
Linker Information
General Information of This Linker
| Linker ID |
LIN00019
|
|||||
|---|---|---|---|---|---|---|
| Linker Name |
Aminooxyacetic acid
|
|||||
| Structure |
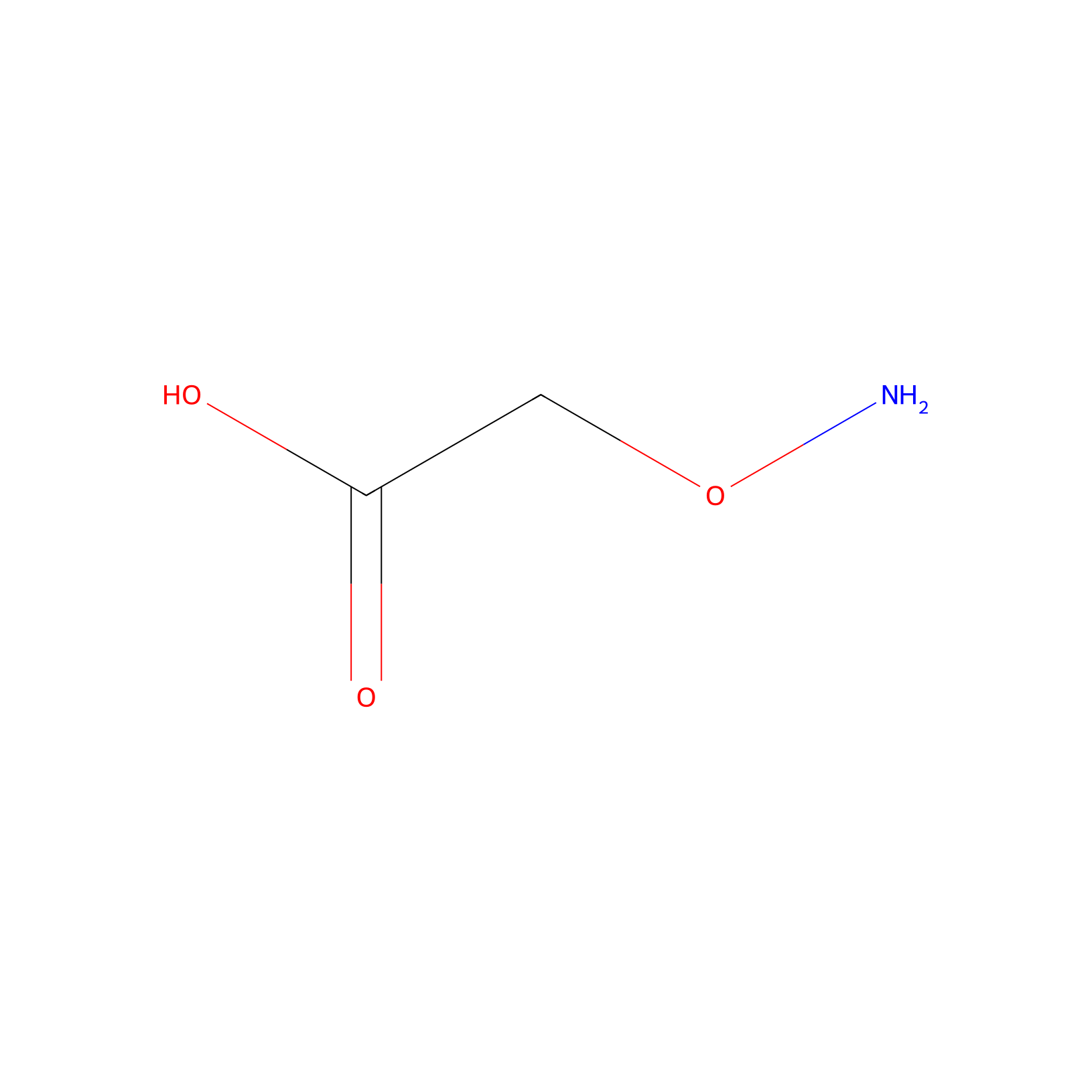
|
|||||
| Formula |
C2H5NO3
|
|||||
| #Ro5 Violations (Lipinski): 1 | Molecular Weight (mw) | 91.07 | ||||
| Lipid-water partition coefficient (xlogp) | -3.4 | |||||
| Hydrogen Bond Donor Count (hbonddonor) | 2 | |||||
| Hydrogen Bond Acceptor Count (hbondacc) | 4 | |||||
| Rotatable Bond Count (rotbonds) | 2 | |||||
| Chemble ID | ||||||
| Chemble ID | ||||||
| PubChem CID | ||||||
| Canonical smiles |
C(C(=O)O)ON
|
|||||
| InChI |
InChI=1S/C2H5NO3/c3-6-1-2(4)5/h1,3H2,(H,4,5)
|
|||||
| InChIKey |
NQRKYASMKDDGHT-UHFFFAOYSA-N
|
|||||
| IUPAC Name |
2-aminooxyacetic acid
|
|||||
Each Peptide-drug Conjugate Related to This Linker
Full Information of The Activity Data of The PDC(s) Related to This Linker
GnRH-III-[2ΔHis, 3D-Tic, 4Lys(Bu), 8Lys(Dau=Aoa)] [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor weight decrease |
27.70%
|
|||
| Administration Dosage | 15 mg/kg Dau content | ||||
| Description |
Based on these tumor weights, we determined that free Dau, 1 and 2 inhibited tumor weight significantly by 40.1, 28.7 and 27.7% in the case of orthotopic human MDA-MB-231 breast tumor model.
|
||||
| In Vivo Model | Orthotopic MDA-MB-231 human breast carcinoma bearing mice model. | ||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Colorectal cancer | ||||
| Efficacy Data | Tumor weight |
87.10%
|
|||
| Administration Dosage | 10 mg/kg Dau content | ||||
| Description |
The obtained data reveal that Dau, 1 and 2 significantly inhibited the tumor growth, whereby the tumor weights were reduced by 84.3, 80.8 and 87.1%, as compared to the control group.
|
||||
| In Vivo Model | Orthotopic HT-29 human colon carcinoma bearing mice model. | ||||
| In Vitro Model | Colon adenocarcinoma | HT-29 cell | CVCL_0320 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor volume decrease |
23.10%
|
|||
| Administration Dosage | 15 mg/kg Dau content | ||||
| Description |
Apart from that, a significant inhibition of the tumor volume was also obtained in groups which were treated with conjugate 1 (34.1%) and 2 (23.1%).
|
||||
| In Vivo Model | Orthotopic MDA-MB-231 human breast carcinoma bearing mice model. | ||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Proliferation index |
25.90%
|
|||
| Administration Dosage | 10 mg/kg Dau content | ||||
| Description |
It was observed that both GnRH-III conjugates 1 and 2 caused a significant decrease of the proliferation index by 16.3 and 25.9%
|
||||
| In Vivo Model | Orthotopic 4T1 mice breast carcinoma bearing mice model. | ||||
| In Vitro Model | Mammary carcinoma | 4T1 cell | CVCL_0125 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Proliferation index |
39.10%
|
|||
| Administration Dosage | 10 mg/kg Dau content | ||||
| Description |
The obtained data revealed that free Dau and both conjugates (1, 2) significantly inhibited the number of micro-metastases in the lung by 33.7, 43.8 and 49.4%, as compared to the control group. The proliferation index of lung metastases was significantly inhibited by 27.8, 37 and 39.1% in groups that were treated with free Dau, 1 and 2.
|
||||
| In Vivo Model | Orthotopic 4T1 mice breast carcinoma bearing mice model. | ||||
| In Vitro Model | Mammary carcinoma | 4T1 cell | CVCL_0125 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Number of macro-metastases decrease |
64.40%
|
|||
| Administration Dosage | 10 mg/kg Dau content | ||||
| Description |
The number of macro-metastases in peripheral organs, such as spleen, lung, liver and kidneys, was counted, in order to determine the anti-metastatic effect of free Dau and the GnRH-III conjugates on aggressive 4T1 BC orthotopic model (Figure 5A). The number of macro-metastases in spleen was significantly decreased in all treated groups (Dau,1and2) by 64.3, 72.8 and 78.1%. In the lung, the number of macro-metastases was also significantly reduced for all treated groups by 55.4, 55.2 and 64.4%, respectively. The numbers of macro-metastases in the liver and kidneys were decreased under treatments, whereby a significant decrease could be only obtained for conjugate2.
Click to Show/Hide
|
||||
| In Vivo Model | Orthotopic 4T1 mice breast carcinoma bearing mice model. | ||||
| In Vitro Model | Mammary carcinoma | 4T1 cell | CVCL_0125 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Number of macro-metastases decrease |
78.10%
|
|||
| Administration Dosage | 10 mg/kg Dau content | ||||
| Description |
The number of macro-metastases in peripheral organs, such as spleen, lung, liver and kidneys, was counted, in order to determine the anti-metastatic effect of free Dau and the GnRH-III conjugates on aggressive 4T1 BC orthotopic model (Figure 5A). The number of macro-metastases in spleen was significantly decreased in all treated groups (Dau, 1 and 2) by 64.3, 72.8 and 78.1%. In the lung, the number of macro-metastases was also significantly reduced for all treated groups by 55.4, 55.2 and 64.4%, respectively. The numbers of macro-metastases in the liver and kidneys were decreased under treatments, whereby a significant decrease could be only obtained for conjugate 2.
Click to Show/Hide
|
||||
| In Vivo Model | Orthotopic 4T1 mice breast carcinoma bearing mice model. | ||||
| In Vitro Model | Mammary carcinoma | 4T1 cell | CVCL_0125 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Micro-metastases decrease |
49.40%
|
|||
| Administration Dosage | 10 mg/kg Dau content | ||||
| Description |
The obtained data revealed that free Dau and both conjugates (1, 2) significantly inhibited the number of micro-metastases in the lung by 33.7, 43.8 and 49.4%, as compared to the control group. The proliferation index of lung metastases was significantly inhibited by 27.8, 37 and 39.1% in groups that were treated with free Dau, 1 and 2.
|
||||
| In Vivo Model | Orthotopic 4T1 mice breast carcinoma bearing mice model. | ||||
| In Vitro Model | Mammary carcinoma | 4T1 cell | CVCL_0125 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Body weigth change |
8.20%
|
|||
| Administration Dosage | 15 mg/kg Dau content | ||||
| Description |
The body weight of the mice in control group was decreased by 2.7%, while in the groups treated with 1 and 2, it was decreased by 10.1 and 8.2%.
|
||||
| In Vivo Model | Orthotopic MDA-MB-231 human breast carcinoma bearing mice model. | ||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
0.14 ± 0.01 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.1 ± 0.2 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | B16 cell | CVCL_F936 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Oral cavity squamous cell carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.7 ± 0.5 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Oral cavity squamous cell carcinoma | PE/CA-PJ41 | CVCL_2680 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.8 ± 0.1 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Mammary carcinoma | 4T1 cell | CVCL_0125 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.9 ± 0.2 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Prostate carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.1 ± 0.2 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | DU145 cell | CVCL_0105 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Ovarian endometrioid adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.1 ± 0.5 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.2 ± 0.7 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.3 ± 0.1 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Lung adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.3 ± 0.7 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Lung adenocarcinoma | H1975 cell | CVCL_1511 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Prostate carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.4 ± 0.6 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.49 ± 0.53 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Normal | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.6 ± 0.7 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | C-26 cell | CVCL_XC68 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.6 ± 0.5 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | A2058 cell | CVCL_1059 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.6 ± 0.6 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | WM983B cell | CVCL_6809 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Amelanotic melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.9 ± 0.6 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | HT168-M1/M9 cell | CVCL_2H39 | ||
| Experiment 17 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Tongue squamous cell carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.9 ± 0.6 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Tongue squamous cell carcinoma | PE/CA-PJ15 cell | CVCL_2678 | ||
| Experiment 18 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.3 ± 0.9 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
The efficacy of current chemotherapeutic treatments against most solid tumors is limited by their systemic toxicity, which is partly associated with the cytotoxic properties of agents such as docetaxel or doxorubicin. To avoid or minimize adverse effects from chemotherapeutic molecules, a promising targeted approach is through peptide-drug conjugates (PDCs) that link anticancer molecules to peptides designed to interact with receptors highly expressed on cancer cells, and which can mediate the molecules rapid internalization within those cells. One such receptor is sortilin (SORT1), also known as neurotensin receptor3, a membranebound receptor that belongs to the VPS10P family of receptors. TH19P01 peptide was recently designed to target and exploit SORT1s ligand internalization function. Studies have confirmed that both TH1902 (a docetaxel-TH19P01 conjugate) and TH1904 (a doxorubicin-TH19P01 conjugate) require a SORT1-dependent mechanism of action to exert anticancer activities. In recent preclinical studies performed in immunocompromised animal models, which are unable to produce mature T-cells, TH1902 was effective against several human SORT1-positive xenograft models including triple-negative breast cancer (TNBC), ovarian cancer, and endometrial cancer.
Click to Show/Hide
|
||||
| Description |
In our experiments, GnRH-III was applied as a targeting peptide. The conjugate [8Lys(Dau=Aoa-GFLG)]-GnRH-III, in which Dau was connected to the side chain of Lys in position 8 through an aminooxyacetylated cathepsin B-labile GFLG spacer, showed significant tumor growth inhibition in s.c.-developed fast-growing C26 murine colon cancer-bearing Balb/c mice. The effect highly depended on the treatment schedule. In the first attempt, the conjugate was administered i.p. at a dose of 8.86 umol (5 mg Dau-content/kg body weight) five times on days 7, 9, 11, 14 and 16 after tumor transplantation (Treatment schedule A). The tumor volume inhibition was only 15.7% on day 26 when the experiment finished. A slight enhancement in inhibition (22.3%) was detected when a dose of conjugate corresponding to 15 mg Dau content/kg body weight (26.6 μmol) was injected only once on day 7 (Treatment schedule B). An additional treatment on day 10 (Treatment schedule C) did not result in any further improvements (21.9% on day 29). However, when the treatment schedule was changed to two treatments with the same dose on days 4 and 7, 46.3% inhibition was observed on day 29 (Treatment schedule D). In contrast, the treatments with free Dau at a dose of 2 mg/kg body weight (3.55 umol) on days 7, 9, 11, 14 and 16 showed only 22.6% inhibition on day 26. The median survival rates of the treated animals in comparison to the control group were 1 (A), 1.23 (B), 1.21 (C) and 1.38 (D), respectively, and 0.81 for free Dau. These results indicate the lower toxicity and the higher tumor volume inhibition effect of the conjugates in comparison with those of free Dau, as well as the importance of the treatment schedule. In another experiment, HT-29 human colon cancer was developed s.c. in immunodeficient SCID mice. Dau and two conjugates (with or without a GFLG spacer between the GnRH-III homing peptide and the payload) were used for the treatment. The first i.p. administrations were performed on day 13 after tumor inoculation. All the mice treated once with 2.5 mg/kg body weight (4.43 umol) free Dau died within 10 days. In contrast, the conjugates at a dose of 15 mg Dau content/kg body weight (26.6 umol) that refers to 52 mg/kg [8Lys(Dau=Aoa)]-GnRH-III and 62.5 mg/kg [8Lys(Dau=Aoa-GFLG)]-GnRH-III conjugates, respectively, did not show significant toxicity. The treatment was repeated on days 23 and 30. Because of the significant weight loss in several mice in the control group, the experiment was terminated on day 35. The tumor growth inhibition could be calculated as reductions in the tumor volume by 44.3% and 57.6% and the tumor weight by 41% and 50%, respectively. Interestingly, the conjugates were poorly effective on orthotopically developed tumors. In the case of the C26 colon tumor-bearing female Balb/c mice, only a 7% reduction in tumor weight was detected on day 13 after the two treatments on days 4 and 7 with [8Lys(Dau=Aoa)]-GnRH-III, at a 26.6 umol/kg (15 mg Dau content) dose. The effect of free Dau (2 mg/kg on days 4 and 7) showed a better inhibitory effect (24.4%). Interestingly, our novel developed GnRH-III derivative, in which Ser in position 4 was replaced by Lys(Ac), was much more potent, with 49.3% inhibition. It is worth mentioning that the rate of cellular uptake of the [4Lys(Ac), 8Lys(Dau=Aoa)]-GnRH-III conjugate by the tumor cells was significantly higher than that of the conjugate with the native GnRH-III sequence.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
| Experiment 19 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.31 ± 0.90 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
| Experiment 20 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Amelanotic melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.5 ± 0.6 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | M24 cell | CVCL_D032 | ||
| Experiment 21 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Invasive breast carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4.0 ± 0.8 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 22 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Minimally invasive lung adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4.0 ± 0.8 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Minimally invasive lung adenocarcinoma | H1650 cell | CVCL_1483 | ||
| Experiment 23 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Lung adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4.3 ± 0.4 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Lung adenocarcinoma | A-549 cell | CVCL_0023 | ||
| Experiment 24 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
7.3 ± 0.3 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
| Experiment 25 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Ovarian serous adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
8.2 ± 0.5 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous adenocarcinoma | OVCAR-3 cell | CVCL_0465 | ||
| Experiment 26 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | High grade ovarian serous adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
9.5 ± 0.8 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | High grade ovarian serous adenocarcinoma | OVCAR-8 cell | CVCL_1629 | ||
| Experiment 27 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Normal | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
19.7 ± 1.2 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MRC-5 cell | CVCL_0440 | ||
| Experiment 28 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Pancreatic ductal adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
56.4 ± 4.5 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
GnRH-III-[4Lys(Bu), 8Lys(Dau=Aoa)] [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor weight decrease |
28.70%
|
|||
| Administration Dosage | 15 mg/kg Dau content | ||||
| Description |
Based on these tumor weights, we determined that free Dau, 1 and 2 inhibited tumor weight significantly by 40.1, 28.7 and 27.7% in the case of orthotopic human MDA-MB-231 breast tumor model.
|
||||
| In Vivo Model | Orthotopic MDA-MB-231 human breast carcinoma bearing mice model. | ||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Colorectal cancer | ||||
| Efficacy Data | Tumor weight |
80.80%
|
|||
| Administration Dosage | 10 mg/kg Dau content | ||||
| Description |
The obtained data reveal that Dau, 1 and 2 significantly inhibited the tumor growth, whereby the tumor weights were reduced by 84.3, 80.8 and 87.1%, as compared to the control group.
|
||||
| In Vivo Model | Orthotopic HT-29 human colon carcinoma bearing mice model. | ||||
| In Vitro Model | Colon adenocarcinoma | HT-29 cell | CVCL_0320 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor volume decrease |
34.10%
|
|||
| Administration Dosage | 15 mg/kg Dau content | ||||
| Description |
Apart from that, a significant inhibition of the tumor volume was also obtained in groups which were treated with conjugate 1 (34.1%) and 2 (23.1%).
|
||||
| In Vivo Model | Orthotopic MDA-MB-231 human breast carcinoma bearing mice model. | ||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Proliferation index |
16.30%
|
|||
| Administration Dosage | 10 mg/kg Dau content | ||||
| Description |
It was observed that both GnRH-III conjugates 1 and 2 caused a significant decrease of the proliferation index by 16.3 and 25.9%
|
||||
| In Vivo Model | Orthotopic 4T1 mice breast carcinoma bearing mice model. | ||||
| In Vitro Model | Mammary carcinoma | 4T1 cell | CVCL_0125 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Proliferation index |
37%
|
|||
| Administration Dosage | 10 mg/kg Dau content | ||||
| Description |
The obtained data revealed that free Dau and both conjugates (1, 2) significantly inhibited the number of micro-metastases in the lung by 33.7, 43.8 and 49.4%, as compared to the control group. The proliferation index of lung metastases was significantly inhibited by 27.8, 37 and 39.1% in groups that were treated with free Dau, 1 and 2.
|
||||
| In Vivo Model | Orthotopic 4T1 mice breast carcinoma bearing mice model. | ||||
| In Vitro Model | Mammary carcinoma | 4T1 cell | CVCL_0125 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Number of macro-metastases decrease |
55.20%
|
|||
| Administration Dosage | 10 mg/kg Dau content | ||||
| Description |
The number of macro-metastases in peripheral organs, such as spleen, lung, liver and kidneys, was counted, in order to determine the anti-metastatic effect of free Dau and the GnRH-III conjugates on aggressive 4T1 BC orthotopic model (Figure 5A). The number of macro-metastases in spleen was significantly decreased in all treated groups (Dau,1and2) by 64.3, 72.8 and 78.1%. In the lung, the number of macro-metastases was also significantly reduced for all treated groups by 55.4, 55.2 and 64.4%, respectively. The numbers of macro-metastases in the liver and kidneys were decreased under treatments, whereby a significant decrease could be only obtained for conjugate2.
Click to Show/Hide
|
||||
| In Vivo Model | Orthotopic 4T1 mice breast carcinoma bearing mice model. | ||||
| In Vitro Model | Mammary carcinoma | 4T1 cell | CVCL_0125 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Number of macro-metastases decrease |
72.80%
|
|||
| Administration Dosage | 10 mg/kg Dau content | ||||
| Description |
The number of macro-metastases in peripheral organs, such as spleen, lung, liver and kidneys, was counted, in order to determine the anti-metastatic effect of free Dau and the GnRH-III conjugates on aggressive 4T1 BC orthotopic model (Figure 5A). The number of macro-metastases in spleen was significantly decreased in all treated groups (Dau, 1 and 2) by 64.3, 72.8 and 78.1%. In the lung, the number of macro-metastases was also significantly reduced for all treated groups by 55.4, 55.2 and 64.4%, respectively. The numbers of macro-metastases in the liver and kidneys were decreased under treatments, whereby a significant decrease could be only obtained for conjugate 2.
Click to Show/Hide
|
||||
| In Vivo Model | Orthotopic 4T1 mice breast carcinoma bearing mice model. | ||||
| In Vitro Model | Mammary carcinoma | 4T1 cell | CVCL_0125 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Micro-metastases decrease |
43.80%
|
|||
| Administration Dosage | 10 mg/kg Dau content | ||||
| Description |
The obtained data revealed that free Dau and both conjugates (1, 2) significantly inhibited the number of micro-metastases in the lung by 33.7, 43.8 and 49.4%, as compared to the control group. The proliferation index of lung metastases was significantly inhibited by 27.8, 37 and 39.1% in groups that were treated with free Dau, 1 and 2.
|
||||
| In Vivo Model | Orthotopic 4T1 mice breast carcinoma bearing mice model. | ||||
| In Vitro Model | Mammary carcinoma | 4T1 cell | CVCL_0125 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Body weigth change |
10.10%
|
|||
| Administration Dosage | 15 mg/kg Dau content | ||||
| Description |
The body weight of the mice in control group was decreased by 2.7%, while in the groups treated with 1 and 2, it was decreased by 10.1 and 8.2%.
|
||||
| In Vivo Model | Orthotopic MDA-MB-231 human breast carcinoma bearing mice model. | ||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Ovarian endometrioid adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.4 ± 1.1 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.2 ± 0.6 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
In our experiments, GnRH-III was applied as a targeting peptide. The conjugate [8Lys(Dau=Aoa-GFLG)]-GnRH-III, in which Dau was connected to the side chain of Lys in position 8 through an aminooxyacetylated cathepsin B-labile GFLG spacer, showed significant tumor growth inhibition in s.c.-developed fast-growing C26 murine colon cancer-bearing Balb/c mice. The effect highly depended on the treatment schedule. In the first attempt, the conjugate was administered i.p. at a dose of 8.86 umol (5 mg Dau-content/kg body weight) five times on days 7, 9, 11, 14 and 16 after tumor transplantation (Treatment schedule A). The tumor volume inhibition was only 15.7% on day 26 when the experiment finished. A slight enhancement in inhibition (22.3%) was detected when a dose of conjugate corresponding to 15 mg Dau content/kg body weight (26.6 μmol) was injected only once on day 7 (Treatment schedule B). An additional treatment on day 10 (Treatment schedule C) did not result in any further improvements (21.9% on day 29). However, when the treatment schedule was changed to two treatments with the same dose on days 4 and 7, 46.3% inhibition was observed on day 29 (Treatment schedule D). In contrast, the treatments with free Dau at a dose of 2 mg/kg body weight (3.55 umol) on days 7, 9, 11, 14 and 16 showed only 22.6% inhibition on day 26. The median survival rates of the treated animals in comparison to the control group were 1 (A), 1.23 (B), 1.21 (C) and 1.38 (D), respectively, and 0.81 for free Dau. These results indicate the lower toxicity and the higher tumor volume inhibition effect of the conjugates in comparison with those of free Dau, as well as the importance of the treatment schedule. In another experiment, HT-29 human colon cancer was developed s.c. in immunodeficient SCID mice. Dau and two conjugates (with or without a GFLG spacer between the GnRH-III homing peptide and the payload) were used for the treatment. The first i.p. administrations were performed on day 13 after tumor inoculation. All the mice treated once with 2.5 mg/kg body weight (4.43 umol) free Dau died within 10 days. In contrast, the conjugates at a dose of 15 mg Dau content/kg body weight (26.6 umol) that refers to 52 mg/kg [8Lys(Dau=Aoa)]-GnRH-III and 62.5 mg/kg [8Lys(Dau=Aoa-GFLG)]-GnRH-III conjugates, respectively, did not show significant toxicity. The treatment was repeated on days 23 and 30. Because of the significant weight loss in several mice in the control group, the experiment was terminated on day 35. The tumor growth inhibition could be calculated as reductions in the tumor volume by 44.3% and 57.6% and the tumor weight by 41% and 50%, respectively. Interestingly, the conjugates were poorly effective on orthotopically developed tumors. In the case of the C26 colon tumor-bearing female Balb/c mice, only a 7% reduction in tumor weight was detected on day 13 after the two treatments on days 4 and 7 with [8Lys(Dau=Aoa)]-GnRH-III, at a 26.6 umol/kg (15 mg Dau content) dose. The effect of free Dau (2 mg/kg on days 4 and 7) showed a better inhibitory effect (24.4%). Interestingly, our novel developed GnRH-III derivative, in which Ser in position 4 was replaced by Lys(Ac), was much more potent, with 49.3% inhibition. It is worth mentioning that the rate of cellular uptake of the [4Lys(Ac), 8Lys(Dau=Aoa)]-GnRH-III conjugate by the tumor cells was significantly higher than that of the conjugate with the native GnRH-III sequence.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.36 ± 0.07 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.2 ± 0.8 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | B16 cell | CVCL_F936 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Lung adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4.1 ± 0.1 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Lung adenocarcinoma | H1975 cell | CVCL_1511 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Oral cavity squamous cell carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4.7 ± 0.8 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Oral cavity squamous cell carcinoma | PE/CA-PJ41 | CVCL_2680 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Prostate carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
5.3 ± 0.4 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | DU145 cell | CVCL_0105 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | High grade ovarian serous adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
5.7 ± 0.8 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | High grade ovarian serous adenocarcinoma | OVCAR-8 cell | CVCL_1629 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
5.8 ± 0.8 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
6.3 ± 0.9 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Mammary carcinoma | 4T1 cell | CVCL_0125 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Prostate carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
6.3 ± 0.3 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
6.8 ± 0.3 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Tongue squamous cell carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
7.4 ± 0.8 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Tongue squamous cell carcinoma | PE/CA-PJ15 cell | CVCL_2678 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
8.4 ± 0.3 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | A2058 cell | CVCL_1059 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
9.0 ± 0.8 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
9.00 ± 1.33µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 17 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Lung adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
9.7 ± 0.6 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Lung adenocarcinoma | A-549 cell | CVCL_0023 | ||
| Experiment 18 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Minimally invasive lung adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
10.5 ± 1.1 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Minimally invasive lung adenocarcinoma | H1650 cell | CVCL_1483 | ||
| Experiment 19 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Normal | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
10.6 ± 0.2 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | C-26 cell | CVCL_XC68 | ||
| Experiment 20 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
12.7 ± 1.5 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | WM983B cell | CVCL_6809 | ||
| Experiment 21 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Amelanotic melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
13.5 ± 1.1 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | HT168-M1/M9 cell | CVCL_2H39 | ||
| Experiment 22 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
15.5 ± 1.7 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
| Experiment 23 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
15.93 ± 0.99 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
| Experiment 24 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Amelanotic melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
16.2 ± 0.2 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | M24 cell | CVCL_D032 | ||
| Experiment 25 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Invasive breast carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
16.5 ± 1.2 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 26 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Normal | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
41.9 ± 3.8 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MRC-5 cell | CVCL_0440 | ||
| Experiment 27 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Ovarian serous adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
46.0 ± 1.3 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous adenocarcinoma | OVCAR-3 cell | CVCL_0465 | ||
| Experiment 28 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Pancreatic ductal adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 100 µM | |||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
Dau=Aoa-LRRYfQWAVGHStaL-NH2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Tumor weight inhibition |
16.60%
|
|||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
Like GnRH and somatostatin, bombesin (BBN) is another example of peptide hor-556 mone which receptors are overexpressed in cancerous tissues. This 14-mer peptide (Glp-Gln-Arg-Leu-Gly-Asn-Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2), first discovered in the skin of the frog Bombina bombina, is associated with the gastrin-releasing peptide (GRP) as a mammalian counterpart. The bombesin receptor family comprises neuromedin receptor B (NMB-R, or BB1), gastrin-releasing peptide receptor (GRP-R, or BB2) and bombesin receptor subtype 3 (BRS-3, or BB3). Among them, GRP-R has been the most investigated so far and has been proven to be upregulated in breast, prostate, pancreas, small-cell lung cancers, among others, hence representing a suitable target for drug delivery to tumors. Many attempts have been made to modify and shorten the sequence of bombesin to tailor its stability, activity (agonist or antagonist), affinity and selectivity towards this receptor. Several research groups have encouraged the use of their optimized structures as putative drug delivery systems, but they were never directly compared. Moreover, the previous examples of conjugates between GRP-R ligands and anthracyclines display a labile ester bond that could cause the early release of the drug in vivo. Therefore, our group took the leap and produced conjugates based on the most promising bombesin analogs as homing devices attached to Dau via an oxime bond and two cathepsin B-cleavable linkers (LRRY or GFLG). Furthermore, a conjugate bearing a novel developed bombesin analog ([6D-Phe, 11-Ala, 13Sta, 14Nle]-BBN(7-14)) was synthesized. The use of oxime-linked Dau as a payload promoted the identification of three conjugates with improved cytostatic activity and cellular uptake by human prostate (PC-3) and breast cancer (MDA-MB-231 and MDA-MB-453) cell lines. The Dau=Aoa-Leu-OH active metabolite was readily released in all the cases in less than 30 min in rat liver lysosomal homogenate, but only L5 (Dau=Aoa-LRRY-[6D-Phe, 13Sta, 14Leu]-BBN(7-14), where Sta is statine) and L6 Dau=Aoa-LRRY-[6D-Phe, 11-Ala, 13Sta, 14Nle]-BBN(7-14) demonstrated a satisfactory stability in the mouse plasma. Therefore, only these two conjugates were further investigated in vivo. As mentioned above, their chronic toxicity was assessed in healthy mice by administering the conjugates doses of 5, 10 and 20 mg Dau content/kg body weight. The PDCs were not critically harmful at any dose, although a relatively high mouse weight loss (10-15%) was induced at the highest dose. The in vivo antitumor efficacy was studied in murine xenograft models bearing s.c.-inoculated PC-3 human prostatic adenocarcinoma. The conjugates were administered intraperitoneally every fifth day starting from day 9 after tumor inoculation, for a total of five treatments, at a dose of 10 mg Dau-content/kg body weight, and their tumor growth inhibition values were compared to those of the mice treated with the maximum tolerated dose of free Dau (1 mg/kg) and 0.9% saline solution (control group). On the last day of the experiment (day 33), L5 and L6 revealed reductions in the tumor size by 21.4% and 31.4% and tumor weights by 16.6% and 33.1% compared to those of the control, respectively. On the other hand, the Dau-treated group had to be terminated on day 26 because of severe toxicity, without showing a significant reduction in the tumor volume and weight. To better understand the long-term effect of the newly developed compounds, the tumor doubling time (DT) was calculated; both the PDCs significantly increased the DT compared to the treatment with free Dau.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Half Maximal Uptake Concentration (UC50) |
12.35 µM
|
|||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
Like GnRH and somatostatin, bombesin (BBN) is another example of peptide hor-556 mone which receptors are overexpressed in cancerous tissues. This 14-mer peptide (Glp-Gln-Arg-Leu-Gly-Asn-Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2), first discovered in the skin of the frog Bombina bombina, is associated with the gastrin-releasing peptide (GRP) as a mammalian counterpart. The bombesin receptor family comprises neuromedin receptor B (NMB-R, or BB1), gastrin-releasing peptide receptor (GRP-R, or BB2) and bombesin receptor subtype 3 (BRS-3, or BB3). Among them, GRP-R has been the most investigated so far and has been proven to be upregulated in breast, prostate, pancreas, small-cell lung cancers, among others, hence representing a suitable target for drug delivery to tumors. Many attempts have been made to modify and shorten the sequence of bombesin to tailor its stability, activity (agonist or antagonist), affinity and selectivity towards this receptor. Several research groups have encouraged the use of their optimized structures as putative drug delivery systems, but they were never directly compared. Moreover, the previous examples of conjugates between GRP-R ligands and anthracyclines display a labile ester bond that could cause the early release of the drug in vivo. Therefore, our group took the leap and produced conjugates based on the most promising bombesin analogs as homing devices attached to Dau via an oxime bond and two cathepsin B-cleavable linkers (LRRY or GFLG). Furthermore, a conjugate bearing a novel developed bombesin analog ([6D-Phe, 11-Ala, 13Sta, 14Nle]-BBN(7-14)) was synthesized. The use of oxime-linked Dau as a payload promoted the identification of three conjugates with improved cytostatic activity and cellular uptake by human prostate (PC-3) and breast cancer (MDA-MB-231 and MDA-MB-453) cell lines. The Dau=Aoa-Leu-OH active metabolite was readily released in all the cases in less than 30 min in rat liver lysosomal homogenate, but only L5 (Dau=Aoa-LRRY-[6D-Phe, 13Sta, 14Leu]-BBN(7-14), where Sta is statine) and L6 Dau=Aoa-LRRY-[6D-Phe, 11-Ala, 13Sta, 14Nle]-BBN(7-14) demonstrated a satisfactory stability in the mouse plasma. Therefore, only these two conjugates were further investigated in vivo. As mentioned above, their chronic toxicity was assessed in healthy mice by administering the conjugates doses of 5, 10 and 20 mg Dau content/kg body weight. The PDCs were not critically harmful at any dose, although a relatively high mouse weight loss (10-15%) was induced at the highest dose. The in vivo antitumor efficacy was studied in murine xenograft models bearing s.c.-inoculated PC-3 human prostatic adenocarcinoma. The conjugates were administered intraperitoneally every fifth day starting from day 9 after tumor inoculation, for a total of five treatments, at a dose of 10 mg Dau-content/kg body weight, and their tumor growth inhibition values were compared to those of the mice treated with the maximum tolerated dose of free Dau (1 mg/kg) and 0.9% saline solution (control group). On the last day of the experiment (day 33), L5 and L6 revealed reductions in the tumor size by 21.4% and 31.4% and tumor weights by 16.6% and 33.1% compared to those of the control, respectively. On the other hand, the Dau-treated group had to be terminated on day 26 because of severe toxicity, without showing a significant reduction in the tumor volume and weight. To better understand the long-term effect of the newly developed compounds, the tumor doubling time (DT) was calculated; both the PDCs significantly increased the DT compared to the treatment with free Dau.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.22 ± 0.19 µM
|
|||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
Like GnRH and somatostatin, bombesin (BBN) is another example of peptide hor-556 mone which receptors are overexpressed in cancerous tissues. This 14-mer peptide (Glp-Gln-Arg-Leu-Gly-Asn-Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2), first discovered in the skin of the frog Bombina bombina, is associated with the gastrin-releasing peptide (GRP) as a mammalian counterpart. The bombesin receptor family comprises neuromedin receptor B (NMB-R, or BB1), gastrin-releasing peptide receptor (GRP-R, or BB2) and bombesin receptor subtype 3 (BRS-3, or BB3). Among them, GRP-R has been the most investigated so far and has been proven to be upregulated in breast, prostate, pancreas, small-cell lung cancers, among others, hence representing a suitable target for drug delivery to tumors. Many attempts have been made to modify and shorten the sequence of bombesin to tailor its stability, activity (agonist or antagonist), affinity and selectivity towards this receptor. Several research groups have encouraged the use of their optimized structures as putative drug delivery systems, but they were never directly compared. Moreover, the previous examples of conjugates between GRP-R ligands and anthracyclines display a labile ester bond that could cause the early release of the drug in vivo. Therefore, our group took the leap and produced conjugates based on the most promising bombesin analogs as homing devices attached to Dau via an oxime bond and two cathepsin B-cleavable linkers (LRRY or GFLG). Furthermore, a conjugate bearing a novel developed bombesin analog ([6D-Phe, 11-Ala, 13Sta, 14Nle]-BBN(7-14)) was synthesized. The use of oxime-linked Dau as a payload promoted the identification of three conjugates with improved cytostatic activity and cellular uptake by human prostate (PC-3) and breast cancer (MDA-MB-231 and MDA-MB-453) cell lines. The Dau=Aoa-Leu-OH active metabolite was readily released in all the cases in less than 30 min in rat liver lysosomal homogenate, but only L5 (Dau=Aoa-LRRY-[6D-Phe, 13Sta, 14Leu]-BBN(7-14), where Sta is statine) and L6 Dau=Aoa-LRRY-[6D-Phe, 11-Ala, 13Sta, 14Nle]-BBN(7-14) demonstrated a satisfactory stability in the mouse plasma. Therefore, only these two conjugates were further investigated in vivo. As mentioned above, their chronic toxicity was assessed in healthy mice by administering the conjugates doses of 5, 10 and 20 mg Dau content/kg body weight. The PDCs were not critically harmful at any dose, although a relatively high mouse weight loss (10-15%) was induced at the highest dose. The in vivo antitumor efficacy was studied in murine xenograft models bearing s.c.-inoculated PC-3 human prostatic adenocarcinoma. The conjugates were administered intraperitoneally every fifth day starting from day 9 after tumor inoculation, for a total of five treatments, at a dose of 10 mg Dau-content/kg body weight, and their tumor growth inhibition values were compared to those of the mice treated with the maximum tolerated dose of free Dau (1 mg/kg) and 0.9% saline solution (control group). On the last day of the experiment (day 33), L5 and L6 revealed reductions in the tumor size by 21.4% and 31.4% and tumor weights by 16.6% and 33.1% compared to those of the control, respectively. On the other hand, the Dau-treated group had to be terminated on day 26 because of severe toxicity, without showing a significant reduction in the tumor volume and weight. To better understand the long-term effect of the newly developed compounds, the tumor doubling time (DT) was calculated; both the PDCs significantly increased the DT compared to the treatment with free Dau.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Drug concentration |
21.40%
|
|||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
Like GnRH and somatostatin, bombesin (BBN) is another example of peptide hor-556 mone which receptors are overexpressed in cancerous tissues. This 14-mer peptide (Glp-Gln-Arg-Leu-Gly-Asn-Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2), first discovered in the skin of the frog Bombina bombina, is associated with the gastrin-releasing peptide (GRP) as a mammalian counterpart. The bombesin receptor family comprises neuromedin receptor B (NMB-R, or BB1), gastrin-releasing peptide receptor (GRP-R, or BB2) and bombesin receptor subtype 3 (BRS-3, or BB3). Among them, GRP-R has been the most investigated so far and has been proven to be upregulated in breast, prostate, pancreas, small-cell lung cancers, among others, hence representing a suitable target for drug delivery to tumors. Many attempts have been made to modify and shorten the sequence of bombesin to tailor its stability, activity (agonist or antagonist), affinity and selectivity towards this receptor. Several research groups have encouraged the use of their optimized structures as putative drug delivery systems, but they were never directly compared. Moreover, the previous examples of conjugates between GRP-R ligands and anthracyclines display a labile ester bond that could cause the early release of the drug in vivo. Therefore, our group took the leap and produced conjugates based on the most promising bombesin analogs as homing devices attached to Dau via an oxime bond and two cathepsin B-cleavable linkers (LRRY or GFLG). Furthermore, a conjugate bearing a novel developed bombesin analog ([6D-Phe, 11-Ala, 13Sta, 14Nle]-BBN(7-14)) was synthesized. The use of oxime-linked Dau as a payload promoted the identification of three conjugates with improved cytostatic activity and cellular uptake by human prostate (PC-3) and breast cancer (MDA-MB-231 and MDA-MB-453) cell lines. The Dau=Aoa-Leu-OH active metabolite was readily released in all the cases in less than 30 min in rat liver lysosomal homogenate, but only L5 (Dau=Aoa-LRRY-[6D-Phe, 13Sta, 14Leu]-BBN(7-14), where Sta is statine) and L6 Dau=Aoa-LRRY-[6D-Phe, 11-Ala, 13Sta, 14Nle]-BBN(7-14) demonstrated a satisfactory stability in the mouse plasma. Therefore, only these two conjugates were further investigated in vivo. As mentioned above, their chronic toxicity was assessed in healthy mice by administering the conjugates doses of 5, 10 and 20 mg Dau content/kg body weight. The PDCs were not critically harmful at any dose, although a relatively high mouse weight loss (10-15%) was induced at the highest dose. The in vivo antitumor efficacy was studied in murine xenograft models bearing s.c.-inoculated PC-3 human prostatic adenocarcinoma. The conjugates were administered intraperitoneally every fifth day starting from day 9 after tumor inoculation, for a total of five treatments, at a dose of 10 mg Dau-content/kg body weight, and their tumor growth inhibition values were compared to those of the mice treated with the maximum tolerated dose of free Dau (1 mg/kg) and 0.9% saline solution (control group). On the last day of the experiment (day 33), L5 and L6 revealed reductions in the tumor size by 21.4% and 31.4% and tumor weights by 16.6% and 33.1% compared to those of the control, respectively. On the other hand, the Dau-treated group had to be terminated on day 26 because of severe toxicity, without showing a significant reduction in the tumor volume and weight. To better understand the long-term effect of the newly developed compounds, the tumor doubling time (DT) was calculated; both the PDCs significantly increased the DT compared to the treatment with free Dau.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
Dau=Aoa-LRRYfQWAVβAlaHStaNle-NH2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Tumor weight inhibition |
33.10%
|
|||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
Like GnRH and somatostatin, bombesin (BBN) is another example of peptide hor-556 mone which receptors are overexpressed in cancerous tissues. This 14-mer peptide (Glp-Gln-Arg-Leu-Gly-Asn-Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2), first discovered in the skin of the frog Bombina bombina, is associated with the gastrin-releasing peptide (GRP) as a mammalian counterpart. The bombesin receptor family comprises neuromedin receptor B (NMB-R, or BB1), gastrin-releasing peptide receptor (GRP-R, or BB2) and bombesin receptor subtype 3 (BRS-3, or BB3). Among them, GRP-R has been the most investigated so far and has been proven to be upregulated in breast, prostate, pancreas, small-cell lung cancers, among others, hence representing a suitable target for drug delivery to tumors. Many attempts have been made to modify and shorten the sequence of bombesin to tailor its stability, activity (agonist or antagonist), affinity and selectivity towards this receptor. Several research groups have encouraged the use of their optimized structures as putative drug delivery systems, but they were never directly compared. Moreover, the previous examples of conjugates between GRP-R ligands and anthracyclines display a labile ester bond that could cause the early release of the drug in vivo. Therefore, our group took the leap and produced conjugates based on the most promising bombesin analogs as homing devices attached to Dau via an oxime bond and two cathepsin B-cleavable linkers (LRRY or GFLG). Furthermore, a conjugate bearing a novel developed bombesin analog ([6D-Phe, 11-Ala, 13Sta, 14Nle]-BBN(7-14)) was synthesized. The use of oxime-linked Dau as a payload promoted the identification of three conjugates with improved cytostatic activity and cellular uptake by human prostate (PC-3) and breast cancer (MDA-MB-231 and MDA-MB-453) cell lines. The Dau=Aoa-Leu-OH active metabolite was readily released in all the cases in less than 30 min in rat liver lysosomal homogenate, but only L5 (Dau=Aoa-LRRY-[6D-Phe, 13Sta, 14Leu]-BBN(7-14), where Sta is statine) and L6 Dau=Aoa-LRRY-[6D-Phe, 11-Ala, 13Sta, 14Nle]-BBN(7-14) demonstrated a satisfactory stability in the mouse plasma. Therefore, only these two conjugates were further investigated in vivo. As mentioned above, their chronic toxicity was assessed in healthy mice by administering the conjugates doses of 5, 10 and 20 mg Dau content/kg body weight. The PDCs were not critically harmful at any dose, although a relatively high mouse weight loss (10-15%) was induced at the highest dose. The in vivo antitumor efficacy was studied in murine xenograft models bearing s.c.-inoculated PC-3 human prostatic adenocarcinoma. The conjugates were administered intraperitoneally every fifth day starting from day 9 after tumor inoculation, for a total of five treatments, at a dose of 10 mg Dau-content/kg body weight, and their tumor growth inhibition values were compared to those of the mice treated with the maximum tolerated dose of free Dau (1 mg/kg) and 0.9% saline solution (control group). On the last day of the experiment (day 33), L5 and L6 revealed reductions in the tumor size by 21.4% and 31.4% and tumor weights by 16.6% and 33.1% compared to those of the control, respectively. On the other hand, the Dau-treated group had to be terminated on day 26 because of severe toxicity, without showing a significant reduction in the tumor volume and weight. To better understand the long-term effect of the newly developed compounds, the tumor doubling time (DT) was calculated; both the PDCs significantly increased the DT compared to the treatment with free Dau.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Half Maximal Uptake Concentration (UC50) |
16.09 µM
|
|||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
Like GnRH and somatostatin, bombesin (BBN) is another example of peptide hor-556 mone which receptors are overexpressed in cancerous tissues. This 14-mer peptide (Glp-Gln-Arg-Leu-Gly-Asn-Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2), first discovered in the skin of the frog Bombina bombina, is associated with the gastrin-releasing peptide (GRP) as a mammalian counterpart. The bombesin receptor family comprises neuromedin receptor B (NMB-R, or BB1), gastrin-releasing peptide receptor (GRP-R, or BB2) and bombesin receptor subtype 3 (BRS-3, or BB3). Among them, GRP-R has been the most investigated so far and has been proven to be upregulated in breast, prostate, pancreas, small-cell lung cancers, among others, hence representing a suitable target for drug delivery to tumors. Many attempts have been made to modify and shorten the sequence of bombesin to tailor its stability, activity (agonist or antagonist), affinity and selectivity towards this receptor. Several research groups have encouraged the use of their optimized structures as putative drug delivery systems, but they were never directly compared. Moreover, the previous examples of conjugates between GRP-R ligands and anthracyclines display a labile ester bond that could cause the early release of the drug in vivo. Therefore, our group took the leap and produced conjugates based on the most promising bombesin analogs as homing devices attached to Dau via an oxime bond and two cathepsin B-cleavable linkers (LRRY or GFLG). Furthermore, a conjugate bearing a novel developed bombesin analog ([6D-Phe, 11-Ala, 13Sta, 14Nle]-BBN(7-14)) was synthesized. The use of oxime-linked Dau as a payload promoted the identification of three conjugates with improved cytostatic activity and cellular uptake by human prostate (PC-3) and breast cancer (MDA-MB-231 and MDA-MB-453) cell lines. The Dau=Aoa-Leu-OH active metabolite was readily released in all the cases in less than 30 min in rat liver lysosomal homogenate, but only L5 (Dau=Aoa-LRRY-[6D-Phe, 13Sta, 14Leu]-BBN(7-14), where Sta is statine) and L6 Dau=Aoa-LRRY-[6D-Phe, 11-Ala, 13Sta, 14Nle]-BBN(7-14) demonstrated a satisfactory stability in the mouse plasma. Therefore, only these two conjugates were further investigated in vivo. As mentioned above, their chronic toxicity was assessed in healthy mice by administering the conjugates doses of 5, 10 and 20 mg Dau content/kg body weight. The PDCs were not critically harmful at any dose, although a relatively high mouse weight loss (10-15%) was induced at the highest dose. The in vivo antitumor efficacy was studied in murine xenograft models bearing s.c.-inoculated PC-3 human prostatic adenocarcinoma. The conjugates were administered intraperitoneally every fifth day starting from day 9 after tumor inoculation, for a total of five treatments, at a dose of 10 mg Dau-content/kg body weight, and their tumor growth inhibition values were compared to those of the mice treated with the maximum tolerated dose of free Dau (1 mg/kg) and 0.9% saline solution (control group). On the last day of the experiment (day 33), L5 and L6 revealed reductions in the tumor size by 21.4% and 31.4% and tumor weights by 16.6% and 33.1% compared to those of the control, respectively. On the other hand, the Dau-treated group had to be terminated on day 26 because of severe toxicity, without showing a significant reduction in the tumor volume and weight. To better understand the long-term effect of the newly developed compounds, the tumor doubling time (DT) was calculated; both the PDCs significantly increased the DT compared to the treatment with free Dau.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
18.04 ± 3.01 µM
|
|||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
Like GnRH and somatostatin, bombesin (BBN) is another example of peptide hor-556 mone which receptors are overexpressed in cancerous tissues. This 14-mer peptide (Glp-Gln-Arg-Leu-Gly-Asn-Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2), first discovered in the skin of the frog Bombina bombina, is associated with the gastrin-releasing peptide (GRP) as a mammalian counterpart. The bombesin receptor family comprises neuromedin receptor B (NMB-R, or BB1), gastrin-releasing peptide receptor (GRP-R, or BB2) and bombesin receptor subtype 3 (BRS-3, or BB3). Among them, GRP-R has been the most investigated so far and has been proven to be upregulated in breast, prostate, pancreas, small-cell lung cancers, among others, hence representing a suitable target for drug delivery to tumors. Many attempts have been made to modify and shorten the sequence of bombesin to tailor its stability, activity (agonist or antagonist), affinity and selectivity towards this receptor. Several research groups have encouraged the use of their optimized structures as putative drug delivery systems, but they were never directly compared. Moreover, the previous examples of conjugates between GRP-R ligands and anthracyclines display a labile ester bond that could cause the early release of the drug in vivo. Therefore, our group took the leap and produced conjugates based on the most promising bombesin analogs as homing devices attached to Dau via an oxime bond and two cathepsin B-cleavable linkers (LRRY or GFLG). Furthermore, a conjugate bearing a novel developed bombesin analog ([6D-Phe, 11-Ala, 13Sta, 14Nle]-BBN(7-14)) was synthesized. The use of oxime-linked Dau as a payload promoted the identification of three conjugates with improved cytostatic activity and cellular uptake by human prostate (PC-3) and breast cancer (MDA-MB-231 and MDA-MB-453) cell lines. The Dau=Aoa-Leu-OH active metabolite was readily released in all the cases in less than 30 min in rat liver lysosomal homogenate, but only L5 (Dau=Aoa-LRRY-[6D-Phe, 13Sta, 14Leu]-BBN(7-14), where Sta is statine) and L6 Dau=Aoa-LRRY-[6D-Phe, 11-Ala, 13Sta, 14Nle]-BBN(7-14) demonstrated a satisfactory stability in the mouse plasma. Therefore, only these two conjugates were further investigated in vivo. As mentioned above, their chronic toxicity was assessed in healthy mice by administering the conjugates doses of 5, 10 and 20 mg Dau content/kg body weight. The PDCs were not critically harmful at any dose, although a relatively high mouse weight loss (10-15%) was induced at the highest dose. The in vivo antitumor efficacy was studied in murine xenograft models bearing s.c.-inoculated PC-3 human prostatic adenocarcinoma. The conjugates were administered intraperitoneally every fifth day starting from day 9 after tumor inoculation, for a total of five treatments, at a dose of 10 mg Dau-content/kg body weight, and their tumor growth inhibition values were compared to those of the mice treated with the maximum tolerated dose of free Dau (1 mg/kg) and 0.9% saline solution (control group). On the last day of the experiment (day 33), L5 and L6 revealed reductions in the tumor size by 21.4% and 31.4% and tumor weights by 16.6% and 33.1% compared to those of the control, respectively. On the other hand, the Dau-treated group had to be terminated on day 26 because of severe toxicity, without showing a significant reduction in the tumor volume and weight. To better understand the long-term effect of the newly developed compounds, the tumor doubling time (DT) was calculated; both the PDCs significantly increased the DT compared to the treatment with free Dau.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Drug concentration |
31.40%
|
|||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
Like GnRH and somatostatin, bombesin (BBN) is another example of peptide hor-556 mone which receptors are overexpressed in cancerous tissues. This 14-mer peptide (Glp-Gln-Arg-Leu-Gly-Asn-Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2), first discovered in the skin of the frog Bombina bombina, is associated with the gastrin-releasing peptide (GRP) as a mammalian counterpart. The bombesin receptor family comprises neuromedin receptor B (NMB-R, or BB1), gastrin-releasing peptide receptor (GRP-R, or BB2) and bombesin receptor subtype 3 (BRS-3, or BB3). Among them, GRP-R has been the most investigated so far and has been proven to be upregulated in breast, prostate, pancreas, small-cell lung cancers, among others, hence representing a suitable target for drug delivery to tumors. Many attempts have been made to modify and shorten the sequence of bombesin to tailor its stability, activity (agonist or antagonist), affinity and selectivity towards this receptor. Several research groups have encouraged the use of their optimized structures as putative drug delivery systems, but they were never directly compared. Moreover, the previous examples of conjugates between GRP-R ligands and anthracyclines display a labile ester bond that could cause the early release of the drug in vivo. Therefore, our group took the leap and produced conjugates based on the most promising bombesin analogs as homing devices attached to Dau via an oxime bond and two cathepsin B-cleavable linkers (LRRY or GFLG). Furthermore, a conjugate bearing a novel developed bombesin analog ([6D-Phe, 11-Ala, 13Sta, 14Nle]-BBN(7-14)) was synthesized. The use of oxime-linked Dau as a payload promoted the identification of three conjugates with improved cytostatic activity and cellular uptake by human prostate (PC-3) and breast cancer (MDA-MB-231 and MDA-MB-453) cell lines. The Dau=Aoa-Leu-OH active metabolite was readily released in all the cases in less than 30 min in rat liver lysosomal homogenate, but only L5 (Dau=Aoa-LRRY-[6D-Phe, 13Sta, 14Leu]-BBN(7-14), where Sta is statine) and L6 Dau=Aoa-LRRY-[6D-Phe, 11-Ala, 13Sta, 14Nle]-BBN(7-14) demonstrated a satisfactory stability in the mouse plasma. Therefore, only these two conjugates were further investigated in vivo. As mentioned above, their chronic toxicity was assessed in healthy mice by administering the conjugates doses of 5, 10 and 20 mg Dau content/kg body weight. The PDCs were not critically harmful at any dose, although a relatively high mouse weight loss (10-15%) was induced at the highest dose. The in vivo antitumor efficacy was studied in murine xenograft models bearing s.c.-inoculated PC-3 human prostatic adenocarcinoma. The conjugates were administered intraperitoneally every fifth day starting from day 9 after tumor inoculation, for a total of five treatments, at a dose of 10 mg Dau-content/kg body weight, and their tumor growth inhibition values were compared to those of the mice treated with the maximum tolerated dose of free Dau (1 mg/kg) and 0.9% saline solution (control group). On the last day of the experiment (day 33), L5 and L6 revealed reductions in the tumor size by 21.4% and 31.4% and tumor weights by 16.6% and 33.1% compared to those of the control, respectively. On the other hand, the Dau-treated group had to be terminated on day 26 because of severe toxicity, without showing a significant reduction in the tumor volume and weight. To better understand the long-term effect of the newly developed compounds, the tumor doubling time (DT) was calculated; both the PDCs significantly increased the DT compared to the treatment with free Dau.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
TKPKG - N-(4-Acetylphenyl)-5-bromo-2-hydroxybenzamide conjugate 2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index |
0.1
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index |
0.3
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
57.9 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
57.9 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
173.1 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
173.1 μM
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
200 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
200 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
23.2 ± 3.7 mM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
53.8 ± 1.1 mM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit | . Not detected | |||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit | . Not detected | |||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
5
|
|||
| Administration Time | 1 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
TKPKGTKPKGTKPKGTKPKG - N-(4-Acetylphenyl)-5-bromo-2-hydroxybenzamide conjugate 4 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index |
0.2
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index |
0.5
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
21.9 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
27.4 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
75.6 μM
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
94.5 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | > 200 μg/mL | |||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | > 250 μg/mL | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
16.8 ± 3.1 mM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
42.9 ± 8.9 mM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit | . Not detected | |||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
TKPKGTKPKGTKPKGTKPKG - N-(4-Acetylphenyl)-5-bromo-2-hydroxybenzamide conjugate 3 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index |
0.2
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index |
0.4
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
24.8 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
31 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
74.3 μM
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
92.9 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | > 200 μg/mL | |||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
250 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
16.5 ± 0.8 mM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
37.6 ± 10.3 mM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit | . Not detected | |||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
15
|
|||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
TKPKGTKPKGTKPKGTKPKG - N-(4-Acetylphenyl)-5-bromo-2-hydroxybenzamide conjugate 5 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index |
0.3
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index |
1.5
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
25.6 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
25.6 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
76.7 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
76.7 μM
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
200 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
200 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
24.9 ± 6.2 mM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
113.3 ± 15.3 mM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit | . Not detected | |||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit | . Not detected | |||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
20
|
|||
| Administration Time | 1 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
TKPKGTKPKGTKPKGTKPKG - N-(4-Acetylphenyl)-5-bromo-2-hydroxybenzamide conjugate 2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index |
0.3
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index |
0.5
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
24.8 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
24.8 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
74.3 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
74.3 μM
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
200 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
200 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
20.9 ± 4.0 mM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
40.4 ± 17.7 mM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit | . Not detected | |||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit | . Not detected | |||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
15
|
|||
| Administration Time | 1 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
TKPKG - N-(4-Acetylphenyl)-5-bromo-2-hydroxybenzamide conjugate 3 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index |
0.3
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index |
0.4
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
62.3 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
62.3 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
186.5 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
186.5 μM
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
200 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
200 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
55.8 ± 3.0 mM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
72.6 ± 6.7 mM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit | . Not detected | |||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit | . Not detected | |||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
0
|
|||
| Administration Time | 1 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
SAL2-Aoa-Thr-OH [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index | > 0.5 | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index | > 0.5 | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
Not detected
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
Not detected
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
Not detected
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
131.5 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
200 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
393.5 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 200 mM | |||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 200 mM | |||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit | . Not detected | |||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit | . Not detected | |||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit | . Not detected | |||
| Administration Time | 1 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
GFLGTKPKGTKPKGTKPKGTKPKG - N-(4-Acetylphenyl)-5-bromo-2-hydroxybenzamide conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index |
0.6
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index |
0.8
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
23.6 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
47.3 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
70.7 μM
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
141.5 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
200 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
400 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
86.6 ± 2.8 mM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
112.4 ± 20.0 mM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit | . Not detected | |||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit | . Not detected | |||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
21
|
|||
| Administration Time | 1 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
TKPKGTKPKGTKPKGTKPKG - N-(4-Acetylphenyl)-5-bromo-2-hydroxybenzamide conjugate 1 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index |
0.9
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index |
1.9
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
28.2 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
42.2 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
47 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
70.4 μM
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
120 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
200 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
39.1 ± 1.6 mM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
79.7 ± 16.7 mM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit | . Not detected | |||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
0
|
|||
| Administration Time | 1 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
20
|
|||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
SAL2-Aoa-Gly-OH [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index | > 0.9 | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index | > 0.9 | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
Not detected
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
Not detected
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
Not detected
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
72 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
100 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
215.4 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 200 mM | |||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 200 mM | |||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit | . Not detected | |||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit | . Not detected | |||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit | . Not detected | |||
| Administration Time | 1 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
TKPKGTKPKGTKPKGTKPKG - N-(4-Acetylphenyl)-5-bromo-2-hydroxybenzamide conjugate 8 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index |
1
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index | > 2.5 | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
13.6 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
27.3 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
40.8 μM
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
81.5 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
100 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
200 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
81.9 ± 4.2 mM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 200 mM | |||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit | . Not detected | |||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
1
|
|||
| Administration Time | 1 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
40
|
|||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
TKPKGTKPKG - N-(4-Acetylphenyl)-5-bromo-2-hydroxybenzamide conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index | > 1.1 | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index | > 1.1 | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
18.7 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
56 μM
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
58.4 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
80 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
174.9 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
250 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 200 mM | |||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 200 mM | |||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit | . Not detected | |||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
0
|
|||
| Administration Time | 1 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
25
|
|||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
TKPKGTKPKGTKPKGTKPKG - N-(4-Acetylphenyl)-5-bromo-2-hydroxybenzamide conjugate 6 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index |
1.3
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index | > 3.2 | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
21.2 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
26.5 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
63.4 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
79.3 μM
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
160 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
200 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
81.6 ± 2.4 mM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 200 mM | |||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
0
|
|||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit | . Not detected | |||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
11
|
|||
| Administration Time | 1 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
GFLGTKPKG - N-(4-Acetylphenyl)-5-bromo-2-hydroxybenzamide conjugate 1 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index |
1.3
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index | > 4.3 | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
15.5 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
25.8 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
46.4 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
51.7 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
60 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
77.3 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
100 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
154.6 μM
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
200 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
58.0 ± 0.1 mM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 200 mM | |||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
0
|
|||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
60
|
|||
| Administration Time | 1 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
TKPKGTKPKGTKPKGTKPKG - N-(4-Acetylphenyl)-5-bromo-2-hydroxybenzamide conjugate 9 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index | > 2.4 | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index | > 2.4 | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
12 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
24.1 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
41.5 μM
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
83 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
100 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
200 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 200 mM | |||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 200 mM | |||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit | . Not detected | |||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
4
|
|||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
60
|
|||
| Administration Time | 1 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
TKPKGTKPKGTKPKGTKPKG - N-(4-Acetylphenyl)-5-bromo-2-hydroxybenzamide conjugate 7 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index | > 2.4 | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index | > 2.4 | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
12.7 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
25.5 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
41.2 μM
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
82.4 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
100 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
200 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 200 mM | |||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 200 mM | |||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit | . Not detected | |||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit | . Not detected | |||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
0
|
|||
| Administration Time | 1 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
TKPRTKPKGTKPKG - N-(4-Acetylphenyl)-5-bromo-2-hydroxybenzamide conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index |
2.7
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index |
2.9
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
23.2 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
34.8 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
58.1 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
80 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
86.9 μM
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
200 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
95.2 ± 4.2 mM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
101.4 ± 5.6 mM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
0
|
|||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit | . Not detected | |||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
8
|
|||
| Administration Time | 1 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
TKPKG - N-(4-Acetylphenyl)-5-bromo-2-hydroxybenzamide conjugate 1 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index |
2.9
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index | > 3.1 | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
21.8 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
36.4 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
60 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
65.3 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
72.7 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
100 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
108.8 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
200 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
217.7 μM
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
191.3 ± 12.4 mM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 200 mM | |||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
10
|
|||
| Administration Time | 1 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
60
|
|||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
GFLGTKPR - N-(4-Acetylphenyl)-5-bromo-2-hydroxybenzamide conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index |
3.6
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index | > 6.3 | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
10.6 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
26.4 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
31.6 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
40 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
52.9 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
79.1 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
100 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
158.2 μM
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
200 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
112.6 ± 26.9 mM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 200 mM | |||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
10
|
|||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
22
|
|||
| Administration Time | 1 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
TKPR - N-(4-Acetylphenyl)-5-bromo-2-hydroxybenzamide conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index | > 4.4 | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index | > 4.4 | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
15 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
37.6 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
40 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
45 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
75.1 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
100 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
112.4 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
200 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
224.8 μM
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 200 mM | |||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 200 mM | |||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
1
|
|||
| Administration Time | 1 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
60
|
|||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
GFLGTKPKG - N-(4-Acetylphenyl)-5-bromo-2-hydroxybenzamide conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index |
5.3
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index | > 6.5 | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
10.3 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
25.9 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
31 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
40 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
51.7 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
77.4 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
100 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
154.8 μM
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
200 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
162.6 ± 17.3 mM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 200 mM | |||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
10
|
|||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
15
|
|||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
50
|
|||
| Administration Time | 1 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
GFLGTKPPR - N-(4-Acetylphenyl)-5-bromo-2-hydroxybenzamide conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index |
5.3
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index | > 6.8 | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
9.8 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
24.6 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
29.4 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
40 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
49.1 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
73.5 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
100 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
146.9 μM
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
200 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
155.6 ± 14.8 mM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 200 mM | |||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
0
|
|||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
11
|
|||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
15
|
|||
| Administration Time | 1 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
TKPKG - N-(4-Acetylphenyl)-5-bromo-2-hydroxybenzamide conjugate 4 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index | > 9.2 | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index | > 9.2 | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
7.3 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
20 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
21.8 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
36.4 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
58.3 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
100 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
108.9 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
160 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
174.3 μM
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 200 mM | |||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 200 mM | |||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
4
|
|||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
12
|
|||
| Administration Time | 1 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
TKPPR - N-(4-Acetylphenyl)-5-bromo-2-hydroxybenzamide conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index | > 9.9 | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Selectivity index | > 9.9 | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis H37Rv | 83332 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
6.8 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
20 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
20.3 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
33.9 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
67.7 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
100 μg/mL
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
101.3 μM
|
|||
| Administration Time | 4 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
The in vitro antimycobacterial activity of the salicylanilides is preserved in the conjugates against M. tuberculosis H37Rv although their MIC values (20.3-186.5 μM) are higher than the MIC of the free salicylanilides (7.5-25.9 μM). There is no difference in the activity between the different salicylanilide containing OT20 or OT20(4-pal) conjugates. In terms of the peptide length, SAL2-Aoa-OT20 showed better activity than the shorter SAL2-Aoa-OT10 and the shortest SAL2-Aoa-T5 showed the best efficacy with very low MIC value (21.8 μM) among these conjugates. The incorporation of the enzyme labile GFLG spacer has no significant benefit in the antimycobacterial activity of the conjugates, the MIC values of the one tuftsin unit containing conjugates with or without spacer is mostly the same. In the case of tetratuftsin derivative containing conjugate the presence of the GFLG spacer reduce its activity (SAL2-Aoa-OT20 with MIC of 81.5 μM vs. SAL2-Aoa-GFLG-OT20 with MIC of 141.5 μM). Among all the conjugates the short tuftsin derivative containing conjugates (namely SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH and SAL2-Aoa-TKPPR-OH) and their analogues with GFLG spacer showed outstanding in vitro antimycobacterial activity against M. tuberculosis H37Rv. The SAL2-Aoa-T5 with amide on the C-terminal was three-times more effective than its analogue SAL2-Aoa-T5-OH with C-terminal carboxyl group. The most effective conjugates are the SAL2-Aoa-TKPPR-OH (MIC value of 20.3 μM) and SAL2-Aoa-T5 (MIC value of 21.8 μM). Their MIC value is only three-times higher than the MIC value of the free salicylanilide SAL2. Both conjugates that contain two molecules of SAL2, i.e., SAL2-Aoa-OT20(4-SAL2-Aoa) and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) were also very effective against M. tuberculosis H37Rv with MIC values of 42.2 and 34.8 μM respectively. From the perspective of the length of the fatty acid, butyric acid and decanoic acid in SAL2-Aoa-OT20(4-but) (MIC of 63.4 μM) and SAL2-Aoa-OT20(4-dec) (MIC of 76.7 μM) slightly improved the activity of SAL2-Aoa-OT20 (MIC of 81.5 μM), their analogue without fatty acid. In the case of the palmitoylated conjugates, a slight influence of the position of palmitic acid side chain can be observed in the activity. SAL2-Aoa-OT20(4-pal) has higher MIC value (92.9 μM) than SAL2-Aoa-OT20, on the contrary SAL2-Aoa-OT20(14-pal) has slightly lower MIC value (74.3 μM) than SAL2-Aoa-OT20 without fatty acid. The presence of the fatty acids has a higher influence on the activity of the shorter conjugates. In the case of SAL2-Aoa-T5(4-dec) (MIC of 186.5 μM) and SAL2-Aoa-T5(4-pal) (MIC of 173.1 μM) the presence of these fatty acids highly reduced their activity compared to their analogue SAL2-Aoa-T5 without fatty acid (MIC of 21.8 μM). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH have a low activity with high MIC values (393.5 and 215.4 μM respectively). In the aspect of the MIC values calculated to the drug contents of the conjugates, most of the OT20 conjugates were effective at 25 μg/mL drug content concentration, which is only 10-times higher than the MIC of the free SAL1 and SAL2 molecules (2.5 μg/mL). However, among the short tuftsin derivative containing conjugates we can find compounds that have even lower MIC values calculated to drug content, between 6.8 and 10.6 μg/mL (SAL2-Aoa-T5, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-TKPPR-OH and SAL2-Aoa-GFLG-TKPPR-OH). The short tuftsin derivative containing conjugates that showed outstanding activity against M. tuberculosis H37Rv sensitive strain were selected to be measured on M. tuberculosis A8 MDR strain. These conjugates can inhibit the resistant bacteria only at the highest concentration tested, at 100 μg/mL (73.5-112.4 μM). These MIC values are 2-5-times higher than their MIC values against the sensitive strain. In the aspect of the drug content, their average MIC value calculated to the drug content is 31 μg/mL, which is 6-times higher than the MIC value of the free drug SAL2 (5 μg/mL).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 1773 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
200 μg/mL
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Mycobacterium abscessus infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
202.7 μM
|
|||
| Administration Time | 1 weeks | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the conjugates were active also against M. abscessus. They possessed MIC values between 40.8 and 224.8 μM. The salicylanilide-OT20 conjugates were the most effective against this strain. The SAL1-Aoa-OT20 (MIC = 41.5 μM) and SAL2-Aoa-OT20 (MIC = 40.8 μM) has similar activity as the free SAL1 (MIC = 34.5 μM) and SAL2 (MIC = 59.8 μM), and interestingly SAL3-Aoa-OT20 (MIC = 41.2 μM) is three-times more effective than the free salicylanilide SAL3 with MIC of 129.4 μM. The other tetratuftsin derivative conjugates with spacer, fatty acid side chain or second salicylanilide (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-OT20(4-but), SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal), SAL2-Aoa-OT20(14-pal) and SAL2-Aoa-OT20(4-SAL2-Aoa)) acted similarly with MICs between 70.4 and 79.3 μM. Having two molecules of salicylanilide in the conjugate showed no significant advantage against M. abscessus. The MIC of SAL2-Aoa-OT20(4-SAL2-Aoa) (70.4 μM) is slightly lower than the MIC of TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) (86.9 μM). The dituftsin derivative containing conjugate SAL2-Aoa-OT10 had fairly good activity (MIC of 56.0 μM). The shortest peptide containing conjugates were the least effective with MICs ranged from 146.9 to 224.8 μM. The incorporation of GFLG spacer made the conjugates slightly more active. The presence of fatty acid side chain in the T5 containing conjugates had no significant influence on the activity against M. abscessus. According to the MIC values calculated to the drug content, the SAL1-3-Aoa-OT20 conjugates (MICdrug content is around 13 μg/mL) were as effective or even more effective than the free SAL1-3 molecules (MIC within the range of 10-40 μg/mL). The other OT20 derivatives with a spacer or fatty acid side chain have MIC values calculated to the drug content around 25 μg/mL and the average value of the short tuftsin derivative containing conjugates is 60 μg/mL.
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 200 mM | |||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Acute monocytic leukemia | MONO-MAC-6 cell | CVCL_1426 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 200 mM | |||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
Most of the acetylated control peptides have no cytostatic activity on MonoMac6 and HepG2 cells at the highest concentration measured (200 μM). Interestingly, among the control peptides with fatty acid modification only the derivatives that contain palmitoyl group have cytostatic effect on both cell types. Ac-OT20(4-pal) showed the lowest IC50 value on MonoMac6 (26.6 μM). Its isomer with palmitic acid on the 14Lys (Ac-OT20(14-pal)) has higher IC50 value (62.8 μM) on MonoMac6. Inversely, the Ac-OT20(14-pal) has lower IC50 value (35.2 μM) than Ac-OT20(4-pal) (77.0 μM) on HepG2 cells. In the case of the shorter Ac-T5(4-pal) it has similar effect like Ac-OT20(14-pal) on both cells. The cytostatic effect of palmitic acid itself and the physical mixture of palmitic acid and Ac-OT20 control peptide was also studied on MonoMac6 cells, and the IC50 values are very similar (178.1 and 175.8 μM respectively). The covalent bond between the palmitic acid and the peptide in the most cytostatic derivative Ac-OT20(4-pal) gives almost 7-times lower IC50 value than in the case of the palmitic acid alone. In the case of the salicylanilide-tuftsin conjugates the results are diverse (Table 3). The conjugates with one tuftsin units (SAL2-Aoa-T5, SAL2-Aoa-T5-OH, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) and their analogues elongated with GFLG spacer have no cytostatic activity on MonoMac6 cells at the highest measured concentration (200 μM). Conjugates with two and four tuftsin units without a side chain modification (SAL2-Aoa-OT10, SAL1-Aoa-OT20, SAL2-Aoa-OT20, SAL3-Aoa-OT20) also does not show cytostatic effects on this cell. The OT20 conjugate elongated with the GFLG spacer (SAL2-Aoa-GFLG-OT20) has a slight cytostatic activity on MonoMac6 (IC50 of 86.6 μM). The presence of two salicylanilide molecules in the conjugate results in the appearance of cytostatic activity, namely SAL2-Aoa-OT20(4-SAL2-Aoa) with 39.1 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with IC50 of 101.4 μM for MonoMac6 cells. The conjugates in which there are fatty acid side chain modification showed a significant cytostatic activity on MonoMac6 cells. The conjugate that have the shortest fatty acid side chain modification (with butanoyl side chain SAL2-Aoa-OT20(4-but)) has no cytostatic activity as an exception. All the palmitoyl side chain containing OT20 conjugates have very low and similar IC50 values (16.5-20.9 μM) and the T5 analogue (SAL2-Aoa-T5(4-pal)) has higher IC50 value (53.8 μM). The two conjugates that have decanoyl side chain modification also have cytostatic activity. The SAL2-Aoa-OT20(4-dec) has IC50 value of 24.9 μM that is comparable with its analogue that have palmitoyl side chain modification (SAL2-Aoa-OT20(4-pal), 16.8 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has higher IC50 value (72.6 μM) than SAL2-Aoa-T5(4-pal) (53.8 μM). The finding that the decanoyl containing conjugates have cytostatic activity is in contrast that we experienced in the case of the control peptides (Ac-OT20(4-dec), Ac-T5(4-dec)) that did not show cytostatic activity nor the decanoic acid itself on MonoMac6 cells. On HepG2 cells one OT20 conjugate (SAL2-Aoa-OT20) has a slight cytostatic activity (81.9 μM), the others (SAL1-Aoa-OT20, SAL3-Aoa-OT20) and the OT10 derivative SAL2-Aoa-OT10 do not exhibited this action at the highest concentration measured. The conjugates with one tuftsin unit (SAL2-Aoa-T5, SAL2-Aoa-TKPR-OH, SAL2-Aoa-TKPPR-OH) do not have cytostatic activity on HepG2 below 200 μM except SAL2-Aoa-T5-OH which has a slight cytostatic effect with IC50 value of 191.3 μM. Interestingly, the presence of the GFLG spacer (SAL2-Aoa-GFLG-OT20, SAL2-Aoa-GFLG-T5, SAL2-Aoa-GFLG-T5-OH, SAL2-Aoa-GFLG-TKPR-OH, SAL2-Aoa-GFLG-TKPPR-OH) makes the conjugates slightly cytostatic on HepG2 cells (IC50 values between 58.0 and 162.6 μM). The conjugates containing two salicylanilide molecules have similar IC50 values to SAL2-Aoa-OT20, SAL2-Aoa-OT20(4-SAL2-Aoa) with 79.7 μM and TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) with 95.2 μM IC50 value. The conjugates with palmitic acid side chain modification showed a significant cytostatic activity on HepG2 cells too. The palmitoyl side chain containing OT20 conjugates have low and similar IC50 values (37.6-42.9 μM). Interestingly, the T5 analogue (SAL2-Aoa-T5(4-pal)) has the lowest IC50 value among all the conjugates (23.2 μM). SAL2-Aoa-OT20 and its analogue with butanoyl side chain (SAL2-Aoa-OT20(4-but)) has the same effect (81.9 and 81.6 μM respectively). On the contrary that we can observe in the case of cytostatic activity on MonoMac6 cells, on HepG2 cells the cytostatic activity of the OT20 conjugate that has decanoyl side chain modification (SAL2-Aoa-OT20(4-dec)) is decreased (113.3 μM). The T5 analogue SAL2-Aoa-T5(4-dec) has a higher cytostatic activity but its IC50 value (55.8 μM) is still lower than the IC50 value of SAL2-Aoa-T5(4-pal) (23.2 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
1
|
|||
| Administration Time | 4 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium tuberculosis infection | Mycobacterium tuberculosis | 83332 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tuberculosis | ||||
| Efficacy Data | Colony forming unit |
13
|
|||
| Administration Time | 1 weeks | ||||
| Administration Dosage | 250 μM | ||||
| MOA of PDC |
INH-tuftsin conjugates GTKPK(INH-CH2-CO)G and [TKPK(INH-CH2-CO)G]4 were effective against M. tuberculosis and the minimal inhibitory concentration values were comparable to the free INH moiety. Enhanced cellular uptake and significant inhibition of intracellular M. tuberculosis was performed by conjugation of new in silico identified drug candidate to [TKPKG]4 tuftsin derivative (TB5-OT20). Tuftsin based conjugates of pyridopyrimidine derivatives with in vitro antitubercular activity presented increased membrane affinity on lipid model systems. Palmitoylated tuftsin conjugate of INH (pal-T5(INH)2, where T5 is TKPKG) and new in silico identified drug candidate (pal-T5(TB820)2) was found to be effective against susceptible and multiresistant M. tuberculosis strains with a relatively high in vitro selectivity. Free INH did not exhibit intracellular antitubercular activity, in contrast, its pal-T5(INH)2 conjugate significantly inhibited the intracellular M. tuberculosis. The palmitoylated conjugates were encapsulated into PLGA (poly(lactide-co-glycolide)) nanoparticles with high encapsulation efficacy and the encapsulated compound showed high in vivo efficacy and low toxicity. In this study various tuftsin derivatives such as tetramer [TKPKG]4 (OT20), heterotrimer TKPR-[TKPKG]2 (TKPR-OT10), dimer [TKPKG]2 (OT10) and monomer tuftsin derivatives TKPKG (T5), TKPR, TKPPR (tuftsin antagonist) were used as carrier peptides. Tuftsin derivatives with modified Lys residues (e.g., introduction of palmitic acid, decanoic acid or butyric acid) were also studied. Modification with fatty acid chains can enhance the lipophilicity, membrane affinity and encapsulation efficacy. Modified conjugable salicylanilides were attached to the tuftsin derivatives via oxime bond directly or by the insertion of a spacer between the drug molecule and the carrier. The oxime bond is chemically stable between pH 3 and 8, the incorporation of an enzymatic cleavable spacer between the drug and the carrier moiety might be necessary for the efficient drug release and activity. As spacer, the enzyme labile GFLG tetrapeptide was applied, which could be cleaved by the lysosomal cysteine protease, cathepsin B. Here we report the synthesis, chemical characterization, in vitro extracellular and intracellular antimycobacterial activity, cytotoxic and cytostatic activity, cellular uptake, membrane integrity studies and lysosomal degradation of salicylanilide-tuftsin conjugates.
Click to Show/Hide
|
||||
| Description |
INH was also tested and did not show antitubercular activity against intracellular bacteria even at 365 μM concentration. The free salicylanilide derivatives did not exhibit intracellular antitubercular activity at none of the used concentrations. In contrast, most of the conjugates significantly reduced the colony forming units (CFU) of intracellular M. tuberculosis bacteria compared to untreated control in a concentration dependent manner. Among the OT20 conjugates SAL1-Aoa-OT20 was more effective than SAL2-Aoa-OT20 and SAL3-Aoa-OT20 at both concentrations. However, in the case of SAL1-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(4-pal) the activity was very similar. In terms of the length of the tuftsin derivatives at 125 μM there is no significant difference, at 250 μM the smallest SAL2-Aoa-T5 was the most effective followed by the longest SAL2-Aoa-OT20. SAL2-Aoa-OT10 resulted in approximately 5-times higher CFU than SAL2-Aoa-T5. The presence of the GFLG spacer in the case of most peptide has no influence on the activity, only in the case of SAL2-Aoa-TKPR-OH, the presence of the spacer reduced its activity 25-times (SAL2-Aoa-GFLG-TKPR-OH). At 250 μM the SAL2-Aoa-T5 with amide on the C-terminal was 10-times more effective than its analogue SAL2-Aoa-T5-OH with carboxyl group on the C-terminal of the peptide. The same tendency can be observed in the case of the derivatives of these peptides with GFLG spacer (SAL2-Aoa-GFLG-T5 vs. SAL2-Aoa-GFLG-T5-OH). The OT20 derivative which contains two molecules of salicylanilides, SAL2-Aoa-OT20(4-SAL2-Aoa) showed a weaker activity than its analogue with only one drug molecule SAL2-Aoa-OT20. The conjugate TKPR-OT10(8-SAL2-Aoa; 13-SAL2-Aoa) that contains also two salicylanilides has better activity than SAL2-Aoa-OT20(4-SAL2-Aoa). It was also more effective than SAL2-Aoa-OT20 and SAL2-Aoa-OT10 at 250 μM concentration, but less effective than SAL2-Aoa-TKPR. SAL2-Aoa-OT20(4-but) has slightly better activity than SAL2-Aoa-OT20 conjugate without fatty acid side chain. The decanoic or palmitic acid side chain containing OT20 derivative (SAL2-Aoa-OT20(4-dec), SAL1-Aoa-OT20(4-pal), SAL2-Aoa-OT20(4-pal) and SAL2-Aoa-OT20(14-pal)) were outstandingly effective both at 125 and 250 μM concentration. At 250 μM SAL2-Aoa-OT20(4-pal) resulted in 5-times lower CFU value than its analogue where the palmitic acid is on the 14th amino acid residue (SAL2-Aoa-OT20(14-pal)). In the case of the T5 derivatives both fatty acid containing conjugates SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal) showed a better activity than SAL2-Aoa-T5 without fatty acid side chain. Interestingly at 125 μM the decanoic acid containing derivative proved to be better than the palmitic acid containing one, but this result is reversed at 250 μM. The OT20 derivatives containing the fatty acids (SAL2-Aoa-OT20(4-dec) and SAL2-Aoa-OT20(4-pal)) showed a more improved effect in comparison with T5 derivatives SAL2-Aoa-T5(4-dec) and SAL2-Aoa-T5(4-pal). The smallest metabolites produced during lysosomal degradation, SAL2-Aoa-Thr-OH and SAL2-Aoa-Gly-OH, also showed an activity against the intracellular bacteria without dependence of concentration. They can reduce the CFU values similarly as e.g., conjugate SAL2-Aoa-OT10, SAL2-Aoa-TKPPR-OH or SAL2-Aoa-GFLG-TKPPR-OH (at 250 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Mycobacterium abscessus infection | Mycobacterium abscessus | 36809 | ||
Dau=Aoa-LRRYQWAVGHLNle-NH2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Half Maximal Uptake Concentration (UC50) |
22.92 µM
|
|||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
Like GnRH and somatostatin, bombesin (BBN) is another example of peptide hor-556 mone which receptors are overexpressed in cancerous tissues. This 14-mer peptide (Glp-Gln-Arg-Leu-Gly-Asn-Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2), first discovered in the skin of the frog Bombina bombina, is associated with the gastrin-releasing peptide (GRP) as a mammalian counterpart. The bombesin receptor family comprises neuromedin receptor B (NMB-R, or BB1), gastrin-releasing peptide receptor (GRP-R, or BB2) and bombesin receptor subtype 3 (BRS-3, or BB3). Among them, GRP-R has been the most investigated so far and has been proven to be upregulated in breast, prostate, pancreas, small-cell lung cancers, among others, hence representing a suitable target for drug delivery to tumors. Many attempts have been made to modify and shorten the sequence of bombesin to tailor its stability, activity (agonist or antagonist), affinity and selectivity towards this receptor. Several research groups have encouraged the use of their optimized structures as putative drug delivery systems, but they were never directly compared. Moreover, the previous examples of conjugates between GRP-R ligands and anthracyclines display a labile ester bond that could cause the early release of the drug in vivo. Therefore, our group took the leap and produced conjugates based on the most promising bombesin analogs as homing devices attached to Dau via an oxime bond and two cathepsin B-cleavable linkers (LRRY or GFLG). Furthermore, a conjugate bearing a novel developed bombesin analog ([6D-Phe, 11-Ala, 13Sta, 14Nle]-BBN(7-14)) was synthesized. The use of oxime-linked Dau as a payload promoted the identification of three conjugates with improved cytostatic activity and cellular uptake by human prostate (PC-3) and breast cancer (MDA-MB-231 and MDA-MB-453) cell lines. The Dau=Aoa-Leu-OH active metabolite was readily released in all the cases in less than 30 min in rat liver lysosomal homogenate, but only L5 (Dau=Aoa-LRRY-[6D-Phe, 13Sta, 14Leu]-BBN(7-14), where Sta is statine) and L6 Dau=Aoa-LRRY-[6D-Phe, 11-Ala, 13Sta, 14Nle]-BBN(7-14) demonstrated a satisfactory stability in the mouse plasma. Therefore, only these two conjugates were further investigated in vivo. As mentioned above, their chronic toxicity was assessed in healthy mice by administering the conjugates doses of 5, 10 and 20 mg Dau content/kg body weight. The PDCs were not critically harmful at any dose, although a relatively high mouse weight loss (10-15%) was induced at the highest dose. The in vivo antitumor efficacy was studied in murine xenograft models bearing s.c.-inoculated PC-3 human prostatic adenocarcinoma. The conjugates were administered intraperitoneally every fifth day starting from day 9 after tumor inoculation, for a total of five treatments, at a dose of 10 mg Dau-content/kg body weight, and their tumor growth inhibition values were compared to those of the mice treated with the maximum tolerated dose of free Dau (1 mg/kg) and 0.9% saline solution (control group). On the last day of the experiment (day 33), L5 and L6 revealed reductions in the tumor size by 21.4% and 31.4% and tumor weights by 16.6% and 33.1% compared to those of the control, respectively. On the other hand, the Dau-treated group had to be terminated on day 26 because of severe toxicity, without showing a significant reduction in the tumor volume and weight. To better understand the long-term effect of the newly developed compounds, the tumor doubling time (DT) was calculated; both the PDCs significantly increased the DT compared to the treatment with free Dau.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4.38 ± 0.33 µM
|
|||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
Like GnRH and somatostatin, bombesin (BBN) is another example of peptide hor-556 mone which receptors are overexpressed in cancerous tissues. This 14-mer peptide (Glp-Gln-Arg-Leu-Gly-Asn-Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2), first discovered in the skin of the frog Bombina bombina, is associated with the gastrin-releasing peptide (GRP) as a mammalian counterpart. The bombesin receptor family comprises neuromedin receptor B (NMB-R, or BB1), gastrin-releasing peptide receptor (GRP-R, or BB2) and bombesin receptor subtype 3 (BRS-3, or BB3). Among them, GRP-R has been the most investigated so far and has been proven to be upregulated in breast, prostate, pancreas, small-cell lung cancers, among others, hence representing a suitable target for drug delivery to tumors. Many attempts have been made to modify and shorten the sequence of bombesin to tailor its stability, activity (agonist or antagonist), affinity and selectivity towards this receptor. Several research groups have encouraged the use of their optimized structures as putative drug delivery systems, but they were never directly compared. Moreover, the previous examples of conjugates between GRP-R ligands and anthracyclines display a labile ester bond that could cause the early release of the drug in vivo. Therefore, our group took the leap and produced conjugates based on the most promising bombesin analogs as homing devices attached to Dau via an oxime bond and two cathepsin B-cleavable linkers (LRRY or GFLG). Furthermore, a conjugate bearing a novel developed bombesin analog ([6D-Phe, 11-Ala, 13Sta, 14Nle]-BBN(7-14)) was synthesized. The use of oxime-linked Dau as a payload promoted the identification of three conjugates with improved cytostatic activity and cellular uptake by human prostate (PC-3) and breast cancer (MDA-MB-231 and MDA-MB-453) cell lines. The Dau=Aoa-Leu-OH active metabolite was readily released in all the cases in less than 30 min in rat liver lysosomal homogenate, but only L5 (Dau=Aoa-LRRY-[6D-Phe, 13Sta, 14Leu]-BBN(7-14), where Sta is statine) and L6 Dau=Aoa-LRRY-[6D-Phe, 11-Ala, 13Sta, 14Nle]-BBN(7-14) demonstrated a satisfactory stability in the mouse plasma. Therefore, only these two conjugates were further investigated in vivo. As mentioned above, their chronic toxicity was assessed in healthy mice by administering the conjugates doses of 5, 10 and 20 mg Dau content/kg body weight. The PDCs were not critically harmful at any dose, although a relatively high mouse weight loss (10-15%) was induced at the highest dose. The in vivo antitumor efficacy was studied in murine xenograft models bearing s.c.-inoculated PC-3 human prostatic adenocarcinoma. The conjugates were administered intraperitoneally every fifth day starting from day 9 after tumor inoculation, for a total of five treatments, at a dose of 10 mg Dau-content/kg body weight, and their tumor growth inhibition values were compared to those of the mice treated with the maximum tolerated dose of free Dau (1 mg/kg) and 0.9% saline solution (control group). On the last day of the experiment (day 33), L5 and L6 revealed reductions in the tumor size by 21.4% and 31.4% and tumor weights by 16.6% and 33.1% compared to those of the control, respectively. On the other hand, the Dau-treated group had to be terminated on day 26 because of severe toxicity, without showing a significant reduction in the tumor volume and weight. To better understand the long-term effect of the newly developed compounds, the tumor doubling time (DT) was calculated; both the PDCs significantly increased the DT compared to the treatment with free Dau.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
Ac-SYSNleEHFRWGK(Dau=Aoa)PV-NH2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
0.13 ± 0.09 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, the native sequence of -MSH was used, in which Met was replaced by Nle (SYSNleEHFRWGKPV). Daunomycin was attached to the amino functional groups via non-cleavable oxime linkage. These types of conjugates have many excellent properties to study and compare the bioactivity of the compounds. One of the developed conjugates contained drug molecules on both conjugation site (N-terminal and the side chain of Lys). The two additional conjugates were modified by the drug, either on theN-terminus or on the Lys side chain. The in vitro cytostatic effect on mouse melanoma cells did not show any significant differences among them. However, results indicated that the conjugates with Dau on the side chain of Lys could enter the cells more rapidly and efficiently. In contrast, the in vivo tumor growth inhibition was the most pronounced in the case of Ac-SYSNleEHFRWGK(Dau=Aoa)PV-NH2(Conj2). It is worth mentioning that the dose was normalized to Dau content, thus the injected dose ofConj3was half that ofConj 2. Nevertheless,Conj2, with one molecule of the drug, might be superior to the conjugate containing two molecules of daunomycin. The higher tumor growth inhibition effect ofConj2overConj1where the Dau is connected to theN-terminus, confirms our previous results with Dau-GnRH-III (Glp-His-Trp-Lys(Bu)-His-Asp-Trp-Lys(Dau=Aoa)-Pro-Gly-NH2), suggesting that H-Lys(Dau=Aoa)-OH can release easily from this position by the dipeptidyl-peptidase activity of cathepsin B.
Click to Show/Hide
|
||||
| Description |
The in vitro cytostatic effects of Dau--MSH conjugates were compared with the PrestoBlue assay, which has a higher specificity and efficacy that of the previously used MTT assay, using the same six human melanoma cell lines used in the real-time qPCR assay. According to our results, free daunomycin exhibited the lowest IC50 values, and therefore the highest cytostatic effect, due to the fact that free daunomycin passively diffuses into cells. Our Dau--MSH conjugates showed similar trends in the effectiveness of the tested cell lines. Conj2 and Conj4 resulted in IC50 values in the low uM range. A calculation of the targeting indices showed that compound Conj2 had an over 2-times higher targeting efficiency in most cell lines compared to the A2058 cell line. Interestingly, WM983A showed the lowest TI among the high-MC1R-expressing cell lines compared to A2058, and WM983B exhibited high TI regardless of the relatively low MC1R expression on the mRNA level. In the case of Conj4, the treatment resulted in the highest TIs with OCM-1 and WM983B cell lines.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | SK-MEL-202 cell | CVCL_6106 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
0.79 ± 0.05 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, the native sequence of -MSH was used, in which Met was replaced by Nle (SYSNleEHFRWGKPV). Daunomycin was attached to the amino functional groups via non-cleavable oxime linkage. These types of conjugates have many excellent properties to study and compare the bioactivity of the compounds. One of the developed conjugates contained drug molecules on both conjugation site (N-terminal and the side chain of Lys). The two additional conjugates were modified by the drug, either on theN-terminus or on the Lys side chain. The in vitro cytostatic effect on mouse melanoma cells did not show any significant differences among them. However, results indicated that the conjugates with Dau on the side chain of Lys could enter the cells more rapidly and efficiently. In contrast, the in vivo tumor growth inhibition was the most pronounced in the case of Ac-SYSNleEHFRWGK(Dau=Aoa)PV-NH2(Conj2). It is worth mentioning that the dose was normalized to Dau content, thus the injected dose ofConj3was half that ofConj 2. Nevertheless,Conj2, with one molecule of the drug, might be superior to the conjugate containing two molecules of daunomycin. The higher tumor growth inhibition effect ofConj2overConj1where the Dau is connected to theN-terminus, confirms our previous results with Dau-GnRH-III (Glp-His-Trp-Lys(Bu)-His-Asp-Trp-Lys(Dau=Aoa)-Pro-Gly-NH2), suggesting that H-Lys(Dau=Aoa)-OH can release easily from this position by the dipeptidyl-peptidase activity of cathepsin B.
Click to Show/Hide
|
||||
| Description |
The in vitro cytostatic effects of Dau--MSH conjugates were compared with the PrestoBlue assay, which has a higher specificity and efficacy that of the previously used MTT assay, using the same six human melanoma cell lines used in the real-time qPCR assay. According to our results, free daunomycin exhibited the lowest IC50 values, and therefore the highest cytostatic effect, due to the fact that free daunomycin passively diffuses into cells. Our Dau--MSH conjugates showed similar trends in the effectiveness of the tested cell lines. Conj2 and Conj4 resulted in IC50 values in the low uM range. A calculation of the targeting indices showed that compound Conj2 had an over 2-times higher targeting efficiency in most cell lines compared to the A2058 cell line. Interestingly, WM983A showed the lowest TI among the high-MC1R-expressing cell lines compared to A2058, and WM983B exhibited high TI regardless of the relatively low MC1R expression on the mRNA level. In the case of Conj4, the treatment resulted in the highest TIs with OCM-1 and WM983B cell lines.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | WM983A cell | CVCL_6808 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
0.95 ± 0.12 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, the native sequence of -MSH was used, in which Met was replaced by Nle (SYSNleEHFRWGKPV). Daunomycin was attached to the amino functional groups via non-cleavable oxime linkage. These types of conjugates have many excellent properties to study and compare the bioactivity of the compounds. One of the developed conjugates contained drug molecules on both conjugation site (N-terminal and the side chain of Lys). The two additional conjugates were modified by the drug, either on theN-terminus or on the Lys side chain. The in vitro cytostatic effect on mouse melanoma cells did not show any significant differences among them. However, results indicated that the conjugates with Dau on the side chain of Lys could enter the cells more rapidly and efficiently. In contrast, the in vivo tumor growth inhibition was the most pronounced in the case of Ac-SYSNleEHFRWGK(Dau=Aoa)PV-NH2(Conj2). It is worth mentioning that the dose was normalized to Dau content, thus the injected dose ofConj3was half that ofConj 2. Nevertheless,Conj2, with one molecule of the drug, might be superior to the conjugate containing two molecules of daunomycin. The higher tumor growth inhibition effect ofConj2overConj1where the Dau is connected to theN-terminus, confirms our previous results with Dau-GnRH-III (Glp-His-Trp-Lys(Bu)-His-Asp-Trp-Lys(Dau=Aoa)-Pro-Gly-NH2), suggesting that H-Lys(Dau=Aoa)-OH can release easily from this position by the dipeptidyl-peptidase activity of cathepsin B.
Click to Show/Hide
|
||||
| Description |
The in vitro cytostatic effects of Dau--MSH conjugates were compared with the PrestoBlue assay, which has a higher specificity and efficacy that of the previously used MTT assay, using the same six human melanoma cell lines used in the real-time qPCR assay. According to our results, free daunomycin exhibited the lowest IC50 values, and therefore the highest cytostatic effect, due to the fact that free daunomycin passively diffuses into cells. Our Dau--MSH conjugates showed similar trends in the effectiveness of the tested cell lines. Conj2 and Conj4 resulted in IC50 values in the low uM range. A calculation of the targeting indices showed that compound Conj2 had an over 2-times higher targeting efficiency in most cell lines compared to the A2058 cell line. Interestingly, WM983A showed the lowest TI among the high-MC1R-expressing cell lines compared to A2058, and WM983B exhibited high TI regardless of the relatively low MC1R expression on the mRNA level. In the case of Conj4, the treatment resulted in the highest TIs with OCM-1 and WM983B cell lines.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | A2058 cell | CVCL_1059 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.00 ± 0.00 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, the native sequence of -MSH was used, in which Met was replaced by Nle (SYSNleEHFRWGKPV). Daunomycin was attached to the amino functional groups via non-cleavable oxime linkage. These types of conjugates have many excellent properties to study and compare the bioactivity of the compounds. One of the developed conjugates contained drug molecules on both conjugation site (N-terminal and the side chain of Lys). The two additional conjugates were modified by the drug, either on theN-terminus or on the Lys side chain. The in vitro cytostatic effect on mouse melanoma cells did not show any significant differences among them. However, results indicated that the conjugates with Dau on the side chain of Lys could enter the cells more rapidly and efficiently. In contrast, the in vivo tumor growth inhibition was the most pronounced in the case of Ac-SYSNleEHFRWGK(Dau=Aoa)PV-NH2(Conj2). It is worth mentioning that the dose was normalized to Dau content, thus the injected dose ofConj3was half that ofConj 2. Nevertheless,Conj2, with one molecule of the drug, might be superior to the conjugate containing two molecules of daunomycin. The higher tumor growth inhibition effect ofConj2overConj1where the Dau is connected to theN-terminus, confirms our previous results with Dau-GnRH-III (Glp-His-Trp-Lys(Bu)-His-Asp-Trp-Lys(Dau=Aoa)-Pro-Gly-NH2), suggesting that H-Lys(Dau=Aoa)-OH can release easily from this position by the dipeptidyl-peptidase activity of cathepsin B.
Click to Show/Hide
|
||||
| Description |
The in vitro cytostatic effects of Dau--MSH conjugates were compared with the PrestoBlue assay, which has a higher specificity and efficacy that of the previously used MTT assay, using the same six human melanoma cell lines used in the real-time qPCR assay. According to our results, free daunomycin exhibited the lowest IC50 values, and therefore the highest cytostatic effect, due to the fact that free daunomycin passively diffuses into cells. Our Dau--MSH conjugates showed similar trends in the effectiveness of the tested cell lines. Conj2 and Conj4 resulted in IC50 values in the low uM range. A calculation of the targeting indices showed that compound Conj2 had an over 2-times higher targeting efficiency in most cell lines compared to the A2058 cell line. Interestingly, WM983A showed the lowest TI among the high-MC1R-expressing cell lines compared to A2058, and WM983B exhibited high TI regardless of the relatively low MC1R expression on the mRNA level. In the case of Conj4, the treatment resulted in the highest TIs with OCM-1 and WM983B cell lines.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | WM983B cell | CVCL_6809 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.39 ± 0.06 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, the native sequence of -MSH was used, in which Met was replaced by Nle (SYSNleEHFRWGKPV). Daunomycin was attached to the amino functional groups via non-cleavable oxime linkage. These types of conjugates have many excellent properties to study and compare the bioactivity of the compounds. One of the developed conjugates contained drug molecules on both conjugation site (N-terminal and the side chain of Lys). The two additional conjugates were modified by the drug, either on theN-terminus or on the Lys side chain. The in vitro cytostatic effect on mouse melanoma cells did not show any significant differences among them. However, results indicated that the conjugates with Dau on the side chain of Lys could enter the cells more rapidly and efficiently. In contrast, the in vivo tumor growth inhibition was the most pronounced in the case of Ac-SYSNleEHFRWGK(Dau=Aoa)PV-NH2(Conj2). It is worth mentioning that the dose was normalized to Dau content, thus the injected dose ofConj3was half that ofConj 2. Nevertheless,Conj2, with one molecule of the drug, might be superior to the conjugate containing two molecules of daunomycin. The higher tumor growth inhibition effect ofConj2overConj1where the Dau is connected to theN-terminus, confirms our previous results with Dau-GnRH-III (Glp-His-Trp-Lys(Bu)-His-Asp-Trp-Lys(Dau=Aoa)-Pro-Gly-NH2), suggesting that H-Lys(Dau=Aoa)-OH can release easily from this position by the dipeptidyl-peptidase activity of cathepsin B.
Click to Show/Hide
|
||||
| Description |
The in vitro cytostatic effects of Dau--MSH conjugates were compared with the PrestoBlue assay, which has a higher specificity and efficacy that of the previously used MTT assay, using the same six human melanoma cell lines used in the real-time qPCR assay. According to our results, free daunomycin exhibited the lowest IC50 values, and therefore the highest cytostatic effect, due to the fact that free daunomycin passively diffuses into cells. Our Dau--MSH conjugates showed similar trends in the effectiveness of the tested cell lines. Conj2 and Conj4 resulted in IC50 values in the low uM range. A calculation of the targeting indices showed that compound Conj2 had an over 2-times higher targeting efficiency in most cell lines compared to the A2058 cell line. Interestingly, WM983A showed the lowest TI among the high-MC1R-expressing cell lines compared to A2058, and WM983B exhibited high TI regardless of the relatively low MC1R expression on the mRNA level. In the case of Conj4, the treatment resulted in the highest TIs with OCM-1 and WM983B cell lines.
Click to Show/Hide
|
||||
| In Vitro Model | Cutaneous melanoma | OCM-3 cell | CVCL_6937 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.51 ± 0.06 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, the native sequence of -MSH was used, in which Met was replaced by Nle (SYSNleEHFRWGKPV). Daunomycin was attached to the amino functional groups via non-cleavable oxime linkage. These types of conjugates have many excellent properties to study and compare the bioactivity of the compounds. One of the developed conjugates contained drug molecules on both conjugation site (N-terminal and the side chain of Lys). The two additional conjugates were modified by the drug, either on theN-terminus or on the Lys side chain. The in vitro cytostatic effect on mouse melanoma cells did not show any significant differences among them. However, results indicated that the conjugates with Dau on the side chain of Lys could enter the cells more rapidly and efficiently. In contrast, the in vivo tumor growth inhibition was the most pronounced in the case of Ac-SYSNleEHFRWGK(Dau=Aoa)PV-NH2(Conj2). It is worth mentioning that the dose was normalized to Dau content, thus the injected dose ofConj3was half that ofConj 2. Nevertheless,Conj2, with one molecule of the drug, might be superior to the conjugate containing two molecules of daunomycin. The higher tumor growth inhibition effect ofConj2overConj1where the Dau is connected to theN-terminus, confirms our previous results with Dau-GnRH-III (Glp-His-Trp-Lys(Bu)-His-Asp-Trp-Lys(Dau=Aoa)-Pro-Gly-NH2), suggesting that H-Lys(Dau=Aoa)-OH can release easily from this position by the dipeptidyl-peptidase activity of cathepsin B.
Click to Show/Hide
|
||||
| Description |
The in vitro cytostatic effects of Dau--MSH conjugates were compared with the PrestoBlue assay, which has a higher specificity and efficacy that of the previously used MTT assay, using the same six human melanoma cell lines used in the real-time qPCR assay. According to our results, free daunomycin exhibited the lowest IC50 values, and therefore the highest cytostatic effect, due to the fact that free daunomycin passively diffuses into cells. Our Dau--MSH conjugates showed similar trends in the effectiveness of the tested cell lines. Conj2 and Conj4 resulted in IC50 values in the low uM range. A calculation of the targeting indices showed that compound Conj2 had an over 2-times higher targeting efficiency in most cell lines compared to the A2058 cell line. Interestingly, WM983A showed the lowest TI among the high-MC1R-expressing cell lines compared to A2058, and WM983B exhibited high TI regardless of the relatively low MC1R expression on the mRNA level. In the case of Conj4, the treatment resulted in the highest TIs with OCM-1 and WM983B cell lines.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | OCM-1 cell | CVCL_6934 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.8 ± 0.7 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, the native sequence of -MSH was used, in which Met was replaced by Nle (SYSNleEHFRWGKPV). Daunomycin was attached to the amino functional groups via non-cleavable oxime linkage. These types of conjugates have many excellent properties to study and compare the bioactivity of the compounds. One of the developed conjugates contained drug molecules on both conjugation site (N-terminal and the side chain of Lys). The two additional conjugates were modified by the drug, either on theN-terminus or on the Lys side chain. The in vitro cytostatic effect on mouse melanoma cells did not show any significant differences among them. However, results indicated that the conjugates with Dau on the side chain of Lys could enter the cells more rapidly and efficiently. In contrast, the in vivo tumor growth inhibition was the most pronounced in the case of Ac-SYSNleEHFRWGK(Dau=Aoa)PV-NH2(Conj2). It is worth mentioning that the dose was normalized to Dau content, thus the injected dose ofConj3was half that ofConj 2. Nevertheless,Conj2, with one molecule of the drug, might be superior to the conjugate containing two molecules of daunomycin. The higher tumor growth inhibition effect ofConj2overConj1where the Dau is connected to theN-terminus, confirms our previous results with Dau-GnRH-III (Glp-His-Trp-Lys(Bu)-His-Asp-Trp-Lys(Dau=Aoa)-Pro-Gly-NH2), suggesting that H-Lys(Dau=Aoa)-OH can release easily from this position by the dipeptidyl-peptidase activity of cathepsin B.
Click to Show/Hide
|
||||
| Description |
In vitro studies of the antiproliferative effect showed that melanoma-targeting Dau-conjugates have significantly higher efficacy on B16 murine melanoma cell lines in comparison to human melanoma cell lines (A2058, M24, and WM983B). The IC50 values were detected between 2 and 2.9 μM on B16 cells, where the highest activity was detected in the case of conjugate Conj3, followed by Conj2 and Conj1. Moreover, the relative potencies of conjugates to free Dau were also calculated as independent values from cell lines. A higher value of relative potency indicated the elevated targeting capacity of the conjugate on particular cell lines. Considering relative potencies, the best targeting capacity was shown to be Conj3 on all four cell lines, followed by Conj2 and Conj1. The highest antitumor activity of Conj3 on cells can be explained by the presence of two Dau molecules in this conjugate in comparison with Conj2 and Conj1, which contain only one drug molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | B16 cell | CVCL_F936 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.2 ± 0.4 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, the native sequence of -MSH was used, in which Met was replaced by Nle (SYSNleEHFRWGKPV). Daunomycin was attached to the amino functional groups via non-cleavable oxime linkage. These types of conjugates have many excellent properties to study and compare the bioactivity of the compounds. One of the developed conjugates contained drug molecules on both conjugation site (N-terminal and the side chain of Lys). The two additional conjugates were modified by the drug, either on theN-terminus or on the Lys side chain. The in vitro cytostatic effect on mouse melanoma cells did not show any significant differences among them. However, results indicated that the conjugates with Dau on the side chain of Lys could enter the cells more rapidly and efficiently. In contrast, the in vivo tumor growth inhibition was the most pronounced in the case of Ac-SYSNleEHFRWGK(Dau=Aoa)PV-NH2(Conj2). It is worth mentioning that the dose was normalized to Dau content, thus the injected dose ofConj3was half that ofConj 2. Nevertheless,Conj2, with one molecule of the drug, might be superior to the conjugate containing two molecules of daunomycin. The higher tumor growth inhibition effect ofConj2overConj1where the Dau is connected to theN-terminus, confirms our previous results with Dau-GnRH-III (Glp-His-Trp-Lys(Bu)-His-Asp-Trp-Lys(Dau=Aoa)-Pro-Gly-NH2), suggesting that H-Lys(Dau=Aoa)-OH can release easily from this position by the dipeptidyl-peptidase activity of cathepsin B.
Click to Show/Hide
|
||||
| Description |
In vitro studies of the antiproliferative effect showed that melanoma-targeting Dau-conjugates have significantly higher efficacy on B16 murine melanoma cell lines in comparison to human melanoma cell lines (A2058, M24, and WM983B). The IC50 values were detected between 2 and 2.9 μM on B16 cells, where the highest activity was detected in the case of conjugate Conj3, followed by Conj2 and Conj1. Moreover, the relative potencies of conjugates to free Dau were also calculated as independent values from cell lines. A higher value of relative potency indicated the elevated targeting capacity of the conjugate on particular cell lines. Considering relative potencies, the best targeting capacity was shown to be Conj3 on all four cell lines, followed by Conj2 and Conj1. The highest antitumor activity of Conj3 on cells can be explained by the presence of two Dau molecules in this conjugate in comparison with Conj2 and Conj1, which contain only one drug molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | A2058 cell | CVCL_1059 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
7.9 ± 0.7 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, the native sequence of -MSH was used, in which Met was replaced by Nle (SYSNleEHFRWGKPV). Daunomycin was attached to the amino functional groups via non-cleavable oxime linkage. These types of conjugates have many excellent properties to study and compare the bioactivity of the compounds. One of the developed conjugates contained drug molecules on both conjugation site (N-terminal and the side chain of Lys). The two additional conjugates were modified by the drug, either on theN-terminus or on the Lys side chain. The in vitro cytostatic effect on mouse melanoma cells did not show any significant differences among them. However, results indicated that the conjugates with Dau on the side chain of Lys could enter the cells more rapidly and efficiently. In contrast, the in vivo tumor growth inhibition was the most pronounced in the case of Ac-SYSNleEHFRWGK(Dau=Aoa)PV-NH2(Conj2). It is worth mentioning that the dose was normalized to Dau content, thus the injected dose ofConj3was half that ofConj 2. Nevertheless,Conj2, with one molecule of the drug, might be superior to the conjugate containing two molecules of daunomycin. The higher tumor growth inhibition effect ofConj2overConj1where the Dau is connected to theN-terminus, confirms our previous results with Dau-GnRH-III (Glp-His-Trp-Lys(Bu)-His-Asp-Trp-Lys(Dau=Aoa)-Pro-Gly-NH2), suggesting that H-Lys(Dau=Aoa)-OH can release easily from this position by the dipeptidyl-peptidase activity of cathepsin B.
Click to Show/Hide
|
||||
| Description |
In vitro studies of the antiproliferative effect showed that melanoma-targeting Dau-conjugates have significantly higher efficacy on B16 murine melanoma cell lines in comparison to human melanoma cell lines (A2058, M24, and WM983B). The IC50 values were detected between 2 and 2.9 μM on B16 cells, where the highest activity was detected in the case of conjugate Conj3, followed by Conj2 and Conj1. Moreover, the relative potencies of conjugates to free Dau were also calculated as independent values from cell lines. A higher value of relative potency indicated the elevated targeting capacity of the conjugate on particular cell lines. Considering relative potencies, the best targeting capacity was shown to be Conj3 on all four cell lines, followed by Conj2 and Conj1. The highest antitumor activity of Conj3 on cells can be explained by the presence of two Dau molecules in this conjugate in comparison with Conj2 and Conj1, which contain only one drug molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | WM983B cell | CVCL_6809 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
11.5 ± 0.4 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, the native sequence of -MSH was used, in which Met was replaced by Nle (SYSNleEHFRWGKPV). Daunomycin was attached to the amino functional groups via non-cleavable oxime linkage. These types of conjugates have many excellent properties to study and compare the bioactivity of the compounds. One of the developed conjugates contained drug molecules on both conjugation site (N-terminal and the side chain of Lys). The two additional conjugates were modified by the drug, either on theN-terminus or on the Lys side chain. The in vitro cytostatic effect on mouse melanoma cells did not show any significant differences among them. However, results indicated that the conjugates with Dau on the side chain of Lys could enter the cells more rapidly and efficiently. In contrast, the in vivo tumor growth inhibition was the most pronounced in the case of Ac-SYSNleEHFRWGK(Dau=Aoa)PV-NH2(Conj2). It is worth mentioning that the dose was normalized to Dau content, thus the injected dose ofConj3was half that ofConj 2. Nevertheless,Conj2, with one molecule of the drug, might be superior to the conjugate containing two molecules of daunomycin. The higher tumor growth inhibition effect ofConj2overConj1where the Dau is connected to theN-terminus, confirms our previous results with Dau-GnRH-III (Glp-His-Trp-Lys(Bu)-His-Asp-Trp-Lys(Dau=Aoa)-Pro-Gly-NH2), suggesting that H-Lys(Dau=Aoa)-OH can release easily from this position by the dipeptidyl-peptidase activity of cathepsin B.
Click to Show/Hide
|
||||
| Description |
In vitro studies of the antiproliferative effect showed that melanoma-targeting Dau-conjugates have significantly higher efficacy on B16 murine melanoma cell lines in comparison to human melanoma cell lines (A2058, M24, and WM983B). The IC50 values were detected between 2 and 2.9 μM on B16 cells, where the highest activity was detected in the case of conjugate Conj3, followed by Conj2 and Conj1. Moreover, the relative potencies of conjugates to free Dau were also calculated as independent values from cell lines. A higher value of relative potency indicated the elevated targeting capacity of the conjugate on particular cell lines. Considering relative potencies, the best targeting capacity was shown to be Conj3 on all four cell lines, followed by Conj2 and Conj1. The highest antitumor activity of Conj3 on cells can be explained by the presence of two Dau molecules in this conjugate in comparison with Conj2 and Conj1, which contain only one drug molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | M24 cell | CVCL_D032 | ||
[8Lys(Dau=Aoa-LRRY)]-GnRH-III [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.8 ± 0.5 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
As a payload for PDCs (and ADCs), analogues of the duocarmycins are attractive. Duocarmycin SA and yatakemycin rank among the most potent natural cytotoxins discovered. The cyclopropyl and prodrug seco forms are both naturally occurring and equipotent in most circumstances. Studies of the binding-driven bonding model of their interaction with DNA suggest that their utility will be enhanced when targeted to tumor cells. In fact, SYD985, an ADC that utilizes a peptide linker for a duocarmycin analogue to trastuzumab, has recently been progressed to phase III clinical trial.
Click to Show/Hide
|
||||
| Description |
The main drug-related toxic side effect of anthracyclines is cardiotoxicity, leading to cardiomyopathy and heart failure. Comparative experiments to determine the cardiotoxicity of peptide-anthracycline conjugates with different linkages might be informative for the other conjugates with different types of drugs as well. For this purpose, human cardiomyocytes (HCM) and human umbilical vein endothelial cells (HUVEC) were used as models. The long-term (0-72 h) cytotoxic effect of sixteen GnRH-based conjugates containing Dox and Dau was determined by real-time impedimetric sensing using the xCELLigence SP system (ACEA Biosciences, San Diego, CA, USA) [75]. The results indicated that the ester-linked GnRH-Dox conjugates, including Zoptarelin Doxorubicin, showed significant toxicity at 100 nM and 1 uM, which was remarkably pronounced on the HCM cells. The cytotoxic effect was comparable to that of the free drug, especially at the highest concentration. In contrast, the conjugates with oxime-linked Dau showed no or only a minor toxicity on both the cell lines (Table 2). These data confirm that the linkage between the payload and homing peptide has a significant influence on early drug release and, consequently, an undesired toxic side effect. We may also conclude that the search for more suitable homing peptides might be more important than the application of cleavable bonds between the drug and the peptide to develop efficient DDSs.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
28.6 ± 5.5 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
The main drug-related toxic side effect of anthracyclines is cardiotoxicity, leading to cardiomyopathy and heart failure. Comparative experiments to determine the cardiotoxicity of peptide-anthracycline conjugates with different linkages might be informative for the other conjugates with different types of drugs as well. For this purpose, human cardiomyocytes (HCM) and human umbilical vein endothelial cells (HUVEC) were used as models. The long-term (0-72 h) cytotoxic effect of sixteen GnRH-based conjugates containing Dox and Dau was determined by real-time impedimetric sensing using the xCELLigence SP system (ACEA Biosciences, San Diego, CA, USA) [75]. The results indicated that the ester-linked GnRH-Dox conjugates, including Zoptarelin Doxorubicin, showed significant toxicity at 100 nM and 1 uM, which was remarkably pronounced on the HCM cells. The cytotoxic effect was comparable to that of the free drug, especially at the highest concentration. In contrast, the conjugates with oxime-linked Dau showed no or only a minor toxicity on both the cell lines (Table 2). These data confirm that the linkage between the payload and homing peptide has a significant influence on early drug release and, consequently, an undesired toxic side effect. We may also conclude that the search for more suitable homing peptides might be more important than the application of cleavable bonds between the drug and the peptide to develop efficient DDSs.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
GnRH-III-[2ΔHis, 3D-Tic, 7D-Trp 8Lys(Dau=Aoa)] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.90 ± 0.58 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
7.88 ± 1.24 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
10.70 ± 0.95 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
Dau=Aoa-SYSNleEHFRWGK(Dau=Aoa)PV-NH2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.0 ± 0.7 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, the native sequence of -MSH was used, in which Met was replaced by Nle (SYSNleEHFRWGKPV). Daunomycin was attached to the amino functional groups via non-cleavable oxime linkage. These types of conjugates have many excellent properties to study and compare the bioactivity of the compounds. One of the developed conjugates contained drug molecules on both conjugation site (N-terminal and the side chain of Lys). The two additional conjugates were modified by the drug, either on theN-terminus or on the Lys side chain. The in vitro cytostatic effect on mouse melanoma cells did not show any significant differences among them. However, results indicated that the conjugates with Dau on the side chain of Lys could enter the cells more rapidly and efficiently. In contrast, the in vivo tumor growth inhibition was the most pronounced in the case of Ac-SYSNleEHFRWGK(Dau=Aoa)PV-NH2(Conj2). It is worth mentioning that the dose was normalized to Dau content, thus the injected dose ofConj3was half that ofConj 2. Nevertheless,Conj2, with one molecule of the drug, might be superior to the conjugate containing two molecules of daunomycin. The higher tumor growth inhibition effect ofConj2overConj1where the Dau is connected to theN-terminus, confirms our previous results with Dau-GnRH-III (Glp-His-Trp-Lys(Bu)-His-Asp-Trp-Lys(Dau=Aoa)-Pro-Gly-NH2), suggesting that H-Lys(Dau=Aoa)-OH can release easily from this position by the dipeptidyl-peptidase activity of cathepsin B.
Click to Show/Hide
|
||||
| Description |
In vitro studies of the antiproliferative effect showed that melanoma-targeting Dau-conjugates have significantly higher efficacy on B16 murine melanoma cell lines in comparison to human melanoma cell lines (A2058, M24, and WM983B). The IC50 values were detected between 2 and 2.9 μM on B16 cells, where the highest activity was detected in the case of conjugate Conj3, followed by Conj2 and Conj1. Moreover, the relative potencies of conjugates to free Dau were also calculated as independent values from cell lines. A higher value of relative potency indicated the elevated targeting capacity of the conjugate on particular cell lines. Considering relative potencies, the best targeting capacity was shown to be Conj3 on all four cell lines, followed by Conj2 and Conj1. The highest antitumor activity of Conj3 on cells can be explained by the presence of two Dau molecules in this conjugate in comparison with Conj2 and Conj1, which contain only one drug molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | B16 cell | CVCL_F936 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.0 ± 0.8 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, the native sequence of -MSH was used, in which Met was replaced by Nle (SYSNleEHFRWGKPV). Daunomycin was attached to the amino functional groups via non-cleavable oxime linkage. These types of conjugates have many excellent properties to study and compare the bioactivity of the compounds. One of the developed conjugates contained drug molecules on both conjugation site (N-terminal and the side chain of Lys). The two additional conjugates were modified by the drug, either on theN-terminus or on the Lys side chain. The in vitro cytostatic effect on mouse melanoma cells did not show any significant differences among them. However, results indicated that the conjugates with Dau on the side chain of Lys could enter the cells more rapidly and efficiently. In contrast, the in vivo tumor growth inhibition was the most pronounced in the case of Ac-SYSNleEHFRWGK(Dau=Aoa)PV-NH2(Conj2). It is worth mentioning that the dose was normalized to Dau content, thus the injected dose ofConj3was half that ofConj 2. Nevertheless,Conj2, with one molecule of the drug, might be superior to the conjugate containing two molecules of daunomycin. The higher tumor growth inhibition effect ofConj2overConj1where the Dau is connected to theN-terminus, confirms our previous results with Dau-GnRH-III (Glp-His-Trp-Lys(Bu)-His-Asp-Trp-Lys(Dau=Aoa)-Pro-Gly-NH2), suggesting that H-Lys(Dau=Aoa)-OH can release easily from this position by the dipeptidyl-peptidase activity of cathepsin B.
Click to Show/Hide
|
||||
| Description |
In vitro studies of the antiproliferative effect showed that melanoma-targeting Dau-conjugates have significantly higher efficacy on B16 murine melanoma cell lines in comparison to human melanoma cell lines (A2058, M24, and WM983B). The IC50 values were detected between 2 and 2.9 μM on B16 cells, where the highest activity was detected in the case of conjugate Conj3, followed by Conj2 and Conj1. Moreover, the relative potencies of conjugates to free Dau were also calculated as independent values from cell lines. A higher value of relative potency indicated the elevated targeting capacity of the conjugate on particular cell lines. Considering relative potencies, the best targeting capacity was shown to be Conj3 on all four cell lines, followed by Conj2 and Conj1. The highest antitumor activity of Conj3 on cells can be explained by the presence of two Dau molecules in this conjugate in comparison with Conj2 and Conj1, which contain only one drug molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | A2058 cell | CVCL_1059 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.6 ± 0.2 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, the native sequence of -MSH was used, in which Met was replaced by Nle (SYSNleEHFRWGKPV). Daunomycin was attached to the amino functional groups via non-cleavable oxime linkage. These types of conjugates have many excellent properties to study and compare the bioactivity of the compounds. One of the developed conjugates contained drug molecules on both conjugation site (N-terminal and the side chain of Lys). The two additional conjugates were modified by the drug, either on theN-terminus or on the Lys side chain. The in vitro cytostatic effect on mouse melanoma cells did not show any significant differences among them. However, results indicated that the conjugates with Dau on the side chain of Lys could enter the cells more rapidly and efficiently. In contrast, the in vivo tumor growth inhibition was the most pronounced in the case of Ac-SYSNleEHFRWGK(Dau=Aoa)PV-NH2(Conj2). It is worth mentioning that the dose was normalized to Dau content, thus the injected dose ofConj3was half that ofConj 2. Nevertheless,Conj2, with one molecule of the drug, might be superior to the conjugate containing two molecules of daunomycin. The higher tumor growth inhibition effect ofConj2overConj1where the Dau is connected to theN-terminus, confirms our previous results with Dau-GnRH-III (Glp-His-Trp-Lys(Bu)-His-Asp-Trp-Lys(Dau=Aoa)-Pro-Gly-NH2), suggesting that H-Lys(Dau=Aoa)-OH can release easily from this position by the dipeptidyl-peptidase activity of cathepsin B.
Click to Show/Hide
|
||||
| Description |
In vitro studies of the antiproliferative effect showed that melanoma-targeting Dau-conjugates have significantly higher efficacy on B16 murine melanoma cell lines in comparison to human melanoma cell lines (A2058, M24, and WM983B). The IC50 values were detected between 2 and 2.9 μM on B16 cells, where the highest activity was detected in the case of conjugate Conj3, followed by Conj2 and Conj1. Moreover, the relative potencies of conjugates to free Dau were also calculated as independent values from cell lines. A higher value of relative potency indicated the elevated targeting capacity of the conjugate on particular cell lines. Considering relative potencies, the best targeting capacity was shown to be Conj3 on all four cell lines, followed by Conj2 and Conj1. The highest antitumor activity of Conj3 on cells can be explained by the presence of two Dau molecules in this conjugate in comparison with Conj2 and Conj1, which contain only one drug molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | WM983B cell | CVCL_6809 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
11.0 ± 0.8 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, the native sequence of -MSH was used, in which Met was replaced by Nle (SYSNleEHFRWGKPV). Daunomycin was attached to the amino functional groups via non-cleavable oxime linkage. These types of conjugates have many excellent properties to study and compare the bioactivity of the compounds. One of the developed conjugates contained drug molecules on both conjugation site (N-terminal and the side chain of Lys). The two additional conjugates were modified by the drug, either on theN-terminus or on the Lys side chain. The in vitro cytostatic effect on mouse melanoma cells did not show any significant differences among them. However, results indicated that the conjugates with Dau on the side chain of Lys could enter the cells more rapidly and efficiently. In contrast, the in vivo tumor growth inhibition was the most pronounced in the case of Ac-SYSNleEHFRWGK(Dau=Aoa)PV-NH2(Conj2). It is worth mentioning that the dose was normalized to Dau content, thus the injected dose ofConj3was half that ofConj 2. Nevertheless,Conj2, with one molecule of the drug, might be superior to the conjugate containing two molecules of daunomycin. The higher tumor growth inhibition effect ofConj2overConj1where the Dau is connected to theN-terminus, confirms our previous results with Dau-GnRH-III (Glp-His-Trp-Lys(Bu)-His-Asp-Trp-Lys(Dau=Aoa)-Pro-Gly-NH2), suggesting that H-Lys(Dau=Aoa)-OH can release easily from this position by the dipeptidyl-peptidase activity of cathepsin B.
Click to Show/Hide
|
||||
| Description |
In vitro studies of the antiproliferative effect showed that melanoma-targeting Dau-conjugates have significantly higher efficacy on B16 murine melanoma cell lines in comparison to human melanoma cell lines (A2058, M24, and WM983B). The IC50 values were detected between 2 and 2.9 μM on B16 cells, where the highest activity was detected in the case of conjugate Conj3, followed by Conj2 and Conj1. Moreover, the relative potencies of conjugates to free Dau were also calculated as independent values from cell lines. A higher value of relative potency indicated the elevated targeting capacity of the conjugate on particular cell lines. Considering relative potencies, the best targeting capacity was shown to be Conj3 on all four cell lines, followed by Conj2 and Conj1. The highest antitumor activity of Conj3 on cells can be explained by the presence of two Dau molecules in this conjugate in comparison with Conj2 and Conj1, which contain only one drug molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | M24 cell | CVCL_D032 | ||
Ac-NleEHfRWGK(Dau=Aoa)-NH2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.06 ± 0.10 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, the native sequence of -MSH was used, in which Met was replaced by Nle (SYSNleEHFRWGKPV). Daunomycin was attached to the amino functional groups via non-cleavable oxime linkage. These types of conjugates have many excellent properties to study and compare the bioactivity of the compounds. One of the developed conjugates contained drug molecules on both conjugation site (N-terminal and the side chain of Lys). The two additional conjugates were modified by the drug, either on theN-terminus or on the Lys side chain. The in vitro cytostatic effect on mouse melanoma cells did not show any significant differences among them. However, results indicated that the conjugates with Dau on the side chain of Lys could enter the cells more rapidly and efficiently. In contrast, the in vivo tumor growth inhibition was the most pronounced in the case of Ac-SYSNleEHFRWGK(Dau=Aoa)PV-NH2(Conj2). It is worth mentioning that the dose was normalized to Dau content, thus the injected dose ofConj3was half that ofConj 2. Nevertheless,Conj2, with one molecule of the drug, might be superior to the conjugate containing two molecules of daunomycin. The higher tumor growth inhibition effect ofConj2overConj1where the Dau is connected to theN-terminus, confirms our previous results with Dau-GnRH-III (Glp-His-Trp-Lys(Bu)-His-Asp-Trp-Lys(Dau=Aoa)-Pro-Gly-NH2), suggesting that H-Lys(Dau=Aoa)-OH can release easily from this position by the dipeptidyl-peptidase activity of cathepsin B.
Click to Show/Hide
|
||||
| Description |
The in vitro cytostatic effects of Dau--MSH conjugates were compared with the PrestoBlue assay, which has a higher specificity and efficacy that of the previously used MTT assay, using the same six human melanoma cell lines used in the real-time qPCR assay. According to our results, free daunomycin exhibited the lowest IC50 values, and therefore the highest cytostatic effect, due to the fact that free daunomycin passively diffuses into cells. Our Dau--MSH conjugates showed similar trends in the effectiveness of the tested cell lines. Conj2 and Conj4 resulted in IC50 values in the low uM range. A calculation of the targeting indices showed that compound Conj2 had an over 2-times higher targeting efficiency in most cell lines compared to the A2058 cell line. Interestingly, WM983A showed the lowest TI among the high-MC1R-expressing cell lines compared to A2058, and WM983B exhibited high TI regardless of the relatively low MC1R expression on the mRNA level. In the case of Conj4, the treatment resulted in the highest TIs with OCM-1 and WM983B cell lines.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | SK-MEL-202 cell | CVCL_6106 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
7.15 ± 0.07 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, the native sequence of -MSH was used, in which Met was replaced by Nle (SYSNleEHFRWGKPV). Daunomycin was attached to the amino functional groups via non-cleavable oxime linkage. These types of conjugates have many excellent properties to study and compare the bioactivity of the compounds. One of the developed conjugates contained drug molecules on both conjugation site (N-terminal and the side chain of Lys). The two additional conjugates were modified by the drug, either on theN-terminus or on the Lys side chain. The in vitro cytostatic effect on mouse melanoma cells did not show any significant differences among them. However, results indicated that the conjugates with Dau on the side chain of Lys could enter the cells more rapidly and efficiently. In contrast, the in vivo tumor growth inhibition was the most pronounced in the case of Ac-SYSNleEHFRWGK(Dau=Aoa)PV-NH2(Conj2). It is worth mentioning that the dose was normalized to Dau content, thus the injected dose ofConj3was half that ofConj 2. Nevertheless,Conj2, with one molecule of the drug, might be superior to the conjugate containing two molecules of daunomycin. The higher tumor growth inhibition effect ofConj2overConj1where the Dau is connected to theN-terminus, confirms our previous results with Dau-GnRH-III (Glp-His-Trp-Lys(Bu)-His-Asp-Trp-Lys(Dau=Aoa)-Pro-Gly-NH2), suggesting that H-Lys(Dau=Aoa)-OH can release easily from this position by the dipeptidyl-peptidase activity of cathepsin B.
Click to Show/Hide
|
||||
| Description |
The in vitro cytostatic effects of Dau--MSH conjugates were compared with the PrestoBlue assay, which has a higher specificity and efficacy that of the previously used MTT assay, using the same six human melanoma cell lines used in the real-time qPCR assay. According to our results, free daunomycin exhibited the lowest IC50 values, and therefore the highest cytostatic effect, due to the fact that free daunomycin passively diffuses into cells. Our Dau--MSH conjugates showed similar trends in the effectiveness of the tested cell lines. Conj2 and Conj4 resulted in IC50 values in the low uM range. A calculation of the targeting indices showed that compound Conj2 had an over 2-times higher targeting efficiency in most cell lines compared to the A2058 cell line. Interestingly, WM983A showed the lowest TI among the high-MC1R-expressing cell lines compared to A2058, and WM983B exhibited high TI regardless of the relatively low MC1R expression on the mRNA level. In the case of Conj4, the treatment resulted in the highest TIs with OCM-1 and WM983B cell lines.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | A2058 cell | CVCL_1059 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
10.18 ± 0.06 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, the native sequence of -MSH was used, in which Met was replaced by Nle (SYSNleEHFRWGKPV). Daunomycin was attached to the amino functional groups via non-cleavable oxime linkage. These types of conjugates have many excellent properties to study and compare the bioactivity of the compounds. One of the developed conjugates contained drug molecules on both conjugation site (N-terminal and the side chain of Lys). The two additional conjugates were modified by the drug, either on theN-terminus or on the Lys side chain. The in vitro cytostatic effect on mouse melanoma cells did not show any significant differences among them. However, results indicated that the conjugates with Dau on the side chain of Lys could enter the cells more rapidly and efficiently. In contrast, the in vivo tumor growth inhibition was the most pronounced in the case of Ac-SYSNleEHFRWGK(Dau=Aoa)PV-NH2(Conj2). It is worth mentioning that the dose was normalized to Dau content, thus the injected dose ofConj3was half that ofConj 2. Nevertheless,Conj2, with one molecule of the drug, might be superior to the conjugate containing two molecules of daunomycin. The higher tumor growth inhibition effect ofConj2overConj1where the Dau is connected to theN-terminus, confirms our previous results with Dau-GnRH-III (Glp-His-Trp-Lys(Bu)-His-Asp-Trp-Lys(Dau=Aoa)-Pro-Gly-NH2), suggesting that H-Lys(Dau=Aoa)-OH can release easily from this position by the dipeptidyl-peptidase activity of cathepsin B.
Click to Show/Hide
|
||||
| Description |
The in vitro cytostatic effects of Dau--MSH conjugates were compared with the PrestoBlue assay, which has a higher specificity and efficacy that of the previously used MTT assay, using the same six human melanoma cell lines used in the real-time qPCR assay. According to our results, free daunomycin exhibited the lowest IC50 values, and therefore the highest cytostatic effect, due to the fact that free daunomycin passively diffuses into cells. Our Dau--MSH conjugates showed similar trends in the effectiveness of the tested cell lines. Conj2 and Conj4 resulted in IC50 values in the low uM range. A calculation of the targeting indices showed that compound Conj2 had an over 2-times higher targeting efficiency in most cell lines compared to the A2058 cell line. Interestingly, WM983A showed the lowest TI among the high-MC1R-expressing cell lines compared to A2058, and WM983B exhibited high TI regardless of the relatively low MC1R expression on the mRNA level. In the case of Conj4, the treatment resulted in the highest TIs with OCM-1 and WM983B cell lines.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | WM983B cell | CVCL_6809 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
13.50 ± 0.06 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, the native sequence of -MSH was used, in which Met was replaced by Nle (SYSNleEHFRWGKPV). Daunomycin was attached to the amino functional groups via non-cleavable oxime linkage. These types of conjugates have many excellent properties to study and compare the bioactivity of the compounds. One of the developed conjugates contained drug molecules on both conjugation site (N-terminal and the side chain of Lys). The two additional conjugates were modified by the drug, either on theN-terminus or on the Lys side chain. The in vitro cytostatic effect on mouse melanoma cells did not show any significant differences among them. However, results indicated that the conjugates with Dau on the side chain of Lys could enter the cells more rapidly and efficiently. In contrast, the in vivo tumor growth inhibition was the most pronounced in the case of Ac-SYSNleEHFRWGK(Dau=Aoa)PV-NH2(Conj2). It is worth mentioning that the dose was normalized to Dau content, thus the injected dose ofConj3was half that ofConj 2. Nevertheless,Conj2, with one molecule of the drug, might be superior to the conjugate containing two molecules of daunomycin. The higher tumor growth inhibition effect ofConj2overConj1where the Dau is connected to theN-terminus, confirms our previous results with Dau-GnRH-III (Glp-His-Trp-Lys(Bu)-His-Asp-Trp-Lys(Dau=Aoa)-Pro-Gly-NH2), suggesting that H-Lys(Dau=Aoa)-OH can release easily from this position by the dipeptidyl-peptidase activity of cathepsin B.
Click to Show/Hide
|
||||
| Description |
The in vitro cytostatic effects of Dau--MSH conjugates were compared with the PrestoBlue assay, which has a higher specificity and efficacy that of the previously used MTT assay, using the same six human melanoma cell lines used in the real-time qPCR assay. According to our results, free daunomycin exhibited the lowest IC50 values, and therefore the highest cytostatic effect, due to the fact that free daunomycin passively diffuses into cells. Our Dau--MSH conjugates showed similar trends in the effectiveness of the tested cell lines. Conj2 and Conj4 resulted in IC50 values in the low uM range. A calculation of the targeting indices showed that compound Conj2 had an over 2-times higher targeting efficiency in most cell lines compared to the A2058 cell line. Interestingly, WM983A showed the lowest TI among the high-MC1R-expressing cell lines compared to A2058, and WM983B exhibited high TI regardless of the relatively low MC1R expression on the mRNA level. In the case of Conj4, the treatment resulted in the highest TIs with OCM-1 and WM983B cell lines.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | WM983A cell | CVCL_6808 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
25.47 ± 0.08 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, the native sequence of -MSH was used, in which Met was replaced by Nle (SYSNleEHFRWGKPV). Daunomycin was attached to the amino functional groups via non-cleavable oxime linkage. These types of conjugates have many excellent properties to study and compare the bioactivity of the compounds. One of the developed conjugates contained drug molecules on both conjugation site (N-terminal and the side chain of Lys). The two additional conjugates were modified by the drug, either on theN-terminus or on the Lys side chain. The in vitro cytostatic effect on mouse melanoma cells did not show any significant differences among them. However, results indicated that the conjugates with Dau on the side chain of Lys could enter the cells more rapidly and efficiently. In contrast, the in vivo tumor growth inhibition was the most pronounced in the case of Ac-SYSNleEHFRWGK(Dau=Aoa)PV-NH2(Conj2). It is worth mentioning that the dose was normalized to Dau content, thus the injected dose ofConj3was half that ofConj 2. Nevertheless,Conj2, with one molecule of the drug, might be superior to the conjugate containing two molecules of daunomycin. The higher tumor growth inhibition effect ofConj2overConj1where the Dau is connected to theN-terminus, confirms our previous results with Dau-GnRH-III (Glp-His-Trp-Lys(Bu)-His-Asp-Trp-Lys(Dau=Aoa)-Pro-Gly-NH2), suggesting that H-Lys(Dau=Aoa)-OH can release easily from this position by the dipeptidyl-peptidase activity of cathepsin B.
Click to Show/Hide
|
||||
| Description |
The in vitro cytostatic effects of Dau--MSH conjugates were compared with the PrestoBlue assay, which has a higher specificity and efficacy that of the previously used MTT assay, using the same six human melanoma cell lines used in the real-time qPCR assay. According to our results, free daunomycin exhibited the lowest IC50 values, and therefore the highest cytostatic effect, due to the fact that free daunomycin passively diffuses into cells. Our Dau--MSH conjugates showed similar trends in the effectiveness of the tested cell lines. Conj2 and Conj4 resulted in IC50 values in the low uM range. A calculation of the targeting indices showed that compound Conj2 had an over 2-times higher targeting efficiency in most cell lines compared to the A2058 cell line. Interestingly, WM983A showed the lowest TI among the high-MC1R-expressing cell lines compared to A2058, and WM983B exhibited high TI regardless of the relatively low MC1R expression on the mRNA level. In the case of Conj4, the treatment resulted in the highest TIs with OCM-1 and WM983B cell lines.
Click to Show/Hide
|
||||
| In Vitro Model | Cutaneous melanoma | OCM-3 cell | CVCL_6937 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
29.68 ± 0.06 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, the native sequence of -MSH was used, in which Met was replaced by Nle (SYSNleEHFRWGKPV). Daunomycin was attached to the amino functional groups via non-cleavable oxime linkage. These types of conjugates have many excellent properties to study and compare the bioactivity of the compounds. One of the developed conjugates contained drug molecules on both conjugation site (N-terminal and the side chain of Lys). The two additional conjugates were modified by the drug, either on theN-terminus or on the Lys side chain. The in vitro cytostatic effect on mouse melanoma cells did not show any significant differences among them. However, results indicated that the conjugates with Dau on the side chain of Lys could enter the cells more rapidly and efficiently. In contrast, the in vivo tumor growth inhibition was the most pronounced in the case of Ac-SYSNleEHFRWGK(Dau=Aoa)PV-NH2(Conj2). It is worth mentioning that the dose was normalized to Dau content, thus the injected dose ofConj3was half that ofConj 2. Nevertheless,Conj2, with one molecule of the drug, might be superior to the conjugate containing two molecules of daunomycin. The higher tumor growth inhibition effect ofConj2overConj1where the Dau is connected to theN-terminus, confirms our previous results with Dau-GnRH-III (Glp-His-Trp-Lys(Bu)-His-Asp-Trp-Lys(Dau=Aoa)-Pro-Gly-NH2), suggesting that H-Lys(Dau=Aoa)-OH can release easily from this position by the dipeptidyl-peptidase activity of cathepsin B.
Click to Show/Hide
|
||||
| Description |
The in vitro cytostatic effects of Dau--MSH conjugates were compared with the PrestoBlue assay, which has a higher specificity and efficacy that of the previously used MTT assay, using the same six human melanoma cell lines used in the real-time qPCR assay. According to our results, free daunomycin exhibited the lowest IC50 values, and therefore the highest cytostatic effect, due to the fact that free daunomycin passively diffuses into cells. Our Dau--MSH conjugates showed similar trends in the effectiveness of the tested cell lines. Conj2 and Conj4 resulted in IC50 values in the low uM range. A calculation of the targeting indices showed that compound Conj2 had an over 2-times higher targeting efficiency in most cell lines compared to the A2058 cell line. Interestingly, WM983A showed the lowest TI among the high-MC1R-expressing cell lines compared to A2058, and WM983B exhibited high TI regardless of the relatively low MC1R expression on the mRNA level. In the case of Conj4, the treatment resulted in the highest TIs with OCM-1 and WM983B cell lines.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | OCM-1 cell | CVCL_6934 | ||
GnRH-III-[4Lys(Bu), 8Lys(Dau=Aoa), 10ΔGly-NH-Et] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.23 ± 0.40 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
12.41 ± 2.30 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
17.84 ± 0.08 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
GnRH-III-[8Lys(Dau=Aoa)] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.54 ± 0.67µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
6.5 ± 1.8 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
The cancer phenotype is commonly associated with aberrant glycosylation patterns. One glycan that is directly linked to cancer is the Thomsen-Friedenreich antigen (TF or CD176). It is a disaccharide composed of a galactose β1-3 N-acetylgalactosamine, O-linked to a glycoprotein through serine or threonine residues and commonly written as Galβ1-3GalNAc--O-Ser/Thr. The TF is therapeutically attractive due to its cryptic nature in normal cells and exposure in embryonic and cancer cells. The expression of the TF has been demonstrated in 90% of primary human carcinomas, including in the lung, the breast, and the pancreas. Additionally, cancer initiating cells or cancer stem cells in the lung, liver, and colon express the TF. The peptide sequence HGRFILPWWYAFSPS (TF-peptide) is known to bind tightly to the TF (Kd = 1.2 M) and has been demonstrated to inhibit processes directly involved in TF accessibility.
Click to Show/Hide
|
||||
| Description |
The main drug-related toxic side effect of anthracyclines is cardiotoxicity, leading to cardiomyopathy and heart failure. Comparative experiments to determine the cardiotoxicity of peptide-anthracycline conjugates with different linkages might be informative for the other conjugates with different types of drugs as well. For this purpose, human cardiomyocytes (HCM) and human umbilical vein endothelial cells (HUVEC) were used as models. The long-term (0-72 h) cytotoxic effect of sixteen GnRH-based conjugates containing Dox and Dau was determined by real-time impedimetric sensing using the xCELLigence SP system (ACEA Biosciences, San Diego, CA, USA) [75]. The results indicated that the ester-linked GnRH-Dox conjugates, including Zoptarelin Doxorubicin, showed significant toxicity at 100 nM and 1 uM, which was remarkably pronounced on the HCM cells. The cytotoxic effect was comparable to that of the free drug, especially at the highest concentration. In contrast, the conjugates with oxime-linked Dau showed no or only a minor toxicity on both the cell lines (Table 2). These data confirm that the linkage between the payload and homing peptide has a significant influence on early drug release and, consequently, an undesired toxic side effect. We may also conclude that the search for more suitable homing peptides might be more important than the application of cleavable bonds between the drug and the peptide to develop efficient DDSs.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
8.22 ± 0.13 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
13.89 ± 3.62 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
| Experiment 5 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
14.2 ± 3.2 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
In our experiments, GnRH-III was applied as a targeting peptide. The conjugate [8Lys(Dau=Aoa-GFLG)]-GnRH-III, in which Dau was connected to the side chain of Lys in position 8 through an aminooxyacetylated cathepsin B-labile GFLG spacer, showed significant tumor growth inhibition in s.c.-developed fast-growing C26 murine colon cancer-bearing Balb/c mice. The effect highly depended on the treatment schedule. In the first attempt, the conjugate was administered i.p. at a dose of 8.86 umol (5 mg Dau-content/kg body weight) five times on days 7, 9, 11, 14 and 16 after tumor transplantation (Treatment schedule A). The tumor volume inhibition was only 15.7% on day 26 when the experiment finished. A slight enhancement in inhibition (22.3%) was detected when a dose of conjugate corresponding to 15 mg Dau content/kg body weight (26.6 μmol) was injected only once on day 7 (Treatment schedule B). An additional treatment on day 10 (Treatment schedule C) did not result in any further improvements (21.9% on day 29). However, when the treatment schedule was changed to two treatments with the same dose on days 4 and 7, 46.3% inhibition was observed on day 29 (Treatment schedule D). In contrast, the treatments with free Dau at a dose of 2 mg/kg body weight (3.55 umol) on days 7, 9, 11, 14 and 16 showed only 22.6% inhibition on day 26. The median survival rates of the treated animals in comparison to the control group were 1 (A), 1.23 (B), 1.21 (C) and 1.38 (D), respectively, and 0.81 for free Dau. These results indicate the lower toxicity and the higher tumor volume inhibition effect of the conjugates in comparison with those of free Dau, as well as the importance of the treatment schedule. In another experiment, HT-29 human colon cancer was developed s.c. in immunodeficient SCID mice. Dau and two conjugates (with or without a GFLG spacer between the GnRH-III homing peptide and the payload) were used for the treatment. The first i.p. administrations were performed on day 13 after tumor inoculation. All the mice treated once with 2.5 mg/kg body weight (4.43 umol) free Dau died within 10 days. In contrast, the conjugates at a dose of 15 mg Dau content/kg body weight (26.6 umol) that refers to 52 mg/kg [8Lys(Dau=Aoa)]-GnRH-III and 62.5 mg/kg [8Lys(Dau=Aoa-GFLG)]-GnRH-III conjugates, respectively, did not show significant toxicity. The treatment was repeated on days 23 and 30. Because of the significant weight loss in several mice in the control group, the experiment was terminated on day 35. The tumor growth inhibition could be calculated as reductions in the tumor volume by 44.3% and 57.6% and the tumor weight by 41% and 50%, respectively. Interestingly, the conjugates were poorly effective on orthotopically developed tumors. In the case of the C26 colon tumor-bearing female Balb/c mice, only a 7% reduction in tumor weight was detected on day 13 after the two treatments on days 4 and 7 with [8Lys(Dau=Aoa)]-GnRH-III, at a 26.6 umol/kg (15 mg Dau content) dose. The effect of free Dau (2 mg/kg on days 4 and 7) showed a better inhibitory effect (24.4%). Interestingly, our novel developed GnRH-III derivative, in which Ser in position 4 was replaced by Lys(Ac), was much more potent, with 49.3% inhibition. It is worth mentioning that the rate of cellular uptake of the [4Lys(Ac), 8Lys(Dau=Aoa)]-GnRH-III conjugate by the tumor cells was significantly higher than that of the conjugate with the native GnRH-III sequence.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
| Experiment 6 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
27.8 ± 4.2 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
The main drug-related toxic side effect of anthracyclines is cardiotoxicity, leading to cardiomyopathy and heart failure. Comparative experiments to determine the cardiotoxicity of peptide-anthracycline conjugates with different linkages might be informative for the other conjugates with different types of drugs as well. For this purpose, human cardiomyocytes (HCM) and human umbilical vein endothelial cells (HUVEC) were used as models. The long-term (0-72 h) cytotoxic effect of sixteen GnRH-based conjugates containing Dox and Dau was determined by real-time impedimetric sensing using the xCELLigence SP system (ACEA Biosciences, San Diego, CA, USA) [75]. The results indicated that the ester-linked GnRH-Dox conjugates, including Zoptarelin Doxorubicin, showed significant toxicity at 100 nM and 1 uM, which was remarkably pronounced on the HCM cells. The cytotoxic effect was comparable to that of the free drug, especially at the highest concentration. In contrast, the conjugates with oxime-linked Dau showed no or only a minor toxicity on both the cell lines (Table 2). These data confirm that the linkage between the payload and homing peptide has a significant influence on early drug release and, consequently, an undesired toxic side effect. We may also conclude that the search for more suitable homing peptides might be more important than the application of cleavable bonds between the drug and the peptide to develop efficient DDSs.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
GnRH-III-[3D-Tic, 4Lys(Bu), 8Lys(Dau=Aoa)] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.56 ± 0.51 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
12.73 ± 3.10 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
GnRH-III-[3D-Tic, 4Lys(Bu), 7D-Trp 8Lys(Dau=Aoa)] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.57 ± 0.47 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
16.83 ± 0.66 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
GnRH-III-[2ΔHis, 3D-Tic, 8Lys(Dau=Aoa)] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.75 ± 0.17 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
9.35 ± 1.93 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
10.32 ± 1.32 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
GnRH-III-[2ΔHis, 3D-Tic, 4Lys(Bu), 7D-Trp 8Lys(Dau=Aoa)] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.81 ± 0.04 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
8.18 ± 0.18 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
16.55 ± 0.30 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
GnRH-III-[3D-Tic, 8Lys(Dau=Aoa)] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.89 ± 0.62 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
8.75 ± 0.86 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
[4Lys(Dau=Aoa),8Lys(Dau=Aoa)]-GnRH-III [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.9 ± 0.9 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
As a payload for PDCs (and ADCs), analogues of the duocarmycins are attractive. Duocarmycin SA and yatakemycin rank among the most potent natural cytotoxins discovered. The cyclopropyl and prodrug seco forms are both naturally occurring and equipotent in most circumstances. Studies of the binding-driven bonding model of their interaction with DNA suggest that their utility will be enhanced when targeted to tumor cells. In fact, SYD985, an ADC that utilizes a peptide linker for a duocarmycin analogue to trastuzumab, has recently been progressed to phase III clinical trial.
Click to Show/Hide
|
||||
| Description |
The main drug-related toxic side effect of anthracyclines is cardiotoxicity, leading to cardiomyopathy and heart failure. Comparative experiments to determine the cardiotoxicity of peptide-anthracycline conjugates with different linkages might be informative for the other conjugates with different types of drugs as well. For this purpose, human cardiomyocytes (HCM) and human umbilical vein endothelial cells (HUVEC) were used as models. The long-term (0-72 h) cytotoxic effect of sixteen GnRH-based conjugates containing Dox and Dau was determined by real-time impedimetric sensing using the xCELLigence SP system (ACEA Biosciences, San Diego, CA, USA) [75]. The results indicated that the ester-linked GnRH-Dox conjugates, including Zoptarelin Doxorubicin, showed significant toxicity at 100 nM and 1 uM, which was remarkably pronounced on the HCM cells. The cytotoxic effect was comparable to that of the free drug, especially at the highest concentration. In contrast, the conjugates with oxime-linked Dau showed no or only a minor toxicity on both the cell lines (Table 2). These data confirm that the linkage between the payload and homing peptide has a significant influence on early drug release and, consequently, an undesired toxic side effect. We may also conclude that the search for more suitable homing peptides might be more important than the application of cleavable bonds between the drug and the peptide to develop efficient DDSs.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
6.8 ± 1.0 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
The main drug-related toxic side effect of anthracyclines is cardiotoxicity, leading to cardiomyopathy and heart failure. Comparative experiments to determine the cardiotoxicity of peptide-anthracycline conjugates with different linkages might be informative for the other conjugates with different types of drugs as well. For this purpose, human cardiomyocytes (HCM) and human umbilical vein endothelial cells (HUVEC) were used as models. The long-term (0-72 h) cytotoxic effect of sixteen GnRH-based conjugates containing Dox and Dau was determined by real-time impedimetric sensing using the xCELLigence SP system (ACEA Biosciences, San Diego, CA, USA) [75]. The results indicated that the ester-linked GnRH-Dox conjugates, including Zoptarelin Doxorubicin, showed significant toxicity at 100 nM and 1 uM, which was remarkably pronounced on the HCM cells. The cytotoxic effect was comparable to that of the free drug, especially at the highest concentration. In contrast, the conjugates with oxime-linked Dau showed no or only a minor toxicity on both the cell lines (Table 2). These data confirm that the linkage between the payload and homing peptide has a significant influence on early drug release and, consequently, an undesired toxic side effect. We may also conclude that the search for more suitable homing peptides might be more important than the application of cleavable bonds between the drug and the peptide to develop efficient DDSs.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
Dau=Aoa-SYSNleEHFRWGKPV-NH2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.9 ± 0.6 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, the native sequence of -MSH was used, in which Met was replaced by Nle (SYSNleEHFRWGKPV). Daunomycin was attached to the amino functional groups via non-cleavable oxime linkage. These types of conjugates have many excellent properties to study and compare the bioactivity of the compounds. One of the developed conjugates contained drug molecules on both conjugation site (N-terminal and the side chain of Lys). The two additional conjugates were modified by the drug, either on theN-terminus or on the Lys side chain. The in vitro cytostatic effect on mouse melanoma cells did not show any significant differences among them. However, results indicated that the conjugates with Dau on the side chain of Lys could enter the cells more rapidly and efficiently. In contrast, the in vivo tumor growth inhibition was the most pronounced in the case of Ac-SYSNleEHFRWGK(Dau=Aoa)PV-NH2(Conj2). It is worth mentioning that the dose was normalized to Dau content, thus the injected dose ofConj3was half that ofConj 2. Nevertheless,Conj2, with one molecule of the drug, might be superior to the conjugate containing two molecules of daunomycin. The higher tumor growth inhibition effect ofConj2overConj1where the Dau is connected to theN-terminus, confirms our previous results with Dau-GnRH-III (Glp-His-Trp-Lys(Bu)-His-Asp-Trp-Lys(Dau=Aoa)-Pro-Gly-NH2), suggesting that H-Lys(Dau=Aoa)-OH can release easily from this position by the dipeptidyl-peptidase activity of cathepsin B.
Click to Show/Hide
|
||||
| Description |
In vitro studies of the antiproliferative effect showed that melanoma-targeting Dau-conjugates have significantly higher efficacy on B16 murine melanoma cell lines in comparison to human melanoma cell lines (A2058, M24, and WM983B). The IC50 values were detected between 2 and 2.9 μM on B16 cells, where the highest activity was detected in the case of conjugate Conj3, followed by Conj2 and Conj1. Moreover, the relative potencies of conjugates to free Dau were also calculated as independent values from cell lines. A higher value of relative potency indicated the elevated targeting capacity of the conjugate on particular cell lines. Considering relative potencies, the best targeting capacity was shown to be Conj3 on all four cell lines, followed by Conj2 and Conj1. The highest antitumor activity of Conj3 on cells can be explained by the presence of two Dau molecules in this conjugate in comparison with Conj2 and Conj1, which contain only one drug molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | B16 cell | CVCL_F936 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
9.8 ± 5.4 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, the native sequence of -MSH was used, in which Met was replaced by Nle (SYSNleEHFRWGKPV). Daunomycin was attached to the amino functional groups via non-cleavable oxime linkage. These types of conjugates have many excellent properties to study and compare the bioactivity of the compounds. One of the developed conjugates contained drug molecules on both conjugation site (N-terminal and the side chain of Lys). The two additional conjugates were modified by the drug, either on theN-terminus or on the Lys side chain. The in vitro cytostatic effect on mouse melanoma cells did not show any significant differences among them. However, results indicated that the conjugates with Dau on the side chain of Lys could enter the cells more rapidly and efficiently. In contrast, the in vivo tumor growth inhibition was the most pronounced in the case of Ac-SYSNleEHFRWGK(Dau=Aoa)PV-NH2(Conj2). It is worth mentioning that the dose was normalized to Dau content, thus the injected dose ofConj3was half that ofConj 2. Nevertheless,Conj2, with one molecule of the drug, might be superior to the conjugate containing two molecules of daunomycin. The higher tumor growth inhibition effect ofConj2overConj1where the Dau is connected to theN-terminus, confirms our previous results with Dau-GnRH-III (Glp-His-Trp-Lys(Bu)-His-Asp-Trp-Lys(Dau=Aoa)-Pro-Gly-NH2), suggesting that H-Lys(Dau=Aoa)-OH can release easily from this position by the dipeptidyl-peptidase activity of cathepsin B.
Click to Show/Hide
|
||||
| Description |
In vitro studies of the antiproliferative effect showed that melanoma-targeting Dau-conjugates have significantly higher efficacy on B16 murine melanoma cell lines in comparison to human melanoma cell lines (A2058, M24, and WM983B). The IC50 values were detected between 2 and 2.9 μM on B16 cells, where the highest activity was detected in the case of conjugate Conj3, followed by Conj2 and Conj1. Moreover, the relative potencies of conjugates to free Dau were also calculated as independent values from cell lines. A higher value of relative potency indicated the elevated targeting capacity of the conjugate on particular cell lines. Considering relative potencies, the best targeting capacity was shown to be Conj3 on all four cell lines, followed by Conj2 and Conj1. The highest antitumor activity of Conj3 on cells can be explained by the presence of two Dau molecules in this conjugate in comparison with Conj2 and Conj1, which contain only one drug molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | A2058 cell | CVCL_1059 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
9.9 ± 1.5 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, the native sequence of -MSH was used, in which Met was replaced by Nle (SYSNleEHFRWGKPV). Daunomycin was attached to the amino functional groups via non-cleavable oxime linkage. These types of conjugates have many excellent properties to study and compare the bioactivity of the compounds. One of the developed conjugates contained drug molecules on both conjugation site (N-terminal and the side chain of Lys). The two additional conjugates were modified by the drug, either on theN-terminus or on the Lys side chain. The in vitro cytostatic effect on mouse melanoma cells did not show any significant differences among them. However, results indicated that the conjugates with Dau on the side chain of Lys could enter the cells more rapidly and efficiently. In contrast, the in vivo tumor growth inhibition was the most pronounced in the case of Ac-SYSNleEHFRWGK(Dau=Aoa)PV-NH2(Conj2). It is worth mentioning that the dose was normalized to Dau content, thus the injected dose ofConj3was half that ofConj 2. Nevertheless,Conj2, with one molecule of the drug, might be superior to the conjugate containing two molecules of daunomycin. The higher tumor growth inhibition effect ofConj2overConj1where the Dau is connected to theN-terminus, confirms our previous results with Dau-GnRH-III (Glp-His-Trp-Lys(Bu)-His-Asp-Trp-Lys(Dau=Aoa)-Pro-Gly-NH2), suggesting that H-Lys(Dau=Aoa)-OH can release easily from this position by the dipeptidyl-peptidase activity of cathepsin B.
Click to Show/Hide
|
||||
| Description |
In vitro studies of the antiproliferative effect showed that melanoma-targeting Dau-conjugates have significantly higher efficacy on B16 murine melanoma cell lines in comparison to human melanoma cell lines (A2058, M24, and WM983B). The IC50 values were detected between 2 and 2.9 μM on B16 cells, where the highest activity was detected in the case of conjugate Conj3, followed by Conj2 and Conj1. Moreover, the relative potencies of conjugates to free Dau were also calculated as independent values from cell lines. A higher value of relative potency indicated the elevated targeting capacity of the conjugate on particular cell lines. Considering relative potencies, the best targeting capacity was shown to be Conj3 on all four cell lines, followed by Conj2 and Conj1. The highest antitumor activity of Conj3 on cells can be explained by the presence of two Dau molecules in this conjugate in comparison with Conj2 and Conj1, which contain only one drug molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | WM983B cell | CVCL_6809 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
12.8 ± 1.6 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In this study, the native sequence of -MSH was used, in which Met was replaced by Nle (SYSNleEHFRWGKPV). Daunomycin was attached to the amino functional groups via non-cleavable oxime linkage. These types of conjugates have many excellent properties to study and compare the bioactivity of the compounds. One of the developed conjugates contained drug molecules on both conjugation site (N-terminal and the side chain of Lys). The two additional conjugates were modified by the drug, either on theN-terminus or on the Lys side chain. The in vitro cytostatic effect on mouse melanoma cells did not show any significant differences among them. However, results indicated that the conjugates with Dau on the side chain of Lys could enter the cells more rapidly and efficiently. In contrast, the in vivo tumor growth inhibition was the most pronounced in the case of Ac-SYSNleEHFRWGK(Dau=Aoa)PV-NH2(Conj2). It is worth mentioning that the dose was normalized to Dau content, thus the injected dose ofConj3was half that ofConj 2. Nevertheless,Conj2, with one molecule of the drug, might be superior to the conjugate containing two molecules of daunomycin. The higher tumor growth inhibition effect ofConj2overConj1where the Dau is connected to theN-terminus, confirms our previous results with Dau-GnRH-III (Glp-His-Trp-Lys(Bu)-His-Asp-Trp-Lys(Dau=Aoa)-Pro-Gly-NH2), suggesting that H-Lys(Dau=Aoa)-OH can release easily from this position by the dipeptidyl-peptidase activity of cathepsin B.
Click to Show/Hide
|
||||
| Description |
In vitro studies of the antiproliferative effect showed that melanoma-targeting Dau-conjugates have significantly higher efficacy on B16 murine melanoma cell lines in comparison to human melanoma cell lines (A2058, M24, and WM983B). The IC50 values were detected between 2 and 2.9 μM on B16 cells, where the highest activity was detected in the case of conjugate Conj3, followed by Conj2 and Conj1. Moreover, the relative potencies of conjugates to free Dau were also calculated as independent values from cell lines. A higher value of relative potency indicated the elevated targeting capacity of the conjugate on particular cell lines. Considering relative potencies, the best targeting capacity was shown to be Conj3 on all four cell lines, followed by Conj2 and Conj1. The highest antitumor activity of Conj3 on cells can be explained by the presence of two Dau molecules in this conjugate in comparison with Conj2 and Conj1, which contain only one drug molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | M24 cell | CVCL_D032 | ||
[8Lys(Dau=Aoa-K(Dau=Aoa))]GnRH-III [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.0 ± 0.4 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
The cancer phenotype is commonly associated with aberrant glycosylation patterns. One glycan that is directly linked to cancer is the Thomsen-Friedenreich antigen (TF or CD176). It is a disaccharide composed of a galactose β1-3 N-acetylgalactosamine, O-linked to a glycoprotein through serine or threonine residues and commonly written as Galβ1-3GalNAc--O-Ser/Thr. The TF is therapeutically attractive due to its cryptic nature in normal cells and exposure in embryonic and cancer cells. The expression of the TF has been demonstrated in 90% of primary human carcinomas, including in the lung, the breast, and the pancreas. Additionally, cancer initiating cells or cancer stem cells in the lung, liver, and colon express the TF. The peptide sequence HGRFILPWWYAFSPS (TF-peptide) is known to bind tightly to the TF (Kd = 1.2 M) and has been demonstrated to inhibit processes directly involved in TF accessibility.
Click to Show/Hide
|
||||
| Description |
The main drug-related toxic side effect of anthracyclines is cardiotoxicity, leading to cardiomyopathy and heart failure. Comparative experiments to determine the cardiotoxicity of peptide-anthracycline conjugates with different linkages might be informative for the other conjugates with different types of drugs as well. For this purpose, human cardiomyocytes (HCM) and human umbilical vein endothelial cells (HUVEC) were used as models. The long-term (0-72 h) cytotoxic effect of sixteen GnRH-based conjugates containing Dox and Dau was determined by real-time impedimetric sensing using the xCELLigence SP system (ACEA Biosciences, San Diego, CA, USA) [75]. The results indicated that the ester-linked GnRH-Dox conjugates, including Zoptarelin Doxorubicin, showed significant toxicity at 100 nM and 1 uM, which was remarkably pronounced on the HCM cells. The cytotoxic effect was comparable to that of the free drug, especially at the highest concentration. In contrast, the conjugates with oxime-linked Dau showed no or only a minor toxicity on both the cell lines (Table 2). These data confirm that the linkage between the payload and homing peptide has a significant influence on early drug release and, consequently, an undesired toxic side effect. We may also conclude that the search for more suitable homing peptides might be more important than the application of cleavable bonds between the drug and the peptide to develop efficient DDSs.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
5.6 ± 2.0 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
The main drug-related toxic side effect of anthracyclines is cardiotoxicity, leading to cardiomyopathy and heart failure. Comparative experiments to determine the cardiotoxicity of peptide-anthracycline conjugates with different linkages might be informative for the other conjugates with different types of drugs as well. For this purpose, human cardiomyocytes (HCM) and human umbilical vein endothelial cells (HUVEC) were used as models. The long-term (0-72 h) cytotoxic effect of sixteen GnRH-based conjugates containing Dox and Dau was determined by real-time impedimetric sensing using the xCELLigence SP system (ACEA Biosciences, San Diego, CA, USA) [75]. The results indicated that the ester-linked GnRH-Dox conjugates, including Zoptarelin Doxorubicin, showed significant toxicity at 100 nM and 1 uM, which was remarkably pronounced on the HCM cells. The cytotoxic effect was comparable to that of the free drug, especially at the highest concentration. In contrast, the conjugates with oxime-linked Dau showed no or only a minor toxicity on both the cell lines (Table 2). These data confirm that the linkage between the payload and homing peptide has a significant influence on early drug release and, consequently, an undesired toxic side effect. We may also conclude that the search for more suitable homing peptides might be more important than the application of cleavable bonds between the drug and the peptide to develop efficient DDSs.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
[4Lys(Ac),8Lys(Dau=Aoa)]-GnRH-III [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.1 ± 1.7 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
The cancer phenotype is commonly associated with aberrant glycosylation patterns. One glycan that is directly linked to cancer is the Thomsen-Friedenreich antigen (TF or CD176). It is a disaccharide composed of a galactose β1-3 N-acetylgalactosamine, O-linked to a glycoprotein through serine or threonine residues and commonly written as Galβ1-3GalNAc--O-Ser/Thr. The TF is therapeutically attractive due to its cryptic nature in normal cells and exposure in embryonic and cancer cells. The expression of the TF has been demonstrated in 90% of primary human carcinomas, including in the lung, the breast, and the pancreas. Additionally, cancer initiating cells or cancer stem cells in the lung, liver, and colon express the TF. The peptide sequence HGRFILPWWYAFSPS (TF-peptide) is known to bind tightly to the TF (Kd = 1.2 M) and has been demonstrated to inhibit processes directly involved in TF accessibility.
Click to Show/Hide
|
||||
| Description |
The main drug-related toxic side effect of anthracyclines is cardiotoxicity, leading to cardiomyopathy and heart failure. Comparative experiments to determine the cardiotoxicity of peptide-anthracycline conjugates with different linkages might be informative for the other conjugates with different types of drugs as well. For this purpose, human cardiomyocytes (HCM) and human umbilical vein endothelial cells (HUVEC) were used as models. The long-term (0-72 h) cytotoxic effect of sixteen GnRH-based conjugates containing Dox and Dau was determined by real-time impedimetric sensing using the xCELLigence SP system (ACEA Biosciences, San Diego, CA, USA) [75]. The results indicated that the ester-linked GnRH-Dox conjugates, including Zoptarelin Doxorubicin, showed significant toxicity at 100 nM and 1 uM, which was remarkably pronounced on the HCM cells. The cytotoxic effect was comparable to that of the free drug, especially at the highest concentration. In contrast, the conjugates with oxime-linked Dau showed no or only a minor toxicity on both the cell lines (Table 2). These data confirm that the linkage between the payload and homing peptide has a significant influence on early drug release and, consequently, an undesired toxic side effect. We may also conclude that the search for more suitable homing peptides might be more important than the application of cleavable bonds between the drug and the peptide to develop efficient DDSs.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
7.4 ± 2.6 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
In our experiments, GnRH-III was applied as a targeting peptide. The conjugate [8Lys(Dau=Aoa-GFLG)]-GnRH-III, in which Dau was connected to the side chain of Lys in position 8 through an aminooxyacetylated cathepsin B-labile GFLG spacer, showed significant tumor growth inhibition in s.c.-developed fast-growing C26 murine colon cancer-bearing Balb/c mice. The effect highly depended on the treatment schedule. In the first attempt, the conjugate was administered i.p. at a dose of 8.86 umol (5 mg Dau-content/kg body weight) five times on days 7, 9, 11, 14 and 16 after tumor transplantation (Treatment schedule A). The tumor volume inhibition was only 15.7% on day 26 when the experiment finished. A slight enhancement in inhibition (22.3%) was detected when a dose of conjugate corresponding to 15 mg Dau content/kg body weight (26.6 μmol) was injected only once on day 7 (Treatment schedule B). An additional treatment on day 10 (Treatment schedule C) did not result in any further improvements (21.9% on day 29). However, when the treatment schedule was changed to two treatments with the same dose on days 4 and 7, 46.3% inhibition was observed on day 29 (Treatment schedule D). In contrast, the treatments with free Dau at a dose of 2 mg/kg body weight (3.55 umol) on days 7, 9, 11, 14 and 16 showed only 22.6% inhibition on day 26. The median survival rates of the treated animals in comparison to the control group were 1 (A), 1.23 (B), 1.21 (C) and 1.38 (D), respectively, and 0.81 for free Dau. These results indicate the lower toxicity and the higher tumor volume inhibition effect of the conjugates in comparison with those of free Dau, as well as the importance of the treatment schedule. In another experiment, HT-29 human colon cancer was developed s.c. in immunodeficient SCID mice. Dau and two conjugates (with or without a GFLG spacer between the GnRH-III homing peptide and the payload) were used for the treatment. The first i.p. administrations were performed on day 13 after tumor inoculation. All the mice treated once with 2.5 mg/kg body weight (4.43 umol) free Dau died within 10 days. In contrast, the conjugates at a dose of 15 mg Dau content/kg body weight (26.6 umol) that refers to 52 mg/kg [8Lys(Dau=Aoa)]-GnRH-III and 62.5 mg/kg [8Lys(Dau=Aoa-GFLG)]-GnRH-III conjugates, respectively, did not show significant toxicity. The treatment was repeated on days 23 and 30. Because of the significant weight loss in several mice in the control group, the experiment was terminated on day 35. The tumor growth inhibition could be calculated as reductions in the tumor volume by 44.3% and 57.6% and the tumor weight by 41% and 50%, respectively. Interestingly, the conjugates were poorly effective on orthotopically developed tumors. In the case of the C26 colon tumor-bearing female Balb/c mice, only a 7% reduction in tumor weight was detected on day 13 after the two treatments on days 4 and 7 with [8Lys(Dau=Aoa)]-GnRH-III, at a 26.6 umol/kg (15 mg Dau content) dose. The effect of free Dau (2 mg/kg on days 4 and 7) showed a better inhibitory effect (24.4%). Interestingly, our novel developed GnRH-III derivative, in which Ser in position 4 was replaced by Lys(Ac), was much more potent, with 49.3% inhibition. It is worth mentioning that the rate of cellular uptake of the [4Lys(Ac), 8Lys(Dau=Aoa)]-GnRH-III conjugate by the tumor cells was significantly higher than that of the conjugate with the native GnRH-III sequence.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
GnRH-III-[3D-Tic, 7D-Trp 8Lys(Dau=Aoa)] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.42 ± 0.39 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
15.34 ± 2.63 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
GnRH-III-[4Lys(Bu), 6Asp(OMe), 8Lys(Dau=Aoa)] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.44 ± 0.51 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
18.00 ± 0.13 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
GnRH-III-[3D-Trp, 8Lys(Dau=Aoa)] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.60 ± 0.28 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
15.25 ± 2.51 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
[8Lys(Dau=Aoa-GFLG)]-GnRH-III [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.9 ± 1.2 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
As a payload for PDCs (and ADCs), analogues of the duocarmycins are attractive. Duocarmycin SA and yatakemycin rank among the most potent natural cytotoxins discovered. The cyclopropyl and prodrug seco forms are both naturally occurring and equipotent in most circumstances. Studies of the binding-driven bonding model of their interaction with DNA suggest that their utility will be enhanced when targeted to tumor cells. In fact, SYD985, an ADC that utilizes a peptide linker for a duocarmycin analogue to trastuzumab, has recently been progressed to phase III clinical trial.
Click to Show/Hide
|
||||
| Description |
The main drug-related toxic side effect of anthracyclines is cardiotoxicity, leading to cardiomyopathy and heart failure. Comparative experiments to determine the cardiotoxicity of peptide-anthracycline conjugates with different linkages might be informative for the other conjugates with different types of drugs as well. For this purpose, human cardiomyocytes (HCM) and human umbilical vein endothelial cells (HUVEC) were used as models. The long-term (0-72 h) cytotoxic effect of sixteen GnRH-based conjugates containing Dox and Dau was determined by real-time impedimetric sensing using the xCELLigence SP system (ACEA Biosciences, San Diego, CA, USA) [75]. The results indicated that the ester-linked GnRH-Dox conjugates, including Zoptarelin Doxorubicin, showed significant toxicity at 100 nM and 1 uM, which was remarkably pronounced on the HCM cells. The cytotoxic effect was comparable to that of the free drug, especially at the highest concentration. In contrast, the conjugates with oxime-linked Dau showed no or only a minor toxicity on both the cell lines (Table 2). These data confirm that the linkage between the payload and homing peptide has a significant influence on early drug release and, consequently, an undesired toxic side effect. We may also conclude that the search for more suitable homing peptides might be more important than the application of cleavable bonds between the drug and the peptide to develop efficient DDSs.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
19.4 ± 3.1 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
In our experiments, GnRH-III was applied as a targeting peptide. The conjugate [8Lys(Dau=Aoa-GFLG)]-GnRH-III, in which Dau was connected to the side chain of Lys in position 8 through an aminooxyacetylated cathepsin B-labile GFLG spacer, showed significant tumor growth inhibition in s.c.-developed fast-growing C26 murine colon cancer-bearing Balb/c mice. The effect highly depended on the treatment schedule. In the first attempt, the conjugate was administered i.p. at a dose of 8.86 umol (5 mg Dau-content/kg body weight) five times on days 7, 9, 11, 14 and 16 after tumor transplantation (Treatment schedule A). The tumor volume inhibition was only 15.7% on day 26 when the experiment finished. A slight enhancement in inhibition (22.3%) was detected when a dose of conjugate corresponding to 15 mg Dau content/kg body weight (26.6 μmol) was injected only once on day 7 (Treatment schedule B). An additional treatment on day 10 (Treatment schedule C) did not result in any further improvements (21.9% on day 29). However, when the treatment schedule was changed to two treatments with the same dose on days 4 and 7, 46.3% inhibition was observed on day 29 (Treatment schedule D). In contrast, the treatments with free Dau at a dose of 2 mg/kg body weight (3.55 umol) on days 7, 9, 11, 14 and 16 showed only 22.6% inhibition on day 26. The median survival rates of the treated animals in comparison to the control group were 1 (A), 1.23 (B), 1.21 (C) and 1.38 (D), respectively, and 0.81 for free Dau. These results indicate the lower toxicity and the higher tumor volume inhibition effect of the conjugates in comparison with those of free Dau, as well as the importance of the treatment schedule. In another experiment, HT-29 human colon cancer was developed s.c. in immunodeficient SCID mice. Dau and two conjugates (with or without a GFLG spacer between the GnRH-III homing peptide and the payload) were used for the treatment. The first i.p. administrations were performed on day 13 after tumor inoculation. All the mice treated once with 2.5 mg/kg body weight (4.43 umol) free Dau died within 10 days. In contrast, the conjugates at a dose of 15 mg Dau content/kg body weight (26.6 umol) that refers to 52 mg/kg [8Lys(Dau=Aoa)]-GnRH-III and 62.5 mg/kg [8Lys(Dau=Aoa-GFLG)]-GnRH-III conjugates, respectively, did not show significant toxicity. The treatment was repeated on days 23 and 30. Because of the significant weight loss in several mice in the control group, the experiment was terminated on day 35. The tumor growth inhibition could be calculated as reductions in the tumor volume by 44.3% and 57.6% and the tumor weight by 41% and 50%, respectively. Interestingly, the conjugates were poorly effective on orthotopically developed tumors. In the case of the C26 colon tumor-bearing female Balb/c mice, only a 7% reduction in tumor weight was detected on day 13 after the two treatments on days 4 and 7 with [8Lys(Dau=Aoa)]-GnRH-III, at a 26.6 umol/kg (15 mg Dau content) dose. The effect of free Dau (2 mg/kg on days 4 and 7) showed a better inhibitory effect (24.4%). Interestingly, our novel developed GnRH-III derivative, in which Ser in position 4 was replaced by Lys(Ac), was much more potent, with 49.3% inhibition. It is worth mentioning that the rate of cellular uptake of the [4Lys(Ac), 8Lys(Dau=Aoa)]-GnRH-III conjugate by the tumor cells was significantly higher than that of the conjugate with the native GnRH-III sequence.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
GnRH-III-[6Asp(OMe), 8Lys(Dau=Aoa)] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4.81 ± 0.72 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
10.66 ± 1.76 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
GnRH-III-[8Lys(Dau=Aoa), 10ΔGly-NH-Et] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4.88 ± 0.01 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
14.18 ± 3.59 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
14.33 ± 1.18 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
GnRH-III-[3D-Trp, 4Lys(Bu), 8Lys(Dau=Aoa)] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
6.64 ± 1.58 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
15.03 ± 2.51 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
Dau=Aoa-TFFYGGSRGK(Dau=Aoa)RNNFKTEEY-OH [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
7.8 ± 6.3 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The blood-brain barrier (BBB) is a semipermeable system, and, therefore, most of the active substances are poorly transported through this barrier, resulting in decreased therapeutic effects. Angiopep-2 (TFFYGGSRGKRNNFKTEEY) is a peptide ligand of low-density lipoprotein receptor-related protein-1 (LRP1), which can cross the BBB via receptor-mediated transcytosis and simultaneously target glioblastomas. Angiopep-2 contains three amino groups that have previously been used to produce drug-peptide conjugates, although the role and importance of each position have not yet been investigated. Thus, we studied the number and position of drug molecules in Angiopep-2 based conjugates. Conjugates containing one, two, and three daunomycin molecules conjugated via oxime linkage in all possible variations were prepared. The in vitro cytostatic effect and cellular uptake of the conjugates were investigated on U87 human glioblastoma cells. Degradation studies in the presence of rat liver lysosomal homogenates were also performed in order for us to better understand the structure-activity relationship and to determine the smallest metabolites. Conjugates with the best cytostatic effects had a drug molecule at the N-terminus. We demonstrated that the increasing number of drug molecules does not necessarily increase the efficacy of the conjugates, and proved that modification of the different conjugation sites results in differing biological effectiveness.
Click to Show/Hide
|
||||
| Description |
The in vitro cytostatic effect of the synthesized peptide (1) and daunomycin-peptide conjugates (2-8) was investigated on U87 human glioblastoma cells. The cells were treated with the peptide and conjugates at different concentrations (0.05-50 uM) for 24 h, and after a washing step, the cells were incubated for another 48 h at 37 C. The cytostatic effect of the compounds was determined using the MTT test. The measured IC50values are shown in. The free Angiopep-2 did not show any effect on tumor cells up to a 50 μM concentration. However, no clear correlation could be obtained between the number of the daunomycin and the cytostatic efficacy. Among the conjugates with only one Dau, compound3,in which Dau was attached to theN-terminus, showed the highest in vitro cytostatic effect on glioblastoma cells. When the Dau was conjugated to the Lys side chain in position 15 (compound2), the effect decreased significantly, and conjugate4was also not significantly better. In case of conjugates with two drug molecules, a similar tendency was observed. Compound7,with Dau at theN-terminus and on the Lys side chain in position 10, showed the highest activity. When the Lys side chains were used as conjugation sites, the formed compound (6) had moderate activity on U87 cells. Surprisingly, conjugate5,with Dau at theN-terminus and Lys side chain in position 15, showed the lowest efficacy on the U87 cells. Compound8,with three Dau, showed moderate activity. Thus, the effectiveness of Angiopep-2-daunomycin conjugates does not primarily depend on the number of drug molecules, but rather on their position within the molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
Dau=Aoa-TFFYGGSRGKRNNFKTEEY-OH [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
10.9 ± 2.8 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The blood-brain barrier (BBB) is a semipermeable system, and, therefore, most of the active substances are poorly transported through this barrier, resulting in decreased therapeutic effects. Angiopep-2 (TFFYGGSRGKRNNFKTEEY) is a peptide ligand of low-density lipoprotein receptor-related protein-1 (LRP1), which can cross the BBB via receptor-mediated transcytosis and simultaneously target glioblastomas. Angiopep-2 contains three amino groups that have previously been used to produce drug-peptide conjugates, although the role and importance of each position have not yet been investigated. Thus, we studied the number and position of drug molecules in Angiopep-2 based conjugates. Conjugates containing one, two, and three daunomycin molecules conjugated via oxime linkage in all possible variations were prepared. The in vitro cytostatic effect and cellular uptake of the conjugates were investigated on U87 human glioblastoma cells. Degradation studies in the presence of rat liver lysosomal homogenates were also performed in order for us to better understand the structure-activity relationship and to determine the smallest metabolites. Conjugates with the best cytostatic effects had a drug molecule at the N-terminus. We demonstrated that the increasing number of drug molecules does not necessarily increase the efficacy of the conjugates, and proved that modification of the different conjugation sites results in differing biological effectiveness.
Click to Show/Hide
|
||||
| Description |
The in vitro cytostatic effect of the synthesized peptide (1) and daunomycin-peptide conjugates (2-8) was investigated on U87 human glioblastoma cells. The cells were treated with the peptide and conjugates at different concentrations (0.05-50 uM) for 24 h, and after a washing step, the cells were incubated for another 48 h at 37 C. The cytostatic effect of the compounds was determined using the MTT test. The measured IC50values are shown in. The free Angiopep-2 did not show any effect on tumor cells up to a 50 μM concentration. However, no clear correlation could be obtained between the number of the daunomycin and the cytostatic efficacy. Among the conjugates with only one Dau, compound3,in which Dau was attached to theN-terminus, showed the highest in vitro cytostatic effect on glioblastoma cells. When the Dau was conjugated to the Lys side chain in position 15 (compound2), the effect decreased significantly, and conjugate4was also not significantly better. In case of conjugates with two drug molecules, a similar tendency was observed. Compound7,with Dau at theN-terminus and on the Lys side chain in position 10, showed the highest activity. When the Lys side chains were used as conjugation sites, the formed compound (6) had moderate activity on U87 cells. Surprisingly, conjugate5,with Dau at theN-terminus and Lys side chain in position 15, showed the lowest efficacy on the U87 cells. Compound8,with three Dau, showed moderate activity. Thus, the effectiveness of Angiopep-2-daunomycin conjugates does not primarily depend on the number of drug molecules, but rather on their position within the molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
H-TFFYGGSRGK(Dau=Aoa)RNNFK(Dau=Aoa)TEEY-OH [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
21.6 ± 5.4 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The blood-brain barrier (BBB) is a semipermeable system, and, therefore, most of the active substances are poorly transported through this barrier, resulting in decreased therapeutic effects. Angiopep-2 (TFFYGGSRGKRNNFKTEEY) is a peptide ligand of low-density lipoprotein receptor-related protein-1 (LRP1), which can cross the BBB via receptor-mediated transcytosis and simultaneously target glioblastomas. Angiopep-2 contains three amino groups that have previously been used to produce drug-peptide conjugates, although the role and importance of each position have not yet been investigated. Thus, we studied the number and position of drug molecules in Angiopep-2 based conjugates. Conjugates containing one, two, and three daunomycin molecules conjugated via oxime linkage in all possible variations were prepared. The in vitro cytostatic effect and cellular uptake of the conjugates were investigated on U87 human glioblastoma cells. Degradation studies in the presence of rat liver lysosomal homogenates were also performed in order for us to better understand the structure-activity relationship and to determine the smallest metabolites. Conjugates with the best cytostatic effects had a drug molecule at the N-terminus. We demonstrated that the increasing number of drug molecules does not necessarily increase the efficacy of the conjugates, and proved that modification of the different conjugation sites results in differing biological effectiveness.
Click to Show/Hide
|
||||
| Description |
The in vitro cytostatic effect of the synthesized peptide (1) and daunomycin-peptide conjugates (2-8) was investigated on U87 human glioblastoma cells. The cells were treated with the peptide and conjugates at different concentrations (0.05-50 uM) for 24 h, and after a washing step, the cells were incubated for another 48 h at 37 C. The cytostatic effect of the compounds was determined using the MTT test. The measured IC50values are shown in. The free Angiopep-2 did not show any effect on tumor cells up to a 50 μM concentration. However, no clear correlation could be obtained between the number of the daunomycin and the cytostatic efficacy. Among the conjugates with only one Dau, compound3,in which Dau was attached to theN-terminus, showed the highest in vitro cytostatic effect on glioblastoma cells. When the Dau was conjugated to the Lys side chain in position 15 (compound2), the effect decreased significantly, and conjugate4was also not significantly better. In case of conjugates with two drug molecules, a similar tendency was observed. Compound7,with Dau at theN-terminus and on the Lys side chain in position 10, showed the highest activity. When the Lys side chains were used as conjugation sites, the formed compound (6) had moderate activity on U87 cells. Surprisingly, conjugate5,with Dau at theN-terminus and Lys side chain in position 15, showed the lowest efficacy on the U87 cells. Compound8,with three Dau, showed moderate activity. Thus, the effectiveness of Angiopep-2-daunomycin conjugates does not primarily depend on the number of drug molecules, but rather on their position within the molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
Dau=Aoa-TFFYGGSRGK(Dau=Aoa)RNNFK(Dau=Aoa)TEEY-OH [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
23.6 ± 6.3 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The blood-brain barrier (BBB) is a semipermeable system, and, therefore, most of the active substances are poorly transported through this barrier, resulting in decreased therapeutic effects. Angiopep-2 (TFFYGGSRGKRNNFKTEEY) is a peptide ligand of low-density lipoprotein receptor-related protein-1 (LRP1), which can cross the BBB via receptor-mediated transcytosis and simultaneously target glioblastomas. Angiopep-2 contains three amino groups that have previously been used to produce drug-peptide conjugates, although the role and importance of each position have not yet been investigated. Thus, we studied the number and position of drug molecules in Angiopep-2 based conjugates. Conjugates containing one, two, and three daunomycin molecules conjugated via oxime linkage in all possible variations were prepared. The in vitro cytostatic effect and cellular uptake of the conjugates were investigated on U87 human glioblastoma cells. Degradation studies in the presence of rat liver lysosomal homogenates were also performed in order for us to better understand the structure-activity relationship and to determine the smallest metabolites. Conjugates with the best cytostatic effects had a drug molecule at the N-terminus. We demonstrated that the increasing number of drug molecules does not necessarily increase the efficacy of the conjugates, and proved that modification of the different conjugation sites results in differing biological effectiveness.
Click to Show/Hide
|
||||
| Description |
The in vitro cytostatic effect of the synthesized peptide (1) and daunomycin-peptide conjugates (2-8) was investigated on U87 human glioblastoma cells. The cells were treated with the peptide and conjugates at different concentrations (0.05-50 uM) for 24 h, and after a washing step, the cells were incubated for another 48 h at 37 C. The cytostatic effect of the compounds was determined using the MTT test. The measured IC50values are shown in. The free Angiopep-2 did not show any effect on tumor cells up to a 50 μM concentration. However, no clear correlation could be obtained between the number of the daunomycin and the cytostatic efficacy. Among the conjugates with only one Dau, compound3,in which Dau was attached to theN-terminus, showed the highest in vitro cytostatic effect on glioblastoma cells. When the Dau was conjugated to the Lys side chain in position 15 (compound2), the effect decreased significantly, and conjugate4was also not significantly better. In case of conjugates with two drug molecules, a similar tendency was observed. Compound7,with Dau at theN-terminus and on the Lys side chain in position 10, showed the highest activity. When the Lys side chains were used as conjugation sites, the formed compound (6) had moderate activity on U87 cells. Surprisingly, conjugate5,with Dau at theN-terminus and Lys side chain in position 15, showed the lowest efficacy on the U87 cells. Compound8,with three Dau, showed moderate activity. Thus, the effectiveness of Angiopep-2-daunomycin conjugates does not primarily depend on the number of drug molecules, but rather on their position within the molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
H-TFFYGGSRGK(Dau=Aoa)RNNFKTEEY-OH [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
24.0 ± 5.9 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The blood-brain barrier (BBB) is a semipermeable system, and, therefore, most of the active substances are poorly transported through this barrier, resulting in decreased therapeutic effects. Angiopep-2 (TFFYGGSRGKRNNFKTEEY) is a peptide ligand of low-density lipoprotein receptor-related protein-1 (LRP1), which can cross the BBB via receptor-mediated transcytosis and simultaneously target glioblastomas. Angiopep-2 contains three amino groups that have previously been used to produce drug-peptide conjugates, although the role and importance of each position have not yet been investigated. Thus, we studied the number and position of drug molecules in Angiopep-2 based conjugates. Conjugates containing one, two, and three daunomycin molecules conjugated via oxime linkage in all possible variations were prepared. The in vitro cytostatic effect and cellular uptake of the conjugates were investigated on U87 human glioblastoma cells. Degradation studies in the presence of rat liver lysosomal homogenates were also performed in order for us to better understand the structure-activity relationship and to determine the smallest metabolites. Conjugates with the best cytostatic effects had a drug molecule at the N-terminus. We demonstrated that the increasing number of drug molecules does not necessarily increase the efficacy of the conjugates, and proved that modification of the different conjugation sites results in differing biological effectiveness.
Click to Show/Hide
|
||||
| Description |
The in vitro cytostatic effect of the synthesized peptide (1) and daunomycin-peptide conjugates (2-8) was investigated on U87 human glioblastoma cells. The cells were treated with the peptide and conjugates at different concentrations (0.05-50 uM) for 24 h, and after a washing step, the cells were incubated for another 48 h at 37 C. The cytostatic effect of the compounds was determined using the MTT test. The measured IC50values are shown in. The free Angiopep-2 did not show any effect on tumor cells up to a 50 μM concentration. However, no clear correlation could be obtained between the number of the daunomycin and the cytostatic efficacy. Among the conjugates with only one Dau, compound3,in which Dau was attached to theN-terminus, showed the highest in vitro cytostatic effect on glioblastoma cells. When the Dau was conjugated to the Lys side chain in position 15 (compound2), the effect decreased significantly, and conjugate4was also not significantly better. In case of conjugates with two drug molecules, a similar tendency was observed. Compound7,with Dau at theN-terminus and on the Lys side chain in position 10, showed the highest activity. When the Lys side chains were used as conjugation sites, the formed compound (6) had moderate activity on U87 cells. Surprisingly, conjugate5,with Dau at theN-terminus and Lys side chain in position 15, showed the lowest efficacy on the U87 cells. Compound8,with three Dau, showed moderate activity. Thus, the effectiveness of Angiopep-2-daunomycin conjugates does not primarily depend on the number of drug molecules, but rather on their position within the molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
Octreotide doxorubicin conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
27.14 ± 2.47 μM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | CellTiter-Glo cell proliferation assay | ||||
| MOA of PDC |
In the present work, we introduce a new approach to overcome all the aforementioned limitations. The cytotoxic drug doxorubicin is coupled to the tumor-targeting vector octreotide via a disulfide-intercalating cross-linking reagent. On the one hand, this reagent creates an oxime bond with the drug, and on the other hand, two disulfides with octreotide to keep the cyclic structure of the peptide. The combination of a hydrolytically stable oxime bond and disulfides leads to the formation of a novel bioconjugate superior to any previous anticancer drug-somatostatin analog hybrid as it allows the efficient release of the toxic cargo within the reducing environment of cancer cells. The versatility of the linker molecule described here will enable its future application not only in targeted drug delivery, but also in the chemical modification of therapeutic proteins.
Click to Show/Hide
|
||||
| Description |
We selected cells, where doxorubicin is typically applied and which overexpress somatostatin receptors, like the human pancreatic carcinoma cell line MIA PaCa-2 or MCF-7. Doxorubicins antiproliferative action on MIA PaCa-2 after 72 h incubation with the drug was as expected very strong (IC50 = 0.80 ± 0.13 μM), whereas octreotide was not able to inhibit cell growth by 50% up to a concentration of 150 μM. The cytotoxic effects of the conjugate expressed, as half maximal inhibitory concentration was much stronger compared to the precursor peptide, but, nevertheless, lower than that of doxorubicin (IC50 = 31.50 ± 1.74 μM). This characteristic feature of 12 is attributed to the different cellular uptake mechanisms of the substances. Doxorubicin is taken up quickly by passive diffusion, while octreotide enters cells by receptor-mediated endocytosis. Furthermore, it needs to be considered that the conjugate releases after cleavage by glutathione a doxorubicin derivative, which is still carrying a small cross-linker residue. Previous studies have shown that these types of molecules are, nevertheless, capable of successfully interacting with DNA, to mediate their cytotoxic properties, but to a lesser extent.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Pancreatic carcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
31.50 ± 1.74 μM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | CellTiter-Glo cell proliferation assay | ||||
| MOA of PDC |
In the present work, we introduce a new approach to overcome all the aforementioned limitations. The cytotoxic drug doxorubicin is coupled to the tumor-targeting vector octreotide via a disulfide-intercalating cross-linking reagent. On the one hand, this reagent creates an oxime bond with the drug, and on the other hand, two disulfides with octreotide to keep the cyclic structure of the peptide. The combination of a hydrolytically stable oxime bond and disulfides leads to the formation of a novel bioconjugate superior to any previous anticancer drug-somatostatin analog hybrid as it allows the efficient release of the toxic cargo within the reducing environment of cancer cells. The versatility of the linker molecule described here will enable its future application not only in targeted drug delivery, but also in the chemical modification of therapeutic proteins.
Click to Show/Hide
|
||||
| Description |
We selected cells, where doxorubicin is typically applied and which overexpress somatostatin receptors, like the human pancreatic carcinoma cell line MIA PaCa-2 or MCF-7. Doxorubicins antiproliferative action on MIA PaCa-2 after 72 h incubation with the drug was as expected very strong (IC50 = 0.80 ± 0.13 μM), whereas octreotide was not able to inhibit cell growth by 50% up to a concentration of 150 μM. The cytotoxic effects of the conjugate expressed, as half maximal inhibitory concentration was much stronger compared to the precursor peptide, but, nevertheless, lower than that of doxorubicin (IC50 = 31.50 ± 1.74 μM). This characteristic feature of 12 is attributed to the different cellular uptake mechanisms of the substances. Doxorubicin is taken up quickly by passive diffusion, while octreotide enters cells by receptor-mediated endocytosis. Furthermore, it needs to be considered that the conjugate releases after cleavage by glutathione a doxorubicin derivative, which is still carrying a small cross-linker residue. Previous studies have shown that these types of molecules are, nevertheless, capable of successfully interacting with DNA, to mediate their cytotoxic properties, but to a lesser extent.
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | MIA PaCa-2 cell | CVCL_0428 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Pancreatic adenocarcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
48.90 ± 5.40 μM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | CellTiter-Glo cell proliferation assay | ||||
| MOA of PDC |
In the present work, we introduce a new approach to overcome all the aforementioned limitations. The cytotoxic drug doxorubicin is coupled to the tumor-targeting vector octreotide via a disulfide-intercalating cross-linking reagent. On the one hand, this reagent creates an oxime bond with the drug, and on the other hand, two disulfides with octreotide to keep the cyclic structure of the peptide. The combination of a hydrolytically stable oxime bond and disulfides leads to the formation of a novel bioconjugate superior to any previous anticancer drug-somatostatin analog hybrid as it allows the efficient release of the toxic cargo within the reducing environment of cancer cells. The versatility of the linker molecule described here will enable its future application not only in targeted drug delivery, but also in the chemical modification of therapeutic proteins.
Click to Show/Hide
|
||||
| Description |
We selected cells, where doxorubicin is typically applied and which overexpress somatostatin receptors, like the human pancreatic carcinoma cell line MIA PaCa-2 or MCF-7. Doxorubicins antiproliferative action on MIA PaCa-2 after 72 h incubation with the drug was as expected very strong (IC50 = 0.80 ± 0.13 μM), whereas octreotide was not able to inhibit cell growth by 50% up to a concentration of 150 μM. The cytotoxic effects of the conjugate expressed, as half maximal inhibitory concentration was much stronger compared to the precursor peptide, but, nevertheless, lower than that of doxorubicin (IC50 = 31.50 ± 1.74 μM). This characteristic feature of 12 is attributed to the different cellular uptake mechanisms of the substances. Doxorubicin is taken up quickly by passive diffusion, while octreotide enters cells by receptor-mediated endocytosis. Furthermore, it needs to be considered that the conjugate releases after cleavage by glutathione a doxorubicin derivative, which is still carrying a small cross-linker residue. Previous studies have shown that these types of molecules are, nevertheless, capable of successfully interacting with DNA, to mediate their cytotoxic properties, but to a lesser extent.
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | Capan-1 cell | CVCL_0237 | ||
H-TFFYGGSRGKRNNFK(Dau=Aoa)TEEY-OH [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
30.2 ± 6.4 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The blood-brain barrier (BBB) is a semipermeable system, and, therefore, most of the active substances are poorly transported through this barrier, resulting in decreased therapeutic effects. Angiopep-2 (TFFYGGSRGKRNNFKTEEY) is a peptide ligand of low-density lipoprotein receptor-related protein-1 (LRP1), which can cross the BBB via receptor-mediated transcytosis and simultaneously target glioblastomas. Angiopep-2 contains three amino groups that have previously been used to produce drug-peptide conjugates, although the role and importance of each position have not yet been investigated. Thus, we studied the number and position of drug molecules in Angiopep-2 based conjugates. Conjugates containing one, two, and three daunomycin molecules conjugated via oxime linkage in all possible variations were prepared. The in vitro cytostatic effect and cellular uptake of the conjugates were investigated on U87 human glioblastoma cells. Degradation studies in the presence of rat liver lysosomal homogenates were also performed in order for us to better understand the structure-activity relationship and to determine the smallest metabolites. Conjugates with the best cytostatic effects had a drug molecule at the N-terminus. We demonstrated that the increasing number of drug molecules does not necessarily increase the efficacy of the conjugates, and proved that modification of the different conjugation sites results in differing biological effectiveness.
Click to Show/Hide
|
||||
| Description |
The in vitro cytostatic effect of the synthesized peptide (1) and daunomycin-peptide conjugates (2-8) was investigated on U87 human glioblastoma cells. The cells were treated with the peptide and conjugates at different concentrations (0.05-50 uM) for 24 h, and after a washing step, the cells were incubated for another 48 h at 37 C. The cytostatic effect of the compounds was determined using the MTT test. The measured IC50values are shown in. The free Angiopep-2 did not show any effect on tumor cells up to a 50 μM concentration. However, no clear correlation could be obtained between the number of the daunomycin and the cytostatic efficacy. Among the conjugates with only one Dau, compound3,in which Dau was attached to theN-terminus, showed the highest in vitro cytostatic effect on glioblastoma cells. When the Dau was conjugated to the Lys side chain in position 15 (compound2), the effect decreased significantly, and conjugate4was also not significantly better. In case of conjugates with two drug molecules, a similar tendency was observed. Compound7,with Dau at theN-terminus and on the Lys side chain in position 10, showed the highest activity. When the Lys side chains were used as conjugation sites, the formed compound (6) had moderate activity on U87 cells. Surprisingly, conjugate5,with Dau at theN-terminus and Lys side chain in position 15, showed the lowest efficacy on the U87 cells. Compound8,with three Dau, showed moderate activity. Thus, the effectiveness of Angiopep-2-daunomycin conjugates does not primarily depend on the number of drug molecules, but rather on their position within the molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
Dau=Aoa-TFFYGGSRGKRNNFK(Dau=Aoa)TEEY-OH [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
32.3 ± 8.1 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The blood-brain barrier (BBB) is a semipermeable system, and, therefore, most of the active substances are poorly transported through this barrier, resulting in decreased therapeutic effects. Angiopep-2 (TFFYGGSRGKRNNFKTEEY) is a peptide ligand of low-density lipoprotein receptor-related protein-1 (LRP1), which can cross the BBB via receptor-mediated transcytosis and simultaneously target glioblastomas. Angiopep-2 contains three amino groups that have previously been used to produce drug-peptide conjugates, although the role and importance of each position have not yet been investigated. Thus, we studied the number and position of drug molecules in Angiopep-2 based conjugates. Conjugates containing one, two, and three daunomycin molecules conjugated via oxime linkage in all possible variations were prepared. The in vitro cytostatic effect and cellular uptake of the conjugates were investigated on U87 human glioblastoma cells. Degradation studies in the presence of rat liver lysosomal homogenates were also performed in order for us to better understand the structure-activity relationship and to determine the smallest metabolites. Conjugates with the best cytostatic effects had a drug molecule at the N-terminus. We demonstrated that the increasing number of drug molecules does not necessarily increase the efficacy of the conjugates, and proved that modification of the different conjugation sites results in differing biological effectiveness.
Click to Show/Hide
|
||||
| Description |
The in vitro cytostatic effect of the synthesized peptide (1) and daunomycin-peptide conjugates (2-8) was investigated on U87 human glioblastoma cells. The cells were treated with the peptide and conjugates at different concentrations (0.05-50 uM) for 24 h, and after a washing step, the cells were incubated for another 48 h at 37 C. The cytostatic effect of the compounds was determined using the MTT test. The measured IC50values are shown in. The free Angiopep-2 did not show any effect on tumor cells up to a 50 μM concentration. However, no clear correlation could be obtained between the number of the daunomycin and the cytostatic efficacy. Among the conjugates with only one Dau, compound3,in which Dau was attached to theN-terminus, showed the highest in vitro cytostatic effect on glioblastoma cells. When the Dau was conjugated to the Lys side chain in position 15 (compound2), the effect decreased significantly, and conjugate4was also not significantly better. In case of conjugates with two drug molecules, a similar tendency was observed. Compound7,with Dau at theN-terminus and on the Lys side chain in position 10, showed the highest activity. When the Lys side chains were used as conjugation sites, the formed compound (6) had moderate activity on U87 cells. Surprisingly, conjugate5,with Dau at theN-terminus and Lys side chain in position 15, showed the lowest efficacy on the U87 cells. Compound8,with three Dau, showed moderate activity. Thus, the effectiveness of Angiopep-2-daunomycin conjugates does not primarily depend on the number of drug molecules, but rather on their position within the molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
Dau=Aoa-GFLG-c[DKP-RGD] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
2.5 μM
|
|||
| Administration Time | 72 h | ||||
| Description |
All dau-loaded conjugates showed high toxicity in all cell lines tested with EC50 values in the lower micromolar range.
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
5.4 μM
|
|||
| Administration Time | 72 h | ||||
| Description |
All dau-loaded conjugates showed high toxicity in all cell lines tested with EC50 values in the lower micromolar range.
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
| Experiment 3 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
14.7 μM
|
|||
| Administration Time | 72 h | ||||
| Description |
All dau-loaded conjugates showed high toxicity in all cell lines tested with EC50 values in the lower micromolar range.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
49.4 μM
|
|||
| Administration Time | 15 min | ||||
| Description |
More specifically, compounds1band3bdemonstrated significantly higher activity against U87 cells compared to MCF-7 and HT-29 cells (Figure6). Importantly,1bwas more active than3bdemonstrating that the attached CPP is indeed necessary to increase the overall cellular uptake and thus cytotoxic activity of the conjugates. Of note is also that2bwas less efficient than1bbut marginally more active than3b, although with no selectivity. For the two conjugates bearing the GFLG motif, it was seen that3cis still significantly more active in U87 cells expressing integrin receptors. By contrast,1ckept its selectivity against HT-29 cells, but was pronouncedly more active in MCF-7 cells compared to1b.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) | > 150 μM | |||
| Administration Time | 15 min | ||||
| Description |
More specifically, compounds1band3bdemonstrated significantly higher activity against U87 cells compared to MCF-7 and HT-29 cells (Figure6). Importantly,1bwas more active than3bdemonstrating that the attached CPP is indeed necessary to increase the overall cellular uptake and thus cytotoxic activity of the conjugates. Of note is also that2bwas less efficient than1bbut marginally more active than3b, although with no selectivity. For the two conjugates bearing the GFLG motif, it was seen that3cis still significantly more active in U87 cells expressing integrin receptors. By contrast,1ckept its selectivity against HT-29 cells, but was pronouncedly more active in MCF-7 cells compared to1b.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
| Experiment 6 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) | > 150 μM | |||
| Administration Time | 15 min | ||||
| Description |
More specifically, compounds1band3bdemonstrated significantly higher activity against U87 cells compared to MCF-7 and HT-29 cells (Figure6). Importantly,1bwas more active than3bdemonstrating that the attached CPP is indeed necessary to increase the overall cellular uptake and thus cytotoxic activity of the conjugates. Of note is also that2bwas less efficient than1bbut marginally more active than3b, although with no selectivity. For the two conjugates bearing the GFLG motif, it was seen that3cis still significantly more active in U87 cells expressing integrin receptors. By contrast,1ckept its selectivity against HT-29 cells, but was pronouncedly more active in MCF-7 cells compared to1b.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
c[DKP-RGD]-PEG4-sC18(dau=Aoa-Lys8) [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
3 μM
|
|||
| Administration Time | 72 h | ||||
| Description |
All dau-loaded conjugates showed high toxicity in all cell lines tested with EC50 values in the lower micromolar range.
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
| Experiment 2 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
5.6 μM
|
|||
| Administration Time | 72 h | ||||
| Description |
All dau-loaded conjugates showed high toxicity in all cell lines tested with EC50 values in the lower micromolar range.
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
9.1 μM
|
|||
| Administration Time | 72 h | ||||
| Description |
All dau-loaded conjugates showed high toxicity in all cell lines tested with EC50 values in the lower micromolar range.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
12.5 μM
|
|||
| Administration Time | 15 min | ||||
| Description |
More specifically, compounds 1b and 3b demonstrated significantly higher activity against U87 cells compared to MCF-7 and HT-29 cells (Figure 6). Importantly, 1b was more active than 3b demonstrating that the attached CPP is indeed necessary to increase the overall cellular uptake and thus cytotoxic activity of the conjugates. Of note is also that 2b was less efficient than 1b but marginally more active than 3b, although with no selectivity. For the two conjugates bearing the GFLG motif, it was seen that 3c is still significantly more active in U87 cells expressing integrin receptors. By contrast, 1c kept its selectivity against HT-29 cells, but was pronouncedly more active in MCF-7 cells compared to 1b.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
50.5 μM
|
|||
| Administration Time | 15 min | ||||
| Description |
More specifically, compounds 1b and 3b demonstrated significantly higher activity against U87 cells compared to MCF-7 and HT-29 cells (Figure 6). Importantly, 1b was more active than 3b demonstrating that the attached CPP is indeed necessary to increase the overall cellular uptake and thus cytotoxic activity of the conjugates. Of note is also that 2b was less efficient than 1b but marginally more active than 3b, although with no selectivity. For the two conjugates bearing the GFLG motif, it was seen that 3c is still significantly more active in U87 cells expressing integrin receptors. By contrast, 1c kept its selectivity against HT-29 cells, but was pronouncedly more active in MCF-7 cells compared to 1b.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
| Experiment 6 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
53.4 μM
|
|||
| Administration Time | 15 min | ||||
| Description |
More specifically, compounds 1b and 3b demonstrated significantly higher activity against U87 cells compared to MCF-7 and HT-29 cells (Figure 6). Importantly, 1b was more active than 3b demonstrating that the attached CPP is indeed necessary to increase the overall cellular uptake and thus cytotoxic activity of the conjugates. Of note is also that 2b was less efficient than 1b but marginally more active than 3b, although with no selectivity. For the two conjugates bearing the GFLG motif, it was seen that 3c is still significantly more active in U87 cells expressing integrin receptors. By contrast, 1c kept its selectivity against HT-29 cells, but was pronouncedly more active in MCF-7 cells compared to 1b.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
c[DKP-RGD]-PEG4-Aoa=dau [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
3 μM
|
|||
| Administration Time | 72 h | ||||
| Description |
All dau-loaded conjugates showed high toxicity in all cell lines tested with EC50 values in the lower micromolar range.
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
9.2 μM
|
|||
| Administration Time | 72 h | ||||
| Description |
All dau-loaded conjugates showed high toxicity in all cell lines tested with EC50 values in the lower micromolar range.
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
| Experiment 3 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
21.7 μM
|
|||
| Administration Time | 72 h | ||||
| Description |
All dau-loaded conjugates showed high toxicity in all cell lines tested with EC50 values in the lower micromolar range.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
56.9 μM
|
|||
| Administration Time | 15 min | ||||
| Description |
More specifically, compounds1band3bdemonstrated significantly higher activity against U87 cells compared to MCF-7 and HT-29 cells (Figure6). Importantly,1bwas more active than3bdemonstrating that the attached CPP is indeed necessary to increase the overall cellular uptake and thus cytotoxic activity of the conjugates. Of note is also that2bwas less efficient than1bbut marginally more active than3b, although with no selectivity. For the two conjugates bearing the GFLG motif, it was seen that3cis still significantly more active in U87 cells expressing integrin receptors. By contrast,1ckept its selectivity against HT-29 cells, but was pronouncedly more active in MCF-7 cells compared to1b.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) | > 150 μM | |||
| Administration Time | 15 min | ||||
| Description |
More specifically, compounds1band3bdemonstrated significantly higher activity against U87 cells compared to MCF-7 and HT-29 cells (Figure6). Importantly,1bwas more active than3bdemonstrating that the attached CPP is indeed necessary to increase the overall cellular uptake and thus cytotoxic activity of the conjugates. Of note is also that2bwas less efficient than1bbut marginally more active than3b, although with no selectivity. For the two conjugates bearing the GFLG motif, it was seen that3cis still significantly more active in U87 cells expressing integrin receptors. By contrast,1ckept its selectivity against HT-29 cells, but was pronouncedly more active in MCF-7 cells compared to1b.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
| Experiment 6 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) | > 150 μM | |||
| Administration Time | 15 min | ||||
| Description |
More specifically, compounds1band3bdemonstrated significantly higher activity against U87 cells compared to MCF-7 and HT-29 cells (Figure6). Importantly,1bwas more active than3bdemonstrating that the attached CPP is indeed necessary to increase the overall cellular uptake and thus cytotoxic activity of the conjugates. Of note is also that2bwas less efficient than1bbut marginally more active than3b, although with no selectivity. For the two conjugates bearing the GFLG motif, it was seen that3cis still significantly more active in U87 cells expressing integrin receptors. By contrast,1ckept its selectivity against HT-29 cells, but was pronouncedly more active in MCF-7 cells compared to1b.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
sC18(dau-Aoa-Lys8) [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
5 μM
|
|||
| Administration Time | 72 h | ||||
| Description |
All dau-loaded conjugates showed high toxicity in all cell lines tested with EC50 values in the lower micromolar range.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
5.8 μM
|
|||
| Administration Time | 72 h | ||||
| Description |
All dau-loaded conjugates showed high toxicity in all cell lines tested with EC50 values in the lower micromolar range.
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
9.2 μM
|
|||
| Administration Time | 72 h | ||||
| Description |
All dau-loaded conjugates showed high toxicity in all cell lines tested with EC50 values in the lower micromolar range.
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
| Experiment 4 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
25.1 μM
|
|||
| Administration Time | 15 min | ||||
| Description |
More specifically, compounds1band3bdemonstrated significantly higher activity against U87 cells compared to MCF-7 and HT-29 cells (Figure6). Importantly,1bwas more active than3bdemonstrating that the attached CPP is indeed necessary to increase the overall cellular uptake and thus cytotoxic activity of the conjugates. Of note is also that2bwas less efficient than1bbut marginally more active than3b, although with no selectivity. For the two conjugates bearing the GFLG motif, it was seen that3cis still significantly more active in U87 cells expressing integrin receptors. By contrast,1ckept its selectivity against HT-29 cells, but was pronouncedly more active in MCF-7 cells compared to3b.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
42.2 μM
|
|||
| Administration Time | 15 min | ||||
| Description |
More specifically, compounds1band3bdemonstrated significantly higher activity against U87 cells compared to MCF-7 and HT-29 cells (Figure6). Importantly,1bwas more active than3bdemonstrating that the attached CPP is indeed necessary to increase the overall cellular uptake and thus cytotoxic activity of the conjugates. Of note is also that2bwas less efficient than1bbut marginally more active than3b, although with no selectivity. For the two conjugates bearing the GFLG motif, it was seen that3cis still significantly more active in U87 cells expressing integrin receptors. By contrast,1ckept its selectivity against HT-29 cells, but was pronouncedly more active in MCF-7 cells compared to1b.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) | > 100 μM | |||
| Administration Time | 15 min | ||||
| Description |
More specifically, compounds1band3bdemonstrated significantly higher activity against U87 cells compared to MCF-7 and HT-29 cells (Figure6). Importantly,1bwas more active than3bdemonstrating that the attached CPP is indeed necessary to increase the overall cellular uptake and thus cytotoxic activity of the conjugates. Of note is also that2bwas less efficient than1bbut marginally more active than3b, although with no selectivity. For the two conjugates bearing the GFLG motif, it was seen that3cis still significantly more active in U87 cells expressing integrin receptors. By contrast,1ckept its selectivity against HT-29 cells, but was pronouncedly more active in MCF-7 cells compared to2b.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
References