
Peptide-drug Conjugate Information
General Information of This Peptide-drug Conjugate (PDC)
| PDC ID |
PDC_00208
|
|||||
|---|---|---|---|---|---|---|
| PDC Name |
GnRH-III-[8Lys(Dau=Aoa)]
|
|||||
| PDC Status |
Investigative
|
|||||
| Indication |
In total 3 Indication(s)
|
|||||
| Structure |
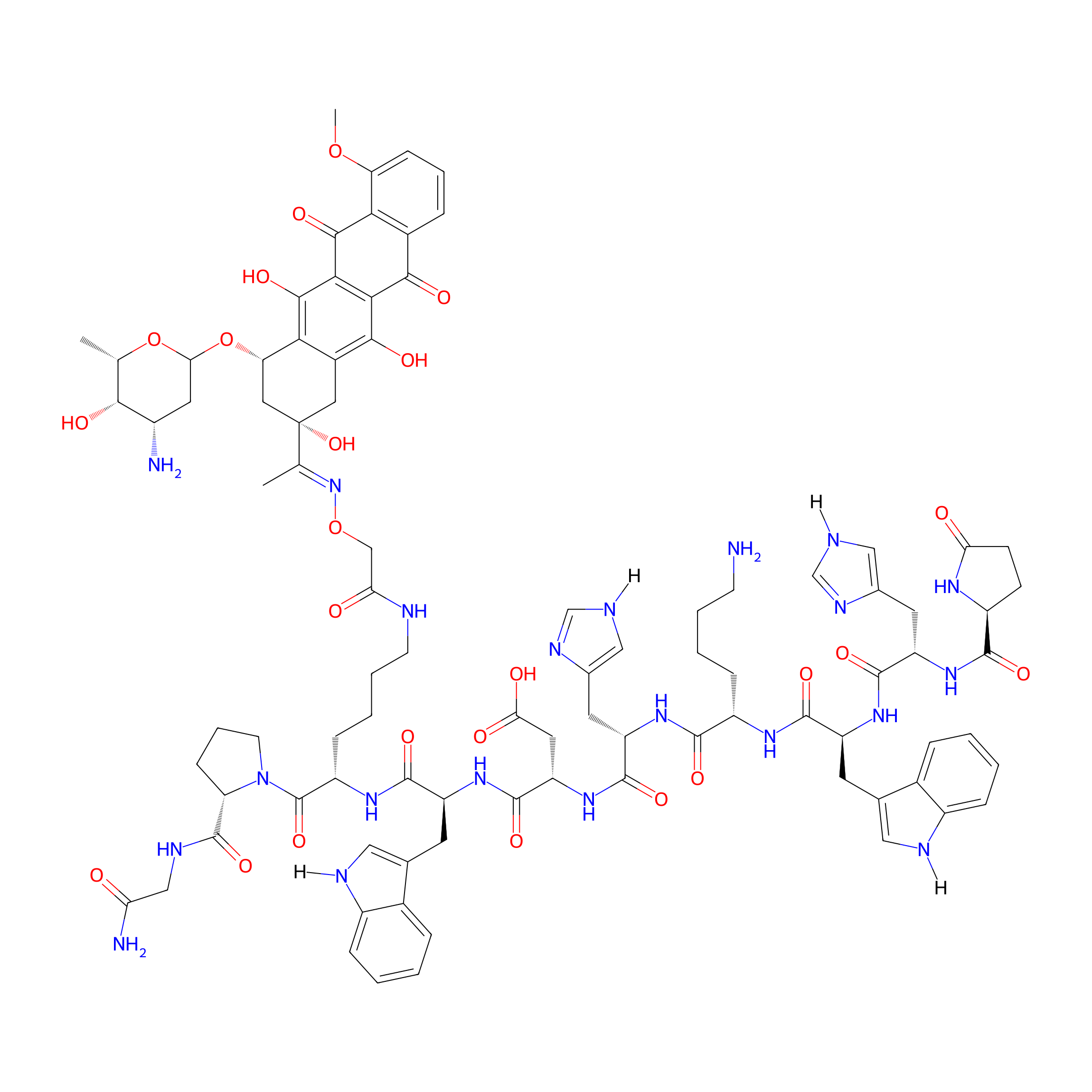
|
|||||
| Peptide Name |
GnRH-III
|
Peptide Info | ||||
| Receptor Name |
Gonadotropin-releasing hormone receptor (GNRHR)
|
Receptor Info | ||||
| Drug Name |
Daunorubicin
|
Drug Info | ||||
| Therapeutic Target |
DNA topoisomerase 2-alpha (TOP2A)
|
Target Info | ||||
| Linker Name |
Aminooxyacetic acid
|
Linker Info | ||||
| Peptide Modified Type |
Amino acid modifications
|
|||||
| Modified Segment |
Incorporation of unnatural amino acids: pyroglutamic acid (
|
|||||
| Ternimal Modification |
N-terminal modification
|
|||||
| Formula |
C91H111N21O24
|
|||||
| #Ro5 Violations (Lipinski): 4 | Molecular Weight | 1883.012 | ||||
| Lipid-water partition coefficient (xlogp) | -1.6331 | |||||
| Hydrogen Bond Donor Count (hbonddonor) | 22 | |||||
| Hydrogen Bond Acceptor Count (hbondacc) | 28 | |||||
| Rotatable Bond Count (rotbonds) | 44 | |||||
Full List of Activity Data of This Peptide-drug Conjugate
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.54 ± 0.67µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
6.5 ± 1.8 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
The cancer phenotype is commonly associated with aberrant glycosylation patterns. One glycan that is directly linked to cancer is the Thomsen-Friedenreich antigen (TF or CD176). It is a disaccharide composed of a galactose β1-3 N-acetylgalactosamine, O-linked to a glycoprotein through serine or threonine residues and commonly written as Galβ1-3GalNAc--O-Ser/Thr. The TF is therapeutically attractive due to its cryptic nature in normal cells and exposure in embryonic and cancer cells. The expression of the TF has been demonstrated in 90% of primary human carcinomas, including in the lung, the breast, and the pancreas. Additionally, cancer initiating cells or cancer stem cells in the lung, liver, and colon express the TF. The peptide sequence HGRFILPWWYAFSPS (TF-peptide) is known to bind tightly to the TF (Kd = 1.2 M) and has been demonstrated to inhibit processes directly involved in TF accessibility.
Click to Show/Hide
|
||||
| Description |
The main drug-related toxic side effect of anthracyclines is cardiotoxicity, leading to cardiomyopathy and heart failure. Comparative experiments to determine the cardiotoxicity of peptide-anthracycline conjugates with different linkages might be informative for the other conjugates with different types of drugs as well. For this purpose, human cardiomyocytes (HCM) and human umbilical vein endothelial cells (HUVEC) were used as models. The long-term (0-72 h) cytotoxic effect of sixteen GnRH-based conjugates containing Dox and Dau was determined by real-time impedimetric sensing using the xCELLigence SP system (ACEA Biosciences, San Diego, CA, USA) [75]. The results indicated that the ester-linked GnRH-Dox conjugates, including Zoptarelin Doxorubicin, showed significant toxicity at 100 nM and 1 uM, which was remarkably pronounced on the HCM cells. The cytotoxic effect was comparable to that of the free drug, especially at the highest concentration. In contrast, the conjugates with oxime-linked Dau showed no or only a minor toxicity on both the cell lines (Table 2). These data confirm that the linkage between the payload and homing peptide has a significant influence on early drug release and, consequently, an undesired toxic side effect. We may also conclude that the search for more suitable homing peptides might be more important than the application of cleavable bonds between the drug and the peptide to develop efficient DDSs.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
8.22 ± 0.13 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
13.89 ± 3.62 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
| Experiment 5 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
14.2 ± 3.2 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
In our experiments, GnRH-III was applied as a targeting peptide. The conjugate [8Lys(Dau=Aoa-GFLG)]-GnRH-III, in which Dau was connected to the side chain of Lys in position 8 through an aminooxyacetylated cathepsin B-labile GFLG spacer, showed significant tumor growth inhibition in s.c.-developed fast-growing C26 murine colon cancer-bearing Balb/c mice. The effect highly depended on the treatment schedule. In the first attempt, the conjugate was administered i.p. at a dose of 8.86 umol (5 mg Dau-content/kg body weight) five times on days 7, 9, 11, 14 and 16 after tumor transplantation (Treatment schedule A). The tumor volume inhibition was only 15.7% on day 26 when the experiment finished. A slight enhancement in inhibition (22.3%) was detected when a dose of conjugate corresponding to 15 mg Dau content/kg body weight (26.6 μmol) was injected only once on day 7 (Treatment schedule B). An additional treatment on day 10 (Treatment schedule C) did not result in any further improvements (21.9% on day 29). However, when the treatment schedule was changed to two treatments with the same dose on days 4 and 7, 46.3% inhibition was observed on day 29 (Treatment schedule D). In contrast, the treatments with free Dau at a dose of 2 mg/kg body weight (3.55 umol) on days 7, 9, 11, 14 and 16 showed only 22.6% inhibition on day 26. The median survival rates of the treated animals in comparison to the control group were 1 (A), 1.23 (B), 1.21 (C) and 1.38 (D), respectively, and 0.81 for free Dau. These results indicate the lower toxicity and the higher tumor volume inhibition effect of the conjugates in comparison with those of free Dau, as well as the importance of the treatment schedule. In another experiment, HT-29 human colon cancer was developed s.c. in immunodeficient SCID mice. Dau and two conjugates (with or without a GFLG spacer between the GnRH-III homing peptide and the payload) were used for the treatment. The first i.p. administrations were performed on day 13 after tumor inoculation. All the mice treated once with 2.5 mg/kg body weight (4.43 umol) free Dau died within 10 days. In contrast, the conjugates at a dose of 15 mg Dau content/kg body weight (26.6 umol) that refers to 52 mg/kg [8Lys(Dau=Aoa)]-GnRH-III and 62.5 mg/kg [8Lys(Dau=Aoa-GFLG)]-GnRH-III conjugates, respectively, did not show significant toxicity. The treatment was repeated on days 23 and 30. Because of the significant weight loss in several mice in the control group, the experiment was terminated on day 35. The tumor growth inhibition could be calculated as reductions in the tumor volume by 44.3% and 57.6% and the tumor weight by 41% and 50%, respectively. Interestingly, the conjugates were poorly effective on orthotopically developed tumors. In the case of the C26 colon tumor-bearing female Balb/c mice, only a 7% reduction in tumor weight was detected on day 13 after the two treatments on days 4 and 7 with [8Lys(Dau=Aoa)]-GnRH-III, at a 26.6 umol/kg (15 mg Dau content) dose. The effect of free Dau (2 mg/kg on days 4 and 7) showed a better inhibitory effect (24.4%). Interestingly, our novel developed GnRH-III derivative, in which Ser in position 4 was replaced by Lys(Ac), was much more potent, with 49.3% inhibition. It is worth mentioning that the rate of cellular uptake of the [4Lys(Ac), 8Lys(Dau=Aoa)]-GnRH-III conjugate by the tumor cells was significantly higher than that of the conjugate with the native GnRH-III sequence.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
| Experiment 6 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
27.8 ± 4.2 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
The main drug-related toxic side effect of anthracyclines is cardiotoxicity, leading to cardiomyopathy and heart failure. Comparative experiments to determine the cardiotoxicity of peptide-anthracycline conjugates with different linkages might be informative for the other conjugates with different types of drugs as well. For this purpose, human cardiomyocytes (HCM) and human umbilical vein endothelial cells (HUVEC) were used as models. The long-term (0-72 h) cytotoxic effect of sixteen GnRH-based conjugates containing Dox and Dau was determined by real-time impedimetric sensing using the xCELLigence SP system (ACEA Biosciences, San Diego, CA, USA) [75]. The results indicated that the ester-linked GnRH-Dox conjugates, including Zoptarelin Doxorubicin, showed significant toxicity at 100 nM and 1 uM, which was remarkably pronounced on the HCM cells. The cytotoxic effect was comparable to that of the free drug, especially at the highest concentration. In contrast, the conjugates with oxime-linked Dau showed no or only a minor toxicity on both the cell lines (Table 2). These data confirm that the linkage between the payload and homing peptide has a significant influence on early drug release and, consequently, an undesired toxic side effect. We may also conclude that the search for more suitable homing peptides might be more important than the application of cleavable bonds between the drug and the peptide to develop efficient DDSs.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
References