
Linker Information
General Information of This Linker
| Linker ID |
LIN00066
|
|||||
|---|---|---|---|---|---|---|
| Linker Name |
N-succinimidyl-3-maleimidopropionate
|
|||||
| Linker Type |
Enzyme-sensitive linkers
|
|||||
| Structure |
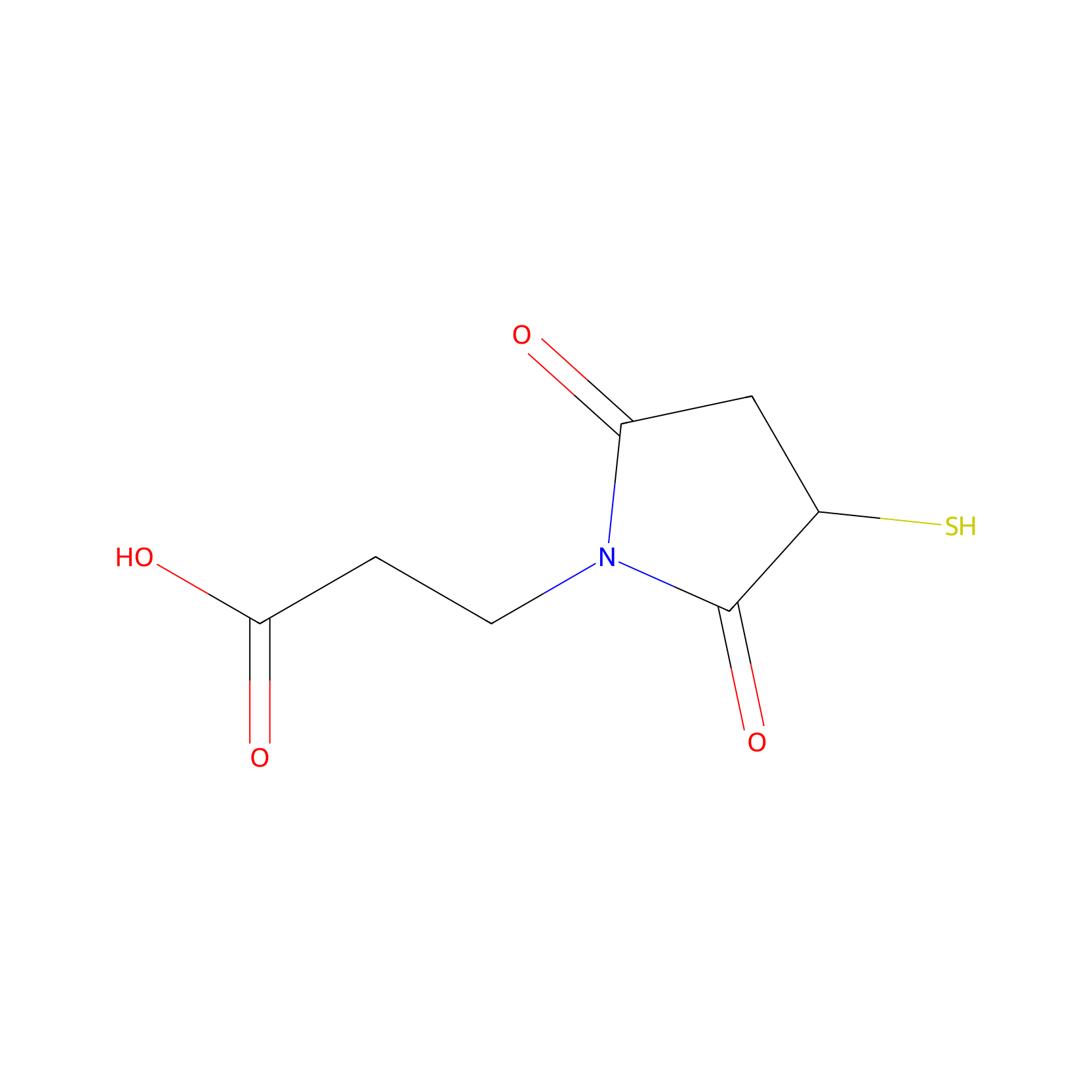
|
|||||
| Formula |
C7H9NO4S
|
|||||
| #Ro5 Violations (Lipinski): 0 | Molecular Weight (mw) | 203.219 | ||||
| Lipid-water partition coefficient (xlogp) | -0.4816 | |||||
| Hydrogen Bond Donor Count (hbonddonor) | 2 | |||||
| Hydrogen Bond Acceptor Count (hbondacc) | 4 | |||||
| Rotatable Bond Count (rotbonds) | 3 | |||||
| Canonical smiles |
O=C(O)CCN1C(=O)CC(S)C1=O
|
|||||
| InChI |
InChI=1S/C7H9NO4S/c9-5-3-4(13)7(12)8(5)2-1-6(10)11/h4,13H,1-3H2,(H,10,11)
|
|||||
| InChIKey |
GSQZMNTYKCPUAA-UHFFFAOYSA-N
|
|||||
Each Peptide-drug Conjugate Related to This Linker
Full Information of The Activity Data of The PDC(s) Related to This Linker
M-DM1 [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
58.30%
|
|||
| MOA of PDC |
Recently, peptides that offer versatility in drug discovery for the successful treatment of cancers have emerged. Peptide-drug conjugates (PDCs) represent an important therapeutic strategy for increasing tumor penetration and selectivity. We previously reported that M, which is extracted from bee venom, targets M2 macrophages and improves tumor treatment in lung cancer. M is an amphipathic peptide with 26 amino acid residues (AIGAVLKVLTTGLPALISWIKRKRQQ) that specifically binds to M2-like TAMs. Mertansine (DM1) is a strong cytotoxic agent that interacts with tubulin and inhibits the assembly of tubulin into microtubules. Because it targets microtubules and inhibits cell cycle, its clinical efficacy as a potential anti-cancer agent has been studied. However, meaningful results have yet to be obtained in clinical trials, with patients also suffering several side effects, such as myelosuppression. Antibody drug conjugates (ADCs) are currently the most successful type of drug conjugates, consisting of antibody (targeting), linker (linking the antibody to the payload), and Payload (killing target cells). Payload is a cytotoxic compound and is divided into microtubule inhibitors, such as DM1, and DNA-damaging agents, such as anthracyclines. DM1 alone has not been developed as a drug but is currently used as an antibody-drug conjugate. It has been reported that the peptide LLC2B combined with DM1 exerts an anticancer effect in breast and esophageal squamous cell carcinoma, suggesting it could be developed as a potential PDC for tumor treatment. In the present study, we investigated the anti-cancer effects of M-DM1, which was synthesized using M as a carrier for targeting TAMs and DM1 as a payload. We found that M-DM1 exerts its therapeutic effects by specifically depleting M2-like TAMs in a melanoma mouse model. Through the regulation of M2-like TAMs, M-DM1 induced a significant increase in the infiltration of effector cells, such as CD8 T cells and natural killer (NK) cells, into the TME. Collectively, our results suggest that depleting M2-like TAMs in the TME by treatment with M-DM1 has immunotherapeutic effects in malignant melanoma.
Click to Show/Hide
|
||||
| Description |
To examine the therapeutic effects of M-DM1 in mice, we used the B16F10 tumor-bearing mouse model. We subcutaneously injected cancer cells into C57BL/6 mice and treated them with 20 nmol/kg M, DM1, or M-DM1 via intraperitoneal injections every 3 days. While slight growth inhibition was observed with M and DM1, M-DM1 was found to significantly suppress tumor growth. None of the mice showed any weight loss until the end of the study period, suggesting that there were no significant toxicities due to the drug treatments. At 21 days after tumor cell inoculation, the mice were sacrificed, and tumor volumes and weights were measured. A marked decrease in the tumor volumes and weights was observed in the M-DM1-treated mice group compared to that in the other groups. In addition, we determined the survival rate of B16-bearing mice following treatment with each peptide. The median survival of the M-DM1 group was significantly higher than that of the other groups. These results demonstrate that M-DM1 is highly effective in inhibiting tumor growth and prolonging survival in melanoma.
Click to Show/Hide
|
||||
| In Vivo Model | B16-F10 tumor-bearing mouse model. | ||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.042 µM
|
|||
| Evaluation Method | MTS assay | ||||
| MOA of PDC |
Recently, peptides that offer versatility in drug discovery for the successful treatment of cancers have emerged. Peptide-drug conjugates (PDCs) represent an important therapeutic strategy for increasing tumor penetration and selectivity. We previously reported that M, which is extracted from bee venom, targets M2 macrophages and improves tumor treatment in lung cancer. M is an amphipathic peptide with 26 amino acid residues (AIGAVLKVLTTGLPALISWIKRKRQQ) that specifically binds to M2-like TAMs. Mertansine (DM1) is a strong cytotoxic agent that interacts with tubulin and inhibits the assembly of tubulin into microtubules. Because it targets microtubules and inhibits cell cycle, its clinical efficacy as a potential anti-cancer agent has been studied. However, meaningful results have yet to be obtained in clinical trials, with patients also suffering several side effects, such as myelosuppression. Antibody drug conjugates (ADCs) are currently the most successful type of drug conjugates, consisting of antibody (targeting), linker (linking the antibody to the payload), and Payload (killing target cells). Payload is a cytotoxic compound and is divided into microtubule inhibitors, such as DM1, and DNA-damaging agents, such as anthracyclines. DM1 alone has not been developed as a drug but is currently used as an antibody-drug conjugate. It has been reported that the peptide LLC2B combined with DM1 exerts an anticancer effect in breast and esophageal squamous cell carcinoma, suggesting it could be developed as a potential PDC for tumor treatment. In the present study, we investigated the anti-cancer effects of M-DM1, which was synthesized using M as a carrier for targeting TAMs and DM1 as a payload. We found that M-DM1 exerts its therapeutic effects by specifically depleting M2-like TAMs in a melanoma mouse model. Through the regulation of M2-like TAMs, M-DM1 induced a significant increase in the infiltration of effector cells, such as CD8 T cells and natural killer (NK) cells, into the TME. Collectively, our results suggest that depleting M2-like TAMs in the TME by treatment with M-DM1 has immunotherapeutic effects in malignant melanoma.
Click to Show/Hide
|
||||
| Description |
To evaluate the selective cytotoxicity of M-DM1 against M0, M1, and M2 macrophages and TAMs, the THP-1-derived macrophages were treated with different concentrations of M, DM1, and M-DM1 (0.05-10 uM) after differentiation into M0, M1, M2, and TAMs. A cytotoxicity assay was performed using the MTS assay. The half-maximal inhibitory concentrations (IC50) of M-DM1 were 2.104, 2.457, 1.488, and 1.042 uM for M0, M1, M2 macrophages, and TAMs, respectively. M-DM1 induced the apoptosis of M2-like TAMs at low concentrations compared to that observed with M2 macrophages, whereas DM1 alone did not have a cytotoxic effect on any of the macrophages.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | Tumor-associated macrophages | Homo sapiens | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.488 µM
|
|||
| Evaluation Method | MTS assay | ||||
| MOA of PDC |
Recently, peptides that offer versatility in drug discovery for the successful treatment of cancers have emerged. Peptide-drug conjugates (PDCs) represent an important therapeutic strategy for increasing tumor penetration and selectivity. We previously reported that M, which is extracted from bee venom, targets M2 macrophages and improves tumor treatment in lung cancer. M is an amphipathic peptide with 26 amino acid residues (AIGAVLKVLTTGLPALISWIKRKRQQ) that specifically binds to M2-like TAMs. Mertansine (DM1) is a strong cytotoxic agent that interacts with tubulin and inhibits the assembly of tubulin into microtubules. Because it targets microtubules and inhibits cell cycle, its clinical efficacy as a potential anti-cancer agent has been studied. However, meaningful results have yet to be obtained in clinical trials, with patients also suffering several side effects, such as myelosuppression. Antibody drug conjugates (ADCs) are currently the most successful type of drug conjugates, consisting of antibody (targeting), linker (linking the antibody to the payload), and Payload (killing target cells). Payload is a cytotoxic compound and is divided into microtubule inhibitors, such as DM1, and DNA-damaging agents, such as anthracyclines. DM1 alone has not been developed as a drug but is currently used as an antibody-drug conjugate. It has been reported that the peptide LLC2B combined with DM1 exerts an anticancer effect in breast and esophageal squamous cell carcinoma, suggesting it could be developed as a potential PDC for tumor treatment. In the present study, we investigated the anti-cancer effects of M-DM1, which was synthesized using M as a carrier for targeting TAMs and DM1 as a payload. We found that M-DM1 exerts its therapeutic effects by specifically depleting M2-like TAMs in a melanoma mouse model. Through the regulation of M2-like TAMs, M-DM1 induced a significant increase in the infiltration of effector cells, such as CD8 T cells and natural killer (NK) cells, into the TME. Collectively, our results suggest that depleting M2-like TAMs in the TME by treatment with M-DM1 has immunotherapeutic effects in malignant melanoma.
Click to Show/Hide
|
||||
| Description |
To evaluate the selective cytotoxicity of M-DM1 against M0, M1, and M2 macrophages and TAMs, the THP-1-derived macrophages were treated with different concentrations of M, DM1, and M-DM1 (0.05-10 uM) after differentiation into M0, M1, M2, and TAMs. A cytotoxicity assay was performed using the MTS assay. The half-maximal inhibitory concentrations (IC50) of M-DM1 were 2.104, 2.457, 1.488, and 1.042 uM for M0, M1, M2 macrophages, and TAMs, respectively. M-DM1 induced the apoptosis of M2-like TAMs at low concentrations compared to that observed with M2 macrophages, whereas DM1 alone did not have a cytotoxic effect on any of the macrophages.
Click to Show/Hide
|
||||
| In Vitro Model | Human monocytic leukemia | THP-1 M2 type macrophages | CVCL_0006 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.104 µM
|
|||
| Evaluation Method | MTS assay | ||||
| MOA of PDC |
Recently, peptides that offer versatility in drug discovery for the successful treatment of cancers have emerged. Peptide-drug conjugates (PDCs) represent an important therapeutic strategy for increasing tumor penetration and selectivity. We previously reported that M, which is extracted from bee venom, targets M2 macrophages and improves tumor treatment in lung cancer. M is an amphipathic peptide with 26 amino acid residues (AIGAVLKVLTTGLPALISWIKRKRQQ) that specifically binds to M2-like TAMs. Mertansine (DM1) is a strong cytotoxic agent that interacts with tubulin and inhibits the assembly of tubulin into microtubules. Because it targets microtubules and inhibits cell cycle, its clinical efficacy as a potential anti-cancer agent has been studied. However, meaningful results have yet to be obtained in clinical trials, with patients also suffering several side effects, such as myelosuppression. Antibody drug conjugates (ADCs) are currently the most successful type of drug conjugates, consisting of antibody (targeting), linker (linking the antibody to the payload), and Payload (killing target cells). Payload is a cytotoxic compound and is divided into microtubule inhibitors, such as DM1, and DNA-damaging agents, such as anthracyclines. DM1 alone has not been developed as a drug but is currently used as an antibody-drug conjugate. It has been reported that the peptide LLC2B combined with DM1 exerts an anticancer effect in breast and esophageal squamous cell carcinoma, suggesting it could be developed as a potential PDC for tumor treatment. In the present study, we investigated the anti-cancer effects of M-DM1, which was synthesized using M as a carrier for targeting TAMs and DM1 as a payload. We found that M-DM1 exerts its therapeutic effects by specifically depleting M2-like TAMs in a melanoma mouse model. Through the regulation of M2-like TAMs, M-DM1 induced a significant increase in the infiltration of effector cells, such as CD8 T cells and natural killer (NK) cells, into the TME. Collectively, our results suggest that depleting M2-like TAMs in the TME by treatment with M-DM1 has immunotherapeutic effects in malignant melanoma.
Click to Show/Hide
|
||||
| Description |
To evaluate the selective cytotoxicity of M-DM1 against M0, M1, and M2 macrophages and TAMs, the THP-1-derived macrophages were treated with different concentrations of M, DM1, and M-DM1 (0.05-10 uM) after differentiation into M0, M1, M2, and TAMs. A cytotoxicity assay was performed using the MTS assay. The half-maximal inhibitory concentrations (IC50) of M-DM1 were 2.104, 2.457, 1.488, and 1.042 uM for M0, M1, M2 macrophages, and TAMs, respectively. M-DM1 induced the apoptosis of M2-like TAMs at low concentrations compared to that observed with M2 macrophages, whereas DM1 alone did not have a cytotoxic effect on any of the macrophages.
Click to Show/Hide
|
||||
| In Vitro Model | Human monocytic leukemia | THP-1 M0 type macrophages | CVCL_0006 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.457 µM
|
|||
| Evaluation Method | MTS assay | ||||
| MOA of PDC |
Recently, peptides that offer versatility in drug discovery for the successful treatment of cancers have emerged. Peptide-drug conjugates (PDCs) represent an important therapeutic strategy for increasing tumor penetration and selectivity. We previously reported that M, which is extracted from bee venom, targets M2 macrophages and improves tumor treatment in lung cancer. M is an amphipathic peptide with 26 amino acid residues (AIGAVLKVLTTGLPALISWIKRKRQQ) that specifically binds to M2-like TAMs. Mertansine (DM1) is a strong cytotoxic agent that interacts with tubulin and inhibits the assembly of tubulin into microtubules. Because it targets microtubules and inhibits cell cycle, its clinical efficacy as a potential anti-cancer agent has been studied. However, meaningful results have yet to be obtained in clinical trials, with patients also suffering several side effects, such as myelosuppression. Antibody drug conjugates (ADCs) are currently the most successful type of drug conjugates, consisting of antibody (targeting), linker (linking the antibody to the payload), and Payload (killing target cells). Payload is a cytotoxic compound and is divided into microtubule inhibitors, such as DM1, and DNA-damaging agents, such as anthracyclines. DM1 alone has not been developed as a drug but is currently used as an antibody-drug conjugate. It has been reported that the peptide LLC2B combined with DM1 exerts an anticancer effect in breast and esophageal squamous cell carcinoma, suggesting it could be developed as a potential PDC for tumor treatment. In the present study, we investigated the anti-cancer effects of M-DM1, which was synthesized using M as a carrier for targeting TAMs and DM1 as a payload. We found that M-DM1 exerts its therapeutic effects by specifically depleting M2-like TAMs in a melanoma mouse model. Through the regulation of M2-like TAMs, M-DM1 induced a significant increase in the infiltration of effector cells, such as CD8 T cells and natural killer (NK) cells, into the TME. Collectively, our results suggest that depleting M2-like TAMs in the TME by treatment with M-DM1 has immunotherapeutic effects in malignant melanoma.
Click to Show/Hide
|
||||
| Description |
To evaluate the selective cytotoxicity of M-DM1 against M0, M1, and M2 macrophages and TAMs, the THP-1-derived macrophages were treated with different concentrations of M, DM1, and M-DM1 (0.05-10 uM) after differentiation into M0, M1, M2, and TAMs. A cytotoxicity assay was performed using the MTS assay. The half-maximal inhibitory concentrations (IC50) of M-DM1 were 2.104, 2.457, 1.488, and 1.042 uM for M0, M1, M2 macrophages, and TAMs, respectively. M-DM1 induced the apoptosis of M2-like TAMs at low concentrations compared to that observed with M2 macrophages, whereas DM1 alone did not have a cytotoxic effect on any of the macrophages.
Click to Show/Hide
|
||||
| In Vitro Model | Human monocytic leukemia | THP-1 M1 type macrophages | CVCL_0006 | ||
BP9a-DOX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
18.5 ± 3.77 µM
|
|||
| Evaluation Method | CCK-8 assay | ||||
| Description |
Its antiproliferative activity against HepG2 cells (IC50 18.5 ± 3.77 uM) was lower than that of free DOX.
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
[d-Cys6-des-Gly10-Pro9-NHEt]-GnRH-Dox [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
10%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 100 µM | ||||
| Evaluation Method | MTT assay | ||||
| Description |
The results showed that all the conjugates had lower antiproliferative effects than Dox at the tested concentrations (Figure 2A); this may be related to inefficient release of Dox from the conjugate caused by the relative stable thioether bond linkage between Dox-SMP and GnRH analog. The antiproliferative effects of the three conjugates were close at 25, 50, and 75 uM. While at 100 uM, conjugate II exhibited higher inhibitory effect than that of I and III.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 7.45 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
28%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 75 µM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Although RNT with 177Lu-DOTATATE/PSMA is known as a novel and effective therapy option for cancer that significantly improves the quality of life and survival of patients, it may have acute or chronic side effects. Therefore, any method that can ameliorate these side effects is useful in the RNT process. For this purpose, a few clinical studies have reported that antioxidants as free radical scavengers such as amifostine and vitamins C and E can reduce radioiodine-related side effects, particularly in salivary glands in thyroid cancer patients.
Click to Show/Hide
|
||||
| Description |
The results showed that all the conjugates had lower antiproliferative effects than Dox at the tested concentrations (Figure 2A); this may be related to inefficient release of Dox from the conjugate caused by the relative stable thioether bond linkage between Dox-SMP and GnRH analog. The antiproliferative effects of the three conjugates were close at 25, 50, and 75 uM. While at 100 uM, conjugate II exhibited higher inhibitory effect than that of I and III.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 7.45 h | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
45%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 50 µM | ||||
| Evaluation Method | MTT assay | ||||
| Description |
The results showed that all the conjugates had lower antiproliferative effects than Dox at the tested concentrations (Figure 2A); this may be related to inefficient release of Dox from the conjugate caused by the relative stable thioether bond linkage between Dox-SMP and GnRH analog. The antiproliferative effects of the three conjugates were close at 25, 50, and 75 uM. While at 100 uM, conjugate II exhibited higher inhibitory effect than that of I and III.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 7.45 h | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
48%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 100 µM; with 10- µM leuprolide for 2 hours | ||||
| Description |
The combinations of conjugate II and leuprolide exhibited lower antiproliferative efficacy on MCF-7 cells than conjugate II individually at all the tested concentrations (Figure 3A).
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 7.45 h | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
50%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 75 µM; with 10- µM leuprolide for 2 hours | ||||
| MOA of PDC |
Vitamin C as a water-soluble vitamin is the reduced form of ascorbic acid. No significant adverse effect of taking high doses of vitamin C (over 2000 mg/day) has been reported due to the water-soluble feature of vitamin C. Vitamin C directly reacts with hydroxy, alkoxyl, and lipid peroxyl radicals and converts them to alcohol, water, and hydroperoxide lipid, respectively. It has been shown that taking vitamin C before radioiodine therapy can ameliorate the oxidative stress effect of radioiodine. The radioprotective effects of vitamin C are mainly due to its free radical scavenging activity.
Click to Show/Hide
|
||||
| Description |
The combinations of conjugate II and leuprolide exhibited lower antiproliferative efficacy on MCF-7 cells than conjugate II individually at all the tested concentrations (Figure 3A).
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 7.45 h | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
52%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 25 µM | ||||
| Evaluation Method | MTT assay | ||||
| Description |
The results showed that all the conjugates had lower antiproliferative effects than Dox at the tested concentrations (Figure 2A); this may be related to inefficient release of Dox from the conjugate caused by the relative stable thioether bond linkage between Dox-SMP and GnRH analog. The antiproliferative effects of the three conjugates were close at 25, 50, and 75 uM. While at 100 uM, conjugate II exhibited higher inhibitory effect than that of I and III.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 7.45 h | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
72%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 50 µM; with 10- µM leuprolide for 2 hours | ||||
| Description |
The combinations of conjugate II and leuprolide exhibited lower antiproliferative efficacy on MCF-7 cells than conjugate II individually at all the tested concentrations (Figure 3A).
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 7.45 h | ||||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
75%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 100 µM | ||||
| Evaluation Method | MTT assay | ||||
| Description |
However, by linking to [d-Cys6-des-Gly10-Pro9-NHEt]-GnRH, the cytotoxicity of Dox against 3T3 cells was reduced because the cell viability was over 87% after treatment with conjugate II in 25, 50, and 75 uM of equivalent concentration of Dox. Even at 100 uM, conjugate II maintained the cell viability to about 74% (Figure 2B).
|
||||
| In Vitro Model | Normal | NIH 3T3 cell | CVCL_0594 | ||
| Half life period | 7.45 h | ||||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
90%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 75 µM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In biological systems, antioxidants such as catalase, superoxide dismutase, glutathione peroxidase, and glutathione reductase are responsible for the elimination or reduction of the adverse effects of ROS, that is, they prevent or reduce ROS generation. Dietary antioxidants, such as vitamins E, A, and C, and anthocyanins and polyphenols have a role in the protection of cells against ROS damage.
Click to Show/Hide
|
||||
| Description |
However, by linking to [d-Cys6-des-Gly10-Pro9-NHEt]-GnRH, the cytotoxicity of Dox against 3T3 cells was reduced because the cell viability was over 87% after treatment with conjugate II in 25, 50, and 75 uM of equivalent concentration of Dox. Even at 100 uM, conjugate II maintained the cell viability to about 74% (Figure 2B).
|
||||
| In Vitro Model | Normal | NIH 3T3 cell | CVCL_0594 | ||
| Half life period | 7.45 h | ||||
| Experiment 10 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
92%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 50 µM | ||||
| Evaluation Method | MTT assay | ||||
| Description |
However, by linking to [d-Cys6-des-Gly10-Pro9-NHEt]-GnRH, the cytotoxicity of Dox against 3T3 cells was reduced because the cell viability was over 87% after treatment with conjugate II in 25, 50, and 75 uM of equivalent concentration of Dox. Even at 100 uM, conjugate II maintained the cell viability to about 74% (Figure 2B).
|
||||
| In Vitro Model | Normal | NIH 3T3 cell | CVCL_0594 | ||
| Half life period | 7.45 h | ||||
| Experiment 11 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
98%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 25 µM | ||||
| Evaluation Method | MTT assay | ||||
| Description |
However, by linking to [d-Cys6-des-Gly10-Pro9-NHEt]-GnRH, the cytotoxicity of Dox against 3T3 cells was reduced because the cell viability was over 87% after treatment with conjugate II in 25, 50, and 75 uM of equivalent concentration of Dox. Even at 100 uM, conjugate II maintained the cell viability to about 74% (Figure 2B).
|
||||
| In Vitro Model | Normal | NIH 3T3 cell | CVCL_0594 | ||
| Half life period | 7.45 h | ||||
| Experiment 12 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
108%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 25 µM; with 10- µM leuprolide for 2 hours | ||||
| Description |
The combinations of conjugate II and leuprolide exhibited lower antiproliferative efficacy on MCF-7 cells than conjugate II individually at all the tested concentrations (Figure 3A).
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 7.45 h | ||||
[d-Cys6-des-Gly10-Pro9-NH2]-GnRH-Dox [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
30%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 100 µM | ||||
| Evaluation Method | MTT assay | ||||
| Description |
The results showed that all the conjugates had lower antiproliferative effects than Dox at the tested concentrations (Figure 2A); this may be related to inefficient release of Dox from the conjugate caused by the relative stable thioether bond linkage between Dox-SMP and GnRH analog. The antiproliferative effects of the three conjugates were close at 25, 50, and 75 uM. While at 100 uM, conjugate II exhibited higher inhibitory effect than that of I and III.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 4.67 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
38%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 75 µM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
lthough RNT with 177Lu-DOTATATE/PSMA is known as a novel and effective therapy option for cancer that significantly improves the quality of life and survival of patients, it may have acute or chronic side effects. Therefore, any method that can ameliorate these side effects is useful in the RNT process. For this purpose, a few clinical studies have reported that antioxidants as free radical scavengers such as amifostine and vitamins C and E can reduce radioiodine-related side effects, particularly in salivary glands in thyroid cancer patients.
Click to Show/Hide
|
||||
| Description |
The results showed that all the conjugates had lower antiproliferative effects than Dox at the tested concentrations (Figure 2A); this may be related to inefficient release of Dox from the conjugate caused by the relative stable thioether bond linkage between Dox-SMP and GnRH analog. The antiproliferative effects of the three conjugates were close at 25, 50, and 75 uM. While at 100 uM, conjugate II exhibited higher inhibitory effect than that of I and III.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 4.67 h | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
50%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 50 µM | ||||
| Evaluation Method | MTT assay | ||||
| Description |
The results showed that all the conjugates had lower antiproliferative effects than Dox at the tested concentrations (Figure 2A); this may be related to inefficient release of Dox from the conjugate caused by the relative stable thioether bond linkage between Dox-SMP and GnRH analog. The antiproliferative effects of the three conjugates were close at 25, 50, and 75 uM. While at 100 uM, conjugate II exhibited higher inhibitory effect than that of I and III.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 4.67 h | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
55%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 25 µM | ||||
| Evaluation Method | MTT assay | ||||
| Description |
The results showed that all the conjugates had lower antiproliferative effects than Dox at the tested concentrations (Figure 2A); this may be related to inefficient release of Dox from the conjugate caused by the relative stable thioether bond linkage between Dox-SMP and GnRH analog. The antiproliferative effects of the three conjugates were close at 25, 50, and 75 uM. While at 100 uM, conjugate II exhibited higher inhibitory effect than that of I and III.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 4.67 h | ||||
[d-Cys6-des-Gly10-Pro9-α-azaGly-NH2]-GnRH-Dox [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
0.3
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 100 µM | ||||
| Evaluation Method | MTT assay | ||||
| Description |
The results showed that all the conjugates had lower antiproliferative effects than Dox at the tested concentrations (Figure 2A); this may be related to inefficient release of Dox from the conjugate caused by the relative stable thioether bond linkage between Dox-SMP and GnRH analog. The antiproliferative effects of the three conjugates were close at 25, 50, and 75 uM. While at 100 uM, conjugate II exhibited higher inhibitory effect than that of I and III.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 5.23 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
0.38
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 75 µM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Vitamin C as a water-soluble vitamin is the reduced form of ascorbic acid. No significant adverse effect of taking high doses of vitamin C (over 2000 mg/day) has been reported due to the water-soluble feature of vitamin C. Vitamin C directly reacts with hydroxy, alkoxyl, and lipid peroxyl radicals and converts them to alcohol, water, and hydroperoxide lipid, respectively. It has been shown that taking vitamin C before radioiodine therapy can ameliorate the oxidative stress effect of radioiodine. The radioprotective effects of vitamin C are mainly due to its free radical scavenging activity.
Click to Show/Hide
|
||||
| Description |
The results showed that all the conjugates had lower antiproliferative effects than Dox at the tested concentrations (Figure 2A); this may be related to inefficient release of Dox from the conjugate caused by the relative stable thioether bond linkage between Dox-SMP and GnRH analog. The antiproliferative effects of the three conjugates were close at 25, 50, and 75 uM. While at 100 uM, conjugate II exhibited higher inhibitory effect than that of I and III.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 5.23 h | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
0.5
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 50 µM | ||||
| Evaluation Method | MTT assay | ||||
| Description |
The results showed that all the conjugates had lower antiproliferative effects than Dox at the tested concentrations (Figure 2A); this may be related to inefficient release of Dox from the conjugate caused by the relative stable thioether bond linkage between Dox-SMP and GnRH analog. The antiproliferative effects of the three conjugates were close at 25, 50, and 75 uM. While at 100 uM, conjugate II exhibited higher inhibitory effect than that of I and III.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 5.23 h | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
0.59
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 25 µM | ||||
| Evaluation Method | MTT assay | ||||
| Description |
The results showed that all the conjugates had lower antiproliferative effects than Dox at the tested concentrations (Figure 2A); this may be related to inefficient release of Dox from the conjugate caused by the relative stable thioether bond linkage between Dox-SMP and GnRH analog. The antiproliferative effects of the three conjugates were close at 25, 50, and 75 uM. While at 100 uM, conjugate II exhibited higher inhibitory effect than that of I and III.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 5.23 h | ||||
References