
Peptide-drug Conjugate Information
General Information of This Peptide-drug Conjugate (PDC)
| PDC ID |
PDC_00480
|
|||||
|---|---|---|---|---|---|---|
| PDC Name |
TKPR - N-(4-Acetylphenyl)-5-bromo-2-hydroxybenzamide conjugate
|
|||||
| PDC Status |
Investigative
|
|||||
| Indication |
In total 3 Indication(s)
|
|||||
| Structure |
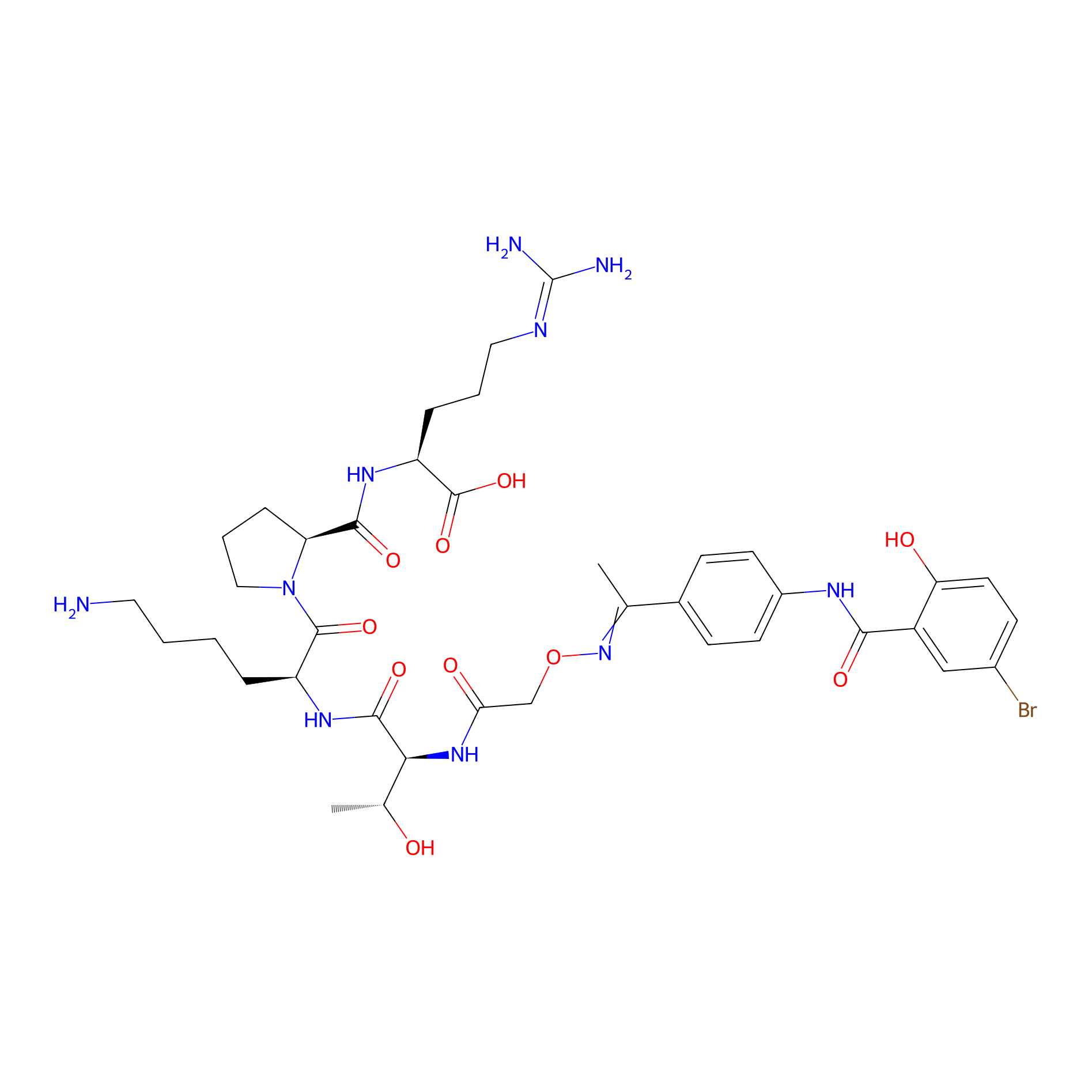
|
|||||
| Peptide Name |
TKPR
|
Peptide Info | ||||
| Drug Name |
N-(4-Acetylphenyl)-5-bromo-2-hydroxybenzamide
|
Drug Info | ||||
| Linker Name |
Aminooxyacetic acid
|
Linker Info | ||||
| Formula |
C38H53BrN10O10
|
|||||
| #Ro5 Violations (Lipinski): 4 | Molecular Weight | 889.806 | ||||
| Lipid-water partition coefficient (xlogp) | 0.241 | |||||
| Hydrogen Bond Donor Count (hbonddonor) | 10 | |||||
| Hydrogen Bond Acceptor Count (hbondacc) | 12 | |||||
| Rotatable Bond Count (rotbonds) | 22 | |||||
Full List of Activity Data of This Peptide-drug Conjugate
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
1
|
|||
| Administration Time | 1 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
60
|
|||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 200 mM | |||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 200 mM | |||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
15 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
37.6 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
40 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
45 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
75.1 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
100 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
112.4 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
200 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
224.8 μM
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index | > 4.4 | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index | > 4.4 | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
References