
Linker Information
General Information of This Linker
| Linker ID |
LIN00022
|
|||||
|---|---|---|---|---|---|---|
| Linker Name |
SPDP
|
|||||
| Linker Type |
GSH concentration-sensitive linkers
|
|||||
| Structure |
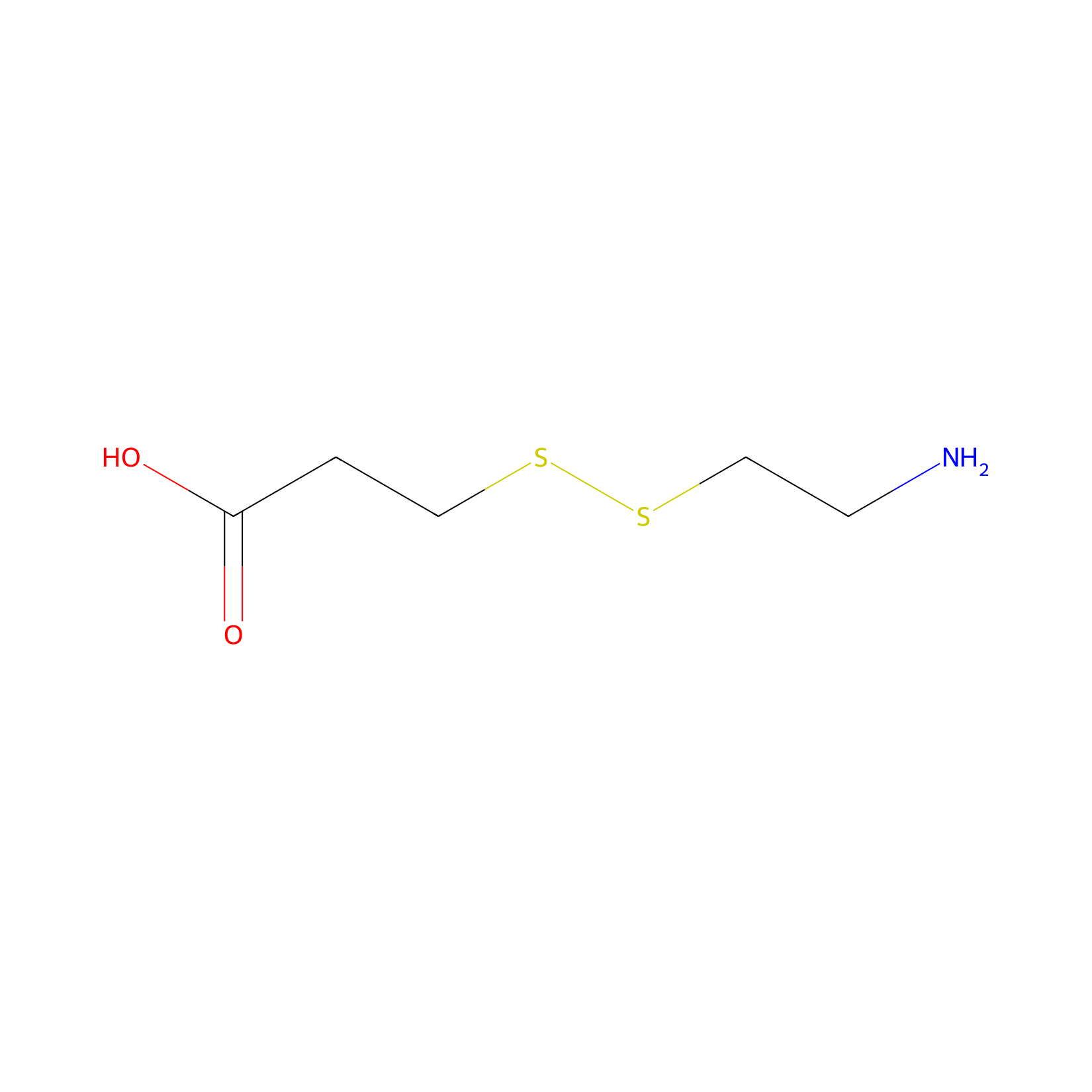
|
|||||
| Formula |
C5H11NO2S2
|
|||||
| #Ro5 Violations (Lipinski): 1 | Molecular Weight (mw) | 181.3 | ||||
| Lipid-water partition coefficient (xlogp) | -2.8 | |||||
| Hydrogen Bond Donor Count (hbonddonor) | 2 | |||||
| Hydrogen Bond Acceptor Count (hbondacc) | 5 | |||||
| Rotatable Bond Count (rotbonds) | 6 | |||||
| PubChem CID | ||||||
| Canonical smiles |
C(CSSCCN)C(=O)O
|
|||||
| InChI |
InChI=1S/C5H11NO2S2/c6-2-4-10-9-3-1-5(7)8/h1-4,6H2,(H,7,8)
|
|||||
| InChIKey |
HMMFDEBVQNRZLJ-UHFFFAOYSA-N
|
|||||
| IUPAC Name |
3-(2-aminoethyldisulfanyl)propanoic acid
|
|||||
Each Peptide-drug Conjugate Related to This Linker
Full Information of The Activity Data of The PDC(s) Related to This Linker
An2-M6G [Preclinical]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Severe pain | ||||
| Efficacy Data | Maximal antinociceptive effect |
34.90%
|
|||
| Administration Time | Intravenous administration 60 min | ||||
| Administration Dosage | 4 mg/kg | ||||
| Evaluation Method | Rat tail-flick test assay | ||||
| MOA of PDC |
Given the high analgesic potency of M6G, without induction of the M3G metabolite that antagonizes the analgesic effect of morphine, M6G could be a promising drug to treat moderate to severe pain. The major issue of systemic use of M6G is its poor BBB permeability. In this study, we proposed to increase the BBB penetration of M6G and morphine by conjugation to the shuttle angiopep-2 peptide (An2). Morphine and M6G were first conjugated to An2, a 19-mer peptide that crosses the BBB by low-density lipoprotein receptor-related protein 1 (LRP1) receptor-mediated transcytosis.
Click to Show/Hide
|
||||
| Description |
Indeed, at an equimolar dose of 3 mg/kg of morphine (i.e., 12 mg/kg), An2-M6G produced a latency to tail withdrawal reaching the cutoff (i.e., 10 seconds) after 30 minutes, an effect lasting at least 3 hours. The %MPE calculated at 60 minutes after the intravenous injection of An2-M6G at 4, 8, and 12 mg/kg (equivalent to 1, 2, and 3 mg/kg of morphine and to 1.5, 3, and 4.5 mg/kg of M6G) was 34.9%, 66.2%, and 100%, respectively.
Click to Show/Hide
|
||||
| In Vivo Model | Rat model. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Severe pain | ||||
| Efficacy Data | Maximal antinociceptive effect |
66.20%
|
|||
| Administration Time | Intravenous administration 60 min | ||||
| Administration Dosage | 8 mg/kg | ||||
| Evaluation Method | Rat tail-flick test assay | ||||
| MOA of PDC |
Given the high analgesic potency of M6G, without induction of the M3G metabolite that antagonizes the analgesic effect of morphine, M6G could be a promising drug to treat moderate to severe pain. The major issue of systemic use of M6G is its poor BBB permeability. In this study, we proposed to increase the BBB penetration of M6G and morphine by conjugation to the shuttle angiopep-2 peptide (An2). Morphine and M6G were first conjugated to An2, a 19-mer peptide that crosses the BBB by low-density lipoprotein receptor-related protein 1 (LRP1) receptor-mediated transcytosis.
Click to Show/Hide
|
||||
| Description |
Indeed, at an equimolar dose of 3 mg/kg of morphine (i.e., 12 mg/kg), An2-M6G produced a latency to tail withdrawal reaching the cutoff (i.e., 10 seconds) after 30 minutes, an effect lasting at least 3 hours. The %MPE calculated at 60 minutes after the intravenous injection of An2-M6G at 4, 8, and 12 mg/kg (equivalent to 1, 2, and 3 mg/kg of morphine and to 1.5, 3, and 4.5 mg/kg of M6G) was 34.9%, 66.2%, and 100%, respectively.
Click to Show/Hide
|
||||
| In Vivo Model | Rat model. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Severe pain | ||||
| Efficacy Data | Maximal antinociceptive effect |
74%
|
|||
| Administration Time | Subcutaneous administration 200 min | ||||
| Administration Dosage | 12 mg/kg | ||||
| Evaluation Method | Rat tail-flick test assay | ||||
| MOA of PDC |
Given the high analgesic potency of M6G, without induction of the M3G metabolite that antagonizes the analgesic effect of morphine, M6G could be a promising drug to treat moderate to severe pain. The major issue of systemic use of M6G is its poor BBB permeability. In this study, we proposed to increase the BBB penetration of M6G and morphine by conjugation to the shuttle angiopep-2 peptide (An2). Morphine and M6G were first conjugated to An2, a 19-mer peptide that crosses the BBB by low-density lipoprotein receptor-related protein 1 (LRP1) receptor-mediated transcytosis.
Click to Show/Hide
|
||||
| Description |
We also measured the analgesic effect of An2-morphine and An2-M6G after subcutaneous injections. Despite similar MPE at the peak effect, subcutaneous injection of 20 mg/kg An2-morphine (equivalent to 5.5 mg/kg of morphine) produced an analgesic effect that was more prolonged over the time than what was observed with an equimolar dose of morphine.
|
||||
| In Vivo Model | Rat model. | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Severe pain | ||||
| Efficacy Data | Maximal antinociceptive effect |
100%
|
|||
| Administration Time | Intravenous administration 60 min | ||||
| Administration Dosage | 12 mg/kg | ||||
| Evaluation Method | Rat tail-flick test assay | ||||
| MOA of PDC |
Given the high analgesic potency of M6G, without induction of the M3G metabolite that antagonizes the analgesic effect of morphine, M6G could be a promising drug to treat moderate to severe pain. The major issue of systemic use of M6G is its poor BBB permeability. In this study, we proposed to increase the BBB penetration of M6G and morphine by conjugation to the shuttle angiopep-2 peptide (An2). Morphine and M6G were first conjugated to An2, a 19-mer peptide that crosses the BBB by low-density lipoprotein receptor-related protein 1 (LRP1) receptor-mediated transcytosis.
Click to Show/Hide
|
||||
| Description |
Indeed, at an equimolar dose of 3 mg/kg of morphine (i.e., 12 mg/kg), An2-M6G produced a latency to tail withdrawal reaching the cutoff (i.e., 10 seconds) after 30 minutes, an effect lasting at least 3 hours. The %MPE calculated at 60 minutes after the intravenous injection of An2-M6G at 4, 8, and 12 mg/kg (equivalent to 1, 2, and 3 mg/kg of morphine and to 1.5, 3, and 4.5 mg/kg of M6G) was 34.9%, 66.2%, and 100%, respectively.
Click to Show/Hide
|
||||
| In Vivo Model | Rat model. | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Severe pain | ||||
| Efficacy Data | Maximal antinociceptive effect |
100%
|
|||
| Administration Time | Intravenous administration 30 min | ||||
| Administration Dosage | 6 mg/kg | ||||
| Evaluation Method | Hot-plate test assay | ||||
| MOA of PDC |
Given the high analgesic potency of M6G, without induction of the M3G metabolite that antagonizes the analgesic effect of morphine, M6G could be a promising drug to treat moderate to severe pain. The major issue of systemic use of M6G is its poor BBB permeability. In this study, we proposed to increase the BBB penetration of M6G and morphine by conjugation to the shuttle angiopep-2 peptide (An2). Morphine and M6G were first conjugated to An2, a 19-mer peptide that crosses the BBB by low-density lipoprotein receptor-related protein 1 (LRP1) receptor-mediated transcytosis.
Click to Show/Hide
|
||||
| Description |
Similar results were also obtained in the hot-plate test using male CD1 mice. Over a 2-hour period, both morphine and An2-morphine caused similar increases in hot-plate latencies. Likewise, mice receiving An2-M6G (6 mg/kg i.v.) also exhibited a sustained and superior analgesic effect compared with equimolar doses of either morphine or M6G.
|
||||
| In Vivo Model | Male CD1 mice. | ||||
LT7-SS-DOX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Proliferation inhibitory activity |
41.00%
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
Both conjugates exhibited targeted antiproliferative effects on TfR overexpressed tumor cells and little toxicity to TfR low-expressed normal cells compared with free DOX. Moreover, the DT7-SS-DOX conjugate possessed higher serum stability, more sustained reduction-triggered drug release characteristics, and stronger in vitro antiproliferative activity as compared to LT7-SS-DOX.
Click to Show/Hide
|
||||
| Description |
Even at an equal DOX concentration of 40 μM, the cell viability of the three types of tumor cells after exposure to this conjugate for 48 h were 41.0%, 61.5%, and 67.2%, respectively.
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Proliferation inhibitory activity |
61.50%
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
Both conjugates exhibited targeted antiproliferative effects on TfR overexpressed tumor cells and little toxicity to TfR low-expressed normal cells compared with free DOX. Moreover, the DT7-SS-DOX conjugate possessed higher serum stability, more sustained reduction-triggered drug release characteristics, and stronger in vitro antiproliferative activity as compared to LT7-SS-DOX.
Click to Show/Hide
|
||||
| Description |
Even at an equal DOX concentration of 40 μM, the cell viability of the three types of tumor cells after exposure to this conjugate for 48 h were 41.0%, 61.5%, and 67.2%, respectively.
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Lung adenocarcinoma | ||||
| Efficacy Data | Proliferation inhibitory activity |
67.20%
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
Both conjugates exhibited targeted antiproliferative effects on TfR overexpressed tumor cells and little toxicity to TfR low-expressed normal cells compared with free DOX. Moreover, the DT7-SS-DOX conjugate possessed higher serum stability, more sustained reduction-triggered drug release characteristics, and stronger in vitro antiproliferative activity as compared to LT7-SS-DOX.
Click to Show/Hide
|
||||
| Description |
Even at an equal DOX concentration of 40 μM, the cell viability of the three types of tumor cells after exposure to this conjugate for 48 h were 41.0%, 61.5%, and 67.2%, respectively.
|
||||
| In Vitro Model | Lung adenocarcinoma | A-549 cell | CVCL_0023 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Proliferation inhibitory activity |
73.10%
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
Both conjugates exhibited targeted antiproliferative effects on TfR overexpressed tumor cells and little toxicity to TfR low-expressed normal cells compared with free DOX. Moreover, the DT7-SS-DOX conjugate possessed higher serum stability, more sustained reduction-triggered drug release characteristics, and stronger in vitro antiproliferative activity as compared to LT7-SS-DOX.
Click to Show/Hide
|
||||
| Description |
The proliferation inhibitory activity of LT7-SS-DOX was the weakest among the three drugs because the cell viabilities of U87, HepG2, and A549 cells after incubation with LT7-SS-DOX (equal DOX concentration of 20 μM) for 48 h were 95.1%, 73.1%, and 83.2%, respectively.
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Lung adenocarcinoma | ||||
| Efficacy Data | Proliferation inhibitory activity |
83.20%
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
Both conjugates exhibited targeted antiproliferative effects on TfR overexpressed tumor cells and little toxicity to TfR low-expressed normal cells compared with free DOX. Moreover, the DT7-SS-DOX conjugate possessed higher serum stability, more sustained reduction-triggered drug release characteristics, and stronger in vitro antiproliferative activity as compared to LT7-SS-DOX.
Click to Show/Hide
|
||||
| Description |
The proliferation inhibitory activity of LT7-SS-DOX was the weakest among the three drugs because the cell viabilities of U87, HepG2, and A549 cells after incubation with LT7-SS-DOX (equal DOX concentration of 20 μM) for 48 h were 95.1%, 73.1%, and 83.2%, respectively.
|
||||
| In Vitro Model | Lung adenocarcinoma | A-549 cell | CVCL_0023 | ||
DT7-SS-DOX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Proliferation inhibitory activity |
95.10%
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
Both conjugates exhibited targeted antiproliferative effects on TfR overexpressed tumor cells and little toxicity to TfR low-expressed normal cells compared with free DOX. Moreover, the DT7-SS-DOX conjugate possessed higher serum stability, more sustained reduction-triggered drug release characteristics, and stronger in vitro antiproliferative activity as compared to LT7-SS-DOX.
Click to Show/Hide
|
||||
| Description |
The proliferation inhibitory activity of LT7-SS-DOX was the weakest among the three drugs because the cell viabilities of U87, HepG2, and A549 cells after incubation with LT7-SS-DOX (equal DOX concentration of 20 μM) for 48 h were 95.1%, 73.1%, and 83.2%, respectively.
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
5.70 ± 0.22 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
Both conjugates exhibited targeted antiproliferative effects on TfR overexpressed tumor cells and little toxicity to TfR low-expressed normal cells compared with free DOX. Moreover, the DT7-SS-DOX conjugate possessed higher serum stability, more sustained reduction-triggered drug release characteristics, and stronger in vitro antiproliferative activity as compared to LT7-SS-DOX.
Click to Show/Hide
|
||||
| Description |
DT7-SS-DOX exhibited good in vitro antiproliferative activity against the three tumor cell lines, with IC50 values of 5.70 ± 0.22 μM (U87), 7.01 ± 1.64 μM (HepG2), and 20.61 ± 4.81 μM (A549), respectively.
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
7.01 ± 1.64 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
Both conjugates exhibited targeted antiproliferative effects on TfR overexpressed tumor cells and little toxicity to TfR low-expressed normal cells compared with free DOX. Moreover, the DT7-SS-DOX conjugate possessed higher serum stability, more sustained reduction-triggered drug release characteristics, and stronger in vitro antiproliferative activity as compared to LT7-SS-DOX.
Click to Show/Hide
|
||||
| Description |
DT7-SS-DOX exhibited good in vitro antiproliferative activity against the three tumor cell lines, with IC50 values of 5.70 ± 0.22 μM (U87), 7.01 ± 1.64 μM (HepG2), and 20.61 ± 4.81 μM (A549), respectively.
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Lung adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
20.61 ± 4.81 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
Both conjugates exhibited targeted antiproliferative effects on TfR overexpressed tumor cells and little toxicity to TfR low-expressed normal cells compared with free DOX. Moreover, the DT7-SS-DOX conjugate possessed higher serum stability, more sustained reduction-triggered drug release characteristics, and stronger in vitro antiproliferative activity as compared to LT7-SS-DOX.
Click to Show/Hide
|
||||
| Description |
DT7-SS-DOX exhibited good in vitro antiproliferative activity against the three tumor cell lines, with IC50 values of 5.70 ± 0.22 μM (U87), 7.01 ± 1.64 μM (HepG2), and 20.61 ± 4.81 μM (A549), respectively.
|
||||
| In Vitro Model | Lung adenocarcinoma | A-549 cell | CVCL_0023 | ||
References