IC50) | 12 nM | N.A. | L1210 cell | CVCL_0382 | [62] | |
| Half Maximal Inhibitory Concentration (IC50) | 12 nM | N.A. | U-373MG ATCC cell | CVCL_2219 | [57] | |
| Half Maximal Inhibitory Concentration (IC50) | 12 nM | N.A. | HL-60 cell | CVCL_0002 | [63] | |
| Half Maximal Inhibitory Concentration (IC50) | 12.6 nM | N.A. | Ehrlich cell | CVCL_3873 | [51] | |
| Half Maximal Inhibitory Concentration (IC50) | 13 nM | N.A. | A-549 cell | CVCL_0023 | [64] | |
| Half Maximal Inhibitory Concentration (IC50) | 13.1 nM | N.A. | SW620 cell | CVCL_0547 | [65] | |
| Half Maximal Inhibitory Concentration (IC50) | 13.5 nM | N.A. | A-549 cell | CVCL_0023 | [66] | |
| Half Maximal Inhibitory Concentration (IC50) | 15 nM | N.A. | SW480 cell | CVCL_0546 | [67] | |
| Half Maximal Inhibitory Concentration (IC50) | 18 nM | N.A. | HT29 cell | CVCL_A8EZ | [67] | |
| Half Maximal Inhibitory Concentration (IC50) | 19 nM | N.A. | KB cell | CVCL_0372 | [68] | |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | KB cell | CVCL_0372 | [53] | |
| Half Maximal Inhibitory Concentration (IC50) | 21 nM | N.A. | IGROV-1 cell | CVCL_1304 | [69] | |
| Half Maximal Inhibitory Concentration (IC50) | 21 nM | N.A. | HeLa cell | CVCL_0030 | [58] | |
| Half Maximal Inhibitory Concentration (IC50) | 22 nM | N.A. | IGROV-1 cell | CVCL_1304 | [69] | |
| Half Maximal Inhibitory Concentration (IC50) | 22.7 nM | N.A. | HL-60 cell | CVCL_0002 | [66] | |
| Half Maximal Inhibitory Concentration (IC50) | 23 nM | N.A. | DU145 cell | CVCL_0105 | [42] | |
| Half Maximal Inhibitory Concentration (IC50) | 23 nM | N.A. | P388 cell | CVCL_7222 | [68] | |
| Half Maximal Inhibitory Concentration (IC50) | 23 nM | N.A. | A-549 cell | CVCL_0023 | [42] | |
| Half Maximal Inhibitory Concentration (IC50) | 24 nM | N.A. | L1210 cell | CVCL_0382 | [59] | |
| Half Maximal Inhibitory Concentration (IC50) | 25 nM | N.A. | WiDr cell | CVCL_2760 | [70] | |
| Half Maximal Inhibitory Concentration (IC50) | 25 nM | N.A. | HL-60 cell | CVCL_0002 | [71] | |
| Half Maximal Inhibitory Concentration (IC50) | 26 nM | N.A. | LOX IMVI cell | CVCL_1381 | [42] | |
| Half Maximal Inhibitory Concentration (IC50) | 27 nM | N.A. | SCC-25 cell | CVCL_1682 | [42] | |
| Half Maximal Inhibitory Concentration (IC50) | 28 nM | N.A. | MOLT-4 cell | CVCL_0013 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | 28 nM | N.A. | UACC-62 cell | CVCL_1780 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | 30 nM | N.A. | L1210 cell | CVCL_0382 | [73] | |
| Half Maximal Inhibitory Concentration (IC50) | 30 nM | N.A. | HCT 15 cell | CVCL_0292 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | 31 nM | N.A. | OVCAR-8 cell | CVCL_1629 | [42] | |
| Half Maximal Inhibitory Concentration (IC50) | 32 nM | N.A. | HT29 cell | CVCL_A8EZ | [42] | |
| Half Maximal Inhibitory Concentration (IC50) | 33 nM | N.A. | RPMI-8226 cell | CVCL_7353 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | 33 nM | N.A. | A-549 cell | CVCL_0023 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | 33 nM | N.A. | KM12 cell | CVCL_1331 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | 33 nM | N.A. | SW620 cell | CVCL_0547 | [42] | |
| Half Maximal Inhibitory Concentration (IC50) | 35 nM | N.A. | SF539 cell | CVCL_1691 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | 36 nM | N.A. | MCF-7 cell | CVCL_0031 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | 40 nM | N.A. | ACHN cell | CVCL_1067 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | 40 nM | N.A. | Raji cell | CVCL_0511 | [74] | |
| Half Maximal Inhibitory Concentration (IC50) | 40 nM | N.A. | RT-4 cell | CVCL_0036 | [75] | |
| Half Maximal Inhibitory Concentration (IC50) | 42 nM | N.A. | KM12 cell | CVCL_1331 | [42] | |
| Half Maximal Inhibitory Concentration (IC50) | 43 nM | N.A. | NCI-H23 cell | CVCL_1547 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | 45 nM | N.A. | DU145 cell | CVCL_0105 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | 50 nM | N.A. | MCF-7 cell | CVCL_0031 | [75] | |
| Half Maximal Inhibitory Concentration (IC50) | 52 nM | N.A. | SF268 cell | CVCL_1689 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | 63 nM | N.A. | U-251MG cell | CVCL_0021 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | 66 nM | N.A. | MC-38 cell | CVCL_B288 | [68] | |
| Half Maximal Inhibitory Concentration (IC50) | 66 nM | N.A. | P388 cell | CVCL_7222 | [76] | |
| Half Maximal Inhibitory Concentration (IC50) | 77 nM | N.A. | DAN-G cell | CVCL_0243 | [75] | |
| Half Maximal Inhibitory Concentration (IC50) | 78 nM | N.A. | NCI-ADR-RES cell | CVCL_1452 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | 87 nM | N.A. | SK-MEL-5 cell | CVCL_0527 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | 100 nM | N.A. | A-549 cell | CVCL_0023 | [77] | |
| Half Maximal Inhibitory Concentration (IC50) | 100 nM | N.A. | HeLa cell | CVCL_0030 | [78] | |
| Half Maximal Inhibitory Concentration (IC50) | 110 nM | N.A. | BGC-823 cell | CVCL_3360 | [78] | |
| Half Maximal Inhibitory Concentration (IC50) | 110 nM | N.A. | HCC 2998 cell | CVCL_1266 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | 120.5 nM | N.A. | R2 cell | CVCL_C5SG | [79] | |
| Half Maximal Inhibitory Concentration (IC50) | 121 nM | N.A. | R2 cell | CVCL_C5SG | [69] | |
| Half Maximal Inhibitory Concentration (IC50) | 150 nM | N.A. | SCC-25 cell | CVCL_1682 | [55] | |
| Half Maximal Inhibitory Concentration (IC50) | 191 nM | N.A. | UO-31 cell | CVCL_1911 | [42] | |
| Half Maximal Inhibitory Concentration (IC50) | 200 nM | N.A. | L1210 cell | CVCL_0382 | [80] | |
| Half Maximal Inhibitory Concentration (IC50) | 216 nM | N.A. | R2 cell | CVCL_C5SG | [69] | |
| Half Maximal Inhibitory Concentration (IC50) | 300 nM | N.A. | LNCaP cell | CVCL_0395 | [81] | |
| Half Maximal Inhibitory Concentration (IC50) | 398 nM | N.A. | OVCAR-3 cell | CVCL_0465 | [42] | |
| Half Maximal Inhibitory Concentration (IC50) | 400 nM | N.A. | OVCAR-3 cell | CVCL_0465 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | 450 nM | N.A. | NCI-H522 cell | CVCL_1567 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | 790 nM | N.A. | UACC-257 cell | CVCL_1779 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | 870 nM | N.A. | COLO205 cell | CVCL_F402 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | 980 nM | N.A. | OVCAR-5 cell | CVCL_1628 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | >1000 nM | N.A. | RXF 393 cell | CVCL_1673 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | >1000 nM | N.A. | OVCAR-4 cell | CVCL_1627 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | >1000 nM | N.A. | R2 cell | CVCL_C5SG | [53] | |
| Half Maximal Inhibitory Concentration (IC50) | >1000 nM | N.A. | HOP-92 cell | CVCL_1286 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | >1000 nM | N.A. | Malme-3M cell | CVCL_1438 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | >1000 nM | N.A. | SK-MEL-2 cell | CVCL_0069 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | 1000 nM | N.A. | L1210 cell | CVCL_0382 | [62] | |
| Half Maximal Inhibitory Concentration (IC50) | >1000 nM | N.A. | T-47D cell | CVCL_0553 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | >1000 nM | N.A. | Hs 578T cell | CVCL_0332 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | >1000 nM | N.A. | SNB-75 cell | CVCL_1706 | [42] | |
| Half Maximal Inhibitory Concentration (IC50) | >1000 nM | N.A. | SK-MEL-28 cell | CVCL_0526 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | >1000 nM | N.A. | BT-549 cell | CVCL_1092 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | >1000 nM | N.A. | MDA-MB-435 cell | CVCL_0417 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | >1000 nM | N.A. | MV4-11 cell | CVCL_0064 | [72] | |
| Half Maximal Inhibitory Concentration (IC50) | 1.3 uM | N.A. | L1210 cell | CVCL_0382 | [82] | |
| Half Maximal Inhibitory Concentration (IC50) | 1.4 uM | N.A. | HT29 cell | CVCL_A8EZ | [81] | |
| Half Maximal Inhibitory Concentration (IC50) | 1.57 uM | N.A. | EL4 cell | CVCL_0255 | [83] | |
| Half Maximal Inhibitory Concentration (IC50) | 2.55 uM | N.A. | CCRF-CEM cell | CVCL_0207 | [52] | |
| Half Maximal Inhibitory Concentration (IC50) | 5.1 uM | N.A. | EL4 cell | CVCL_0255 | [84] | |
| Half Maximal Inhibitory Concentration (IC50) | 5.52 uM | N.A. | A-427 cell | CVCL_1055 | [75] | |
| Half Maximal Inhibitory Concentration (IC50) | 60 uM | N.A. | L1210 cell | CVCL_0382 | [85] | |
| Half Maximal Inhibitory Concentration (IC50) | 82.3 uM | N.A. | Bel-7402 cell | CVCL_5492 | [78] | |
| Half Maximal Inhibitory Concentration (IC50) | >100 uM | N.A. | HeLa cell | CVCL_0030 | [66] | |
| Half Maximal Inhibitory Concentration (IC50) | >100 uM | N.A. | MDA-MB-231 cell | CVCL_0062 | [86] | |
| Half Maximal Inhibitory Concentration (IC50) | 186 uM | N.A. | L1210 cell | CVCL_0382 | [48] | |
| Half Maximal Inhibitory Concentration (IC50) | 197 uM | N.A. | L1210 ( R81) cell | CVCL_N028 | [49] | |
| Half Maximal Inhibitory Concentration (IC50) | 220 uM | N.A. | L1210 cell | CVCL_0382 | [87] | |
| Half Maximal Inhibitory Concentration (IC50) | 419 uM | N.A. | K562 cell | CVCL_0004 | [88] | |
| Half Maximal Lethal Concentration (IC50) | 40 nM | N.A. | MCF-7 cell | CVCL_0031 | [89] | |
Each Peptide-drug Conjugate Related to This Drug
Full Information of The Activity Data of The PDC(s) Related to This Drug
STRAP-4-MTX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Cervical cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.01 ± 0.22 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
Electrostatics modulates the interactions of CPPs with the corresponding cell surface. Therefore, for the design of the amino acid side chain sequences, we reverse-engineered the spatial electrostatic potential distribution of the previously designed peptides with cell penetration ability. The spatial electrostatic fingerprints of the earlier designed peptides were generated as described previously. The reverse-engineered sequences with a shorter (7-mer) chain length were compared to the previous designs through multiple iterations of amino acid sequences. Their respective electrostatic profiles were used for the design of four syndiotactic, cationic, amphipathic peptides with optimal sequence selection. These syndiotactic re-engineered amphipathic peptides (STRAPs) were tested for their ability to penetrate cells and deliver a small molecule (methotrexate). The amino acid constitutions of STRAP-1, STRAP-2, STRAP-3, and STRAP-4 are the same. However, STRAP-2 is the stereochemically reversed STRAP-1; similarly, STRAP-4 is the stereochemically reversed STRAP-3. The use of LDLD and DLDL stereochemical sequences in the design of STRAPs resulted in the differential electrostatic signatures for the same amino acid sequences. The designed peptides have a higher cellular uptake in TNBC cells (MDA-MB-231) than the standard control TAT peptide. They could penetrate cells by both active and passive processes, and their activity is not reduced in biological fluids (human plasma and bovine serum). Furthermore, the delivery of MTX as STRAP-MTX conjugates helped to overcome the drug resistance of the MDA-MB-231 cells under in vitro conditions. The delivery of the STARP-4-MTX conjugate in TNBC xenograft tumors was more effective in the reduction of both the tumor size and metastasis to the lungs, liver, spleen, and lymph nodes.
Click to Show/Hide
|
||||
| Description |
To deliver the functional molecule into the cells, we attached an anticancer drug, methotrexate (MTX), to the N-terminus of STRAPs. The peptides were tested for their capability to deliver the active drug molecule in breast and cervical cancer cells. MDA-MB-231 and HeLa cells were treated with varying concentrations of MTX and STRAP-MTX conjugates for 72 h. Cell viability was assessed using the MTT assay. The cytotoxicity of MTX increased when delivered as peptide-MTX conjugates to the cells. This resulted in an overall reduction in the inhibitory concentration cytotoxic to 50% of cells (IC50) for MTX. The MTX resistance for MDA-MB-231 cells is well-established. The IC50 for MTX in MDA-MB-231 cells was minimum when delivered as a STRAP-4-MTX conjugate. Similar lowering of IC50 values in HeLa cells was observed. We also treated HEK-293 cells with MTX-STRAP conjugates. The peptide-drug conjugates showed no significant toxicity to the HEK-293 cells under the tested conditions, thereby confirming the earlier reported observation of higher uptake rates in cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.34 ± 0.19 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
Electrostatics modulates the interactions of CPPs with the corresponding cell surface. Therefore, for the design of the amino acid side chain sequences, we reverse-engineered the spatial electrostatic potential distribution of the previously designed peptides with cell penetration ability. The spatial electrostatic fingerprints of the earlier designed peptides were generated as described previously. The reverse-engineered sequences with a shorter (7-mer) chain length were compared to the previous designs through multiple iterations of amino acid sequences. Their respective electrostatic profiles were used for the design of four syndiotactic, cationic, amphipathic peptides with optimal sequence selection. These syndiotactic re-engineered amphipathic peptides (STRAPs) were tested for their ability to penetrate cells and deliver a small molecule (methotrexate). The amino acid constitutions of STRAP-1, STRAP-2, STRAP-3, and STRAP-4 are the same. However, STRAP-2 is the stereochemically reversed STRAP-1; similarly, STRAP-4 is the stereochemically reversed STRAP-3. The use of LDLD and DLDL stereochemical sequences in the design of STRAPs resulted in the differential electrostatic signatures for the same amino acid sequences. The designed peptides have a higher cellular uptake in TNBC cells (MDA-MB-231) than the standard control TAT peptide. They could penetrate cells by both active and passive processes, and their activity is not reduced in biological fluids (human plasma and bovine serum). Furthermore, the delivery of MTX as STRAP-MTX conjugates helped to overcome the drug resistance of the MDA-MB-231 cells under in vitro conditions. The delivery of the STARP-4-MTX conjugate in TNBC xenograft tumors was more effective in the reduction of both the tumor size and metastasis to the lungs, liver, spleen, and lymph nodes.
Click to Show/Hide
|
||||
| Description |
To deliver the functional molecule into the cells, we attached an anticancer drug, methotrexate (MTX), to the N-terminus of STRAPs. The peptides were tested for their capability to deliver the active drug molecule in breast and cervical cancer cells. MDA-MB-231 and HeLa cells were treated with varying concentrations of MTX and STRAP-MTX conjugates for 72 h. Cell viability was assessed using the MTT assay. The cytotoxicity of MTX increased when delivered as peptide-MTX conjugates to the cells. This resulted in an overall reduction in the inhibitory concentration cytotoxic to 50% of cells (IC50) for MTX. The MTX resistance for MDA-MB-231 cells is well-established. The IC50 for MTX in MDA-MB-231 cells was minimum when delivered as a STRAP-4-MTX conjugate. Similar lowering of IC50 values in HeLa cells was observed. We also treated HEK-293 cells with MTX-STRAP conjugates. The peptide-drug conjugates showed no significant toxicity to the HEK-293 cells under the tested conditions, thereby confirming the earlier reported observation of higher uptake rates in cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Renal cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
Electrostatics modulates the interactions of CPPs with the corresponding cell surface. Therefore, for the design of the amino acid side chain sequences, we reverse-engineered the spatial electrostatic potential distribution of the previously designed peptides with cell penetration ability. The spatial electrostatic fingerprints of the earlier designed peptides were generated as described previously. The reverse-engineered sequences with a shorter (7-mer) chain length were compared to the previous designs through multiple iterations of amino acid sequences. Their respective electrostatic profiles were used for the design of four syndiotactic, cationic, amphipathic peptides with optimal sequence selection. These syndiotactic re-engineered amphipathic peptides (STRAPs) were tested for their ability to penetrate cells and deliver a small molecule (methotrexate). The amino acid constitutions of STRAP-1, STRAP-2, STRAP-3, and STRAP-4 are the same. However, STRAP-2 is the stereochemically reversed STRAP-1; similarly, STRAP-4 is the stereochemically reversed STRAP-3. The use of LDLD and DLDL stereochemical sequences in the design of STRAPs resulted in the differential electrostatic signatures for the same amino acid sequences. The designed peptides have a higher cellular uptake in TNBC cells (MDA-MB-231) than the standard control TAT peptide. They could penetrate cells by both active and passive processes, and their activity is not reduced in biological fluids (human plasma and bovine serum). Furthermore, the delivery of MTX as STRAP-MTX conjugates helped to overcome the drug resistance of the MDA-MB-231 cells under in vitro conditions. The delivery of the STARP-4-MTX conjugate in TNBC xenograft tumors was more effective in the reduction of both the tumor size and metastasis to the lungs, liver, spleen, and lymph nodes.
Click to Show/Hide
|
||||
| Description |
To deliver the functional molecule into the cells, we attached an anticancer drug, methotrexate (MTX), to the N-terminus of STRAPs. The peptides were tested for their capability to deliver the active drug molecule in breast and cervical cancer cells. MDA-MB-231 and HeLa cells were treated with varying concentrations of MTX and STRAP-MTX conjugates for 72 h. Cell viability was assessed using the MTT assay. The cytotoxicity of MTX increased when delivered as peptide-MTX conjugates to the cells. This resulted in an overall reduction in the inhibitory concentration cytotoxic to 50% of cells (IC50) for MTX. The MTX resistance for MDA-MB-231 cells is well-established. The IC50 for MTX in MDA-MB-231 cells was minimum when delivered as a STRAP-4-MTX conjugate. Similar lowering of IC50 values in HeLa cells was observed. We also treated HEK-293 cells with MTX-STRAP conjugates. The peptide-drug conjugates showed no significant toxicity to the HEK-293 cells under the tested conditions, thereby confirming the earlier reported observation of higher uptake rates in cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | HEK293 cell | CVCL_0045 | ||
STRAP-3-MTX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Cervical cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.22 ± 0.18 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
Electrostatics modulates the interactions of CPPs with the corresponding cell surface. Therefore, for the design of the amino acid side chain sequences, we reverse-engineered the spatial electrostatic potential distribution of the previously designed peptides with cell penetration ability. The spatial electrostatic fingerprints of the earlier designed peptides were generated as described previously. The reverse-engineered sequences with a shorter (7-mer) chain length were compared to the previous designs through multiple iterations of amino acid sequences. Their respective electrostatic profiles were used for the design of four syndiotactic, cationic, amphipathic peptides with optimal sequence selection. These syndiotactic re-engineered amphipathic peptides (STRAPs) were tested for their ability to penetrate cells and deliver a small molecule (methotrexate). The amino acid constitutions of STRAP-1, STRAP-2, STRAP-3, and STRAP-4 are the same. However, STRAP-2 is the stereochemically reversed STRAP-1; similarly, STRAP-4 is the stereochemically reversed STRAP-3. The use of LDLD and DLDL stereochemical sequences in the design of STRAPs resulted in the differential electrostatic signatures for the same amino acid sequences. The designed peptides have a higher cellular uptake in TNBC cells (MDA-MB-231) than the standard control TAT peptide. They could penetrate cells by both active and passive processes, and their activity is not reduced in biological fluids (human plasma and bovine serum). Furthermore, the delivery of MTX as STRAP-MTX conjugates helped to overcome the drug resistance of the MDA-MB-231 cells under in vitro conditions. The delivery of the STARP-4-MTX conjugate in TNBC xenograft tumors was more effective in the reduction of both the tumor size and metastasis to the lungs, liver, spleen, and lymph nodes.
Click to Show/Hide
|
||||
| Description |
To deliver the functional molecule into the cells, we attached an anticancer drug, methotrexate (MTX), to the N-terminus of STRAPs. The peptides were tested for their capability to deliver the active drug molecule in breast and cervical cancer cells. MDA-MB-231 and HeLa cells were treated with varying concentrations of MTX and STRAP-MTX conjugates for 72 h. Cell viability was assessed using the MTT assay. The cytotoxicity of MTX increased when delivered as peptide-MTX conjugates to the cells. This resulted in an overall reduction in the inhibitory concentration cytotoxic to 50% of cells (IC50) for MTX. The MTX resistance for MDA-MB-231 cells is well-established. The IC50 for MTX in MDA-MB-231 cells was minimum when delivered as a STRAP-4-MTX conjugate. Similar lowering of IC50 values in HeLa cells was observed. We also treated HEK-293 cells with MTX-STRAP conjugates. The peptide-drug conjugates showed no significant toxicity to the HEK-293 cells under the tested conditions, thereby confirming the earlier reported observation of higher uptake rates in cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.79 ± 0.31 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
Electrostatics modulates the interactions of CPPs with the corresponding cell surface. Therefore, for the design of the amino acid side chain sequences, we reverse-engineered the spatial electrostatic potential distribution of the previously designed peptides with cell penetration ability. The spatial electrostatic fingerprints of the earlier designed peptides were generated as described previously. The reverse-engineered sequences with a shorter (7-mer) chain length were compared to the previous designs through multiple iterations of amino acid sequences. Their respective electrostatic profiles were used for the design of four syndiotactic, cationic, amphipathic peptides with optimal sequence selection. These syndiotactic re-engineered amphipathic peptides (STRAPs) were tested for their ability to penetrate cells and deliver a small molecule (methotrexate). The amino acid constitutions of STRAP-1, STRAP-2, STRAP-3, and STRAP-4 are the same. However, STRAP-2 is the stereochemically reversed STRAP-1; similarly, STRAP-4 is the stereochemically reversed STRAP-3. The use of LDLD and DLDL stereochemical sequences in the design of STRAPs resulted in the differential electrostatic signatures for the same amino acid sequences. The designed peptides have a higher cellular uptake in TNBC cells (MDA-MB-231) than the standard control TAT peptide. They could penetrate cells by both active and passive processes, and their activity is not reduced in biological fluids (human plasma and bovine serum). Furthermore, the delivery of MTX as STRAP-MTX conjugates helped to overcome the drug resistance of the MDA-MB-231 cells under in vitro conditions. The delivery of the STARP-4-MTX conjugate in TNBC xenograft tumors was more effective in the reduction of both the tumor size and metastasis to the lungs, liver, spleen, and lymph nodes.
Click to Show/Hide
|
||||
| Description |
To deliver the functional molecule into the cells, we attached an anticancer drug, methotrexate (MTX), to the N-terminus of STRAPs. The peptides were tested for their capability to deliver the active drug molecule in breast and cervical cancer cells. MDA-MB-231 and HeLa cells were treated with varying concentrations of MTX and STRAP-MTX conjugates for 72 h. Cell viability was assessed using the MTT assay. The cytotoxicity of MTX increased when delivered as peptide-MTX conjugates to the cells. This resulted in an overall reduction in the inhibitory concentration cytotoxic to 50% of cells (IC50) for MTX. The MTX resistance for MDA-MB-231 cells is well-established. The IC50 for MTX in MDA-MB-231 cells was minimum when delivered as a STRAP-4-MTX conjugate. Similar lowering of IC50 values in HeLa cells was observed. We also treated HEK-293 cells with MTX-STRAP conjugates. The peptide-drug conjugates showed no significant toxicity to the HEK-293 cells under the tested conditions, thereby confirming the earlier reported observation of higher uptake rates in cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Renal cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
Electrostatics modulates the interactions of CPPs with the corresponding cell surface. Therefore, for the design of the amino acid side chain sequences, we reverse-engineered the spatial electrostatic potential distribution of the previously designed peptides with cell penetration ability. The spatial electrostatic fingerprints of the earlier designed peptides were generated as described previously. The reverse-engineered sequences with a shorter (7-mer) chain length were compared to the previous designs through multiple iterations of amino acid sequences. Their respective electrostatic profiles were used for the design of four syndiotactic, cationic, amphipathic peptides with optimal sequence selection. These syndiotactic re-engineered amphipathic peptides (STRAPs) were tested for their ability to penetrate cells and deliver a small molecule (methotrexate). The amino acid constitutions of STRAP-1, STRAP-2, STRAP-3, and STRAP-4 are the same. However, STRAP-2 is the stereochemically reversed STRAP-1; similarly, STRAP-4 is the stereochemically reversed STRAP-3. The use of LDLD and DLDL stereochemical sequences in the design of STRAPs resulted in the differential electrostatic signatures for the same amino acid sequences. The designed peptides have a higher cellular uptake in TNBC cells (MDA-MB-231) than the standard control TAT peptide. They could penetrate cells by both active and passive processes, and their activity is not reduced in biological fluids (human plasma and bovine serum). Furthermore, the delivery of MTX as STRAP-MTX conjugates helped to overcome the drug resistance of the MDA-MB-231 cells under in vitro conditions. The delivery of the STARP-4-MTX conjugate in TNBC xenograft tumors was more effective in the reduction of both the tumor size and metastasis to the lungs, liver, spleen, and lymph nodes.
Click to Show/Hide
|
||||
| Description |
To deliver the functional molecule into the cells, we attached an anticancer drug, methotrexate (MTX), to the N-terminus of STRAPs. The peptides were tested for their capability to deliver the active drug molecule in breast and cervical cancer cells. MDA-MB-231 and HeLa cells were treated with varying concentrations of MTX and STRAP-MTX conjugates for 72 h. Cell viability was assessed using the MTT assay. The cytotoxicity of MTX increased when delivered as peptide-MTX conjugates to the cells. This resulted in an overall reduction in the inhibitory concentration cytotoxic to 50% of cells (IC50) for MTX. The MTX resistance for MDA-MB-231 cells is well-established. The IC50 for MTX in MDA-MB-231 cells was minimum when delivered as a STRAP-4-MTX conjugate. Similar lowering of IC50 values in HeLa cells was observed. We also treated HEK-293 cells with MTX-STRAP conjugates. The peptide-drug conjugates showed no significant toxicity to the HEK-293 cells under the tested conditions, thereby confirming the earlier reported observation of higher uptake rates in cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | HEK293 cell | CVCL_0045 | ||
STRAP-1-MTX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Cervical cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.63 ± 0.28 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
Electrostatics modulates the interactions of CPPs with the corresponding cell surface. Therefore, for the design of the amino acid side chain sequences, we reverse-engineered the spatial electrostatic potential distribution of the previously designed peptides with cell penetration ability. The spatial electrostatic fingerprints of the earlier designed peptides were generated as described previously. The reverse-engineered sequences with a shorter (7-mer) chain length were compared to the previous designs through multiple iterations of amino acid sequences. Their respective electrostatic profiles were used for the design of four syndiotactic, cationic, amphipathic peptides with optimal sequence selection. These syndiotactic re-engineered amphipathic peptides (STRAPs) were tested for their ability to penetrate cells and deliver a small molecule (methotrexate). The amino acid constitutions of STRAP-1, STRAP-2, STRAP-3, and STRAP-4 are the same. However, STRAP-2 is the stereochemically reversed STRAP-1; similarly, STRAP-4 is the stereochemically reversed STRAP-3. The use of LDLD and DLDL stereochemical sequences in the design of STRAPs resulted in the differential electrostatic signatures for the same amino acid sequences. The designed peptides have a higher cellular uptake in TNBC cells (MDA-MB-231) than the standard control TAT peptide. They could penetrate cells by both active and passive processes, and their activity is not reduced in biological fluids (human plasma and bovine serum). Furthermore, the delivery of MTX as STRAP-MTX conjugates helped to overcome the drug resistance of the MDA-MB-231 cells under in vitro conditions. The delivery of the STARP-4-MTX conjugate in TNBC xenograft tumors was more effective in the reduction of both the tumor size and metastasis to the lungs, liver, spleen, and lymph nodes.
Click to Show/Hide
|
||||
| Description |
To deliver the functional molecule into the cells, we attached an anticancer drug, methotrexate (MTX), to the N-terminus of STRAPs. The peptides were tested for their capability to deliver the active drug molecule in breast and cervical cancer cells. MDA-MB-231 and HeLa cells were treated with varying concentrations of MTX and STRAP-MTX conjugates for 72 h. Cell viability was assessed using the MTT assay. The cytotoxicity of MTX increased when delivered as peptide-MTX conjugates to the cells. This resulted in an overall reduction in the inhibitory concentration cytotoxic to 50% of cells (IC50) for MTX. The MTX resistance for MDA-MB-231 cells is well-established. The IC50 for MTX in MDA-MB-231 cells was minimum when delivered as a STRAP-4-MTX conjugate. Similar lowering of IC50 values in HeLa cells was observed. We also treated HEK-293 cells with MTX-STRAP conjugates. The peptide-drug conjugates showed no significant toxicity to the HEK-293 cells under the tested conditions, thereby confirming the earlier reported observation of higher uptake rates in cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 2.26 ± 0.31 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
Electrostatics modulates the interactions of CPPs with the corresponding cell surface. Therefore, for the design of the amino acid side chain sequences, we reverse-engineered the spatial electrostatic potential distribution of the previously designed peptides with cell penetration ability. The spatial electrostatic fingerprints of the earlier designed peptides were generated as described previously. The reverse-engineered sequences with a shorter (7-mer) chain length were compared to the previous designs through multiple iterations of amino acid sequences. Their respective electrostatic profiles were used for the design of four syndiotactic, cationic, amphipathic peptides with optimal sequence selection. These syndiotactic re-engineered amphipathic peptides (STRAPs) were tested for their ability to penetrate cells and deliver a small molecule (methotrexate). The amino acid constitutions of STRAP-1, STRAP-2, STRAP-3, and STRAP-4 are the same. However, STRAP-2 is the stereochemically reversed STRAP-1; similarly, STRAP-4 is the stereochemically reversed STRAP-3. The use of LDLD and DLDL stereochemical sequences in the design of STRAPs resulted in the differential electrostatic signatures for the same amino acid sequences. The designed peptides have a higher cellular uptake in TNBC cells (MDA-MB-231) than the standard control TAT peptide. They could penetrate cells by both active and passive processes, and their activity is not reduced in biological fluids (human plasma and bovine serum). Furthermore, the delivery of MTX as STRAP-MTX conjugates helped to overcome the drug resistance of the MDA-MB-231 cells under in vitro conditions. The delivery of the STARP-4-MTX conjugate in TNBC xenograft tumors was more effective in the reduction of both the tumor size and metastasis to the lungs, liver, spleen, and lymph nodes.
Click to Show/Hide
|
||||
| Description |
To deliver the functional molecule into the cells, we attached an anticancer drug, methotrexate (MTX), to the N-terminus of STRAPs. The peptides were tested for their capability to deliver the active drug molecule in breast and cervical cancer cells. MDA-MB-231 and HeLa cells were treated with varying concentrations of MTX and STRAP-MTX conjugates for 72 h. Cell viability was assessed using the MTT assay. The cytotoxicity of MTX increased when delivered as peptide-MTX conjugates to the cells. This resulted in an overall reduction in the inhibitory concentration cytotoxic to 50% of cells (IC50) for MTX. The MTX resistance for MDA-MB-231 cells is well-established. The IC50 for MTX in MDA-MB-231 cells was minimum when delivered as a STRAP-4-MTX conjugate. Similar lowering of IC50 values in HeLa cells was observed. We also treated HEK-293 cells with MTX-STRAP conjugates. The peptide-drug conjugates showed no significant toxicity to the HEK-293 cells under the tested conditions, thereby confirming the earlier reported observation of higher uptake rates in cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Renal cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
Electrostatics modulates the interactions of CPPs with the corresponding cell surface. Therefore, for the design of the amino acid side chain sequences, we reverse-engineered the spatial electrostatic potential distribution of the previously designed peptides with cell penetration ability. The spatial electrostatic fingerprints of the earlier designed peptides were generated as described previously. The reverse-engineered sequences with a shorter (7-mer) chain length were compared to the previous designs through multiple iterations of amino acid sequences. Their respective electrostatic profiles were used for the design of four syndiotactic, cationic, amphipathic peptides with optimal sequence selection. These syndiotactic re-engineered amphipathic peptides (STRAPs) were tested for their ability to penetrate cells and deliver a small molecule (methotrexate). The amino acid constitutions of STRAP-1, STRAP-2, STRAP-3, and STRAP-4 are the same. However, STRAP-2 is the stereochemically reversed STRAP-1; similarly, STRAP-4 is the stereochemically reversed STRAP-3. The use of LDLD and DLDL stereochemical sequences in the design of STRAPs resulted in the differential electrostatic signatures for the same amino acid sequences. The designed peptides have a higher cellular uptake in TNBC cells (MDA-MB-231) than the standard control TAT peptide. They could penetrate cells by both active and passive processes, and their activity is not reduced in biological fluids (human plasma and bovine serum). Furthermore, the delivery of MTX as STRAP-MTX conjugates helped to overcome the drug resistance of the MDA-MB-231 cells under in vitro conditions. The delivery of the STARP-4-MTX conjugate in TNBC xenograft tumors was more effective in the reduction of both the tumor size and metastasis to the lungs, liver, spleen, and lymph nodes.
Click to Show/Hide
|
||||
| Description |
To deliver the functional molecule into the cells, we attached an anticancer drug, methotrexate (MTX), to the N-terminus of STRAPs. The peptides were tested for their capability to deliver the active drug molecule in breast and cervical cancer cells. MDA-MB-231 and HeLa cells were treated with varying concentrations of MTX and STRAP-MTX conjugates for 72 h. Cell viability was assessed using the MTT assay. The cytotoxicity of MTX increased when delivered as peptide-MTX conjugates to the cells. This resulted in an overall reduction in the inhibitory concentration cytotoxic to 50% of cells (IC50) for MTX. The MTX resistance for MDA-MB-231 cells is well-established. The IC50 for MTX in MDA-MB-231 cells was minimum when delivered as a STRAP-4-MTX conjugate. Similar lowering of IC50 values in HeLa cells was observed. We also treated HEK-293 cells with MTX-STRAP conjugates. The peptide-drug conjugates showed no significant toxicity to the HEK-293 cells under the tested conditions, thereby confirming the earlier reported observation of higher uptake rates in cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | HEK293 cell | CVCL_0045 | ||
STRAP-2-MTX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.82 ± 0.24 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
Electrostatics modulates the interactions of CPPs with the corresponding cell surface. Therefore, for the design of the amino acid side chain sequences, we reverse-engineered the spatial electrostatic potential distribution of the previously designed peptides with cell penetration ability. The spatial electrostatic fingerprints of the earlier designed peptides were generated as described previously. The reverse-engineered sequences with a shorter (7-mer) chain length were compared to the previous designs through multiple iterations of amino acid sequences. Their respective electrostatic profiles were used for the design of four syndiotactic, cationic, amphipathic peptides with optimal sequence selection. These syndiotactic re-engineered amphipathic peptides (STRAPs) were tested for their ability to penetrate cells and deliver a small molecule (methotrexate). The amino acid constitutions of STRAP-1, STRAP-2, STRAP-3, and STRAP-4 are the same. However, STRAP-2 is the stereochemically reversed STRAP-1; similarly, STRAP-4 is the stereochemically reversed STRAP-3. The use of LDLD and DLDL stereochemical sequences in the design of STRAPs resulted in the differential electrostatic signatures for the same amino acid sequences. The designed peptides have a higher cellular uptake in TNBC cells (MDA-MB-231) than the standard control TAT peptide. They could penetrate cells by both active and passive processes, and their activity is not reduced in biological fluids (human plasma and bovine serum). Furthermore, the delivery of MTX as STRAP-MTX conjugates helped to overcome the drug resistance of the MDA-MB-231 cells under in vitro conditions. The delivery of the STARP-4-MTX conjugate in TNBC xenograft tumors was more effective in the reduction of both the tumor size and metastasis to the lungs, liver, spleen, and lymph nodes.
Click to Show/Hide
|
||||
| Description |
To deliver the functional molecule into the cells, we attached an anticancer drug, methotrexate (MTX), to the N-terminus of STRAPs. The peptides were tested for their capability to deliver the active drug molecule in breast and cervical cancer cells. MDA-MB-231 and HeLa cells were treated with varying concentrations of MTX and STRAP-MTX conjugates for 72 h. Cell viability was assessed using the MTT assay. The cytotoxicity of MTX increased when delivered as peptide-MTX conjugates to the cells. This resulted in an overall reduction in the inhibitory concentration cytotoxic to 50% of cells (IC50) for MTX. The MTX resistance for MDA-MB-231 cells is well-established. The IC50 for MTX in MDA-MB-231 cells was minimum when delivered as a STRAP-4-MTX conjugate. Similar lowering of IC50 values in HeLa cells was observed. We also treated HEK-293 cells with MTX-STRAP conjugates. The peptide-drug conjugates showed no significant toxicity to the HEK-293 cells under the tested conditions, thereby confirming the earlier reported observation of higher uptake rates in cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Cervical cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 11.8 ± 2.28 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
Electrostatics modulates the interactions of CPPs with the corresponding cell surface. Therefore, for the design of the amino acid side chain sequences, we reverse-engineered the spatial electrostatic potential distribution of the previously designed peptides with cell penetration ability. The spatial electrostatic fingerprints of the earlier designed peptides were generated as described previously. The reverse-engineered sequences with a shorter (7-mer) chain length were compared to the previous designs through multiple iterations of amino acid sequences. Their respective electrostatic profiles were used for the design of four syndiotactic, cationic, amphipathic peptides with optimal sequence selection. These syndiotactic re-engineered amphipathic peptides (STRAPs) were tested for their ability to penetrate cells and deliver a small molecule (methotrexate). The amino acid constitutions of STRAP-1, STRAP-2, STRAP-3, and STRAP-4 are the same. However, STRAP-2 is the stereochemically reversed STRAP-1; similarly, STRAP-4 is the stereochemically reversed STRAP-3. The use of LDLD and DLDL stereochemical sequences in the design of STRAPs resulted in the differential electrostatic signatures for the same amino acid sequences. The designed peptides have a higher cellular uptake in TNBC cells (MDA-MB-231) than the standard control TAT peptide. They could penetrate cells by both active and passive processes, and their activity is not reduced in biological fluids (human plasma and bovine serum). Furthermore, the delivery of MTX as STRAP-MTX conjugates helped to overcome the drug resistance of the MDA-MB-231 cells under in vitro conditions. The delivery of the STARP-4-MTX conjugate in TNBC xenograft tumors was more effective in the reduction of both the tumor size and metastasis to the lungs, liver, spleen, and lymph nodes.
Click to Show/Hide
|
||||
| Description |
To deliver the functional molecule into the cells, we attached an anticancer drug, methotrexate (MTX), to the N-terminus of STRAPs. The peptides were tested for their capability to deliver the active drug molecule in breast and cervical cancer cells. MDA-MB-231 and HeLa cells were treated with varying concentrations of MTX and STRAP-MTX conjugates for 72 h. Cell viability was assessed using the MTT assay. The cytotoxicity of MTX increased when delivered as peptide-MTX conjugates to the cells. This resulted in an overall reduction in the inhibitory concentration cytotoxic to 50% of cells (IC50) for MTX. The MTX resistance for MDA-MB-231 cells is well-established. The IC50 for MTX in MDA-MB-231 cells was minimum when delivered as a STRAP-4-MTX conjugate. Similar lowering of IC50 values in HeLa cells was observed. We also treated HEK-293 cells with MTX-STRAP conjugates. The peptide-drug conjugates showed no significant toxicity to the HEK-293 cells under the tested conditions, thereby confirming the earlier reported observation of higher uptake rates in cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Renal cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
Electrostatics modulates the interactions of CPPs with the corresponding cell surface. Therefore, for the design of the amino acid side chain sequences, we reverse-engineered the spatial electrostatic potential distribution of the previously designed peptides with cell penetration ability. The spatial electrostatic fingerprints of the earlier designed peptides were generated as described previously. The reverse-engineered sequences with a shorter (7-mer) chain length were compared to the previous designs through multiple iterations of amino acid sequences. Their respective electrostatic profiles were used for the design of four syndiotactic, cationic, amphipathic peptides with optimal sequence selection. These syndiotactic re-engineered amphipathic peptides (STRAPs) were tested for their ability to penetrate cells and deliver a small molecule (methotrexate). The amino acid constitutions of STRAP-1, STRAP-2, STRAP-3, and STRAP-4 are the same. However, STRAP-2 is the stereochemically reversed STRAP-1; similarly, STRAP-4 is the stereochemically reversed STRAP-3. The use of LDLD and DLDL stereochemical sequences in the design of STRAPs resulted in the differential electrostatic signatures for the same amino acid sequences. The designed peptides have a higher cellular uptake in TNBC cells (MDA-MB-231) than the standard control TAT peptide. They could penetrate cells by both active and passive processes, and their activity is not reduced in biological fluids (human plasma and bovine serum). Furthermore, the delivery of MTX as STRAP-MTX conjugates helped to overcome the drug resistance of the MDA-MB-231 cells under in vitro conditions. The delivery of the STARP-4-MTX conjugate in TNBC xenograft tumors was more effective in the reduction of both the tumor size and metastasis to the lungs, liver, spleen, and lymph nodes.
Click to Show/Hide
|
||||
| Description |
To deliver the functional molecule into the cells, we attached an anticancer drug, methotrexate (MTX), to the N-terminus of STRAPs. The peptides were tested for their capability to deliver the active drug molecule in breast and cervical cancer cells. MDA-MB-231 and HeLa cells were treated with varying concentrations of MTX and STRAP-MTX conjugates for 72 h. Cell viability was assessed using the MTT assay. The cytotoxicity of MTX increased when delivered as peptide-MTX conjugates to the cells. This resulted in an overall reduction in the inhibitory concentration cytotoxic to 50% of cells (IC50) for MTX. The MTX resistance for MDA-MB-231 cells is well-established. The IC50 for MTX in MDA-MB-231 cells was minimum when delivered as a STRAP-4-MTX conjugate. Similar lowering of IC50 values in HeLa cells was observed. We also treated HEK-293 cells with MTX-STRAP conjugates. The peptide-drug conjugates showed no significant toxicity to the HEK-293 cells under the tested conditions, thereby confirming the earlier reported observation of higher uptake rates in cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | HEK293 cell | CVCL_0045 | ||
PDC-5d [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [90] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 2.0 (5.7 ± 0.1) μM | |||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-468 cell | CVCL_0419 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [90] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 4.2 (5.4 ± 0.1) μM | |||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [90] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 5.0 (5.3 ± 0.1) μM | |||
| In Vitro Model | Invasive breast carcinoma | T-47D cell | CVCL_0553 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [90] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) | 8.9 nM | |||
| In Vitro Model | Normal | COS-7 cell | CVCL_0224 | ||
PDC-5b [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [90] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 3.6 (5.4 ± 0.1) μM | |||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-468 cell | CVCL_0419 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [90] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 7.7 (5.1 ± 0.1) μM | |||
| In Vitro Model | Invasive breast carcinoma | T-47D cell | CVCL_0553 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [90] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 8.1 (5.1 ± 0.1) μM | |||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [90] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) | 3.0 (8.5 ± 0.1) nM | |||
| In Vitro Model | Normal | COS-7 cell | CVCL_0224 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [90] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) | 226 (6.6 ± 0.1) nM | |||
| In Vitro Model | Normal | COS-7 cell | CVCL_0224 | ||
PDC-5c [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [90] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 3.8 (5.4 ± 0.1) μM | |||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [90] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 4.0 (5.4 ± 0.1) μM | |||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-468 cell | CVCL_0419 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [90] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 10 µM | |||
| In Vitro Model | Invasive breast carcinoma | T-47D cell | CVCL_0553 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [90] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) | 2.8 (8.5 ± 0.1) nM | |||
| In Vitro Model | Normal | COS-7 cell | CVCL_0224 | ||
PDC-5a [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [90] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 18 (4.8 ± 0.1) μM | |||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-468 cell | CVCL_0419 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [90] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 25 µM | |||
| In Vitro Model | Invasive breast carcinoma | T-47D cell | CVCL_0553 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [90] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 25 µM | |||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [90] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) | 2.9 (8.5 ± 0.1) nM | |||
| In Vitro Model | Normal | COS-7 cell | CVCL_0224 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [90] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) | 209 (6.7 ± 0.1) nM | |||
| In Vitro Model | Normal | COS-7 cell | CVCL_0224 | ||
MTX-cLABL [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Inflammation | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 100 µM | |||
| Evaluation Method | Propidium iodide (PI) assay | ||||
| Administration Time | 1 h | ||||
| MOA of PDC |
In this study, cLABL and cLBEL peptides were linked to methotrexate (MTX) to produce MTX-cLABL and MTX-cLBEL conjugates. The goal was to target MTX to human coronary artery endothelial cells (HCAEC) via the ICAM-1 receptor to lower MTX toxicity and side effects. The biological abilities of MTX-cLABL, MTX-cLBEL, cLABL, cLBEL, and MTX were compared by their activities to inhibit binding of anti-ICAM-1 mAb to ICAM-1 on the surface of HCAEC. In addition, these molecules were compared in inhibiting T cell adhesion to HCAEC monolayers. Finally, their activities in suppressing IL-6 and IL-8 production as inflammatory cytokines were determined. The toxicities of MTX-cLABL and MTX-cLBEL conjugates were also determined relative to MTX alone as well as cLABL and cLBEL peptides.
Click to Show/Hide
|
||||
| Description |
We next determined whether treatment of HCAEC and Molt-3 T cells with peptides, MTX, and MTX-peptide conjugates resulted in inhibition of cell proliferation. Both HCAEC and Molt-3 T cells were affected by test compound in different levels. None of the molecules caused growth stimulation or total culture extinction. A net cell killing of HCAEC was observed upon treatment with MTX at all test concentrations while MTX affected net killing at ≥1.0 uM in Molt-3 T cells. The MTX-peptide conjugates were less toxic than MTX. In HCAEC, the net cell killing was at lower concentration for MTX at ≥0.1 uM compared to MTX-peptide conjugates at ≥500 uM. The net cell killing of Molt-3 T cells was found at ≥1.0 uM for MTX and ≥50 uM for MTX-peptide conjugates. For all test concentrations, the conjugates only resulted in HCAEC partial growth inhibition. For Molt-3 T cells, a total growth inhibition emerged at 100 uM for cLABL and cLBEL; however, 500 uM cLABL and cLBEL did not cause total cell killing for T cells.
Click to Show/Hide
|
||||
| In Vitro Model | human coronary artery endothelial cell | Human coronary artery endothelial cell | Homo sapiens | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Inflammation | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 500 µM | |||
| Evaluation Method | Propidium iodide (PI) assay | ||||
| Administration Time | 1 h | ||||
| MOA of PDC |
In this study, cLABL and cLBEL peptides were linked to methotrexate (MTX) to produce MTX-cLABL and MTX-cLBEL conjugates. The goal was to target MTX to human coronary artery endothelial cells (HCAEC) via the ICAM-1 receptor to lower MTX toxicity and side effects. The biological abilities of MTX-cLABL, MTX-cLBEL, cLABL, cLBEL, and MTX were compared by their activities to inhibit binding of anti-ICAM-1 mAb to ICAM-1 on the surface of HCAEC. In addition, these molecules were compared in inhibiting T cell adhesion to HCAEC monolayers. Finally, their activities in suppressing IL-6 and IL-8 production as inflammatory cytokines were determined. The toxicities of MTX-cLABL and MTX-cLBEL conjugates were also determined relative to MTX alone as well as cLABL and cLBEL peptides.
Click to Show/Hide
|
||||
| Description |
We next determined whether treatment of HCAEC and Molt-3 T cells with peptides, MTX, and MTX-peptide conjugates resulted in inhibition of cell proliferation. Both HCAEC and Molt-3 T cells were affected by test compound in different levels. None of the molecules caused growth stimulation or total culture extinction. A net cell killing of HCAEC was observed upon treatment with MTX at all test concentrations while MTX affected net killing at ≥1.0 uM in Molt-3 T cells. The MTX-peptide conjugates were less toxic than MTX. In HCAEC, the net cell killing was at lower concentration for MTX at ≥0.1 uM compared to MTX-peptide conjugates at ≥500 uM. The net cell killing of Molt-3 T cells was found at ≥1.0 uM for MTX and ≥50 uM for MTX-peptide conjugates. For all test concentrations, the conjugates only resulted in HCAEC partial growth inhibition. For Molt-3 T cells, a total growth inhibition emerged at 100 uM for cLABL and cLBEL; however, 500 uM cLABL and cLBEL did not cause total cell killing for T cells.
Click to Show/Hide
|
||||
| In Vitro Model | Adult T acute lymphoblastic leukemia | Molt-3 T cell | CVCL_0624 | ||
MTX-cLBEL [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Inflammation | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 100 µM | |||
| Evaluation Method | Propidium iodide (PI) assay | ||||
| Administration Time | 1 h | ||||
| MOA of PDC |
In this study, cLABL and cLBEL peptides were linked to methotrexate (MTX) to produce MTX-cLABL and MTX-cLBEL conjugates. The goal was to target MTX to human coronary artery endothelial cells (HCAEC) via the ICAM-1 receptor to lower MTX toxicity and side effects. The biological abilities of MTX-cLABL, MTX-cLBEL, cLABL, cLBEL, and MTX were compared by their activities to inhibit binding of anti-ICAM-1 mAb to ICAM-1 on the surface of HCAEC. In addition, these molecules were compared in inhibiting T cell adhesion to HCAEC monolayers. Finally, their activities in suppressing IL-6 and IL-8 production as inflammatory cytokines were determined. The toxicities of MTX-cLABL and MTX-cLBEL conjugates were also determined relative to MTX alone as well as cLABL and cLBEL peptides.
Click to Show/Hide
|
||||
| Description |
We next determined whether treatment of HCAEC and Molt-3 T cells with peptides, MTX, and MTX-peptide conjugates resulted in inhibition of cell proliferation. Both HCAEC and Molt-3 T cells were affected by test compound in different levels. None of the molecules caused growth stimulation or total culture extinction. A net cell killing of HCAEC was observed upon treatment with MTX at all test concentrations while MTX affected net killing at ≥1.0 uM in Molt-3 T cells. The MTX-peptide conjugates were less toxic than MTX. In HCAEC, the net cell killing was at lower concentration for MTX at ≥0.1 uM compared to MTX-peptide conjugates at ≥500 uM. The net cell killing of Molt-3 T cells was found at ≥1.0 uM for MTX and ≥50 uM for MTX-peptide conjugates. For all test concentrations, the conjugates only resulted in HCAEC partial growth inhibition. For Molt-3 T cells, a total growth inhibition emerged at 100 uM for cLABL and cLBEL; however, 500 uM cLABL and cLBEL did not cause total cell killing for T cells.
Click to Show/Hide
|
||||
| In Vitro Model | human coronary artery endothelial cell | Human coronary artery endothelial cell | Homo sapiens | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Inflammation | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 500 µM | |||
| Evaluation Method | Propidium iodide (PI) assay | ||||
| Administration Time | 1 h | ||||
| MOA of PDC |
In this study, cLABL and cLBEL peptides were linked to methotrexate (MTX) to produce MTX-cLABL and MTX-cLBEL conjugates. The goal was to target MTX to human coronary artery endothelial cells (HCAEC) via the ICAM-1 receptor to lower MTX toxicity and side effects. The biological abilities of MTX-cLABL, MTX-cLBEL, cLABL, cLBEL, and MTX were compared by their activities to inhibit binding of anti-ICAM-1 mAb to ICAM-1 on the surface of HCAEC. In addition, these molecules were compared in inhibiting T cell adhesion to HCAEC monolayers. Finally, their activities in suppressing IL-6 and IL-8 production as inflammatory cytokines were determined. The toxicities of MTX-cLABL and MTX-cLBEL conjugates were also determined relative to MTX alone as well as cLABL and cLBEL peptides.
Click to Show/Hide
|
||||
| Description |
We next determined whether treatment of HCAEC and Molt-3 T cells with peptides, MTX, and MTX-peptide conjugates resulted in inhibition of cell proliferation. Both HCAEC and Molt-3 T cells were affected by test compound in different levels. None of the molecules caused growth stimulation or total culture extinction. A net cell killing of HCAEC was observed upon treatment with MTX at all test concentrations while MTX affected net killing at ≥1.0 uM in Molt-3 T cells. The MTX-peptide conjugates were less toxic than MTX. In HCAEC, the net cell killing was at lower concentration for MTX at ≥0.1 uM compared to MTX-peptide conjugates at ≥500 uM. The net cell killing of Molt-3 T cells was found at ≥1.0 uM for MTX and ≥50 uM for MTX-peptide conjugates. For all test concentrations, the conjugates only resulted in HCAEC partial growth inhibition. For Molt-3 T cells, a total growth inhibition emerged at 100 uM for cLABL and cLBEL; however, 500 uM cLABL and cLBEL did not cause total cell killing for T cells.
Click to Show/Hide
|
||||
| In Vitro Model | Adult T acute lymphoblastic leukemia | Molt-3 T cell | CVCL_0624 | ||
References