
Peptide Information
General Information of This Peptide
| Peptide ID |
PEP00168
|
|||||
|---|---|---|---|---|---|---|
| Peptide Name |
GnRH-III
|
|||||
| Structure |
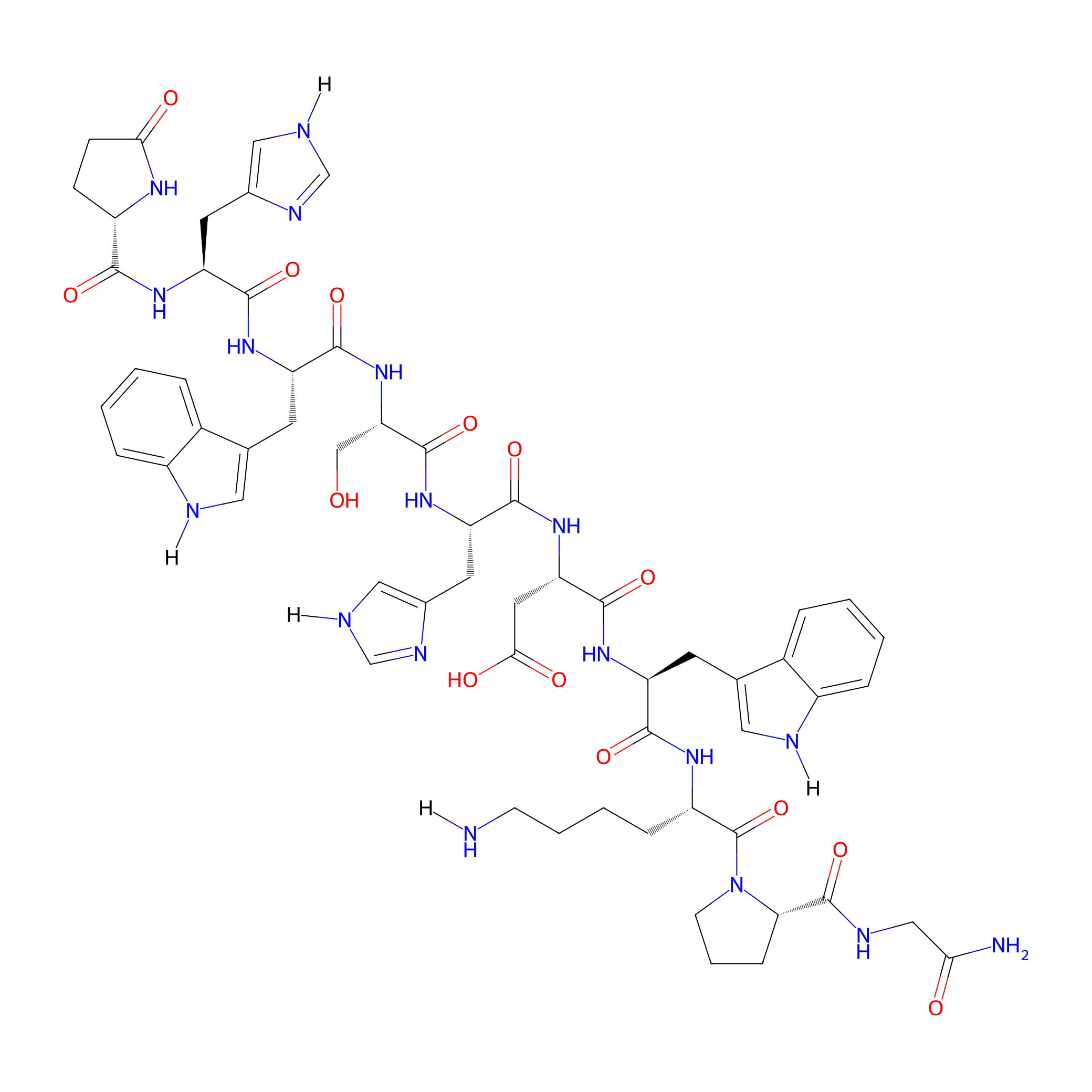
|
|||||
| Sequence |
EHWSHDWKPG-NH2
|
|||||
| Peptide Type |
Linear
|
|||||
| Receptor Name |
Gonadotropin-releasing hormone receptor (GNRHR)
|
Receptor Info | ||||
| PDC Transmembrane Types | Cell targeting peptides (CTPs) | |||||
| Distribution |
LHRH-III (pGlu-His-Trp-Ser-His-Asp-Trp-Lys-Pro-Gly-NH2) is an isoform of human LHRH isolated from sea lamprey. This decapeptide isoform can specifically bind to LHRH-R in cancer cells with strong antiproliferative effect, whereas the potency in its release of LH and FSH is insignificant in mammals.
|
|||||
| Formula |
C59H74N18O14
|
|||||
| Isosmiles |
[H]NCCCC[C@H](NC(=O)[C@H](Cc1cn([H])c2ccccc12)NC(=O)[C@H](CC(=O)O)NC(=O)[C@H](Cc1cn([H])cn1)NC(=O)[C@H](CO)NC(=O)[C@H](Cc1cn([H])c2ccccc12)NC(=O)[C@H](Cc1cn([H])cn1)NC(=O)[C@@H]1CCC(=O)N1)C(=O)N1CCC[C@H]1C(=O)NCC(N)=O
|
|||||
| InChI |
InChI=1S/C59H74N18O14/c60-16-6-5-12-40(59(91)77-17-7-13-47(77)58(90)66-27-48(61)79)70-52(84)41(18-31-23-64-37-10-3-1-8-35(31)37)72-56(88)45(22-50(81)82)75-55(87)44(21-34-26-63-30-68-34)74-57(89)46(28-78)76-53(85)42(19-32-24-65-38-11-4-2-9-36(32)38)71-54(86)43(20-33-25-62-29-67-33)73-51(83)39-14-15-49(80)69-39/h1-4,8-11,23-26,29-30,39-47,64-65,78H,5-7,12-22,27-28,60H2,(H2,61,79)(H,62,67)(H,63,68)(H,66,90)(H,69,80)(H,70,84)(H,71,86)(H,72,88)(H,73,83)(H,74,89)(H,75,87)(H,76,85)(H,81,82)/t39-,40-,41-,42-,43-,44-,45-,46-,47-/m0/s1
|
|||||
| InChIKey |
RTASYRSYWSLWJV-CSYZDTNESA-N
|
|||||
| Pharmaceutical Properties |
Molecule Weight
|
1259.353
|
Polar area
|
497.79
|
||
|
Complexity
|
1258.563189
|
xlogp Value
|
-3.7733
|
|||
|
Heavy Count
|
91
|
Rot Bonds
|
34
|
|||
|
Hbond acc
|
16
|
Hbond Donor
|
17
|
|||
Each Peptide-drug Conjugate Related to This Peptide
Full Information of The Activity Data of The PDC(s) Related to This Peptide
GnRH-III-[2ΔHis, 3D-Tic, 4Lys(Bu), 8Lys(Dau=Aoa)] [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor weight decrease |
27.70%
|
|||
| Administration Dosage | 15 mg/kg Dau content | ||||
| Description |
Based on these tumor weights, we determined that free Dau, 1 and 2 inhibited tumor weight significantly by 40.1, 28.7 and 27.7% in the case of orthotopic human MDA-MB-231 breast tumor model.
|
||||
| In Vivo Model | Orthotopic MDA-MB-231 human breast carcinoma bearing mice model. | ||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Colorectal cancer | ||||
| Efficacy Data | Tumor weight |
87.10%
|
|||
| Administration Dosage | 10 mg/kg Dau content | ||||
| Description |
The obtained data reveal that Dau, 1 and 2 significantly inhibited the tumor growth, whereby the tumor weights were reduced by 84.3, 80.8 and 87.1%, as compared to the control group.
|
||||
| In Vivo Model | Orthotopic HT-29 human colon carcinoma bearing mice model. | ||||
| In Vitro Model | Colon adenocarcinoma | HT-29 cell | CVCL_0320 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor volume decrease |
23.10%
|
|||
| Administration Dosage | 15 mg/kg Dau content | ||||
| Description |
Apart from that, a significant inhibition of the tumor volume was also obtained in groups which were treated with conjugate 1 (34.1%) and 2 (23.1%).
|
||||
| In Vivo Model | Orthotopic MDA-MB-231 human breast carcinoma bearing mice model. | ||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Proliferation index |
25.90%
|
|||
| Administration Dosage | 10 mg/kg Dau content | ||||
| Description |
It was observed that both GnRH-III conjugates 1 and 2 caused a significant decrease of the proliferation index by 16.3 and 25.9%
|
||||
| In Vivo Model | Orthotopic 4T1 mice breast carcinoma bearing mice model. | ||||
| In Vitro Model | Mammary carcinoma | 4T1 cell | CVCL_0125 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Proliferation index |
39.10%
|
|||
| Administration Dosage | 10 mg/kg Dau content | ||||
| Description |
The obtained data revealed that free Dau and both conjugates (1, 2) significantly inhibited the number of micro-metastases in the lung by 33.7, 43.8 and 49.4%, as compared to the control group. The proliferation index of lung metastases was significantly inhibited by 27.8, 37 and 39.1% in groups that were treated with free Dau, 1 and 2.
|
||||
| In Vivo Model | Orthotopic 4T1 mice breast carcinoma bearing mice model. | ||||
| In Vitro Model | Mammary carcinoma | 4T1 cell | CVCL_0125 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Number of macro-metastases decrease |
64.40%
|
|||
| Administration Dosage | 10 mg/kg Dau content | ||||
| Description |
The number of macro-metastases in peripheral organs, such as spleen, lung, liver and kidneys, was counted, in order to determine the anti-metastatic effect of free Dau and the GnRH-III conjugates on aggressive 4T1 BC orthotopic model (Figure 5A). The number of macro-metastases in spleen was significantly decreased in all treated groups (Dau,1and2) by 64.3, 72.8 and 78.1%. In the lung, the number of macro-metastases was also significantly reduced for all treated groups by 55.4, 55.2 and 64.4%, respectively. The numbers of macro-metastases in the liver and kidneys were decreased under treatments, whereby a significant decrease could be only obtained for conjugate2.
Click to Show/Hide
|
||||
| In Vivo Model | Orthotopic 4T1 mice breast carcinoma bearing mice model. | ||||
| In Vitro Model | Mammary carcinoma | 4T1 cell | CVCL_0125 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Number of macro-metastases decrease |
78.10%
|
|||
| Administration Dosage | 10 mg/kg Dau content | ||||
| Description |
The number of macro-metastases in peripheral organs, such as spleen, lung, liver and kidneys, was counted, in order to determine the anti-metastatic effect of free Dau and the GnRH-III conjugates on aggressive 4T1 BC orthotopic model (Figure 5A). The number of macro-metastases in spleen was significantly decreased in all treated groups (Dau, 1 and 2) by 64.3, 72.8 and 78.1%. In the lung, the number of macro-metastases was also significantly reduced for all treated groups by 55.4, 55.2 and 64.4%, respectively. The numbers of macro-metastases in the liver and kidneys were decreased under treatments, whereby a significant decrease could be only obtained for conjugate 2.
Click to Show/Hide
|
||||
| In Vivo Model | Orthotopic 4T1 mice breast carcinoma bearing mice model. | ||||
| In Vitro Model | Mammary carcinoma | 4T1 cell | CVCL_0125 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Micro-metastases decrease |
49.40%
|
|||
| Administration Dosage | 10 mg/kg Dau content | ||||
| Description |
The obtained data revealed that free Dau and both conjugates (1, 2) significantly inhibited the number of micro-metastases in the lung by 33.7, 43.8 and 49.4%, as compared to the control group. The proliferation index of lung metastases was significantly inhibited by 27.8, 37 and 39.1% in groups that were treated with free Dau, 1 and 2.
|
||||
| In Vivo Model | Orthotopic 4T1 mice breast carcinoma bearing mice model. | ||||
| In Vitro Model | Mammary carcinoma | 4T1 cell | CVCL_0125 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Body weigth change |
8.20%
|
|||
| Administration Dosage | 15 mg/kg Dau content | ||||
| Description |
The body weight of the mice in control group was decreased by 2.7%, while in the groups treated with 1 and 2, it was decreased by 10.1 and 8.2%.
|
||||
| In Vivo Model | Orthotopic MDA-MB-231 human breast carcinoma bearing mice model. | ||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
0.14 ± 0.01 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.1 ± 0.2 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | B16 cell | CVCL_F936 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Oral cavity squamous cell carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.7 ± 0.5 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Oral cavity squamous cell carcinoma | PE/CA-PJ41 | CVCL_2680 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.8 ± 0.1 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Mammary carcinoma | 4T1 cell | CVCL_0125 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.9 ± 0.2 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Prostate carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.1 ± 0.2 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | DU145 cell | CVCL_0105 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Ovarian endometrioid adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.1 ± 0.5 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.2 ± 0.7 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.3 ± 0.1 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Lung adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.3 ± 0.7 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Lung adenocarcinoma | H1975 cell | CVCL_1511 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Prostate carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.4 ± 0.6 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.49 ± 0.53 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Normal | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.6 ± 0.7 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | C-26 cell | CVCL_XC68 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.6 ± 0.5 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | A2058 cell | CVCL_1059 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.6 ± 0.6 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | WM983B cell | CVCL_6809 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Amelanotic melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.9 ± 0.6 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | HT168-M1/M9 cell | CVCL_2H39 | ||
| Experiment 17 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Tongue squamous cell carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.9 ± 0.6 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Tongue squamous cell carcinoma | PE/CA-PJ15 cell | CVCL_2678 | ||
| Experiment 18 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.3 ± 0.9 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
The efficacy of current chemotherapeutic treatments against most solid tumors is limited by their systemic toxicity, which is partly associated with the cytotoxic properties of agents such as docetaxel or doxorubicin. To avoid or minimize adverse effects from chemotherapeutic molecules, a promising targeted approach is through peptide-drug conjugates (PDCs) that link anticancer molecules to peptides designed to interact with receptors highly expressed on cancer cells, and which can mediate the molecules rapid internalization within those cells. One such receptor is sortilin (SORT1), also known as neurotensin receptor3, a membranebound receptor that belongs to the VPS10P family of receptors. TH19P01 peptide was recently designed to target and exploit SORT1s ligand internalization function. Studies have confirmed that both TH1902 (a docetaxel-TH19P01 conjugate) and TH1904 (a doxorubicin-TH19P01 conjugate) require a SORT1-dependent mechanism of action to exert anticancer activities. In recent preclinical studies performed in immunocompromised animal models, which are unable to produce mature T-cells, TH1902 was effective against several human SORT1-positive xenograft models including triple-negative breast cancer (TNBC), ovarian cancer, and endometrial cancer.
Click to Show/Hide
|
||||
| Description |
In our experiments, GnRH-III was applied as a targeting peptide. The conjugate [8Lys(Dau=Aoa-GFLG)]-GnRH-III, in which Dau was connected to the side chain of Lys in position 8 through an aminooxyacetylated cathepsin B-labile GFLG spacer, showed significant tumor growth inhibition in s.c.-developed fast-growing C26 murine colon cancer-bearing Balb/c mice. The effect highly depended on the treatment schedule. In the first attempt, the conjugate was administered i.p. at a dose of 8.86 umol (5 mg Dau-content/kg body weight) five times on days 7, 9, 11, 14 and 16 after tumor transplantation (Treatment schedule A). The tumor volume inhibition was only 15.7% on day 26 when the experiment finished. A slight enhancement in inhibition (22.3%) was detected when a dose of conjugate corresponding to 15 mg Dau content/kg body weight (26.6 μmol) was injected only once on day 7 (Treatment schedule B). An additional treatment on day 10 (Treatment schedule C) did not result in any further improvements (21.9% on day 29). However, when the treatment schedule was changed to two treatments with the same dose on days 4 and 7, 46.3% inhibition was observed on day 29 (Treatment schedule D). In contrast, the treatments with free Dau at a dose of 2 mg/kg body weight (3.55 umol) on days 7, 9, 11, 14 and 16 showed only 22.6% inhibition on day 26. The median survival rates of the treated animals in comparison to the control group were 1 (A), 1.23 (B), 1.21 (C) and 1.38 (D), respectively, and 0.81 for free Dau. These results indicate the lower toxicity and the higher tumor volume inhibition effect of the conjugates in comparison with those of free Dau, as well as the importance of the treatment schedule. In another experiment, HT-29 human colon cancer was developed s.c. in immunodeficient SCID mice. Dau and two conjugates (with or without a GFLG spacer between the GnRH-III homing peptide and the payload) were used for the treatment. The first i.p. administrations were performed on day 13 after tumor inoculation. All the mice treated once with 2.5 mg/kg body weight (4.43 umol) free Dau died within 10 days. In contrast, the conjugates at a dose of 15 mg Dau content/kg body weight (26.6 umol) that refers to 52 mg/kg [8Lys(Dau=Aoa)]-GnRH-III and 62.5 mg/kg [8Lys(Dau=Aoa-GFLG)]-GnRH-III conjugates, respectively, did not show significant toxicity. The treatment was repeated on days 23 and 30. Because of the significant weight loss in several mice in the control group, the experiment was terminated on day 35. The tumor growth inhibition could be calculated as reductions in the tumor volume by 44.3% and 57.6% and the tumor weight by 41% and 50%, respectively. Interestingly, the conjugates were poorly effective on orthotopically developed tumors. In the case of the C26 colon tumor-bearing female Balb/c mice, only a 7% reduction in tumor weight was detected on day 13 after the two treatments on days 4 and 7 with [8Lys(Dau=Aoa)]-GnRH-III, at a 26.6 umol/kg (15 mg Dau content) dose. The effect of free Dau (2 mg/kg on days 4 and 7) showed a better inhibitory effect (24.4%). Interestingly, our novel developed GnRH-III derivative, in which Ser in position 4 was replaced by Lys(Ac), was much more potent, with 49.3% inhibition. It is worth mentioning that the rate of cellular uptake of the [4Lys(Ac), 8Lys(Dau=Aoa)]-GnRH-III conjugate by the tumor cells was significantly higher than that of the conjugate with the native GnRH-III sequence.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
| Experiment 19 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.31 ± 0.90 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
| Experiment 20 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Amelanotic melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.5 ± 0.6 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | M24 cell | CVCL_D032 | ||
| Experiment 21 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Invasive breast carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4.0 ± 0.8 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 22 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Minimally invasive lung adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4.0 ± 0.8 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Minimally invasive lung adenocarcinoma | H1650 cell | CVCL_1483 | ||
| Experiment 23 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Lung adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4.3 ± 0.4 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Lung adenocarcinoma | A-549 cell | CVCL_0023 | ||
| Experiment 24 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
7.3 ± 0.3 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
| Experiment 25 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Ovarian serous adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
8.2 ± 0.5 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous adenocarcinoma | OVCAR-3 cell | CVCL_0465 | ||
| Experiment 26 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | High grade ovarian serous adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
9.5 ± 0.8 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | High grade ovarian serous adenocarcinoma | OVCAR-8 cell | CVCL_1629 | ||
| Experiment 27 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Normal | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
19.7 ± 1.2 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MRC-5 cell | CVCL_0440 | ||
| Experiment 28 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Pancreatic ductal adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
56.4 ± 4.5 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
GnRH-III-[4Lys(Bu), 8Lys(Dau=Aoa)] [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor weight decrease |
28.70%
|
|||
| Administration Dosage | 15 mg/kg Dau content | ||||
| Description |
Based on these tumor weights, we determined that free Dau, 1 and 2 inhibited tumor weight significantly by 40.1, 28.7 and 27.7% in the case of orthotopic human MDA-MB-231 breast tumor model.
|
||||
| In Vivo Model | Orthotopic MDA-MB-231 human breast carcinoma bearing mice model. | ||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Colorectal cancer | ||||
| Efficacy Data | Tumor weight |
80.80%
|
|||
| Administration Dosage | 10 mg/kg Dau content | ||||
| Description |
The obtained data reveal that Dau, 1 and 2 significantly inhibited the tumor growth, whereby the tumor weights were reduced by 84.3, 80.8 and 87.1%, as compared to the control group.
|
||||
| In Vivo Model | Orthotopic HT-29 human colon carcinoma bearing mice model. | ||||
| In Vitro Model | Colon adenocarcinoma | HT-29 cell | CVCL_0320 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor volume decrease |
34.10%
|
|||
| Administration Dosage | 15 mg/kg Dau content | ||||
| Description |
Apart from that, a significant inhibition of the tumor volume was also obtained in groups which were treated with conjugate 1 (34.1%) and 2 (23.1%).
|
||||
| In Vivo Model | Orthotopic MDA-MB-231 human breast carcinoma bearing mice model. | ||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Proliferation index |
16.30%
|
|||
| Administration Dosage | 10 mg/kg Dau content | ||||
| Description |
It was observed that both GnRH-III conjugates 1 and 2 caused a significant decrease of the proliferation index by 16.3 and 25.9%
|
||||
| In Vivo Model | Orthotopic 4T1 mice breast carcinoma bearing mice model. | ||||
| In Vitro Model | Mammary carcinoma | 4T1 cell | CVCL_0125 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Proliferation index |
37%
|
|||
| Administration Dosage | 10 mg/kg Dau content | ||||
| Description |
The obtained data revealed that free Dau and both conjugates (1, 2) significantly inhibited the number of micro-metastases in the lung by 33.7, 43.8 and 49.4%, as compared to the control group. The proliferation index of lung metastases was significantly inhibited by 27.8, 37 and 39.1% in groups that were treated with free Dau, 1 and 2.
|
||||
| In Vivo Model | Orthotopic 4T1 mice breast carcinoma bearing mice model. | ||||
| In Vitro Model | Mammary carcinoma | 4T1 cell | CVCL_0125 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Number of macro-metastases decrease |
55.20%
|
|||
| Administration Dosage | 10 mg/kg Dau content | ||||
| Description |
The number of macro-metastases in peripheral organs, such as spleen, lung, liver and kidneys, was counted, in order to determine the anti-metastatic effect of free Dau and the GnRH-III conjugates on aggressive 4T1 BC orthotopic model (Figure 5A). The number of macro-metastases in spleen was significantly decreased in all treated groups (Dau,1and2) by 64.3, 72.8 and 78.1%. In the lung, the number of macro-metastases was also significantly reduced for all treated groups by 55.4, 55.2 and 64.4%, respectively. The numbers of macro-metastases in the liver and kidneys were decreased under treatments, whereby a significant decrease could be only obtained for conjugate2.
Click to Show/Hide
|
||||
| In Vivo Model | Orthotopic 4T1 mice breast carcinoma bearing mice model. | ||||
| In Vitro Model | Mammary carcinoma | 4T1 cell | CVCL_0125 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Number of macro-metastases decrease |
72.80%
|
|||
| Administration Dosage | 10 mg/kg Dau content | ||||
| Description |
The number of macro-metastases in peripheral organs, such as spleen, lung, liver and kidneys, was counted, in order to determine the anti-metastatic effect of free Dau and the GnRH-III conjugates on aggressive 4T1 BC orthotopic model (Figure 5A). The number of macro-metastases in spleen was significantly decreased in all treated groups (Dau, 1 and 2) by 64.3, 72.8 and 78.1%. In the lung, the number of macro-metastases was also significantly reduced for all treated groups by 55.4, 55.2 and 64.4%, respectively. The numbers of macro-metastases in the liver and kidneys were decreased under treatments, whereby a significant decrease could be only obtained for conjugate 2.
Click to Show/Hide
|
||||
| In Vivo Model | Orthotopic 4T1 mice breast carcinoma bearing mice model. | ||||
| In Vitro Model | Mammary carcinoma | 4T1 cell | CVCL_0125 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Micro-metastases decrease |
43.80%
|
|||
| Administration Dosage | 10 mg/kg Dau content | ||||
| Description |
The obtained data revealed that free Dau and both conjugates (1, 2) significantly inhibited the number of micro-metastases in the lung by 33.7, 43.8 and 49.4%, as compared to the control group. The proliferation index of lung metastases was significantly inhibited by 27.8, 37 and 39.1% in groups that were treated with free Dau, 1 and 2.
|
||||
| In Vivo Model | Orthotopic 4T1 mice breast carcinoma bearing mice model. | ||||
| In Vitro Model | Mammary carcinoma | 4T1 cell | CVCL_0125 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Body weigth change |
10.10%
|
|||
| Administration Dosage | 15 mg/kg Dau content | ||||
| Description |
The body weight of the mice in control group was decreased by 2.7%, while in the groups treated with 1 and 2, it was decreased by 10.1 and 8.2%.
|
||||
| In Vivo Model | Orthotopic MDA-MB-231 human breast carcinoma bearing mice model. | ||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Ovarian endometrioid adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.4 ± 1.1 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.2 ± 0.6 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
In our experiments, GnRH-III was applied as a targeting peptide. The conjugate [8Lys(Dau=Aoa-GFLG)]-GnRH-III, in which Dau was connected to the side chain of Lys in position 8 through an aminooxyacetylated cathepsin B-labile GFLG spacer, showed significant tumor growth inhibition in s.c.-developed fast-growing C26 murine colon cancer-bearing Balb/c mice. The effect highly depended on the treatment schedule. In the first attempt, the conjugate was administered i.p. at a dose of 8.86 umol (5 mg Dau-content/kg body weight) five times on days 7, 9, 11, 14 and 16 after tumor transplantation (Treatment schedule A). The tumor volume inhibition was only 15.7% on day 26 when the experiment finished. A slight enhancement in inhibition (22.3%) was detected when a dose of conjugate corresponding to 15 mg Dau content/kg body weight (26.6 μmol) was injected only once on day 7 (Treatment schedule B). An additional treatment on day 10 (Treatment schedule C) did not result in any further improvements (21.9% on day 29). However, when the treatment schedule was changed to two treatments with the same dose on days 4 and 7, 46.3% inhibition was observed on day 29 (Treatment schedule D). In contrast, the treatments with free Dau at a dose of 2 mg/kg body weight (3.55 umol) on days 7, 9, 11, 14 and 16 showed only 22.6% inhibition on day 26. The median survival rates of the treated animals in comparison to the control group were 1 (A), 1.23 (B), 1.21 (C) and 1.38 (D), respectively, and 0.81 for free Dau. These results indicate the lower toxicity and the higher tumor volume inhibition effect of the conjugates in comparison with those of free Dau, as well as the importance of the treatment schedule. In another experiment, HT-29 human colon cancer was developed s.c. in immunodeficient SCID mice. Dau and two conjugates (with or without a GFLG spacer between the GnRH-III homing peptide and the payload) were used for the treatment. The first i.p. administrations were performed on day 13 after tumor inoculation. All the mice treated once with 2.5 mg/kg body weight (4.43 umol) free Dau died within 10 days. In contrast, the conjugates at a dose of 15 mg Dau content/kg body weight (26.6 umol) that refers to 52 mg/kg [8Lys(Dau=Aoa)]-GnRH-III and 62.5 mg/kg [8Lys(Dau=Aoa-GFLG)]-GnRH-III conjugates, respectively, did not show significant toxicity. The treatment was repeated on days 23 and 30. Because of the significant weight loss in several mice in the control group, the experiment was terminated on day 35. The tumor growth inhibition could be calculated as reductions in the tumor volume by 44.3% and 57.6% and the tumor weight by 41% and 50%, respectively. Interestingly, the conjugates were poorly effective on orthotopically developed tumors. In the case of the C26 colon tumor-bearing female Balb/c mice, only a 7% reduction in tumor weight was detected on day 13 after the two treatments on days 4 and 7 with [8Lys(Dau=Aoa)]-GnRH-III, at a 26.6 umol/kg (15 mg Dau content) dose. The effect of free Dau (2 mg/kg on days 4 and 7) showed a better inhibitory effect (24.4%). Interestingly, our novel developed GnRH-III derivative, in which Ser in position 4 was replaced by Lys(Ac), was much more potent, with 49.3% inhibition. It is worth mentioning that the rate of cellular uptake of the [4Lys(Ac), 8Lys(Dau=Aoa)]-GnRH-III conjugate by the tumor cells was significantly higher than that of the conjugate with the native GnRH-III sequence.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.36 ± 0.07 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.2 ± 0.8 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | B16 cell | CVCL_F936 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Lung adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4.1 ± 0.1 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Lung adenocarcinoma | H1975 cell | CVCL_1511 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Oral cavity squamous cell carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4.7 ± 0.8 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Oral cavity squamous cell carcinoma | PE/CA-PJ41 | CVCL_2680 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Prostate carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
5.3 ± 0.4 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | DU145 cell | CVCL_0105 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | High grade ovarian serous adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
5.7 ± 0.8 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | High grade ovarian serous adenocarcinoma | OVCAR-8 cell | CVCL_1629 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
5.8 ± 0.8 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
6.3 ± 0.9 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Mammary carcinoma | 4T1 cell | CVCL_0125 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Prostate carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
6.3 ± 0.3 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
6.8 ± 0.3 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Tongue squamous cell carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
7.4 ± 0.8 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Tongue squamous cell carcinoma | PE/CA-PJ15 cell | CVCL_2678 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
8.4 ± 0.3 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | A2058 cell | CVCL_1059 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
9.0 ± 0.8 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
9.00 ± 1.33µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 17 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Lung adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
9.7 ± 0.6 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Lung adenocarcinoma | A-549 cell | CVCL_0023 | ||
| Experiment 18 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Minimally invasive lung adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
10.5 ± 1.1 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Minimally invasive lung adenocarcinoma | H1650 cell | CVCL_1483 | ||
| Experiment 19 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Normal | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
10.6 ± 0.2 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | C-26 cell | CVCL_XC68 | ||
| Experiment 20 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
12.7 ± 1.5 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | WM983B cell | CVCL_6809 | ||
| Experiment 21 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Amelanotic melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
13.5 ± 1.1 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | HT168-M1/M9 cell | CVCL_2H39 | ||
| Experiment 22 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
15.5 ± 1.7 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
| Experiment 23 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
15.93 ± 0.99 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
| Experiment 24 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Amelanotic melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
16.2 ± 0.2 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | M24 cell | CVCL_D032 | ||
| Experiment 25 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Invasive breast carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
16.5 ± 1.2 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 26 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Normal | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
41.9 ± 3.8 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MRC-5 cell | CVCL_0440 | ||
| Experiment 27 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Ovarian serous adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
46.0 ± 1.3 µM
|
|||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous adenocarcinoma | OVCAR-3 cell | CVCL_0465 | ||
| Experiment 28 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Pancreatic ductal adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 100 µM | |||
| Administration Time | 24 h | ||||
| Description |
The anti-proliferative effect of the GnRH-III conjugates 1 and 2, as well as free Dau, was tested on wide range of cancer cell lines from different origin and also on MRC-5 (human fibroblast) as non-cancerous control cell line. The data showed that both conjugates possess an anti-proliferative effect on all cell types (Table 1). Except for the ovarian cancer cell lines A2780 and OVCAR-8, conjugate 2 displayed higher anti-proliferative activity than conjugate 1, depending on the type of cancer cells. The lowest activity was measured on PANC-1 pancreatic cancer cells, whereby a high IC50 value was also obtained on MRC-5 cells, showing selectivity of the conjugates for cancer cell lines. The obtained IC50 values of the conjugates vary mostly in the low micromolar range and were one to two order of magnitude higher when compared to free Dau that can enter cells non-specifically by passive diffusion. Moreover, the relative potency was calculated as a ratio of conjugates IC50 and free Daus IC50 in order to show the potency of the conjugates independently from the cell line, due to different activity of free Dau. A lower value of relative potency indicates that the conjugates IC50 value is closer to the free Daus IC50 value, which implies that the targeting capacity of the conjugate as well as its anti-tumor effect is stronger on a particular cell line, as compared to a cell line with higher relative potency. The BC cell lines showed good response to the conjugates by IC50 values, as well as by relative potency. Besides, the conjugates showed high anti-proliferative activity on mice CRC cell line C26, while the conjugates showed a moderate anti-proliferative activity on HT-29 human colon adenocarcinoma, but the relative potency was in the same range as for the BC cells.
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
[8Lys(Dox-O-glut)]-GnRH-III [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
0.1 ± 0.1 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
The efficacy of current chemotherapeutic treatments against most solid tumors is limited by their systemic toxicity, which is partly associated with the cytotoxic properties of agents such as docetaxel or doxorubicin. To avoid or minimize adverse effects from chemotherapeutic molecules, a promising targeted approach is through peptide-drug conjugates (PDCs) that link anticancer molecules to peptides designed to interact with receptors highly expressed on cancer cells, and which can mediate the molecules rapid internalization within those cells. One such receptor is sortilin (SORT1), also known as neurotensin receptor3, a membranebound receptor that belongs to the VPS10P family of receptors. TH19P01 peptide was recently designed to target and exploit SORT1s ligand internalization function. Studies have confirmed that both TH1902 (a docetaxel-TH19P01 conjugate) and TH1904 (a doxorubicin-TH19P01 conjugate) require a SORT1-dependent mechanism of action to exert anticancer activities. In recent preclinical studies performed in immunocompromised animal models, which are unable to produce mature T-cells, TH1902 was effective against several human SORT1-positive xenograft models including triple-negative breast cancer (TNBC), ovarian cancer, and endometrial cancer.
Click to Show/Hide
|
||||
| Description |
The main drug-related toxic side effect of anthracyclines is cardiotoxicity, leading to cardiomyopathy and heart failure. Comparative experiments to determine the cardiotoxicity of peptide-anthracycline conjugates with different linkages might be informative for the other conjugates with different types of drugs as well. For this purpose, human cardiomyocytes (HCM) and human umbilical vein endothelial cells (HUVEC) were used as models. The long-term (0-72 h) cytotoxic effect of sixteen GnRH-based conjugates containing Dox and Dau was determined by real-time impedimetric sensing using the xCELLigence SP system (ACEA Biosciences, San Diego, CA, USA) [75]. The results indicated that the ester-linked GnRH-Dox conjugates, including Zoptarelin Doxorubicin, showed significant toxicity at 100 nM and 1 uM, which was remarkably pronounced on the HCM cells. The cytotoxic effect was comparable to that of the free drug, especially at the highest concentration. In contrast, the conjugates with oxime-linked Dau showed no or only a minor toxicity on both the cell lines (Table 2). These data confirm that the linkage between the payload and homing peptide has a significant influence on early drug release and, consequently, an undesired toxic side effect. We may also conclude that the search for more suitable homing peptides might be more important than the application of cleavable bonds between the drug and the peptide to develop efficient DDSs.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.4 ± 0.2 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
The main drug-related toxic side effect of anthracyclines is cardiotoxicity, leading to cardiomyopathy and heart failure. Comparative experiments to determine the cardiotoxicity of peptide-anthracycline conjugates with different linkages might be informative for the other conjugates with different types of drugs as well. For this purpose, human cardiomyocytes (HCM) and human umbilical vein endothelial cells (HUVEC) were used as models. The long-term (0-72 h) cytotoxic effect of sixteen GnRH-based conjugates containing Dox and Dau was determined by real-time impedimetric sensing using the xCELLigence SP system (ACEA Biosciences, San Diego, CA, USA) [75]. The results indicated that the ester-linked GnRH-Dox conjugates, including Zoptarelin Doxorubicin, showed significant toxicity at 100 nM and 1 uM, which was remarkably pronounced on the HCM cells. The cytotoxic effect was comparable to that of the free drug, especially at the highest concentration. In contrast, the conjugates with oxime-linked Dau showed no or only a minor toxicity on both the cell lines (Table 2). These data confirm that the linkage between the payload and homing peptide has a significant influence on early drug release and, consequently, an undesired toxic side effect. We may also conclude that the search for more suitable homing peptides might be more important than the application of cleavable bonds between the drug and the peptide to develop efficient DDSs.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
GnRH-III-[2ΔHis-3D-Tic, 8Lys(glutaryl-Val-Ala-PABC-diamine-PTX)] conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
0.51 ± 0.11 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
0.67 ± 0.07 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
5.03 ± 1.91 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
6.44 ± 1.22 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
10.54 ± 2.01 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Normal human pituitary cell | Homo sapiens | ||
| Experiment 6 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
10.65 ± 1.82 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate cancer | Human prostate cancer cells | Homo sapiens | ||
| Experiment 7 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
14.88 ± 0.33 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate cancer | Human prostate cancer cells | Homo sapiens | ||
| Experiment 8 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
18.49 ± 2.72 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Normal human pituitary cell | Homo sapiens | ||
GnRH-III-[2His-3Trp,8Lys(glutaryl-Dau)] conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
0.66 ± 0.18 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4.89 ± 1.08 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
8.47 ± 1.06 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate cancer | Human prostate cancer cells | Homo sapiens | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
9.86 ± 0.82 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Normal human pituitary cell | Homo sapiens | ||
GnRH-III-[2ΔHis-3D-Tic,8Lys(glutaryl-Dau)] conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
0.77 ± 0.08 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
8.15 ± 3.22 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
10.82 ± 1.98 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Normal human pituitary cell | Homo sapiens | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
12.73 ± 2.23 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate cancer | Human prostate cancer cells | Homo sapiens | ||
[8Lys(Dau=Aoa-LRRY)]-GnRH-III [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.8 ± 0.5 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
As a payload for PDCs (and ADCs), analogues of the duocarmycins are attractive. Duocarmycin SA and yatakemycin rank among the most potent natural cytotoxins discovered. The cyclopropyl and prodrug seco forms are both naturally occurring and equipotent in most circumstances. Studies of the binding-driven bonding model of their interaction with DNA suggest that their utility will be enhanced when targeted to tumor cells. In fact, SYD985, an ADC that utilizes a peptide linker for a duocarmycin analogue to trastuzumab, has recently been progressed to phase III clinical trial.
Click to Show/Hide
|
||||
| Description |
The main drug-related toxic side effect of anthracyclines is cardiotoxicity, leading to cardiomyopathy and heart failure. Comparative experiments to determine the cardiotoxicity of peptide-anthracycline conjugates with different linkages might be informative for the other conjugates with different types of drugs as well. For this purpose, human cardiomyocytes (HCM) and human umbilical vein endothelial cells (HUVEC) were used as models. The long-term (0-72 h) cytotoxic effect of sixteen GnRH-based conjugates containing Dox and Dau was determined by real-time impedimetric sensing using the xCELLigence SP system (ACEA Biosciences, San Diego, CA, USA) [75]. The results indicated that the ester-linked GnRH-Dox conjugates, including Zoptarelin Doxorubicin, showed significant toxicity at 100 nM and 1 uM, which was remarkably pronounced on the HCM cells. The cytotoxic effect was comparable to that of the free drug, especially at the highest concentration. In contrast, the conjugates with oxime-linked Dau showed no or only a minor toxicity on both the cell lines (Table 2). These data confirm that the linkage between the payload and homing peptide has a significant influence on early drug release and, consequently, an undesired toxic side effect. We may also conclude that the search for more suitable homing peptides might be more important than the application of cleavable bonds between the drug and the peptide to develop efficient DDSs.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
28.6 ± 5.5 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
The main drug-related toxic side effect of anthracyclines is cardiotoxicity, leading to cardiomyopathy and heart failure. Comparative experiments to determine the cardiotoxicity of peptide-anthracycline conjugates with different linkages might be informative for the other conjugates with different types of drugs as well. For this purpose, human cardiomyocytes (HCM) and human umbilical vein endothelial cells (HUVEC) were used as models. The long-term (0-72 h) cytotoxic effect of sixteen GnRH-based conjugates containing Dox and Dau was determined by real-time impedimetric sensing using the xCELLigence SP system (ACEA Biosciences, San Diego, CA, USA) [75]. The results indicated that the ester-linked GnRH-Dox conjugates, including Zoptarelin Doxorubicin, showed significant toxicity at 100 nM and 1 uM, which was remarkably pronounced on the HCM cells. The cytotoxic effect was comparable to that of the free drug, especially at the highest concentration. In contrast, the conjugates with oxime-linked Dau showed no or only a minor toxicity on both the cell lines (Table 2). These data confirm that the linkage between the payload and homing peptide has a significant influence on early drug release and, consequently, an undesired toxic side effect. We may also conclude that the search for more suitable homing peptides might be more important than the application of cleavable bonds between the drug and the peptide to develop efficient DDSs.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
GnRH-III-[2ΔHis, 3D-Tic, 7D-Trp 8Lys(Dau=Aoa)] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.90 ± 0.58 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
7.88 ± 1.24 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
10.70 ± 0.95 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
GnRH-III-[4Lys(Bu), 8Lys(Dau=Aoa), 10ΔGly-NH-Et] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.23 ± 0.40 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
12.41 ± 2.30 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
17.84 ± 0.08 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
GnRH-III-[8Lys(Dau=Aoa)] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.54 ± 0.67µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
6.5 ± 1.8 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
The cancer phenotype is commonly associated with aberrant glycosylation patterns. One glycan that is directly linked to cancer is the Thomsen-Friedenreich antigen (TF or CD176). It is a disaccharide composed of a galactose β1-3 N-acetylgalactosamine, O-linked to a glycoprotein through serine or threonine residues and commonly written as Galβ1-3GalNAc--O-Ser/Thr. The TF is therapeutically attractive due to its cryptic nature in normal cells and exposure in embryonic and cancer cells. The expression of the TF has been demonstrated in 90% of primary human carcinomas, including in the lung, the breast, and the pancreas. Additionally, cancer initiating cells or cancer stem cells in the lung, liver, and colon express the TF. The peptide sequence HGRFILPWWYAFSPS (TF-peptide) is known to bind tightly to the TF (Kd = 1.2 M) and has been demonstrated to inhibit processes directly involved in TF accessibility.
Click to Show/Hide
|
||||
| Description |
The main drug-related toxic side effect of anthracyclines is cardiotoxicity, leading to cardiomyopathy and heart failure. Comparative experiments to determine the cardiotoxicity of peptide-anthracycline conjugates with different linkages might be informative for the other conjugates with different types of drugs as well. For this purpose, human cardiomyocytes (HCM) and human umbilical vein endothelial cells (HUVEC) were used as models. The long-term (0-72 h) cytotoxic effect of sixteen GnRH-based conjugates containing Dox and Dau was determined by real-time impedimetric sensing using the xCELLigence SP system (ACEA Biosciences, San Diego, CA, USA) [75]. The results indicated that the ester-linked GnRH-Dox conjugates, including Zoptarelin Doxorubicin, showed significant toxicity at 100 nM and 1 uM, which was remarkably pronounced on the HCM cells. The cytotoxic effect was comparable to that of the free drug, especially at the highest concentration. In contrast, the conjugates with oxime-linked Dau showed no or only a minor toxicity on both the cell lines (Table 2). These data confirm that the linkage between the payload and homing peptide has a significant influence on early drug release and, consequently, an undesired toxic side effect. We may also conclude that the search for more suitable homing peptides might be more important than the application of cleavable bonds between the drug and the peptide to develop efficient DDSs.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
8.22 ± 0.13 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
13.89 ± 3.62 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
| Experiment 5 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
14.2 ± 3.2 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
In our experiments, GnRH-III was applied as a targeting peptide. The conjugate [8Lys(Dau=Aoa-GFLG)]-GnRH-III, in which Dau was connected to the side chain of Lys in position 8 through an aminooxyacetylated cathepsin B-labile GFLG spacer, showed significant tumor growth inhibition in s.c.-developed fast-growing C26 murine colon cancer-bearing Balb/c mice. The effect highly depended on the treatment schedule. In the first attempt, the conjugate was administered i.p. at a dose of 8.86 umol (5 mg Dau-content/kg body weight) five times on days 7, 9, 11, 14 and 16 after tumor transplantation (Treatment schedule A). The tumor volume inhibition was only 15.7% on day 26 when the experiment finished. A slight enhancement in inhibition (22.3%) was detected when a dose of conjugate corresponding to 15 mg Dau content/kg body weight (26.6 μmol) was injected only once on day 7 (Treatment schedule B). An additional treatment on day 10 (Treatment schedule C) did not result in any further improvements (21.9% on day 29). However, when the treatment schedule was changed to two treatments with the same dose on days 4 and 7, 46.3% inhibition was observed on day 29 (Treatment schedule D). In contrast, the treatments with free Dau at a dose of 2 mg/kg body weight (3.55 umol) on days 7, 9, 11, 14 and 16 showed only 22.6% inhibition on day 26. The median survival rates of the treated animals in comparison to the control group were 1 (A), 1.23 (B), 1.21 (C) and 1.38 (D), respectively, and 0.81 for free Dau. These results indicate the lower toxicity and the higher tumor volume inhibition effect of the conjugates in comparison with those of free Dau, as well as the importance of the treatment schedule. In another experiment, HT-29 human colon cancer was developed s.c. in immunodeficient SCID mice. Dau and two conjugates (with or without a GFLG spacer between the GnRH-III homing peptide and the payload) were used for the treatment. The first i.p. administrations were performed on day 13 after tumor inoculation. All the mice treated once with 2.5 mg/kg body weight (4.43 umol) free Dau died within 10 days. In contrast, the conjugates at a dose of 15 mg Dau content/kg body weight (26.6 umol) that refers to 52 mg/kg [8Lys(Dau=Aoa)]-GnRH-III and 62.5 mg/kg [8Lys(Dau=Aoa-GFLG)]-GnRH-III conjugates, respectively, did not show significant toxicity. The treatment was repeated on days 23 and 30. Because of the significant weight loss in several mice in the control group, the experiment was terminated on day 35. The tumor growth inhibition could be calculated as reductions in the tumor volume by 44.3% and 57.6% and the tumor weight by 41% and 50%, respectively. Interestingly, the conjugates were poorly effective on orthotopically developed tumors. In the case of the C26 colon tumor-bearing female Balb/c mice, only a 7% reduction in tumor weight was detected on day 13 after the two treatments on days 4 and 7 with [8Lys(Dau=Aoa)]-GnRH-III, at a 26.6 umol/kg (15 mg Dau content) dose. The effect of free Dau (2 mg/kg on days 4 and 7) showed a better inhibitory effect (24.4%). Interestingly, our novel developed GnRH-III derivative, in which Ser in position 4 was replaced by Lys(Ac), was much more potent, with 49.3% inhibition. It is worth mentioning that the rate of cellular uptake of the [4Lys(Ac), 8Lys(Dau=Aoa)]-GnRH-III conjugate by the tumor cells was significantly higher than that of the conjugate with the native GnRH-III sequence.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
| Experiment 6 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
27.8 ± 4.2 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
The main drug-related toxic side effect of anthracyclines is cardiotoxicity, leading to cardiomyopathy and heart failure. Comparative experiments to determine the cardiotoxicity of peptide-anthracycline conjugates with different linkages might be informative for the other conjugates with different types of drugs as well. For this purpose, human cardiomyocytes (HCM) and human umbilical vein endothelial cells (HUVEC) were used as models. The long-term (0-72 h) cytotoxic effect of sixteen GnRH-based conjugates containing Dox and Dau was determined by real-time impedimetric sensing using the xCELLigence SP system (ACEA Biosciences, San Diego, CA, USA) [75]. The results indicated that the ester-linked GnRH-Dox conjugates, including Zoptarelin Doxorubicin, showed significant toxicity at 100 nM and 1 uM, which was remarkably pronounced on the HCM cells. The cytotoxic effect was comparable to that of the free drug, especially at the highest concentration. In contrast, the conjugates with oxime-linked Dau showed no or only a minor toxicity on both the cell lines (Table 2). These data confirm that the linkage between the payload and homing peptide has a significant influence on early drug release and, consequently, an undesired toxic side effect. We may also conclude that the search for more suitable homing peptides might be more important than the application of cleavable bonds between the drug and the peptide to develop efficient DDSs.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
GnRH-III-[3D-Tic, 4Lys(Bu), 8Lys(Dau=Aoa)] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.56 ± 0.51 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
12.73 ± 3.10 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
GnRH-III-[3D-Tic, 4Lys(Bu), 7D-Trp 8Lys(Dau=Aoa)] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.57 ± 0.47 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
16.83 ± 0.66 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
GnRH-III-[2ΔHis, 3D-Tic, 8Lys(Dau=Aoa)] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.75 ± 0.17 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
9.35 ± 1.93 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
10.32 ± 1.32 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
GnRH-III-[2ΔHis, 3D-Tic, 4Lys(Bu), 7D-Trp 8Lys(Dau=Aoa)] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.81 ± 0.04 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
8.18 ± 0.18 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
16.55 ± 0.30 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
GnRH-III-[2ΔHis-3D-Tic, 8Lys(glutaryl-Val-Ala-PABC-Dau conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.85 ± 0.90 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
20.54 ± 1.46 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate cancer | Human prostate cancer cells | Homo sapiens | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
24.77 ± 1.73 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Normal human pituitary cell | Homo sapiens | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 100 µM | |||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
GnRH-III-[3D-Tic, 8Lys(Dau=Aoa)] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.89 ± 0.62 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
8.75 ± 0.86 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
[4Lys(Dau=Aoa),8Lys(Dau=Aoa)]-GnRH-III [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.9 ± 0.9 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
As a payload for PDCs (and ADCs), analogues of the duocarmycins are attractive. Duocarmycin SA and yatakemycin rank among the most potent natural cytotoxins discovered. The cyclopropyl and prodrug seco forms are both naturally occurring and equipotent in most circumstances. Studies of the binding-driven bonding model of their interaction with DNA suggest that their utility will be enhanced when targeted to tumor cells. In fact, SYD985, an ADC that utilizes a peptide linker for a duocarmycin analogue to trastuzumab, has recently been progressed to phase III clinical trial.
Click to Show/Hide
|
||||
| Description |
The main drug-related toxic side effect of anthracyclines is cardiotoxicity, leading to cardiomyopathy and heart failure. Comparative experiments to determine the cardiotoxicity of peptide-anthracycline conjugates with different linkages might be informative for the other conjugates with different types of drugs as well. For this purpose, human cardiomyocytes (HCM) and human umbilical vein endothelial cells (HUVEC) were used as models. The long-term (0-72 h) cytotoxic effect of sixteen GnRH-based conjugates containing Dox and Dau was determined by real-time impedimetric sensing using the xCELLigence SP system (ACEA Biosciences, San Diego, CA, USA) [75]. The results indicated that the ester-linked GnRH-Dox conjugates, including Zoptarelin Doxorubicin, showed significant toxicity at 100 nM and 1 uM, which was remarkably pronounced on the HCM cells. The cytotoxic effect was comparable to that of the free drug, especially at the highest concentration. In contrast, the conjugates with oxime-linked Dau showed no or only a minor toxicity on both the cell lines (Table 2). These data confirm that the linkage between the payload and homing peptide has a significant influence on early drug release and, consequently, an undesired toxic side effect. We may also conclude that the search for more suitable homing peptides might be more important than the application of cleavable bonds between the drug and the peptide to develop efficient DDSs.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
6.8 ± 1.0 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
The main drug-related toxic side effect of anthracyclines is cardiotoxicity, leading to cardiomyopathy and heart failure. Comparative experiments to determine the cardiotoxicity of peptide-anthracycline conjugates with different linkages might be informative for the other conjugates with different types of drugs as well. For this purpose, human cardiomyocytes (HCM) and human umbilical vein endothelial cells (HUVEC) were used as models. The long-term (0-72 h) cytotoxic effect of sixteen GnRH-based conjugates containing Dox and Dau was determined by real-time impedimetric sensing using the xCELLigence SP system (ACEA Biosciences, San Diego, CA, USA) [75]. The results indicated that the ester-linked GnRH-Dox conjugates, including Zoptarelin Doxorubicin, showed significant toxicity at 100 nM and 1 uM, which was remarkably pronounced on the HCM cells. The cytotoxic effect was comparable to that of the free drug, especially at the highest concentration. In contrast, the conjugates with oxime-linked Dau showed no or only a minor toxicity on both the cell lines (Table 2). These data confirm that the linkage between the payload and homing peptide has a significant influence on early drug release and, consequently, an undesired toxic side effect. We may also conclude that the search for more suitable homing peptides might be more important than the application of cleavable bonds between the drug and the peptide to develop efficient DDSs.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
[8Lys(Dau=Aoa-K(Dau=Aoa))]GnRH-III [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.0 ± 0.4 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
The cancer phenotype is commonly associated with aberrant glycosylation patterns. One glycan that is directly linked to cancer is the Thomsen-Friedenreich antigen (TF or CD176). It is a disaccharide composed of a galactose β1-3 N-acetylgalactosamine, O-linked to a glycoprotein through serine or threonine residues and commonly written as Galβ1-3GalNAc--O-Ser/Thr. The TF is therapeutically attractive due to its cryptic nature in normal cells and exposure in embryonic and cancer cells. The expression of the TF has been demonstrated in 90% of primary human carcinomas, including in the lung, the breast, and the pancreas. Additionally, cancer initiating cells or cancer stem cells in the lung, liver, and colon express the TF. The peptide sequence HGRFILPWWYAFSPS (TF-peptide) is known to bind tightly to the TF (Kd = 1.2 M) and has been demonstrated to inhibit processes directly involved in TF accessibility.
Click to Show/Hide
|
||||
| Description |
The main drug-related toxic side effect of anthracyclines is cardiotoxicity, leading to cardiomyopathy and heart failure. Comparative experiments to determine the cardiotoxicity of peptide-anthracycline conjugates with different linkages might be informative for the other conjugates with different types of drugs as well. For this purpose, human cardiomyocytes (HCM) and human umbilical vein endothelial cells (HUVEC) were used as models. The long-term (0-72 h) cytotoxic effect of sixteen GnRH-based conjugates containing Dox and Dau was determined by real-time impedimetric sensing using the xCELLigence SP system (ACEA Biosciences, San Diego, CA, USA) [75]. The results indicated that the ester-linked GnRH-Dox conjugates, including Zoptarelin Doxorubicin, showed significant toxicity at 100 nM and 1 uM, which was remarkably pronounced on the HCM cells. The cytotoxic effect was comparable to that of the free drug, especially at the highest concentration. In contrast, the conjugates with oxime-linked Dau showed no or only a minor toxicity on both the cell lines (Table 2). These data confirm that the linkage between the payload and homing peptide has a significant influence on early drug release and, consequently, an undesired toxic side effect. We may also conclude that the search for more suitable homing peptides might be more important than the application of cleavable bonds between the drug and the peptide to develop efficient DDSs.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
5.6 ± 2.0 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
The main drug-related toxic side effect of anthracyclines is cardiotoxicity, leading to cardiomyopathy and heart failure. Comparative experiments to determine the cardiotoxicity of peptide-anthracycline conjugates with different linkages might be informative for the other conjugates with different types of drugs as well. For this purpose, human cardiomyocytes (HCM) and human umbilical vein endothelial cells (HUVEC) were used as models. The long-term (0-72 h) cytotoxic effect of sixteen GnRH-based conjugates containing Dox and Dau was determined by real-time impedimetric sensing using the xCELLigence SP system (ACEA Biosciences, San Diego, CA, USA) [75]. The results indicated that the ester-linked GnRH-Dox conjugates, including Zoptarelin Doxorubicin, showed significant toxicity at 100 nM and 1 uM, which was remarkably pronounced on the HCM cells. The cytotoxic effect was comparable to that of the free drug, especially at the highest concentration. In contrast, the conjugates with oxime-linked Dau showed no or only a minor toxicity on both the cell lines (Table 2). These data confirm that the linkage between the payload and homing peptide has a significant influence on early drug release and, consequently, an undesired toxic side effect. We may also conclude that the search for more suitable homing peptides might be more important than the application of cleavable bonds between the drug and the peptide to develop efficient DDSs.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
[4Lys(Ac),8Lys(Dau=Aoa)]-GnRH-III [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.1 ± 1.7 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
The cancer phenotype is commonly associated with aberrant glycosylation patterns. One glycan that is directly linked to cancer is the Thomsen-Friedenreich antigen (TF or CD176). It is a disaccharide composed of a galactose β1-3 N-acetylgalactosamine, O-linked to a glycoprotein through serine or threonine residues and commonly written as Galβ1-3GalNAc--O-Ser/Thr. The TF is therapeutically attractive due to its cryptic nature in normal cells and exposure in embryonic and cancer cells. The expression of the TF has been demonstrated in 90% of primary human carcinomas, including in the lung, the breast, and the pancreas. Additionally, cancer initiating cells or cancer stem cells in the lung, liver, and colon express the TF. The peptide sequence HGRFILPWWYAFSPS (TF-peptide) is known to bind tightly to the TF (Kd = 1.2 M) and has been demonstrated to inhibit processes directly involved in TF accessibility.
Click to Show/Hide
|
||||
| Description |
The main drug-related toxic side effect of anthracyclines is cardiotoxicity, leading to cardiomyopathy and heart failure. Comparative experiments to determine the cardiotoxicity of peptide-anthracycline conjugates with different linkages might be informative for the other conjugates with different types of drugs as well. For this purpose, human cardiomyocytes (HCM) and human umbilical vein endothelial cells (HUVEC) were used as models. The long-term (0-72 h) cytotoxic effect of sixteen GnRH-based conjugates containing Dox and Dau was determined by real-time impedimetric sensing using the xCELLigence SP system (ACEA Biosciences, San Diego, CA, USA) [75]. The results indicated that the ester-linked GnRH-Dox conjugates, including Zoptarelin Doxorubicin, showed significant toxicity at 100 nM and 1 uM, which was remarkably pronounced on the HCM cells. The cytotoxic effect was comparable to that of the free drug, especially at the highest concentration. In contrast, the conjugates with oxime-linked Dau showed no or only a minor toxicity on both the cell lines (Table 2). These data confirm that the linkage between the payload and homing peptide has a significant influence on early drug release and, consequently, an undesired toxic side effect. We may also conclude that the search for more suitable homing peptides might be more important than the application of cleavable bonds between the drug and the peptide to develop efficient DDSs.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
7.4 ± 2.6 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
In our experiments, GnRH-III was applied as a targeting peptide. The conjugate [8Lys(Dau=Aoa-GFLG)]-GnRH-III, in which Dau was connected to the side chain of Lys in position 8 through an aminooxyacetylated cathepsin B-labile GFLG spacer, showed significant tumor growth inhibition in s.c.-developed fast-growing C26 murine colon cancer-bearing Balb/c mice. The effect highly depended on the treatment schedule. In the first attempt, the conjugate was administered i.p. at a dose of 8.86 umol (5 mg Dau-content/kg body weight) five times on days 7, 9, 11, 14 and 16 after tumor transplantation (Treatment schedule A). The tumor volume inhibition was only 15.7% on day 26 when the experiment finished. A slight enhancement in inhibition (22.3%) was detected when a dose of conjugate corresponding to 15 mg Dau content/kg body weight (26.6 μmol) was injected only once on day 7 (Treatment schedule B). An additional treatment on day 10 (Treatment schedule C) did not result in any further improvements (21.9% on day 29). However, when the treatment schedule was changed to two treatments with the same dose on days 4 and 7, 46.3% inhibition was observed on day 29 (Treatment schedule D). In contrast, the treatments with free Dau at a dose of 2 mg/kg body weight (3.55 umol) on days 7, 9, 11, 14 and 16 showed only 22.6% inhibition on day 26. The median survival rates of the treated animals in comparison to the control group were 1 (A), 1.23 (B), 1.21 (C) and 1.38 (D), respectively, and 0.81 for free Dau. These results indicate the lower toxicity and the higher tumor volume inhibition effect of the conjugates in comparison with those of free Dau, as well as the importance of the treatment schedule. In another experiment, HT-29 human colon cancer was developed s.c. in immunodeficient SCID mice. Dau and two conjugates (with or without a GFLG spacer between the GnRH-III homing peptide and the payload) were used for the treatment. The first i.p. administrations were performed on day 13 after tumor inoculation. All the mice treated once with 2.5 mg/kg body weight (4.43 umol) free Dau died within 10 days. In contrast, the conjugates at a dose of 15 mg Dau content/kg body weight (26.6 umol) that refers to 52 mg/kg [8Lys(Dau=Aoa)]-GnRH-III and 62.5 mg/kg [8Lys(Dau=Aoa-GFLG)]-GnRH-III conjugates, respectively, did not show significant toxicity. The treatment was repeated on days 23 and 30. Because of the significant weight loss in several mice in the control group, the experiment was terminated on day 35. The tumor growth inhibition could be calculated as reductions in the tumor volume by 44.3% and 57.6% and the tumor weight by 41% and 50%, respectively. Interestingly, the conjugates were poorly effective on orthotopically developed tumors. In the case of the C26 colon tumor-bearing female Balb/c mice, only a 7% reduction in tumor weight was detected on day 13 after the two treatments on days 4 and 7 with [8Lys(Dau=Aoa)]-GnRH-III, at a 26.6 umol/kg (15 mg Dau content) dose. The effect of free Dau (2 mg/kg on days 4 and 7) showed a better inhibitory effect (24.4%). Interestingly, our novel developed GnRH-III derivative, in which Ser in position 4 was replaced by Lys(Ac), was much more potent, with 49.3% inhibition. It is worth mentioning that the rate of cellular uptake of the [4Lys(Ac), 8Lys(Dau=Aoa)]-GnRH-III conjugate by the tumor cells was significantly higher than that of the conjugate with the native GnRH-III sequence.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
GnRH-III-[3D-Tic, 7D-Trp 8Lys(Dau=Aoa)] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.42 ± 0.39 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
15.34 ± 2.63 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
GnRH-III-[4Lys(Bu), 6Asp(OMe), 8Lys(Dau=Aoa)] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.44 ± 0.51 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
18.00 ± 0.13 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
GnRH-III-[3D-Trp, 8Lys(Dau=Aoa)] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.60 ± 0.28 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
15.25 ± 2.51 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
[8Lys(Dau=Aoa-GFLG)]-GnRH-III [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.9 ± 1.2 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
As a payload for PDCs (and ADCs), analogues of the duocarmycins are attractive. Duocarmycin SA and yatakemycin rank among the most potent natural cytotoxins discovered. The cyclopropyl and prodrug seco forms are both naturally occurring and equipotent in most circumstances. Studies of the binding-driven bonding model of their interaction with DNA suggest that their utility will be enhanced when targeted to tumor cells. In fact, SYD985, an ADC that utilizes a peptide linker for a duocarmycin analogue to trastuzumab, has recently been progressed to phase III clinical trial.
Click to Show/Hide
|
||||
| Description |
The main drug-related toxic side effect of anthracyclines is cardiotoxicity, leading to cardiomyopathy and heart failure. Comparative experiments to determine the cardiotoxicity of peptide-anthracycline conjugates with different linkages might be informative for the other conjugates with different types of drugs as well. For this purpose, human cardiomyocytes (HCM) and human umbilical vein endothelial cells (HUVEC) were used as models. The long-term (0-72 h) cytotoxic effect of sixteen GnRH-based conjugates containing Dox and Dau was determined by real-time impedimetric sensing using the xCELLigence SP system (ACEA Biosciences, San Diego, CA, USA) [75]. The results indicated that the ester-linked GnRH-Dox conjugates, including Zoptarelin Doxorubicin, showed significant toxicity at 100 nM and 1 uM, which was remarkably pronounced on the HCM cells. The cytotoxic effect was comparable to that of the free drug, especially at the highest concentration. In contrast, the conjugates with oxime-linked Dau showed no or only a minor toxicity on both the cell lines (Table 2). These data confirm that the linkage between the payload and homing peptide has a significant influence on early drug release and, consequently, an undesired toxic side effect. We may also conclude that the search for more suitable homing peptides might be more important than the application of cleavable bonds between the drug and the peptide to develop efficient DDSs.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
19.4 ± 3.1 µM
|
|||
| Administration Time | 72 h | ||||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
In our experiments, GnRH-III was applied as a targeting peptide. The conjugate [8Lys(Dau=Aoa-GFLG)]-GnRH-III, in which Dau was connected to the side chain of Lys in position 8 through an aminooxyacetylated cathepsin B-labile GFLG spacer, showed significant tumor growth inhibition in s.c.-developed fast-growing C26 murine colon cancer-bearing Balb/c mice. The effect highly depended on the treatment schedule. In the first attempt, the conjugate was administered i.p. at a dose of 8.86 umol (5 mg Dau-content/kg body weight) five times on days 7, 9, 11, 14 and 16 after tumor transplantation (Treatment schedule A). The tumor volume inhibition was only 15.7% on day 26 when the experiment finished. A slight enhancement in inhibition (22.3%) was detected when a dose of conjugate corresponding to 15 mg Dau content/kg body weight (26.6 μmol) was injected only once on day 7 (Treatment schedule B). An additional treatment on day 10 (Treatment schedule C) did not result in any further improvements (21.9% on day 29). However, when the treatment schedule was changed to two treatments with the same dose on days 4 and 7, 46.3% inhibition was observed on day 29 (Treatment schedule D). In contrast, the treatments with free Dau at a dose of 2 mg/kg body weight (3.55 umol) on days 7, 9, 11, 14 and 16 showed only 22.6% inhibition on day 26. The median survival rates of the treated animals in comparison to the control group were 1 (A), 1.23 (B), 1.21 (C) and 1.38 (D), respectively, and 0.81 for free Dau. These results indicate the lower toxicity and the higher tumor volume inhibition effect of the conjugates in comparison with those of free Dau, as well as the importance of the treatment schedule. In another experiment, HT-29 human colon cancer was developed s.c. in immunodeficient SCID mice. Dau and two conjugates (with or without a GFLG spacer between the GnRH-III homing peptide and the payload) were used for the treatment. The first i.p. administrations were performed on day 13 after tumor inoculation. All the mice treated once with 2.5 mg/kg body weight (4.43 umol) free Dau died within 10 days. In contrast, the conjugates at a dose of 15 mg Dau content/kg body weight (26.6 umol) that refers to 52 mg/kg [8Lys(Dau=Aoa)]-GnRH-III and 62.5 mg/kg [8Lys(Dau=Aoa-GFLG)]-GnRH-III conjugates, respectively, did not show significant toxicity. The treatment was repeated on days 23 and 30. Because of the significant weight loss in several mice in the control group, the experiment was terminated on day 35. The tumor growth inhibition could be calculated as reductions in the tumor volume by 44.3% and 57.6% and the tumor weight by 41% and 50%, respectively. Interestingly, the conjugates were poorly effective on orthotopically developed tumors. In the case of the C26 colon tumor-bearing female Balb/c mice, only a 7% reduction in tumor weight was detected on day 13 after the two treatments on days 4 and 7 with [8Lys(Dau=Aoa)]-GnRH-III, at a 26.6 umol/kg (15 mg Dau content) dose. The effect of free Dau (2 mg/kg on days 4 and 7) showed a better inhibitory effect (24.4%). Interestingly, our novel developed GnRH-III derivative, in which Ser in position 4 was replaced by Lys(Ac), was much more potent, with 49.3% inhibition. It is worth mentioning that the rate of cellular uptake of the [4Lys(Ac), 8Lys(Dau=Aoa)]-GnRH-III conjugate by the tumor cells was significantly higher than that of the conjugate with the native GnRH-III sequence.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
GnRH-III-[2ΔHis-3D-Tic,8Lys(glutaryl-Val-Cit-PABC-Dau)] conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4.24 ± 1.09 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
10.97 ± 1.23 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate cancer | Human prostate cancer cells | Homo sapiens | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
11.70 ± 0.42 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Normal human pituitary cell | Homo sapiens | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 100 µM | |||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
GnRH-III-[6Asp(OMe), 8Lys(Dau=Aoa)] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4.81 ± 0.72 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
10.66 ± 1.76 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
GnRH-III-[8Lys(Dau=Aoa), 10ΔGly-NH-Et] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4.88 ± 0.01 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
14.18 ± 3.59 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
14.33 ± 1.18 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
GnRH-III-[3D-Trp, 4Lys(Bu), 8Lys(Dau=Aoa)] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
6.64 ± 1.58 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
15.03 ± 2.51 µM
|
|||
| Description |
We obtained IC50 values in the low micromolar range on MCF-7 cells varying between 0.14 and 6.64 μM. In the case of HT-29 colon cancer cells, the determined IC50 values were slightly higher and within a range of 3.31-19.10 μM. With exception of compound 10, no significant difference of the cancer cell growth inhibitory effect of the novel bioconjugates and the control conjugates (K1 and K2) could be detected. However, the replacement of 3Trp by 3d-Tic in connection with the deletion of 2His led to an increased cytostatic effect of 10 on both of the analyzed cell lines. The IC50 value of bioconjugate 10 was more than 15-times lower on ER+ breast cancer MCF-7 cells and 5-times lower on colon cancer cells HT-29 compared to the control compound K2. Based on these promising findings, the growth inhibitory effects of conjugate 10 and the related 2His-3d-Tic (3, 5 and 12) conjugates, as well as the 10Gly-NH-Et (7 and 14) containing compounds, were studied on estrogen receptor negative (ER-) MDA-MB-231 breast cancer cells. The corresponding IC50 values are shown in Table 2. The GnRH-III-Dau conjugate 10 revealed also on this cell line the highest anticancer activity with an IC50 value of 2.49 μM. The comparison of the dose-dependent growth inhibitory effect of 10 and K2 on the three cancer cell lines is shown in Figure 1. Considering the results of all three cell lines, only compound 10 which contains the N-terminal modification 2His-d-Tic-Lys(Bu), displayed clearly a reduced cell viability, while the conjugates bearing other substitutions yielded IC50 values which vary only slightly in comparison to the controls.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
GnRH-III-[2His-3Trp,8Lys(glutaryl-Val-Ala-PABC-Dau)] conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
7.48 ± 0.66 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
15.46 ± 0.83 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate cancer | Human prostate cancer cells | Homo sapiens | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
17.04 ± 1.04 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Normal human pituitary cell | Homo sapiens | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
56.19 ± 17.28 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
GnRH-III-[2His-3Trp,8Lys(glutaryl-Val-Cit-PABC-diamine-PTX)] conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
8.61 ± 1.08 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate cancer | Human prostate cancer cells | Homo sapiens | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
9.70 ± 1.07 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Normal human pituitary cell | Homo sapiens | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
67.88 ± 25.36 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 100 µM | |||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
GnRH-III-[2His-3Trp,8Lys(glutaryl-Val-Cit-PABC-Dau)] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
11.18 ± 0.38 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
13.66 ± 1.87 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate cancer | Human prostate cancer cells | Homo sapiens | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
16.12 ± 2.03 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Normal human pituitary cell | Homo sapiens | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
85.57 ± 24.33 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
GnRH-III-[2ΔHis-3D-Tic, 8Lys(glutaryl-diamine-PTX)] conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
22.21 ± 0.96 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate cancer | Human prostate cancer cells | Homo sapiens | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
23.43 ± 1.67 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Normal human pituitary cell | Homo sapiens | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 100 µM | |||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 100 µM | |||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
GnRH-III-[2ΔHis-3D-Tic,8Lys(glutaryl-Val-Cit-PABC-diamine-PTX)] conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
36.29 ± 3.17 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Normal human pituitary cell | Homo sapiens | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
42.67 ± 7.04 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate cancer | Human prostate cancer cells | Homo sapiens | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
48.14 ± 0.47 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 100 µM | |||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
GnRH-III-[2His-3Trp,8Lys(glutaryl-diamine-PTX)] conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
41.52 ± 9.83 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 100 µM | |||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
References