
Linker Information
General Information of This Linker
| Linker ID |
LIN00039
|
|||||
|---|---|---|---|---|---|---|
| Linker Name |
2-(Disulfanyl)ethyl hydrogen carbonate
|
|||||
| Linker Type |
GSH concentration-sensitive linkers
|
|||||
| Structure |
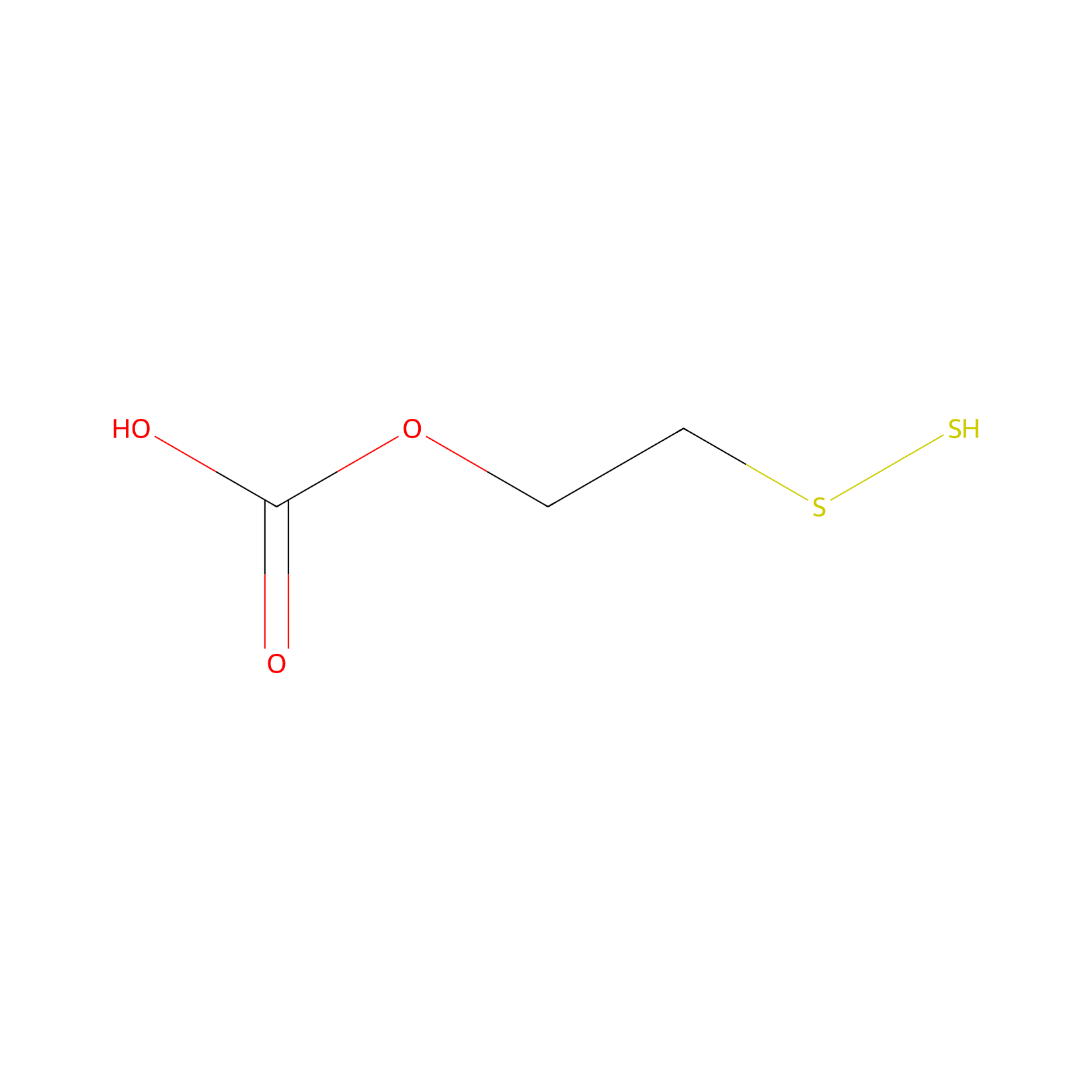
|
|||||
| Formula |
C3H6O3S2
|
|||||
| #Ro5 Violations (Lipinski): 0 | Molecular Weight (mw) | 154.21 | ||||
| Lipid-water partition coefficient (xlogp) | 0.9 | |||||
| Hydrogen Bond Donor Count (hbonddonor) | 2 | |||||
| Hydrogen Bond Acceptor Count (hbondacc) | 5 | |||||
| Rotatable Bond Count (rotbonds) | 4 | |||||
| PubChem CID | ||||||
| Canonical smiles |
C(CSS)OC(=O)O
|
|||||
| InChI |
InChI=1S/C3H6O3S2/c4-3(5)6-1-2-8-7/h7H,1-2H2,(H,4,5)
|
|||||
| InChIKey |
GIRKPUUKWCSTPG-UHFFFAOYSA-N
|
|||||
| IUPAC Name |
2-(disulfanyl)ethyl hydrogen carbonate
|
|||||
Each Peptide-drug Conjugate Related to This Linker
Full Information of The Activity Data of The PDC(s) Related to This Linker
diCPTiRGD [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Tumer volume |
0 mm3
|
|||
| Administration Time | 30 days | ||||
| MOA of PDC |
In this context, we developed a drug-bearing supramolecular hydrogel system to intratumourally (i.t.) deliver CDNs against malignant tumours to achieve cancer chemoimmunotherapy. Our strategy was to chemically conjugate the hydrophilic pep-tide moiety iRGD (a tumour-penetrating peptide that can bind to neuropilin-1 (NRP-1) and trigger tumour tissue penetration) to the hydrophobic anticancer drug CPT to form a self-assembling and self-formulating peptide-drug conjugate (diCPT-iRGD). In aqueous solution, the designed drug amphiphile spontaneously assembles into supramolecular nanotubes (NTs). The negatively charged STING agonist (c-di-AMP (CDA)) can condense on the surface of these positively charged NTs through electrostatic complexations. After injection into the tumour site, the CDA-NT solution can immediately form a hydrogel, functioning as a local reservoir for extended localized release of CDA and CPT to awaken both the innate and adaptive immune systems.
Click to Show/Hide
|
||||
| Description |
When using the diCPT-iRGD NT hydrogel, the CDA-NT treatment led to substantial tumour regression (Fig. 3c-e) and demonstrated a 100% survival rate in mice (Fig. 3f).
|
||||
| In Vivo Model | GL-261 brain cancer C57BL/6 mice. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Survival rate |
100.00%
|
|||
| Administration Time | 30 days | ||||
| MOA of PDC |
In this context, we developed a drug-bearing supramolecular hydrogel system to intratumourally (i.t.) deliver CDNs against malignant tumours to achieve cancer chemoimmunotherapy. Our strategy was to chemically conjugate the hydrophilic pep-tide moiety iRGD (a tumour-penetrating peptide that can bind to neuropilin-1 (NRP-1) and trigger tumour tissue penetration) to the hydrophobic anticancer drug CPT to form a self-assembling and self-formulating peptide-drug conjugate (diCPT-iRGD). In aqueous solution, the designed drug amphiphile spontaneously assembles into supramolecular nanotubes (NTs). The negatively charged STING agonist (c-di-AMP (CDA)) can condense on the surface of these positively charged NTs through electrostatic complexations. After injection into the tumour site, the CDA-NT solution can immediately form a hydrogel, functioning as a local reservoir for extended localized release of CDA and CPT to awaken both the innate and adaptive immune systems.
Click to Show/Hide
|
||||
| Description |
When using the diCPT-iRGD NT hydrogel, the CDA-NT treatment led to substantial tumour regression (Fig. 3c-e) and demonstrated a 100% survival rate in mice (Fig. 3f).
|
||||
| In Vivo Model | GL-261 brain cancer C57BL/6 mice. | ||||
I-4 (NPNWGRSWYNQRFKGC=(-SS-O-COO-CPT)GC=(-SS-O-COO-CPT)GRIKPRKGYTR) [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
0.12 ± 0.07 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.27 ± 0.38 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.78 ± 0.47 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
7.72 ± 0.92 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
17.14 ± 2.42 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
SMAC-P1 CPT conjugates 4 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
0.53 ± 0.17 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.58 ± 0.41 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.50 ± 0.78 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
17.76 ± 1.35 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
18.44 ± 2.09 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
SMAC-P1 CPT conjugates 7 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
0.58 ± 0.21 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.86 ± 1.84 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4.73 ± 1.55 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
6.17 ± 1.21 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
15.15 ± 1.44 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
SMAC-P1 CPT conjugates 1 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
0.85 ± 0.23 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.04 ± 0.75 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4.03 ± 0.25 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
6.43 ± 1.20 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
18.23 ± 2.06 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In this study, a series of conjugates were designed and synthesized by fusing the SMAC peptide and P1 target peptide, and coupling with CPT, with the hope that they can play a synergistic role in promoting cell apoptosis. The in vitro anti-tumor activity study found that the better conjugate 4 had the best activity on HER2-positive cells, while it was less toxic to HER2-negative cells. From the experimental results, the coupling position of CPT affected the activity of the conjugates. Conjugates 1, 4 and 7, which connected the load in the same way, had different inhibitory activities on three HER2-positive cells, and among them, conjugate 4 showed the best activity. We speculated it is related to the releasing efficiency of free SMAC peptide and CPT. The subsequent biological activity evaluations of conjugate 4 confirmed that the introduction of the SMAC peptide sequence can enhance the activity of Caspase 3 and indeed play a role in promoting apoptosis. Conjugate 4 reduced the expression of XIAP and cIAP1 in SK-BR-3 cells in a concentration dependent manner. The in vivo activity study also fully proved that the antitumor activity as well as safety of conjugate 4 was superior to CPT. The pro-apoptotic effect of conjugate 4 is significantly superior to the C4 group without fused SMAC peptide and the C4+SMAC combination drug group. This indicates that that the introduced SMAC peptide exerts a synergistic anti-tumor effect. When CPT is engineered into PDCs molecules, its in vitro activity related to CPT can be retained but is somewhat diminished. This attenuation may be attributed to the fact that PDCs must undergo cellular internalization and release of CPT or its derivatives in the intracellular environment to exert anti-tumor activity.
Click to Show/Hide
|
||||
| Description |
HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, and HER2 negative MDA-MB-231 and MCF-10A cells were selected to determine the cell activity of the target conjugates. From Table 3, most conjugates showed certain cytotoxicity to three HER2 positive cell lines. Among the three connection sites (S1-S3), compounds 1, 4, and 7 with the first connection mode showed better activity. In the three HER2 positive SK-BR-3, NCI-N87 and SK-OV-3 cells, conjugate 4 exhibited superior activity (IC50s of 0.53 ± 0.17 uM, 1.58 ± 0.41 uM, 2.50 ± 0.78 uM, respectively), which almost equal to that of CPT (IC50s of 0.38 ± 0.11 uM, 2.07 ± 0.67 uM, 4.07 ± 1.82 uM, respectively). The activity of conjugate 4 on HER2 positive cells was superior to that of conjugate 1 and 7. In MDA-MB-231 and MCF-10A cells, IC50s of conjugate 4 were 18.44 ± 2.09 uM and 17.76 ± 1.35 uM, respectively, which showed great tumor cell selectivity. Based on the above, conjugate 4 with better in vitro antitumor activity was selected for subsequent biological study.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
I-1 (NPNWGRSWYNQRFKGC=(-SS-O-COO-CPT)GRIKPRKGYTR) [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.47 ± 0.54 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.29 ± 0.67 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4.29 ± 1.11 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
7.60 ± 1.23 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
23.90 ± 1.58 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
To enhance the affinity of the human epidermal growth receptor 2 (HER2) targeted peptide developed previously, bispecific fusion peptidesP1GCGT1andP1GCGCGT1were designed using anin silicoapproach. Molecular dynamic simulation showed that both peptides strongly interacted with HER2 domains II and IV. Compared with peptides targeting each single domain,P1GCGT1andP1GCGCGT1could bind to HER2 more significantly and targeted HER2-positive cells specifically. Additionally, both peptides were used to generate peptide-drug conjugates with camptothecin (CPT), among whichI-1andI-4were screened for enhanced cellular activity and selectivity. Biological evaluation demonstrated thatI-1andI-4induced cell apoptosis, promoted cell cycle arrestin S-phase, and inhibited Topo I activity. The binding affinity assay and confocal analysis revealed thatI-1andI-4were effective at targeting HER2. Moreover,I-1andI-4showed better stability than single targeting peptide and presented enhanced antitumor activity and safety than CPT in tumor-bearing mice.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity of conjugates was evaluated on cell lines with different expression levels of HER2. The results showed that among all conjugates with a single CPT molecule (I-1 to I-3), I-1 was the most cytotoxic against three HER2-positive cell lines (SK-BR-3, NCI-N87, and SK-OV-3 cells), and the calculated IC50 values for the three cells were 1.47 ± 0.54, 3.29 ± 0.67, and 4.29 ± 1.11 uM, respectively. Similarly, among all conjugates with two CPT molecules (I-4 to I-6), I-4 showed the highest cytotoxicity toward HER2-positive cells, and the IC50 values for SK-BR-3, NCI-N87, and SK-OV-3 cells were 0.12 ± 0.07, 1.78 ± 0.47, and 1.27 ± 0.38 uM, respectively. The activity of I-4 against SK-BR-3 cells was also slightly better than that of the positive control CPT (IC50 = 0.21 ± 0.04 uM). The cytotoxicity of I-1 and I-4 against HER2-negative MDA-MB-231 cells was comparatively lower (calculated IC50s were 7.60 ± 1.23 and 7.72 ± 0.92 uM, respectively). In contrast, CPT itself showed higher cytotoxicity against MDA-MB-231 (IC50 = 1.06 ± 0.42 uM), indicating that conjugates showed higher specificity in cell targeting. Furthermore, the cytotoxicity of I-1 and I-4 against normal cells MCF-10A (IC50 = 23.90 ± 1.58 and 17.14 ± 2.42 uM, respectively) was significantly lower than that of CPT alone (2.58 ± 0.77 uM). Based on these results, I-1 and I-4 were selected for subsequent biological evaluation.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
CPT-AAM-1 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Cell survival rate |
43%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 10 µM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, we developed novel melittin analogs with pH-responsive cell-penetrating and membrane-lytic activities by replacing arginines and lysines with histidines. Importantly, we found that the conjugation of cargoes to the N-terminus of melittin analogs decreased their cell-penetrating and membrane-lytic activity compared to the C-terminus, implying that the C-terminus of analogs is more suitable for cargo conjugation. After the attachment of CPT, CPT-AAM-1 and CPT-AAM-2 displayed obvious pH-responsive antitumor activity. CPT-AAM-1 and CPT-AAM-2 destroyed tumor cells through the release of CPT and membrane disruption. Compared with CPT-AAM-2, CPT-AAM-1 showed stronger antitumor activity under acidic conditions. Notably, CPT-AAM-1 significantly inhibited the tumor growth in vivo compared with AAM, AAM-1, and CPT. In addition, CPT-AAM-1 showed relatively low toxicity compared with melittin and CPT. Taken together, our results demonstrate that CPT-AAM-1, with efficient pH-responsive cell-penetrating and membrane-lytic activities, possesses significant therapeutic potential for tumor therapy. This study provides a novel strategy for the development of PDCs based on pH-responsive AMPs for oncology therapeutics.
Click to Show/Hide
|
||||
| Description |
Subsequently, the cytotoxicity of these conjugates against B16-F10 cells was determined. As shown in Fig. 4B, CPT-AAM-1 and CPT-AAM-2 displayed significant pH-dependent antitumor activity after 72 h of treatment, whereas the cytotoxicity of CPT showed no obvious difference at pH 7.4 and 5.5. Notably, CPT-AAM-1 and CPT-AAM-2 exhibited greater cytotoxicity than free CPT under acidic conditions, particularly at high concentrations. Compared to CPT-AAM-2, CPT-AAM-1 displayed strong antitumor activity, suggesting that AAM-1 could deliver more CPT molecules into cells. In addition, this result further demonstrates that the C-terminus of AAM is more suitable for drug conjugation than the N-terminus.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Cell survival rate |
60%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 5 µM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, we developed novel melittin analogs with pH-responsive cell-penetrating and membrane-lytic activities by replacing arginines and lysines with histidines. Importantly, we found that the conjugation of cargoes to the N-terminus of melittin analogs decreased their cell-penetrating and membrane-lytic activity compared to the C-terminus, implying that the C-terminus of analogs is more suitable for cargo conjugation. After the attachment of CPT, CPT-AAM-1 and CPT-AAM-2 displayed obvious pH-responsive antitumor activity. CPT-AAM-1 and CPT-AAM-2 destroyed tumor cells through the release of CPT and membrane disruption. Compared with CPT-AAM-2, CPT-AAM-1 showed stronger antitumor activity under acidic conditions. Notably, CPT-AAM-1 significantly inhibited the tumor growth in vivo compared with AAM, AAM-1, and CPT. In addition, CPT-AAM-1 showed relatively low toxicity compared with melittin and CPT. Taken together, our results demonstrate that CPT-AAM-1, with efficient pH-responsive cell-penetrating and membrane-lytic activities, possesses significant therapeutic potential for tumor therapy. This study provides a novel strategy for the development of PDCs based on pH-responsive AMPs for oncology therapeutics.
Click to Show/Hide
|
||||
| Description |
Subsequently, the cytotoxicity of these conjugates against B16-F10 cells was determined. As shown in Fig. 4B, CPT-AAM-1 and CPT-AAM-2 displayed significant pH-dependent antitumor activity after 72 h of treatment, whereas the cytotoxicity of CPT showed no obvious difference at pH 7.4 and 5.5. Notably, CPT-AAM-1 and CPT-AAM-2 exhibited greater cytotoxicity than free CPT under acidic conditions, particularly at high concentrations. Compared to CPT-AAM-2, CPT-AAM-1 displayed strong antitumor activity, suggesting that AAM-1 could deliver more CPT molecules into cells. In addition, this result further demonstrates that the C-terminus of AAM is more suitable for drug conjugation than the N-terminus.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Cell survival rate |
62%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 2.5 µM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, we developed novel melittin analogs with pH-responsive cell-penetrating and membrane-lytic activities by replacing arginines and lysines with histidines. Importantly, we found that the conjugation of cargoes to the N-terminus of melittin analogs decreased their cell-penetrating and membrane-lytic activity compared to the C-terminus, implying that the C-terminus of analogs is more suitable for cargo conjugation. After the attachment of CPT, CPT-AAM-1 and CPT-AAM-2 displayed obvious pH-responsive antitumor activity. CPT-AAM-1 and CPT-AAM-2 destroyed tumor cells through the release of CPT and membrane disruption. Compared with CPT-AAM-2, CPT-AAM-1 showed stronger antitumor activity under acidic conditions. Notably, CPT-AAM-1 significantly inhibited the tumor growth in vivo compared with AAM, AAM-1, and CPT. In addition, CPT-AAM-1 showed relatively low toxicity compared with melittin and CPT. Taken together, our results demonstrate that CPT-AAM-1, with efficient pH-responsive cell-penetrating and membrane-lytic activities, possesses significant therapeutic potential for tumor therapy. This study provides a novel strategy for the development of PDCs based on pH-responsive AMPs for oncology therapeutics.
Click to Show/Hide
|
||||
| Description |
Subsequently, the cytotoxicity of these conjugates against B16-F10 cells was determined. As shown in Fig. 4B, CPT-AAM-1 and CPT-AAM-2 displayed significant pH-dependent antitumor activity after 72 h of treatment, whereas the cytotoxicity of CPT showed no obvious difference at pH 7.4 and 5.5. Notably, CPT-AAM-1 and CPT-AAM-2 exhibited greater cytotoxicity than free CPT under acidic conditions, particularly at high concentrations. Compared to CPT-AAM-2, CPT-AAM-1 displayed strong antitumor activity, suggesting that AAM-1 could deliver more CPT molecules into cells. In addition, this result further demonstrates that the C-terminus of AAM is more suitable for drug conjugation than the N-terminus.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Cell survival rate |
72%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 1.25 µM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, we developed novel melittin analogs with pH-responsive cell-penetrating and membrane-lytic activities by replacing arginines and lysines with histidines. Importantly, we found that the conjugation of cargoes to the N-terminus of melittin analogs decreased their cell-penetrating and membrane-lytic activity compared to the C-terminus, implying that the C-terminus of analogs is more suitable for cargo conjugation. After the attachment of CPT, CPT-AAM-1 and CPT-AAM-2 displayed obvious pH-responsive antitumor activity. CPT-AAM-1 and CPT-AAM-2 destroyed tumor cells through the release of CPT and membrane disruption. Compared with CPT-AAM-2, CPT-AAM-1 showed stronger antitumor activity under acidic conditions. Notably, CPT-AAM-1 significantly inhibited the tumor growth in vivo compared with AAM, AAM-1, and CPT. In addition, CPT-AAM-1 showed relatively low toxicity compared with melittin and CPT. Taken together, our results demonstrate that CPT-AAM-1, with efficient pH-responsive cell-penetrating and membrane-lytic activities, possesses significant therapeutic potential for tumor therapy. This study provides a novel strategy for the development of PDCs based on pH-responsive AMPs for oncology therapeutics.
Click to Show/Hide
|
||||
| Description |
Subsequently, the cytotoxicity of these conjugates against B16-F10 cells was determined. As shown in Fig. 4B, CPT-AAM-1 and CPT-AAM-2 displayed significant pH-dependent antitumor activity after 72 h of treatment, whereas the cytotoxicity of CPT showed no obvious difference at pH 7.4 and 5.5. Notably, CPT-AAM-1 and CPT-AAM-2 exhibited greater cytotoxicity than free CPT under acidic conditions, particularly at high concentrations. Compared to CPT-AAM-2, CPT-AAM-1 displayed strong antitumor activity, suggesting that AAM-1 could deliver more CPT molecules into cells. In addition, this result further demonstrates that the C-terminus of AAM is more suitable for drug conjugation than the N-terminus.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Cell survival rate |
88%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 0.625 µM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, we developed novel melittin analogs with pH-responsive cell-penetrating and membrane-lytic activities by replacing arginines and lysines with histidines. Importantly, we found that the conjugation of cargoes to the N-terminus of melittin analogs decreased their cell-penetrating and membrane-lytic activity compared to the C-terminus, implying that the C-terminus of analogs is more suitable for cargo conjugation. After the attachment of CPT, CPT-AAM-1 and CPT-AAM-2 displayed obvious pH-responsive antitumor activity. CPT-AAM-1 and CPT-AAM-2 destroyed tumor cells through the release of CPT and membrane disruption. Compared with CPT-AAM-2, CPT-AAM-1 showed stronger antitumor activity under acidic conditions. Notably, CPT-AAM-1 significantly inhibited the tumor growth in vivo compared with AAM, AAM-1, and CPT. In addition, CPT-AAM-1 showed relatively low toxicity compared with melittin and CPT. Taken together, our results demonstrate that CPT-AAM-1, with efficient pH-responsive cell-penetrating and membrane-lytic activities, possesses significant therapeutic potential for tumor therapy. This study provides a novel strategy for the development of PDCs based on pH-responsive AMPs for oncology therapeutics.
Click to Show/Hide
|
||||
| Description |
Subsequently, the cytotoxicity of these conjugates against B16-F10 cells was determined. As shown in Fig. 4B, CPT-AAM-1 and CPT-AAM-2 displayed significant pH-dependent antitumor activity after 72 h of treatment, whereas the cytotoxicity of CPT showed no obvious difference at pH 7.4 and 5.5. Notably, CPT-AAM-1 and CPT-AAM-2 exhibited greater cytotoxicity than free CPT under acidic conditions, particularly at high concentrations. Compared to CPT-AAM-2, CPT-AAM-1 displayed strong antitumor activity, suggesting that AAM-1 could deliver more CPT molecules into cells. In addition, this result further demonstrates that the C-terminus of AAM is more suitable for drug conjugation than the N-terminus.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
CPT-AAM-2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Cell survival rate |
55%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 10 µM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, we developed novel melittin analogs with pH-responsive cell-penetrating and membrane-lytic activities by replacing arginines and lysines with histidines. Importantly, we found that the conjugation of cargoes to the N-terminus of melittin analogs decreased their cell-penetrating and membrane-lytic activity compared to the C-terminus, implying that the C-terminus of analogs is more suitable for cargo conjugation. After the attachment of CPT, CPT-AAM-1 and CPT-AAM-2 displayed obvious pH-responsive antitumor activity. CPT-AAM-1 and CPT-AAM-2 destroyed tumor cells through the release of CPT and membrane disruption. Compared with CPT-AAM-2, CPT-AAM-1 showed stronger antitumor activity under acidic conditions. Notably, CPT-AAM-1 significantly inhibited the tumor growth in vivo compared with AAM, AAM-1, and CPT. In addition, CPT-AAM-1 showed relatively low toxicity compared with melittin and CPT. Taken together, our results demonstrate that CPT-AAM-1, with efficient pH-responsive cell-penetrating and membrane-lytic activities, possesses significant therapeutic potential for tumor therapy. This study provides a novel strategy for the development of PDCs based on pH-responsive AMPs for oncology therapeutics.
Click to Show/Hide
|
||||
| Description |
Subsequently, the cytotoxicity of these conjugates against B16-F10 cells was determined. As shown in Fig. 4B, CPT-AAM-1 and CPT-AAM-2 displayed significant pH-dependent antitumor activity after 72 h of treatment, whereas the cytotoxicity of CPT showed no obvious difference at pH 7.4 and 5.5. Notably, CPT-AAM-1 and CPT-AAM-2 exhibited greater cytotoxicity than free CPT under acidic conditions, particularly at high concentrations. Compared to CPT-AAM-2, CPT-AAM-1 displayed strong antitumor activity, suggesting that AAM-1 could deliver more CPT molecules into cells. In addition, this result further demonstrates that the C-terminus of AAM is more suitable for drug conjugation than the N-terminus.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Cell survival rate |
57%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 5 µM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, we developed novel melittin analogs with pH-responsive cell-penetrating and membrane-lytic activities by replacing arginines and lysines with histidines. Importantly, we found that the conjugation of cargoes to the N-terminus of melittin analogs decreased their cell-penetrating and membrane-lytic activity compared to the C-terminus, implying that the C-terminus of analogs is more suitable for cargo conjugation. After the attachment of CPT, CPT-AAM-1 and CPT-AAM-2 displayed obvious pH-responsive antitumor activity. CPT-AAM-1 and CPT-AAM-2 destroyed tumor cells through the release of CPT and membrane disruption. Compared with CPT-AAM-2, CPT-AAM-1 showed stronger antitumor activity under acidic conditions. Notably, CPT-AAM-1 significantly inhibited the tumor growth in vivo compared with AAM, AAM-1, and CPT. In addition, CPT-AAM-1 showed relatively low toxicity compared with melittin and CPT. Taken together, our results demonstrate that CPT-AAM-1, with efficient pH-responsive cell-penetrating and membrane-lytic activities, possesses significant therapeutic potential for tumor therapy. This study provides a novel strategy for the development of PDCs based on pH-responsive AMPs for oncology therapeutics.
Click to Show/Hide
|
||||
| Description |
Subsequently, the cytotoxicity of these conjugates against B16-F10 cells was determined. As shown in Fig. 4B, CPT-AAM-1 and CPT-AAM-2 displayed significant pH-dependent antitumor activity after 72 h of treatment, whereas the cytotoxicity of CPT showed no obvious difference at pH 7.4 and 5.5. Notably, CPT-AAM-1 and CPT-AAM-2 exhibited greater cytotoxicity than free CPT under acidic conditions, particularly at high concentrations. Compared to CPT-AAM-2, CPT-AAM-1 displayed strong antitumor activity, suggesting that AAM-1 could deliver more CPT molecules into cells. In addition, this result further demonstrates that the C-terminus of AAM is more suitable for drug conjugation than the N-terminus.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Cell survival rate |
74%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 2.5 µM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, we developed novel melittin analogs with pH-responsive cell-penetrating and membrane-lytic activities by replacing arginines and lysines with histidines. Importantly, we found that the conjugation of cargoes to the N-terminus of melittin analogs decreased their cell-penetrating and membrane-lytic activity compared to the C-terminus, implying that the C-terminus of analogs is more suitable for cargo conjugation. After the attachment of CPT, CPT-AAM-1 and CPT-AAM-2 displayed obvious pH-responsive antitumor activity. CPT-AAM-1 and CPT-AAM-2 destroyed tumor cells through the release of CPT and membrane disruption. Compared with CPT-AAM-2, CPT-AAM-1 showed stronger antitumor activity under acidic conditions. Notably, CPT-AAM-1 significantly inhibited the tumor growth in vivo compared with AAM, AAM-1, and CPT. In addition, CPT-AAM-1 showed relatively low toxicity compared with melittin and CPT. Taken together, our results demonstrate that CPT-AAM-1, with efficient pH-responsive cell-penetrating and membrane-lytic activities, possesses significant therapeutic potential for tumor therapy. This study provides a novel strategy for the development of PDCs based on pH-responsive AMPs for oncology therapeutics.
Click to Show/Hide
|
||||
| Description |
Subsequently, the cytotoxicity of these conjugates against B16-F10 cells was determined. As shown in Fig. 4B, CPT-AAM-1 and CPT-AAM-2 displayed significant pH-dependent antitumor activity after 72 h of treatment, whereas the cytotoxicity of CPT showed no obvious difference at pH 7.4 and 5.5. Notably, CPT-AAM-1 and CPT-AAM-2 exhibited greater cytotoxicity than free CPT under acidic conditions, particularly at high concentrations. Compared to CPT-AAM-2, CPT-AAM-1 displayed strong antitumor activity, suggesting that AAM-1 could deliver more CPT molecules into cells. In addition, this result further demonstrates that the C-terminus of AAM is more suitable for drug conjugation than the N-terminus.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Cell survival rate |
86%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 1.25 µM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, we developed novel melittin analogs with pH-responsive cell-penetrating and membrane-lytic activities by replacing arginines and lysines with histidines. Importantly, we found that the conjugation of cargoes to the N-terminus of melittin analogs decreased their cell-penetrating and membrane-lytic activity compared to the C-terminus, implying that the C-terminus of analogs is more suitable for cargo conjugation. After the attachment of CPT, CPT-AAM-1 and CPT-AAM-2 displayed obvious pH-responsive antitumor activity. CPT-AAM-1 and CPT-AAM-2 destroyed tumor cells through the release of CPT and membrane disruption. Compared with CPT-AAM-2, CPT-AAM-1 showed stronger antitumor activity under acidic conditions. Notably, CPT-AAM-1 significantly inhibited the tumor growth in vivo compared with AAM, AAM-1, and CPT. In addition, CPT-AAM-1 showed relatively low toxicity compared with melittin and CPT. Taken together, our results demonstrate that CPT-AAM-1, with efficient pH-responsive cell-penetrating and membrane-lytic activities, possesses significant therapeutic potential for tumor therapy. This study provides a novel strategy for the development of PDCs based on pH-responsive AMPs for oncology therapeutics.
Click to Show/Hide
|
||||
| Description |
Subsequently, the cytotoxicity of these conjugates against B16-F10 cells was determined. As shown in Fig. 4B, CPT-AAM-1 and CPT-AAM-2 displayed significant pH-dependent antitumor activity after 72 h of treatment, whereas the cytotoxicity of CPT showed no obvious difference at pH 7.4 and 5.5. Notably, CPT-AAM-1 and CPT-AAM-2 exhibited greater cytotoxicity than free CPT under acidic conditions, particularly at high concentrations. Compared to CPT-AAM-2, CPT-AAM-1 displayed strong antitumor activity, suggesting that AAM-1 could deliver more CPT molecules into cells. In addition, this result further demonstrates that the C-terminus of AAM is more suitable for drug conjugation than the N-terminus.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Cell survival rate |
94%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 0.625 µM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, we developed novel melittin analogs with pH-responsive cell-penetrating and membrane-lytic activities by replacing arginines and lysines with histidines. Importantly, we found that the conjugation of cargoes to the N-terminus of melittin analogs decreased their cell-penetrating and membrane-lytic activity compared to the C-terminus, implying that the C-terminus of analogs is more suitable for cargo conjugation. After the attachment of CPT, CPT-AAM-1 and CPT-AAM-2 displayed obvious pH-responsive antitumor activity. CPT-AAM-1 and CPT-AAM-2 destroyed tumor cells through the release of CPT and membrane disruption. Compared with CPT-AAM-2, CPT-AAM-1 showed stronger antitumor activity under acidic conditions. Notably, CPT-AAM-1 significantly inhibited the tumor growth in vivo compared with AAM, AAM-1, and CPT. In addition, CPT-AAM-1 showed relatively low toxicity compared with melittin and CPT. Taken together, our results demonstrate that CPT-AAM-1, with efficient pH-responsive cell-penetrating and membrane-lytic activities, possesses significant therapeutic potential for tumor therapy. This study provides a novel strategy for the development of PDCs based on pH-responsive AMPs for oncology therapeutics.
Click to Show/Hide
|
||||
| Description |
Subsequently, the cytotoxicity of these conjugates against B16-F10 cells was determined. As shown in Fig. 4B, CPT-AAM-1 and CPT-AAM-2 displayed significant pH-dependent antitumor activity after 72 h of treatment, whereas the cytotoxicity of CPT showed no obvious difference at pH 7.4 and 5.5. Notably, CPT-AAM-1 and CPT-AAM-2 exhibited greater cytotoxicity than free CPT under acidic conditions, particularly at high concentrations. Compared to CPT-AAM-2, CPT-AAM-1 displayed strong antitumor activity, suggesting that AAM-1 could deliver more CPT molecules into cells. In addition, this result further demonstrates that the C-terminus of AAM is more suitable for drug conjugation than the N-terminus.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
References