
Linker Information
General Information of This Linker
| Linker ID |
LIN00100
|
|||||
|---|---|---|---|---|---|---|
| Linker Name |
Hydrazone bond
|
|||||
| Linker Type |
pH-Sensitive linkers
|
|||||
| Structure |
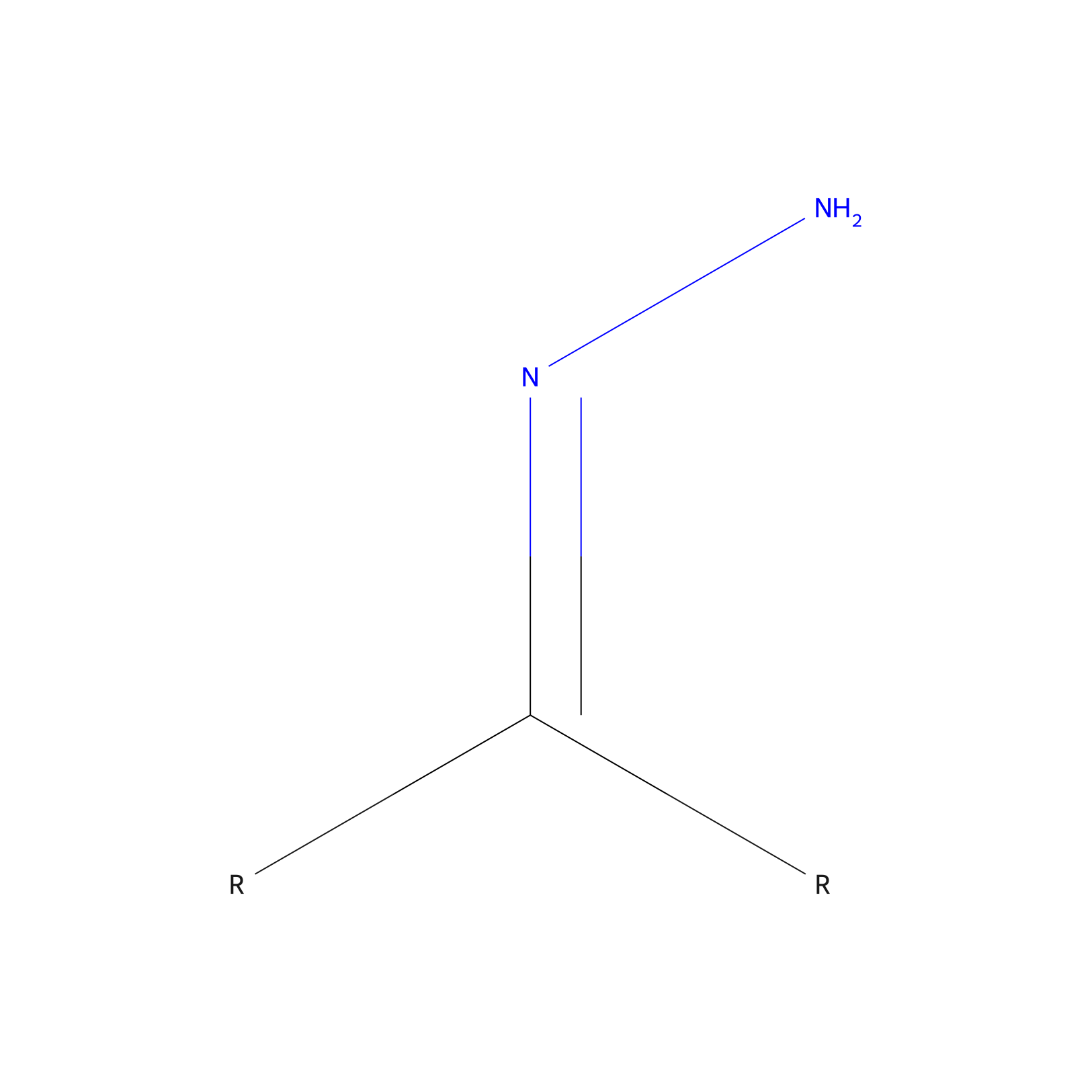
|
|||||
| Formula |
CH2*2N2
|
|||||
| #Ro5 Violations (Lipinski): 0 | Molecular Weight (mw) | 42.041 | ||||
| Lipid-water partition coefficient (xlogp) | -0.6853 | |||||
| Hydrogen Bond Donor Count (hbonddonor) | 1 | |||||
| Hydrogen Bond Acceptor Count (hbondacc) | 2 | |||||
| Rotatable Bond Count (rotbonds) | 0 | |||||
| Canonical smiles |
*C(*)=NN
|
|||||
Each Peptide-drug Conjugate Related to This Linker
Full Information of The Activity Data of The PDC(s) Related to This Linker
TPP-DOX-AP2H [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Average tumor volume shrunk |
55%
|
|||
| Administration Time | 18 days | ||||
| Administration Dosage | 20 µM | ||||
| MOA of PDC |
Initiated by the interaction between AP2H and membrane-anchored LAPTM4B protein, PDC specifically recognized and bound cancer cells. The receptor-mediated endocytic pathway then led to the delivery of PDC to lysosomes. The low pH environment in lysosomes efficiently cut the hydrazone bond and released TPP-DOX. The successful escape from lysosomes enabled further transportation of TPP-DOX to mitochondria directed by targeting group TPP. Because TPP-DOX is highly active in producing ROS and mitochondria are prone to oxidative damage, the disruption in the electron transport chain and ATP synthesis finally lead to mitochondrial dysfunction and cell death. With dual-targeting ability, PDC can effectively bypass the interaction with P-gp. Meanwhile, due to the energy-dependent expression feature of P-gp, the damage to mitochondria inhibit P-gp expression, thus inhibit the efflux. (9) The targeted and mitochondria interrupted cell damaging pathway allow PDC to exert toxicity both wild type and drug resistant
Click to Show/Hide
|
||||
| Description |
The average tumor volume shrunk by 55% at day 18 compared with that of the control group (Figure 4c).
|
||||
| In Vivo Model | HepG2 tumor xenograft model. | ||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
15 µM
|
|||
| MOA of PDC |
Initiated by the interaction between AP2H and membrane-anchored LAPTM4B protein, PDC specifically recognized and bound cancer cells. The receptor-mediated endocytic pathway then led to the delivery of PDC to lysosomes. The low pH environment in lysosomes efficiently cut the hydrazone bond and released TPP-DOX. The successful escape from lysosomes enabled further transportation of TPP-DOX to mitochondria directed by targeting group TPP. Because TPP-DOX is highly active in producing ROS and mitochondria are prone to oxidative damage, the disruption in the electron transport chain and ATP synthesis finally lead to mitochondrial dysfunction and cell death. With dual-targeting ability, PDC can effectively bypass the interaction with P-gp. Meanwhile, due to the energy-dependent expression feature of P-gp, the damage to mitochondria inhibit P-gp expression, thus inhibit the efflux. (9) The targeted and mitochondria interrupted cell damaging pathway allow PDC to exert toxicity both wild type and drug resistant
Click to Show/Hide
|
||||
| Description |
For PDC, its IC50 values against MCF-7/WT (15 uM) and MCF-7/ADR (18 uM) are almost the same, confirming its effectiveness in bypass drug resistance.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
18 µM
|
|||
| MOA of PDC |
Initiated by the interaction between AP2H and membrane-anchored LAPTM4B protein, PDC specifically recognized and bound cancer cells. The receptor-mediated endocytic pathway then led to the delivery of PDC to lysosomes. The low pH environment in lysosomes efficiently cut the hydrazone bond and released TPP-DOX. The successful escape from lysosomes enabled further transportation of TPP-DOX to mitochondria directed by targeting group TPP. Because TPP-DOX is highly active in producing ROS and mitochondria are prone to oxidative damage, the disruption in the electron transport chain and ATP synthesis finally lead to mitochondrial dysfunction and cell death. With dual-targeting ability, PDC can effectively bypass the interaction with P-gp. Meanwhile, due to the energy-dependent expression feature of P-gp, the damage to mitochondria inhibit P-gp expression, thus inhibit the efflux. (9) The targeted and mitochondria interrupted cell damaging pathway allow PDC to exert toxicity both wild type and drug resistant
Click to Show/Hide
|
||||
| Description |
For PDC, its IC50 values against MCF-7/WT (15 uM) and MCF-7/ADR (18 uM) are almost the same, confirming its effectiveness in bypass drug resistance.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF7/ADR cell | CVCL_0031 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
17%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 20 µM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Initiated by the interaction between AP2H and membrane-anchored LAPTM4B protein, PDC specifically recognized and bound cancer cells. The receptor-mediated endocytic pathway then led to the delivery of PDC to lysosomes. The low pH environment in lysosomes efficiently cut the hydrazone bond and released TPP-DOX. The successful escape from lysosomes enabled further transportation of TPP-DOX to mitochondria directed by targeting group TPP. Because TPP-DOX is highly active in producing ROS and mitochondria are prone to oxidative damage, the disruption in the electron transport chain and ATP synthesis finally lead to mitochondrial dysfunction and cell death. With dual-targeting ability, PDC can effectively bypass the interaction with P-gp. Meanwhile, due to the energy-dependent expression feature of P-gp, the damage to mitochondria inhibit P-gp expression, thus inhibit the efflux. (9) The targeted and mitochondria interrupted cell damaging pathway allow PDC to exert toxicity both wild type and drug resistant
Click to Show/Hide
|
||||
| Description |
PDC killed most HepG2 cells with a high efficiency of 83% and left HEK293 cells unaffected.
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
40%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 20 µM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Initiated by the interaction between AP2H and membrane-anchored LAPTM4B protein, PDC specifically recognized and bound cancer cells. The receptor-mediated endocytic pathway then led to the delivery of PDC to lysosomes. The low pH environment in lysosomes efficiently cut the hydrazone bond and released TPP-DOX. The successful escape from lysosomes enabled further transportation of TPP-DOX to mitochondria directed by targeting group TPP. Because TPP-DOX is highly active in producing ROS and mitochondria are prone to oxidative damage, the disruption in the electron transport chain and ATP synthesis finally lead to mitochondrial dysfunction and cell death. With dual-targeting ability, PDC can effectively bypass the interaction with P-gp. Meanwhile, due to the energy-dependent expression feature of P-gp, the damage to mitochondria inhibit P-gp expression, thus inhibit the efflux. (9) The targeted and mitochondria interrupted cell damaging pathway allow PDC to exert toxicity both wild type and drug resistant
Click to Show/Hide
|
||||
| Description |
PDC maintained almost the same cytotoxicity against MCF-7/ADR cells and MCF-7/WT cells
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
45%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 20 µM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Initiated by the interaction between AP2H and membrane-anchored LAPTM4B protein, PDC specifically recognized and bound cancer cells. The receptor-mediated endocytic pathway then led to the delivery of PDC to lysosomes. The low pH environment in lysosomes efficiently cut the hydrazone bond and released TPP-DOX. The successful escape from lysosomes enabled further transportation of TPP-DOX to mitochondria directed by targeting group TPP. Because TPP-DOX is highly active in producing ROS and mitochondria are prone to oxidative damage, the disruption in the electron transport chain and ATP synthesis finally lead to mitochondrial dysfunction and cell death. With dual-targeting ability, PDC can effectively bypass the interaction with P-gp. Meanwhile, due to the energy-dependent expression feature of P-gp, the damage to mitochondria inhibit P-gp expression, thus inhibit the efflux. (9) The targeted and mitochondria interrupted cell damaging pathway allow PDC to exert toxicity both wild type and drug resistant
Click to Show/Hide
|
||||
| Description |
PDC maintained almost the same cytotoxicity against MCF-7/ADR cells and MCF-7/WT cells
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF7/ADR cell | CVCL_0031 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
100%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 20 µM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Initiated by the interaction between AP2H and membrane-anchored LAPTM4B protein, PDC specifically recognized and bound cancer cells. The receptor-mediated endocytic pathway then led to the delivery of PDC to lysosomes. The low pH environment in lysosomes efficiently cut the hydrazone bond and released TPP-DOX. The successful escape from lysosomes enabled further transportation of TPP-DOX to mitochondria directed by targeting group TPP. Because TPP-DOX is highly active in producing ROS and mitochondria are prone to oxidative damage, the disruption in the electron transport chain and ATP synthesis finally lead to mitochondrial dysfunction and cell death. With dual-targeting ability, PDC can effectively bypass the interaction with P-gp. Meanwhile, due to the energy-dependent expression feature of P-gp, the damage to mitochondria inhibit P-gp expression, thus inhibit the efflux. (9) The targeted and mitochondria interrupted cell damaging pathway allow PDC to exert toxicity both wild type and drug resistant
Click to Show/Hide
|
||||
| Description |
PDC killed most HepG2 cells with a high efficiency of 83% and left HEK293 cells unaffected.
|
||||
| In Vitro Model | Normal | HEK293 cell | CVCL_0045 | ||
Omi-hyd-Dex [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
8 µg/mL
|
|||
| Administration Time | 18 h | ||||
| Evaluation Method | MIC assay | ||||
| MOA of PDC |
To minimize the toxic side effect of omiganan and optimize its anti-inflammatory capability, we designed an IMEs-responsive self-delivery nanosystem consisting of omiganan, an anti-inflammatory agent, and natural polysaccharide to achieve on-demand degradation and responsive drug release. Based on the cationic hydrophilic fragments on omiganan, we chose dexamethasone (Dex, a hydrophobic anti-inflammatory drug) to link with this peptide via a hydrazone bond to construct an amphiphilic conjugate (Omi-hyd-Dex). With the assistance of PLGA, this conjugate could self-assemble into nanoparticles (Omi-hyd-Dex NPs) in an aqueous solution without introducing any other hazardous materials. Then, the negatively-charged hyaluronic acid (HA, a natural ligand of ICAM-1 and CD44) was used to coat Omi-hyd-Dex NPs to form a core-shell structural formulation (Omi-hyd-Dex@HA NPs). This HA coating could not only eliminate the hemolytic activity of omiganan to reduce side effects but also act as the IMEs targeting molecule through interaction with intercellular adhesion molecule-1 (ICAM-1) on inflamed endothelial cells. After entering IMEs, the HA coating would be degraded and detached to expose the cationic surface of the Omi-hyd-Dex core and enable it to accumulate in IMEs. Meanwhile, the hydrazone bond between omiganan and Dex could be cleaved in response to the acidic condition of IMEs, thereby releasing the cationic peptide and anti-inflammatory agent that would concurrently inhibit the infection and inflammation precisely.
Click to Show/Hide
|
||||
| Description |
To verify whether Omi-hyd-Dex@HA NPs inherited the antimicrobial activity of omiganan, the MICs were tested by the standardized agar doubling-dilution method as shown in Table 1. Omi-hyd-Dex@HA NPs and free omiganan showed similar extended broad-spectrum antibacterial activity for G+ bacteria, G- bacteria and fungi pathogens, including drug-sensitive strains (S. aureus, P. aeruginosa, K.P., Candida albicans) and drug-resistant strains (MRSA, MDR-K.P.). The MICs of Omi-hyd-Dex@HA NPs showed none or only a one-fold increase compared with the free Omiganan-NHNH2 group. In addition, free Dex did not show any antimicrobial activity for the tested pathogens, indicating that Dex moieties of Omi-hyd-Dex@HA NPs did not contribute to the bacterial killing effect. In contrast, vancomycin, as a standard drug regimen for drug-resistant G+ strains, showed inefficacious for G- strains and fungus. Similar results were also observed in oxacillin-treated groups. Therefore, Omi-hyd-Dex@HA NPs retained the broad-spectrum antimicrobial activity of omiganan.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus infection strain | 1280 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
16 µg/mL
|
|||
| Administration Time | 18 h | ||||
| Evaluation Method | MIC assay | ||||
| MOA of PDC |
To minimize the toxic side effect of omiganan and optimize its anti-inflammatory capability, we designed an IMEs-responsive self-delivery nanosystem consisting of omiganan, an anti-inflammatory agent, and natural polysaccharide to achieve on-demand degradation and responsive drug release. Based on the cationic hydrophilic fragments on omiganan, we chose dexamethasone (Dex, a hydrophobic anti-inflammatory drug) to link with this peptide via a hydrazone bond to construct an amphiphilic conjugate (Omi-hyd-Dex). With the assistance of PLGA, this conjugate could self-assemble into nanoparticles (Omi-hyd-Dex NPs) in an aqueous solution without introducing any other hazardous materials. Then, the negatively-charged hyaluronic acid (HA, a natural ligand of ICAM-1 and CD44) was used to coat Omi-hyd-Dex NPs to form a core-shell structural formulation (Omi-hyd-Dex@HA NPs). This HA coating could not only eliminate the hemolytic activity of omiganan to reduce side effects but also act as the IMEs targeting molecule through interaction with intercellular adhesion molecule-1 (ICAM-1) on inflamed endothelial cells. After entering IMEs, the HA coating would be degraded and detached to expose the cationic surface of the Omi-hyd-Dex core and enable it to accumulate in IMEs. Meanwhile, the hydrazone bond between omiganan and Dex could be cleaved in response to the acidic condition of IMEs, thereby releasing the cationic peptide and anti-inflammatory agent that would concurrently inhibit the infection and inflammation precisely.
Click to Show/Hide
|
||||
| Description |
To verify whether Omi-hyd-Dex@HA NPs inherited the antimicrobial activity of omiganan, the MICs were tested by the standardized agar doubling-dilution method as shown in Table 1. Omi-hyd-Dex@HA NPs and free omiganan showed similar extended broad-spectrum antibacterial activity for G+ bacteria, G- bacteria and fungi pathogens, including drug-sensitive strains (S. aureus, P. aeruginosa, K.P., Candida albicans) and drug-resistant strains (MRSA, MDR-K.P.). The MICs of Omi-hyd-Dex@HA NPs showed none or only a one-fold increase compared with the free Omiganan-NHNH2 group. In addition, free Dex did not show any antimicrobial activity for the tested pathogens, indicating that Dex moieties of Omi-hyd-Dex@HA NPs did not contribute to the bacterial killing effect. In contrast, vancomycin, as a standard drug regimen for drug-resistant G+ strains, showed inefficacious for G- strains and fungus. Similar results were also observed in oxacillin-treated groups. Therefore, Omi-hyd-Dex@HA NPs retained the broad-spectrum antimicrobial activity of omiganan.
Click to Show/Hide
|
||||
| In Vitro Model | Multiple-resistant Staphylococcus aureus infection | Multiple-resistant staphylococcus aureus infection strain | 1280 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
16 µg/mL
|
|||
| Administration Time | 18 h | ||||
| Evaluation Method | MIC assay | ||||
| MOA of PDC |
To minimize the toxic side effect of omiganan and optimize its anti-inflammatory capability, we designed an IMEs-responsive self-delivery nanosystem consisting of omiganan, an anti-inflammatory agent, and natural polysaccharide to achieve on-demand degradation and responsive drug release. Based on the cationic hydrophilic fragments on omiganan, we chose dexamethasone (Dex, a hydrophobic anti-inflammatory drug) to link with this peptide via a hydrazone bond to construct an amphiphilic conjugate (Omi-hyd-Dex). With the assistance of PLGA, this conjugate could self-assemble into nanoparticles (Omi-hyd-Dex NPs) in an aqueous solution without introducing any other hazardous materials. Then, the negatively-charged hyaluronic acid (HA, a natural ligand of ICAM-1 and CD44) was used to coat Omi-hyd-Dex NPs to form a core-shell structural formulation (Omi-hyd-Dex@HA NPs). This HA coating could not only eliminate the hemolytic activity of omiganan to reduce side effects but also act as the IMEs targeting molecule through interaction with intercellular adhesion molecule-1 (ICAM-1) on inflamed endothelial cells. After entering IMEs, the HA coating would be degraded and detached to expose the cationic surface of the Omi-hyd-Dex core and enable it to accumulate in IMEs. Meanwhile, the hydrazone bond between omiganan and Dex could be cleaved in response to the acidic condition of IMEs, thereby releasing the cationic peptide and anti-inflammatory agent that would concurrently inhibit the infection and inflammation precisely.
Click to Show/Hide
|
||||
| Description |
To verify whether Omi-hyd-Dex@HA NPs inherited the antimicrobial activity of omiganan, the MICs were tested by the standardized agar doubling-dilution method as shown in Table 1. Omi-hyd-Dex@HA NPs and free omiganan showed similar extended broad-spectrum antibacterial activity for G+ bacteria, G- bacteria and fungi pathogens, including drug-sensitive strains (S. aureus, P. aeruginosa, K.P., Candida albicans) and drug-resistant strains (MRSA, MDR-K.P.). The MICs of Omi-hyd-Dex@HA NPs showed none or only a one-fold increase compared with the free Omiganan-NHNH2 group. In addition, free Dex did not show any antimicrobial activity for the tested pathogens, indicating that Dex moieties of Omi-hyd-Dex@HA NPs did not contribute to the bacterial killing effect. In contrast, vancomycin, as a standard drug regimen for drug-resistant G+ strains, showed inefficacious for G- strains and fungus. Similar results were also observed in oxacillin-treated groups. Therefore, Omi-hyd-Dex@HA NPs retained the broad-spectrum antimicrobial activity of omiganan.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae strain | 573 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
16 µg/mL
|
|||
| Administration Time | 18 h | ||||
| Evaluation Method | MIC assay | ||||
| MOA of PDC |
To minimize the toxic side effect of omiganan and optimize its anti-inflammatory capability, we designed an IMEs-responsive self-delivery nanosystem consisting of omiganan, an anti-inflammatory agent, and natural polysaccharide to achieve on-demand degradation and responsive drug release. Based on the cationic hydrophilic fragments on omiganan, we chose dexamethasone (Dex, a hydrophobic anti-inflammatory drug) to link with this peptide via a hydrazone bond to construct an amphiphilic conjugate (Omi-hyd-Dex). With the assistance of PLGA, this conjugate could self-assemble into nanoparticles (Omi-hyd-Dex NPs) in an aqueous solution without introducing any other hazardous materials. Then, the negatively-charged hyaluronic acid (HA, a natural ligand of ICAM-1 and CD44) was used to coat Omi-hyd-Dex NPs to form a core-shell structural formulation (Omi-hyd-Dex@HA NPs). This HA coating could not only eliminate the hemolytic activity of omiganan to reduce side effects but also act as the IMEs targeting molecule through interaction with intercellular adhesion molecule-1 (ICAM-1) on inflamed endothelial cells. After entering IMEs, the HA coating would be degraded and detached to expose the cationic surface of the Omi-hyd-Dex core and enable it to accumulate in IMEs. Meanwhile, the hydrazone bond between omiganan and Dex could be cleaved in response to the acidic condition of IMEs, thereby releasing the cationic peptide and anti-inflammatory agent that would concurrently inhibit the infection and inflammation precisely.
Click to Show/Hide
|
||||
| Description |
To verify whether Omi-hyd-Dex@HA NPs inherited the antimicrobial activity of omiganan, the MICs were tested by the standardized agar doubling-dilution method as shown in Table 1. Omi-hyd-Dex@HA NPs and free omiganan showed similar extended broad-spectrum antibacterial activity for G+ bacteria, G- bacteria and fungi pathogens, including drug-sensitive strains (S. aureus, P. aeruginosa, K.P., Candida albicans) and drug-resistant strains (MRSA, MDR-K.P.). The MICs of Omi-hyd-Dex@HA NPs showed none or only a one-fold increase compared with the free Omiganan-NHNH2 group. In addition, free Dex did not show any antimicrobial activity for the tested pathogens, indicating that Dex moieties of Omi-hyd-Dex@HA NPs did not contribute to the bacterial killing effect. In contrast, vancomycin, as a standard drug regimen for drug-resistant G+ strains, showed inefficacious for G- strains and fungus. Similar results were also observed in oxacillin-treated groups. Therefore, Omi-hyd-Dex@HA NPs retained the broad-spectrum antimicrobial activity of omiganan.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | MDR Klebsiella pneumoniae strain | 573 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
32 µg/mL
|
|||
| Administration Time | 18 h | ||||
| Evaluation Method | MIC assay | ||||
| MOA of PDC |
To minimize the toxic side effect of omiganan and optimize its anti-inflammatory capability, we designed an IMEs-responsive self-delivery nanosystem consisting of omiganan, an anti-inflammatory agent, and natural polysaccharide to achieve on-demand degradation and responsive drug release. Based on the cationic hydrophilic fragments on omiganan, we chose dexamethasone (Dex, a hydrophobic anti-inflammatory drug) to link with this peptide via a hydrazone bond to construct an amphiphilic conjugate (Omi-hyd-Dex). With the assistance of PLGA, this conjugate could self-assemble into nanoparticles (Omi-hyd-Dex NPs) in an aqueous solution without introducing any other hazardous materials. Then, the negatively-charged hyaluronic acid (HA, a natural ligand of ICAM-1 and CD44) was used to coat Omi-hyd-Dex NPs to form a core-shell structural formulation (Omi-hyd-Dex@HA NPs). This HA coating could not only eliminate the hemolytic activity of omiganan to reduce side effects but also act as the IMEs targeting molecule through interaction with intercellular adhesion molecule-1 (ICAM-1) on inflamed endothelial cells. After entering IMEs, the HA coating would be degraded and detached to expose the cationic surface of the Omi-hyd-Dex core and enable it to accumulate in IMEs. Meanwhile, the hydrazone bond between omiganan and Dex could be cleaved in response to the acidic condition of IMEs, thereby releasing the cationic peptide and anti-inflammatory agent that would concurrently inhibit the infection and inflammation precisely.
Click to Show/Hide
|
||||
| Description |
To verify whether Omi-hyd-Dex@HA NPs inherited the antimicrobial activity of omiganan, the MICs were tested by the standardized agar doubling-dilution method as shown in Table 1. Omi-hyd-Dex@HA NPs and free omiganan showed similar extended broad-spectrum antibacterial activity for G+ bacteria, G- bacteria and fungi pathogens, including drug-sensitive strains (S. aureus, P. aeruginosa, K.P., Candida albicans) and drug-resistant strains (MRSA, MDR-K.P.). The MICs of Omi-hyd-Dex@HA NPs showed none or only a one-fold increase compared with the free Omiganan-NHNH2 group. In addition, free Dex did not show any antimicrobial activity for the tested pathogens, indicating that Dex moieties of Omi-hyd-Dex@HA NPs did not contribute to the bacterial killing effect. In contrast, vancomycin, as a standard drug regimen for drug-resistant G+ strains, showed inefficacious for G- strains and fungus. Similar results were also observed in oxacillin-treated groups. Therefore, Omi-hyd-Dex@HA NPs retained the broad-spectrum antimicrobial activity of omiganan.
Click to Show/Hide
|
||||
| In Vitro Model | Pseudomonas aeruginosa strain infection | Pseudomonas aeruginosa strain | 287 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
32 µg/mL
|
|||
| Administration Time | 18 h | ||||
| Evaluation Method | MIC assay | ||||
| MOA of PDC |
To minimize the toxic side effect of omiganan and optimize its anti-inflammatory capability, we designed an IMEs-responsive self-delivery nanosystem consisting of omiganan, an anti-inflammatory agent, and natural polysaccharide to achieve on-demand degradation and responsive drug release. Based on the cationic hydrophilic fragments on omiganan, we chose dexamethasone (Dex, a hydrophobic anti-inflammatory drug) to link with this peptide via a hydrazone bond to construct an amphiphilic conjugate (Omi-hyd-Dex). With the assistance of PLGA, this conjugate could self-assemble into nanoparticles (Omi-hyd-Dex NPs) in an aqueous solution without introducing any other hazardous materials. Then, the negatively-charged hyaluronic acid (HA, a natural ligand of ICAM-1 and CD44) was used to coat Omi-hyd-Dex NPs to form a core-shell structural formulation (Omi-hyd-Dex@HA NPs). This HA coating could not only eliminate the hemolytic activity of omiganan to reduce side effects but also act as the IMEs targeting molecule through interaction with intercellular adhesion molecule-1 (ICAM-1) on inflamed endothelial cells. After entering IMEs, the HA coating would be degraded and detached to expose the cationic surface of the Omi-hyd-Dex core and enable it to accumulate in IMEs. Meanwhile, the hydrazone bond between omiganan and Dex could be cleaved in response to the acidic condition of IMEs, thereby releasing the cationic peptide and anti-inflammatory agent that would concurrently inhibit the infection and inflammation precisely.
Click to Show/Hide
|
||||
| Description |
To verify whether Omi-hyd-Dex@HA NPs inherited the antimicrobial activity of omiganan, the MICs were tested by the standardized agar doubling-dilution method as shown in Table 1. Omi-hyd-Dex@HA NPs and free omiganan showed similar extended broad-spectrum antibacterial activity for G+ bacteria, G- bacteria and fungi pathogens, including drug-sensitive strains (S. aureus, P. aeruginosa, K.P., Candida albicans) and drug-resistant strains (MRSA, MDR-K.P.). The MICs of Omi-hyd-Dex@HA NPs showed none or only a one-fold increase compared with the free Omiganan-NHNH2 group. In addition, free Dex did not show any antimicrobial activity for the tested pathogens, indicating that Dex moieties of Omi-hyd-Dex@HA NPs did not contribute to the bacterial killing effect. In contrast, vancomycin, as a standard drug regimen for drug-resistant G+ strains, showed inefficacious for G- strains and fungus. Similar results were also observed in oxacillin-treated groups. Therefore, Omi-hyd-Dex@HA NPs retained the broad-spectrum antimicrobial activity of omiganan.
Click to Show/Hide
|
||||
| In Vitro Model | Candida albicans infection | Candida albicans fungus strains | 5476 | ||
References