
Linker Information
General Information of This Linker
| Linker ID |
LIN00078
|
|||||
|---|---|---|---|---|---|---|
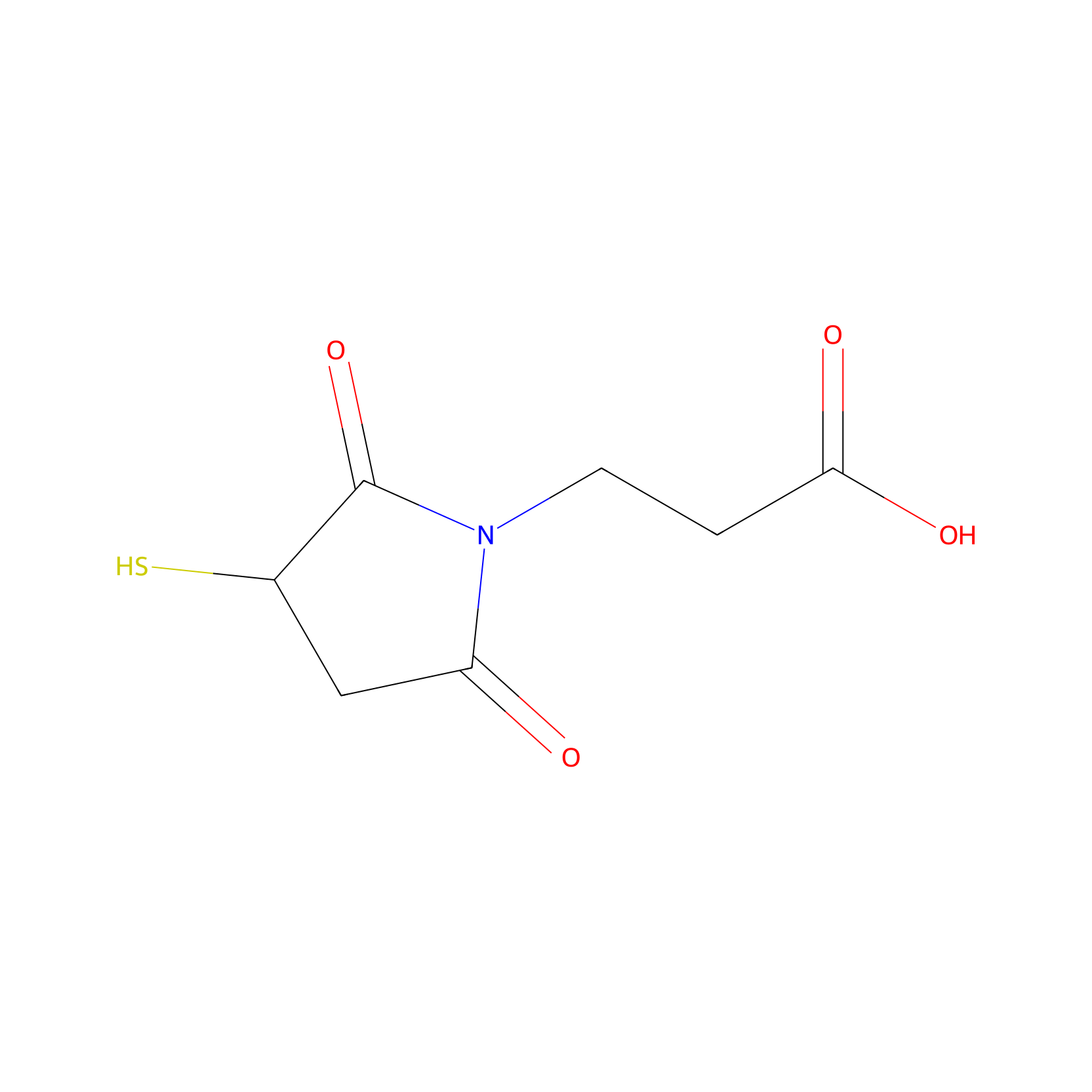
| Linker Name |
Thiol group (SH) and maleimide
|
|||||
| Structure |
|
|||||
| Formula |
C7H9NO4S
|
|||||
| #Ro5 Violations (Lipinski): 0 | Molecular Weight (mw) | 203.219 | ||||
| Lipid-water partition coefficient (xlogp) | -0.4816 | |||||
| Hydrogen Bond Donor Count (hbonddonor) | 2 | |||||
| Hydrogen Bond Acceptor Count (hbondacc) | 4 | |||||
| Rotatable Bond Count (rotbonds) | 3 | |||||
| Canonical smiles |
O=C(O)CCN1C(=O)CC(S)C1=O
|
|||||
| InChI |
InChI=1S/C7H9NO4S/c9-5-3-4(13)7(12)8(5)2-1-6(10)11/h4,13H,1-3H2,(H,10,11)
|
|||||
| InChIKey |
GSQZMNTYKCPUAA-UHFFFAOYSA-N
|
|||||
Each Peptide-drug Conjugate Related to This Linker
Full Information of The Activity Data of The PDC(s) Related to This Linker
LLC2B-Mal-DM1 [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
28.66%
|
|||
| Administration Time | 6 d | ||||
| Administration Dosage | 4.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
34.15%
|
|||
| Administration Time | 6 d | ||||
| Administration Dosage | 2.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
36.81%
|
|||
| Administration Time | 12 d | ||||
| Administration Dosage | 4.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
37.21%
|
|||
| Administration Time | 2 d | ||||
| Administration Dosage | 4.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
38.55%
|
|||
| Administration Time | 10 d | ||||
| Administration Dosage | 4.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
38.73%
|
|||
| Administration Time | 8 d | ||||
| Administration Dosage | 4.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
39.16%
|
|||
| Administration Time | Once a week for 3 weeks | ||||
| Administration Dosage | 4.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
44.93%
|
|||
| Administration Time | 8 d | ||||
| Administration Dosage | 2.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
46.77%
|
|||
| Administration Time | 18 d | ||||
| Administration Dosage | 1.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
47.60%
|
|||
| Administration Time | 16 d | ||||
| Administration Dosage | 4.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
47.63%
|
|||
| Administration Time | Once a week for 3 weeks | ||||
| Administration Dosage | 2.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
48.73%
|
|||
| Administration Time | 16 d | ||||
| Administration Dosage | 2.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
49.13%
|
|||
| Administration Time | 14 d | ||||
| Administration Dosage | 4.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
49.33%
|
|||
| Administration Time | 2 d | ||||
| Administration Dosage | 2.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
50.36%
|
|||
| Administration Time | 18 d | ||||
| Administration Dosage | 4.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
50.81%
|
|||
| Administration Time | 12 d | ||||
| Administration Dosage | 2.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 17 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
51.89%
|
|||
| Administration Time | 18 d | ||||
| Administration Dosage | 2.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 18 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
53.10%
|
|||
| Administration Time | 14 d | ||||
| Administration Dosage | 2.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 19 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
54.75%
|
|||
| Administration Time | 12 d | ||||
| Administration Dosage | 1.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 20 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
54.77%
|
|||
| Administration Time | 16 d | ||||
| Administration Dosage | 1.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 21 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
57.18%
|
|||
| Administration Time | 10 d | ||||
| Administration Dosage | 1.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 22 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
57.94%
|
|||
| Administration Time | 10 d | ||||
| Administration Dosage | 2.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 23 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
59.26%
|
|||
| Administration Time | 14 d | ||||
| Administration Dosage | 1.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 24 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
60.37%
|
|||
| Administration Time | 8 d | ||||
| Administration Dosage | 1.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 25 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
63.86%
|
|||
| Administration Time | 6 d | ||||
| Administration Dosage | 1.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 26 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
65.78%
|
|||
| Administration Time | Once a week for 3 weeks | ||||
| Administration Dosage | 1.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 27 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
71.61%
|
|||
| Administration Time | 2 d | ||||
| Administration Dosage | 1.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Completely suppress concentration |
1 μg DM1 equiv./mL
|
|||
| Administration Time | 12 h | ||||
| Evaluation Method | Clone formation experiment | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
The results show that DM1 could completely suppress the clone formation of MCF-7 cells at 0.001 μg/mL, while LLC2B-SS-DM1 could completely suppress the clone formation of MCF-7 cells at 0.1 μg DM1 equiv./mL. A concentration of 1 μg DM1 equiv./mL was required for LLC2B-Mal-DM1 to achieve the same suppression effect.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
CTCE-DTX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
99.2 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
To increase the DTX solubility and avoid the DTX therapy induced metastasis, we designed a peptide-drug conjugate (CTCE-DTX) by coupling two functional molecules (CXCR4 antagonist peptide and chemotherapeutic agent DTX) through a flexible linker. The amphiphilic monomer of CTCE-DTX could self-assemble into nanoparticles (CTCE-DTX NPs) in the water environment (without the participation of toxic ethanol and Tween 80 as excipients). The CXCR4 antagonist component could target CXCR4 upregulated tumor cells, which not only enhanced the efficacy of DTX but also inhibited the bone and lung metastasis of TNBC.
Click to Show/Hide
|
||||
| Description |
Besides, the combination of DTX and CTCE showed a substantially greater cytotoxicity, with an approximate 2 folds reduction (67.9 nmol/L) in half-maximal inhibitory concentration (IC50) compared to DTX alone. What's more, CTCE-DTX NPs retained a greater cytotoxicity (IC50 = 88.5 nmol/L) than Sc-DTX NPs (IC50 = 169.5 nmol/L), which meant CXCR4 antagonist peptide could work as a sensitizer to DTX. The slightly higher IC50 value of CTCE-DTX NPs than that of DTX + CTCE group may attribute to the incomplete disassembly of nanostructures. Meanwhile, consistent with previous reports14, the CXCR4 antagonist peptide and scramble peptide didn't not show any cytotoxicity to 4T1 cell at low concentration. We also tested the cytotoxicity of the formulations to another CXCR4 high expression (MDA-MB-231) and CXCR4 low expression (HepG2) cell lines and calculated the IC50. In MDA-MB-231 cell line, the IC50 of CTCE-DTX NPs (99.2 nmol/L) was significantly lower than that of DTX (135.8 nmol/L) and Sc-DTX NPs (250.1 nmol/L), which was consist with the tendency of formulations to 4T1 cell line. In comparison, in HepG2 cell line, the IC50 of CTCE-DTX NPs (166.8 nmol/L) was higher than that of DTX (109.5 nmol/L). The results further proved that the recognition of CTCE-DTX NPs with CXCR4 could enhance the cytotoxicity to tumor cells. These results demonstrated that CTCE peptide enhanced the chemosensitivity of cancer cell toward DTX but possessed with no direct toxicity.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Half life period | 25.7 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Liver cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
166.8 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
To increase the DTX solubility and avoid the DTX therapy induced metastasis, we designed a peptide-drug conjugate (CTCE-DTX) by coupling two functional molecules (CXCR4 antagonist peptide and chemotherapeutic agent DTX) through a flexible linker. The amphiphilic monomer of CTCE-DTX could self-assemble into nanoparticles (CTCE-DTX NPs) in the water environment (without the participation of toxic ethanol and Tween 80 as excipients). The CXCR4 antagonist component could target CXCR4 upregulated tumor cells, which not only enhanced the efficacy of DTX but also inhibited the bone and lung metastasis of TNBC.
Click to Show/Hide
|
||||
| Description |
Besides, the combination of DTX and CTCE showed a substantially greater cytotoxicity, with an approximate 2 folds reduction (67.9 nmol/L) in half-maximal inhibitory concentration (IC50) compared to DTX alone. What's more, CTCE-DTX NPs retained a greater cytotoxicity (IC50 = 88.5 nmol/L) than Sc-DTX NPs (IC50 = 169.5 nmol/L), which meant CXCR4 antagonist peptide could work as a sensitizer to DTX. The slightly higher IC50 value of CTCE-DTX NPs than that of DTX + CTCE group may attribute to the incomplete disassembly of nanostructures. Meanwhile, consistent with previous reports14, the CXCR4 antagonist peptide and scramble peptide didn't not show any cytotoxicity to 4T1 cell at low concentration. We also tested the cytotoxicity of the formulations to another CXCR4 high expression (MDA-MB-231) and CXCR4 low expression (HepG2) cell lines and calculated the IC50. In MDA-MB-231 cell line, the IC50 of CTCE-DTX NPs (99.2 nmol/L) was significantly lower than that of DTX (135.8 nmol/L) and Sc-DTX NPs (250.1 nmol/L), which was consist with the tendency of formulations to 4T1 cell line. In comparison, in HepG2 cell line, the IC50 of CTCE-DTX NPs (166.8 nmol/L) was higher than that of DTX (109.5 nmol/L). The results further proved that the recognition of CTCE-DTX NPs with CXCR4 could enhance the cytotoxicity to tumor cells. These results demonstrated that CTCE peptide enhanced the chemosensitivity of cancer cell toward DTX but possessed with no direct toxicity.
Click to Show/Hide
|
||||
| In Vitro Model | Hepatoblastoma | L-O2 cell line | CVCL_0027 | ||
| Half life period | 25.7 h | ||||
SC-CTCE-DTX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Liver cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
206.0 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
To increase the DTX solubility and avoid the DTX therapy induced metastasis, we designed a peptide-drug conjugate (CTCE-DTX) by coupling two functional molecules (CXCR4 antagonist peptide and chemotherapeutic agent DTX) through a flexible linker. The amphiphilic monomer of CTCE-DTX could self-assemble into nanoparticles (CTCE-DTX NPs) in the water environment (without the participation of toxic ethanol and Tween 80 as excipients). The CXCR4 antagonist component could target CXCR4 upregulated tumor cells, which not only enhanced the efficacy of DTX but also inhibited the bone and lung metastasis of TNBC.
Click to Show/Hide
|
||||
| Description |
Besides, the combination of DTX and CTCE showed a substantially greater cytotoxicity, with an approximate 2 folds reduction (67.9 nmol/L) in half-maximal inhibitory concentration (IC50) compared to DTX alone. What's more, CTCE-DTX NPs retained a greater cytotoxicity (IC50 = 88.5 nmol/L) than Sc-DTX NPs (IC50 = 169.5 nmol/L), which meant CXCR4 antagonist peptide could work as a sensitizer to DTX. The slightly higher IC50 value of CTCE-DTX NPs than that of DTX + CTCE group may attribute to the incomplete disassembly of nanostructures. Meanwhile, consistent with previous reports14, the CXCR4 antagonist peptide and scramble peptide didn't not show any cytotoxicity to 4T1 cell at low concentration. We also tested the cytotoxicity of the formulations to another CXCR4 high expression (MDA-MB-231) and CXCR4 low expression (HepG2) cell lines and calculated the IC50. In MDA-MB-231 cell line, the IC50 of CTCE-DTX NPs (99.2 nmol/L) was significantly lower than that of DTX (135.8 nmol/L) and Sc-DTX NPs (250.1 nmol/L), which was consist with the tendency of formulations to 4T1 cell line. In comparison, in HepG2 cell line, the IC50 of CTCE-DTX NPs (166.8 nmol/L) was higher than that of DTX (109.5 nmol/L). The results further proved that the recognition of CTCE-DTX NPs with CXCR4 could enhance the cytotoxicity to tumor cells. These results demonstrated that CTCE peptide enhanced the chemosensitivity of cancer cell toward DTX but possessed with no direct toxicity.
Click to Show/Hide
|
||||
| In Vitro Model | Hepatoblastoma | L-O2 cell line | CVCL_0027 | ||
| Half life period | 29.3 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
250.1 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
To increase the DTX solubility and avoid the DTX therapy induced metastasis, we designed a peptide-drug conjugate (CTCE-DTX) by coupling two functional molecules (CXCR4 antagonist peptide and chemotherapeutic agent DTX) through a flexible linker. The amphiphilic monomer of CTCE-DTX could self-assemble into nanoparticles (CTCE-DTX NPs) in the water environment (without the participation of toxic ethanol and Tween 80 as excipients). The CXCR4 antagonist component could target CXCR4 upregulated tumor cells, which not only enhanced the efficacy of DTX but also inhibited the bone and lung metastasis of TNBC.
Click to Show/Hide
|
||||
| Description |
Besides, the combination of DTX and CTCE showed a substantially greater cytotoxicity, with an approximate 2 folds reduction (67.9 nmol/L) in half-maximal inhibitory concentration (IC50) compared to DTX alone. What's more, CTCE-DTX NPs retained a greater cytotoxicity (IC50 = 88.5 nmol/L) than Sc-DTX NPs (IC50 = 169.5 nmol/L), which meant CXCR4 antagonist peptide could work as a sensitizer to DTX. The slightly higher IC50 value of CTCE-DTX NPs than that of DTX + CTCE group may attribute to the incomplete disassembly of nanostructures. Meanwhile, consistent with previous reports14, the CXCR4 antagonist peptide and scramble peptide didn't not show any cytotoxicity to 4T1 cell at low concentration. We also tested the cytotoxicity of the formulations to another CXCR4 high expression (MDA-MB-231) and CXCR4 low expression (HepG2) cell lines and calculated the IC50. In MDA-MB-231 cell line, the IC50 of CTCE-DTX NPs (99.2 nmol/L) was significantly lower than that of DTX (135.8 nmol/L) and Sc-DTX NPs (250.1 nmol/L), which was consist with the tendency of formulations to 4T1 cell line. In comparison, in HepG2 cell line, the IC50 of CTCE-DTX NPs (166.8 nmol/L) was higher than that of DTX (109.5 nmol/L). The results further proved that the recognition of CTCE-DTX NPs with CXCR4 could enhance the cytotoxicity to tumor cells. These results demonstrated that CTCE peptide enhanced the chemosensitivity of cancer cell toward DTX but possessed with no direct toxicity.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Half life period | 29.3 h | ||||
References