
Peptide-drug Conjugate Information
General Information of This Peptide-drug Conjugate (PDC)
| PDC ID |
PDC_00194
|
|||||
|---|---|---|---|---|---|---|
| PDC Name |
GnRH-III-[2His-3Trp,8Lys(glutaryl-Val-Cit-PABC-Dau)]
|
|||||
| PDC Status |
Investigative
|
|||||
| Indication |
In total 2 Indication(s)
|
|||||
| Structure |
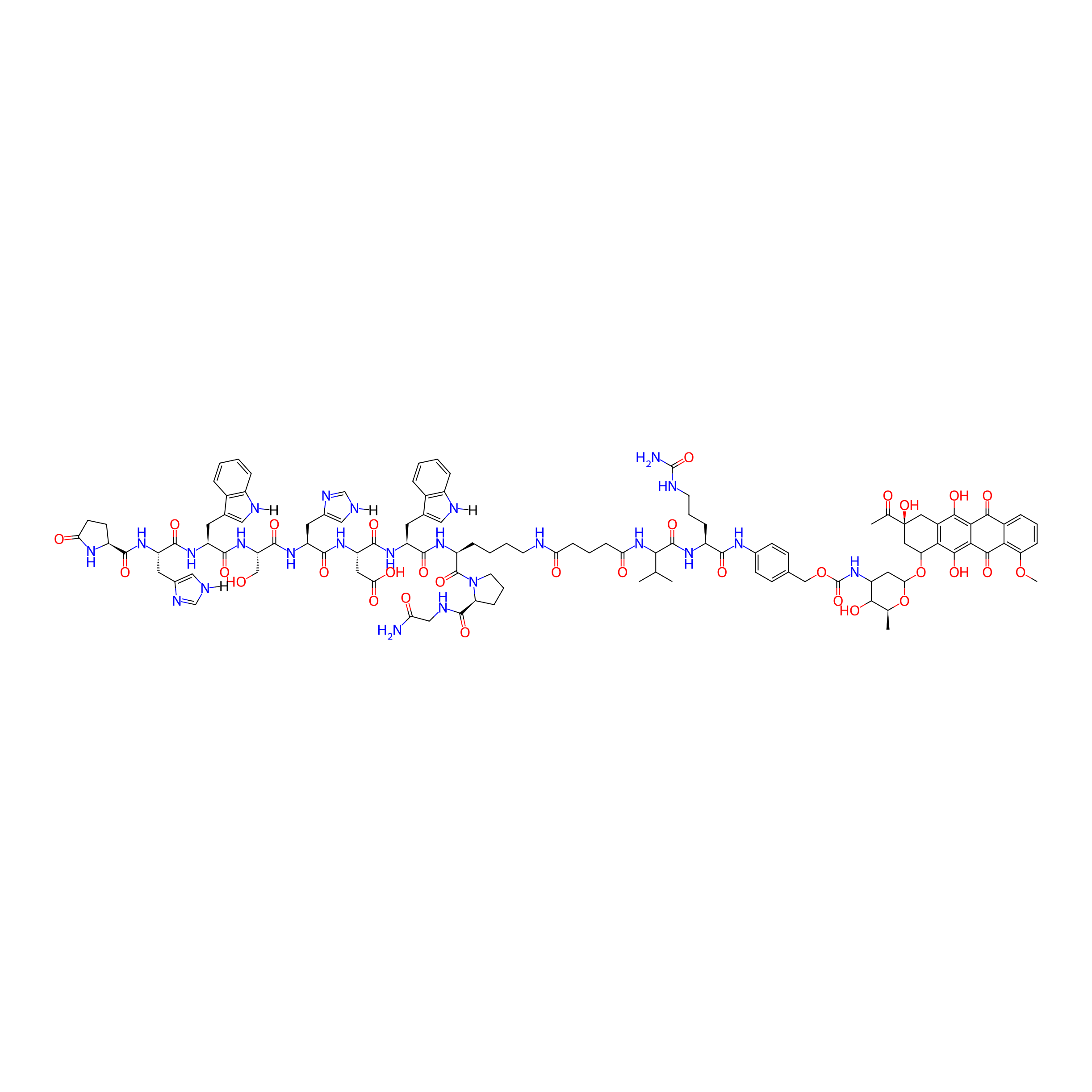
|
|||||
| Peptide Name |
GnRH-III
|
Peptide Info | ||||
| Receptor Name |
Gonadotropin-releasing hormone receptor (GNRHR)
|
Receptor Info | ||||
| Drug Name |
Daunorubicin
|
Drug Info | ||||
| Therapeutic Target |
DNA topoisomerase 2-alpha (TOP2A)
|
Target Info | ||||
| Linker Name |
Val-Cit
|
Linker Info | ||||
| Peptide Modified Type |
Amino acid modifications
|
|||||
| Modified Segment |
Incorporation of unnatural amino acids: pyroglutamic acid (
|
|||||
| Ternimal Modification |
N-terminal modification
|
|||||
| Formula |
C110H134N24O31
|
|||||
| #Ro5 Violations (Lipinski): 4 | Molecular Weight | 2288.419 | ||||
| Lipid-water partition coefficient (xlogp) | -1.4227 | |||||
| Hydrogen Bond Donor Count (hbonddonor) | 27 | |||||
| Hydrogen Bond Acceptor Count (hbondacc) | 32 | |||||
| Rotatable Bond Count (rotbonds) | 55 | |||||
Full List of Activity Data of This Peptide-drug Conjugate
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
11.18 ± 0.38 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
13.66 ± 1.87 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate cancer | Human prostate cancer cells | Homo sapiens | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
16.12 ± 2.03 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Normal human pituitary cell | Homo sapiens | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
85.57 ± 24.33 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
References