
Linker Information
General Information of This Linker
| Linker ID |
LIN00005
|
|||||
|---|---|---|---|---|---|---|
| Linker Name |
Val-Cit
|
|||||
| Linker Type |
Enzyme-sensitive linkers
|
|||||
| Structure |
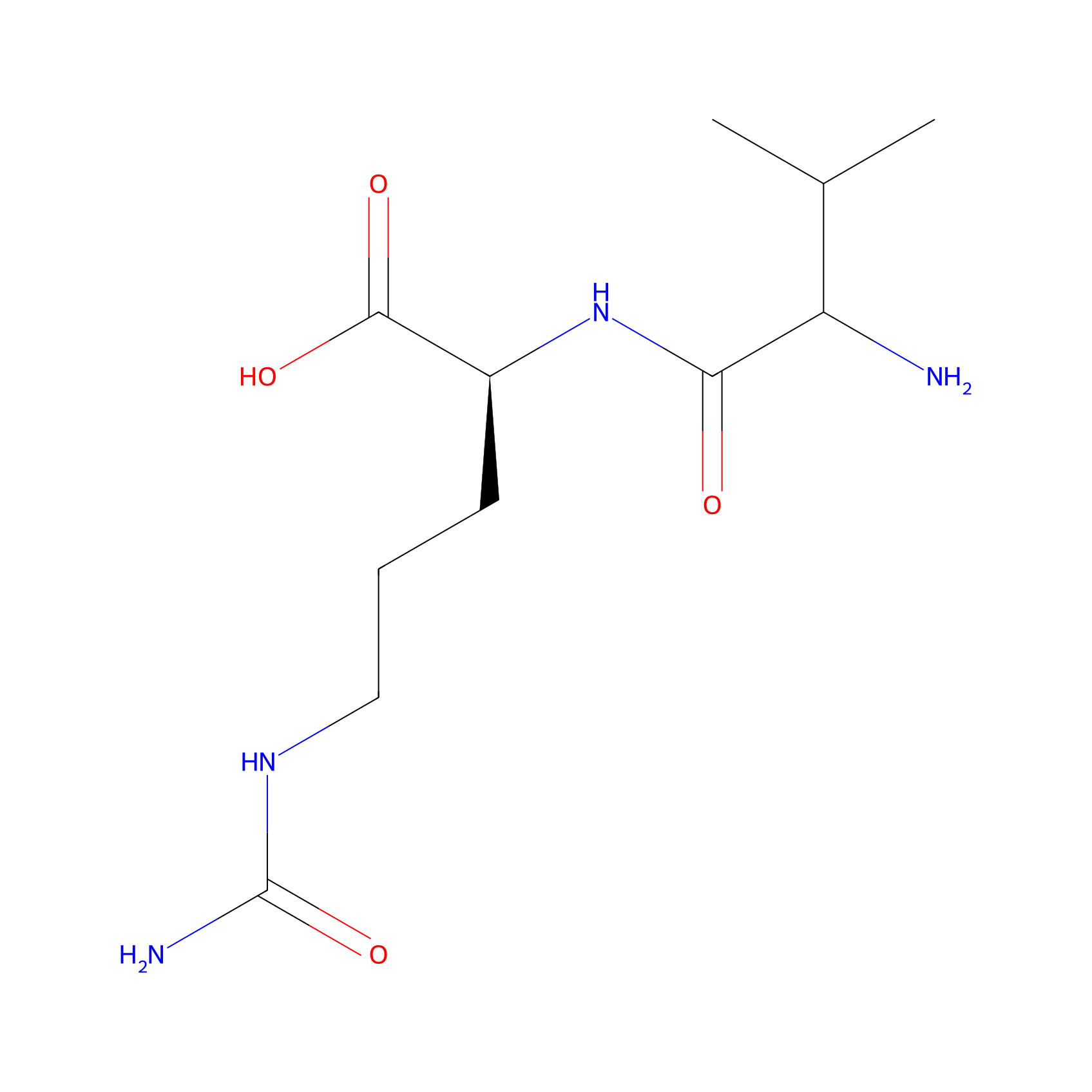
|
|||||
| Formula |
C11H22N4O4
|
|||||
| #Ro5 Violations (Lipinski): 1 | Molecular Weight (mw) | 274.32 | ||||
| Lipid-water partition coefficient (xlogp) | -4.3 | |||||
| Hydrogen Bond Donor Count (hbonddonor) | 5 | |||||
| Hydrogen Bond Acceptor Count (hbondacc) | 5 | |||||
| Rotatable Bond Count (rotbonds) | 8 | |||||
| PubChem CID | ||||||
| Canonical smiles |
CC(C)C(C(=O)NC(CCCNC(=O)N)C(=O)O)N
|
|||||
| InChI |
InChI=1S/C11H22N4O4/c1-6(2)8(12)9(16)15-7(10(17)18)4-3-5-14-11(13)19/h6-8H,3-5,12H2,1-2H3,(H,15,16)(H,17,18)(H3,13,14,19)/t7-,8?/m0/s1
|
|||||
| InChIKey |
AGGWFDNPHKLBBV-JAMMHHFISA-N
|
|||||
| IUPAC Name |
(2S)-2-[(2-amino-3-methylbutanoyl)amino]-5-(carbamoylamino)pentanoic acid
|
|||||
Each Peptide-drug Conjugate Related to This Linker
Full Information of The Activity Data of The PDC(s) Related to This Linker
BT8009 [Phase 2/3]
Identified from the Human Clinical Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
23%
|
|||
| Administration Time | 28 days | ||||
| Administration Dosage | 1 mg/kg | ||||
| MOA of PDC |
The cell adhesion molecule Nectin-4 shows elevated expression in multiple tumor types correlated with poor prognosis. Nectin-4-directed ADCs show efficacy in multiple xenograft tumor models. The ADC enfortumab vedotin (EV) delivers the antimitotic toxin MMAE to Nectin-4-expressing cells via internalization and cleavage of a valine-citrulline dipeptide linker component. Clinical validation of Nectin-4 as a target in urothelial cancer has been demonstrated with EV. In 2021, the FDA-approved EV for patients with locally advanced or metastatic urothelial cancer who had previously received a PD-1 or PD-L1 inhibitor and platinum-containing chemotherapy or were ineligible for cisplatin containing chemotherapy and had received one or more prior lines of therapy. Bicycle toxin conjugates (BTC) are structurally constrained bicyclic peptides conjugated through cleavable linkers to a toxin. Various, well-described linker toxin combinations can be incorporated into the BTC molecule (e.g., BT1718 and BT5528; refs.). They are of low molecular weight (approximately 4-4.5 kDa) and being chemically synthesized can be optimized for appropriate affinity, stability, and solubility relatively simply. Through intravenous administration, high systemic Cmax values can be attained, which, along with BTCs relatively small size, helps drive rapid diffusion into extra-vascular compartment, as reflected in a volume of distribution similar to extracellular fluid. We believe that delivery of a high number of BTCs, each carrying a reduced toxin load (peptide toxin ratio of 1:1) should improve tumor penetration and reduce the impact of the binding site barrier. BTCs show moderate clearance from the systemic vasculature, predominantly by the renal route. This overall profile marks them out from most ADCs and provides the possibility of enhanced clinical efficacy with a wider therapeutic index. BTCs are new therapeutic modality that shows a very different pharmacokinetic and structural profile to classic ADCs, whereas possessing robust tumoricidal properties. BTCs targeting MT1-MMP and EphA2 are currently in clinical trials. BT8009 is the most recent BTC to enter clinical trial. BT8009 is highly selective for Nectin-4 over other nectin family members and an extensive range of cell membrane expressed proteins. It shares the same cleavable linker and toxin combination as EV and can be cleaved by proteases in the TME, releasing cell penetrant MMAE that diffuses into tumor cells, or bystander stromal-supportive cells, evoking cell death, and tumor regression. It shows a robust and dose-dependent antitumor activity in multiple CDX and PDX models, representing lung, breast, bladder, head and neck cancers. Optimal tumor regression is associated with membrane expression of Nectin-4, in conjunction with MMAE sensitivity. Tumor regression rates are comparable for small and large tumors, indicative of deep and rapid penetration throughout the tumor. BT8009 provides tumoricidal activity with several regimens, potentially allowing for clinical titration of dose and dose interval if required.
Click to Show/Hide
|
||||
| Description |
BT8009 demonstrated dose-related effects on tumor growth in CDX and PDX models over the range 1-3 mg/kg when administered qw. Full tumor regressions were routinely achieved in both models with 3 mg/kg, with no tumor regrowth in subsequent weeks off treatment. Stable disease was delivered by 2 mg/kg and at this dose, in the PDX model, tumor growth resumed after drug cessation. In both models animals from the vehicle treated group were treated with 3 mg/kg BT8009 when tumors reached approximately 800 mm3or 1,000 mm3. Profound tumor regression was rapidly initiated in response. In the majority of studies 3 mg/kg, qw, was adopted as the standard dosing regimen.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice non-small cell lung cell-derived xenograft model. | ||||
| Half life period | 1-2 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
60%
|
|||
| Administration Time | 28 days | ||||
| Administration Dosage | 1 mg/kg | ||||
| MOA of PDC |
The cell adhesion molecule Nectin-4 shows elevated expression in multiple tumor types correlated with poor prognosis. Nectin-4-directed ADCs show efficacy in multiple xenograft tumor models. The ADC enfortumab vedotin (EV) delivers the antimitotic toxin MMAE to Nectin-4-expressing cells via internalization and cleavage of a valine-citrulline dipeptide linker component. Clinical validation of Nectin-4 as a target in urothelial cancer has been demonstrated with EV. In 2021, the FDA-approved EV for patients with locally advanced or metastatic urothelial cancer who had previously received a PD-1 or PD-L1 inhibitor and platinum-containing chemotherapy or were ineligible for cisplatin containing chemotherapy and had received one or more prior lines of therapy. Bicycle toxin conjugates (BTC) are structurally constrained bicyclic peptides conjugated through cleavable linkers to a toxin. Various, well-described linker toxin combinations can be incorporated into the BTC molecule (e.g., BT1718 and BT5528; refs.). They are of low molecular weight (approximately 4-4.5 kDa) and being chemically synthesized can be optimized for appropriate affinity, stability, and solubility relatively simply. Through intravenous administration, high systemic Cmax values can be attained, which, along with BTCs relatively small size, helps drive rapid diffusion into extra-vascular compartment, as reflected in a volume of distribution similar to extracellular fluid. We believe that delivery of a high number of BTCs, each carrying a reduced toxin load (peptide toxin ratio of 1:1) should improve tumor penetration and reduce the impact of the binding site barrier. BTCs show moderate clearance from the systemic vasculature, predominantly by the renal route. This overall profile marks them out from most ADCs and provides the possibility of enhanced clinical efficacy with a wider therapeutic index. BTCs are new therapeutic modality that shows a very different pharmacokinetic and structural profile to classic ADCs, whereas possessing robust tumoricidal properties. BTCs targeting MT1-MMP and EphA2 are currently in clinical trials. BT8009 is the most recent BTC to enter clinical trial. BT8009 is highly selective for Nectin-4 over other nectin family members and an extensive range of cell membrane expressed proteins. It shares the same cleavable linker and toxin combination as EV and can be cleaved by proteases in the TME, releasing cell penetrant MMAE that diffuses into tumor cells, or bystander stromal-supportive cells, evoking cell death, and tumor regression. It shows a robust and dose-dependent antitumor activity in multiple CDX and PDX models, representing lung, breast, bladder, head and neck cancers. Optimal tumor regression is associated with membrane expression of Nectin-4, in conjunction with MMAE sensitivity. Tumor regression rates are comparable for small and large tumors, indicative of deep and rapid penetration throughout the tumor. BT8009 provides tumoricidal activity with several regimens, potentially allowing for clinical titration of dose and dose interval if required.
Click to Show/Hide
|
||||
| Description |
BT8009 demonstrated dose-related effects on tumor growth in CDX and PDX models over the range 1-3 mg/kg when administered qw. Full tumor regressions were routinely achieved in both models with 3 mg/kg, with no tumor regrowth in subsequent weeks off treatment. Stable disease was delivered by 2 mg/kg and at this dose, in the PDX model, tumor growth resumed after drug cessation. In both models animals from the vehicle treated group were treated with 3 mg/kg BT8009 when tumors reached approximately 800 mm3or 1,000 mm3. Profound tumor regression was rapidly initiated in response. In the majority of studies 3 mg/kg, qw, was adopted as the standard dosing regimen.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice triple-negative breast cancer cell-derived xenograft model. | ||||
| Half life period | 1-2 h | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
74%
|
|||
| Administration Time | 28 days | ||||
| Administration Dosage | 2 mg/kg | ||||
| MOA of PDC |
The cell adhesion molecule Nectin-4 shows elevated expression in multiple tumor types correlated with poor prognosis. Nectin-4-directed ADCs show efficacy in multiple xenograft tumor models. The ADC enfortumab vedotin (EV) delivers the antimitotic toxin MMAE to Nectin-4-expressing cells via internalization and cleavage of a valine-citrulline dipeptide linker component. Clinical validation of Nectin-4 as a target in urothelial cancer has been demonstrated with EV. In 2021, the FDA-approved EV for patients with locally advanced or metastatic urothelial cancer who had previously received a PD-1 or PD-L1 inhibitor and platinum-containing chemotherapy or were ineligible for cisplatin containing chemotherapy and had received one or more prior lines of therapy. Bicycle toxin conjugates (BTC) are structurally constrained bicyclic peptides conjugated through cleavable linkers to a toxin. Various, well-described linker toxin combinations can be incorporated into the BTC molecule (e.g., BT1718 and BT5528; refs.). They are of low molecular weight (approximately 4-4.5 kDa) and being chemically synthesized can be optimized for appropriate affinity, stability, and solubility relatively simply. Through intravenous administration, high systemic Cmax values can be attained, which, along with BTCs relatively small size, helps drive rapid diffusion into extra-vascular compartment, as reflected in a volume of distribution similar to extracellular fluid. We believe that delivery of a high number of BTCs, each carrying a reduced toxin load (peptide toxin ratio of 1:1) should improve tumor penetration and reduce the impact of the binding site barrier. BTCs show moderate clearance from the systemic vasculature, predominantly by the renal route. This overall profile marks them out from most ADCs and provides the possibility of enhanced clinical efficacy with a wider therapeutic index. BTCs are new therapeutic modality that shows a very different pharmacokinetic and structural profile to classic ADCs, whereas possessing robust tumoricidal properties. BTCs targeting MT1-MMP and EphA2 are currently in clinical trials. BT8009 is the most recent BTC to enter clinical trial. BT8009 is highly selective for Nectin-4 over other nectin family members and an extensive range of cell membrane expressed proteins. It shares the same cleavable linker and toxin combination as EV and can be cleaved by proteases in the TME, releasing cell penetrant MMAE that diffuses into tumor cells, or bystander stromal-supportive cells, evoking cell death, and tumor regression. It shows a robust and dose-dependent antitumor activity in multiple CDX and PDX models, representing lung, breast, bladder, head and neck cancers. Optimal tumor regression is associated with membrane expression of Nectin-4, in conjunction with MMAE sensitivity. Tumor regression rates are comparable for small and large tumors, indicative of deep and rapid penetration throughout the tumor. BT8009 provides tumoricidal activity with several regimens, potentially allowing for clinical titration of dose and dose interval if required.
Click to Show/Hide
|
||||
| Description |
BT8009 demonstrated dose-related effects on tumor growth in CDX and PDX models over the range 1-3 mg/kg when administered qw. Full tumor regressions were routinely achieved in both models with 3 mg/kg, with no tumor regrowth in subsequent weeks off treatment. Stable disease was delivered by 2 mg/kg and at this dose, in the PDX model, tumor growth resumed after drug cessation. In both models animals from the vehicle treated group were treated with 3 mg/kg BT8009 when tumors reached approximately 800 mm3or 1,000 mm3. Profound tumor regression was rapidly initiated in response. In the majority of studies 3 mg/kg, qw, was adopted as the standard dosing regimen.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice triple-negative breast cancer cell-derived xenograft model. | ||||
| Half life period | 1-2 h | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
77%
|
|||
| Administration Time | 28 days | ||||
| Administration Dosage | 2 mg/kg | ||||
| MOA of PDC |
The cell adhesion molecule Nectin-4 shows elevated expression in multiple tumor types correlated with poor prognosis. Nectin-4-directed ADCs show efficacy in multiple xenograft tumor models. The ADC enfortumab vedotin (EV) delivers the antimitotic toxin MMAE to Nectin-4-expressing cells via internalization and cleavage of a valine-citrulline dipeptide linker component. Clinical validation of Nectin-4 as a target in urothelial cancer has been demonstrated with EV. In 2021, the FDA-approved EV for patients with locally advanced or metastatic urothelial cancer who had previously received a PD-1 or PD-L1 inhibitor and platinum-containing chemotherapy or were ineligible for cisplatin containing chemotherapy and had received one or more prior lines of therapy. Bicycle toxin conjugates (BTC) are structurally constrained bicyclic peptides conjugated through cleavable linkers to a toxin. Various, well-described linker toxin combinations can be incorporated into the BTC molecule (e.g., BT1718 and BT5528; refs.). They are of low molecular weight (approximately 4-4.5 kDa) and being chemically synthesized can be optimized for appropriate affinity, stability, and solubility relatively simply. Through intravenous administration, high systemic Cmax values can be attained, which, along with BTCs relatively small size, helps drive rapid diffusion into extra-vascular compartment, as reflected in a volume of distribution similar to extracellular fluid. We believe that delivery of a high number of BTCs, each carrying a reduced toxin load (peptide toxin ratio of 1:1) should improve tumor penetration and reduce the impact of the binding site barrier. BTCs show moderate clearance from the systemic vasculature, predominantly by the renal route. This overall profile marks them out from most ADCs and provides the possibility of enhanced clinical efficacy with a wider therapeutic index. BTCs are new therapeutic modality that shows a very different pharmacokinetic and structural profile to classic ADCs, whereas possessing robust tumoricidal properties. BTCs targeting MT1-MMP and EphA2 are currently in clinical trials. BT8009 is the most recent BTC to enter clinical trial. BT8009 is highly selective for Nectin-4 over other nectin family members and an extensive range of cell membrane expressed proteins. It shares the same cleavable linker and toxin combination as EV and can be cleaved by proteases in the TME, releasing cell penetrant MMAE that diffuses into tumor cells, or bystander stromal-supportive cells, evoking cell death, and tumor regression. It shows a robust and dose-dependent antitumor activity in multiple CDX and PDX models, representing lung, breast, bladder, head and neck cancers. Optimal tumor regression is associated with membrane expression of Nectin-4, in conjunction with MMAE sensitivity. Tumor regression rates are comparable for small and large tumors, indicative of deep and rapid penetration throughout the tumor. BT8009 provides tumoricidal activity with several regimens, potentially allowing for clinical titration of dose and dose interval if required.
Click to Show/Hide
|
||||
| Description |
BT8009 demonstrated dose-related effects on tumor growth in CDX and PDX models over the range 1-3 mg/kg when administered qw. Full tumor regressions were routinely achieved in both models with 3 mg/kg, with no tumor regrowth in subsequent weeks off treatment. Stable disease was delivered by 2 mg/kg and at this dose, in the PDX model, tumor growth resumed after drug cessation. In both models animals from the vehicle treated group were treated with 3 mg/kg BT8009 when tumors reached approximately 800 mm3or 1,000 mm3. Profound tumor regression was rapidly initiated in response. In the majority of studies 3 mg/kg, qw, was adopted as the standard dosing regimen.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice non-small cell lung cell-derived xenograft model. | ||||
| Half life period | 1-2 h | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
96%
|
|||
| Administration Time | 28 days | ||||
| Administration Dosage | 3 mg/kg | ||||
| MOA of PDC |
The cell adhesion molecule Nectin-4 shows elevated expression in multiple tumor types correlated with poor prognosis. Nectin-4-directed ADCs show efficacy in multiple xenograft tumor models. The ADC enfortumab vedotin (EV) delivers the antimitotic toxin MMAE to Nectin-4-expressing cells via internalization and cleavage of a valine-citrulline dipeptide linker component. Clinical validation of Nectin-4 as a target in urothelial cancer has been demonstrated with EV. In 2021, the FDA-approved EV for patients with locally advanced or metastatic urothelial cancer who had previously received a PD-1 or PD-L1 inhibitor and platinum-containing chemotherapy or were ineligible for cisplatin containing chemotherapy and had received one or more prior lines of therapy. Bicycle toxin conjugates (BTC) are structurally constrained bicyclic peptides conjugated through cleavable linkers to a toxin. Various, well-described linker toxin combinations can be incorporated into the BTC molecule (e.g., BT1718 and BT5528; refs.). They are of low molecular weight (approximately 4-4.5 kDa) and being chemically synthesized can be optimized for appropriate affinity, stability, and solubility relatively simply. Through intravenous administration, high systemic Cmax values can be attained, which, along with BTCs relatively small size, helps drive rapid diffusion into extra-vascular compartment, as reflected in a volume of distribution similar to extracellular fluid. We believe that delivery of a high number of BTCs, each carrying a reduced toxin load (peptide toxin ratio of 1:1) should improve tumor penetration and reduce the impact of the binding site barrier. BTCs show moderate clearance from the systemic vasculature, predominantly by the renal route. This overall profile marks them out from most ADCs and provides the possibility of enhanced clinical efficacy with a wider therapeutic index. BTCs are new therapeutic modality that shows a very different pharmacokinetic and structural profile to classic ADCs, whereas possessing robust tumoricidal properties. BTCs targeting MT1-MMP and EphA2 are currently in clinical trials. BT8009 is the most recent BTC to enter clinical trial. BT8009 is highly selective for Nectin-4 over other nectin family members and an extensive range of cell membrane expressed proteins. It shares the same cleavable linker and toxin combination as EV and can be cleaved by proteases in the TME, releasing cell penetrant MMAE that diffuses into tumor cells, or bystander stromal-supportive cells, evoking cell death, and tumor regression. It shows a robust and dose-dependent antitumor activity in multiple CDX and PDX models, representing lung, breast, bladder, head and neck cancers. Optimal tumor regression is associated with membrane expression of Nectin-4, in conjunction with MMAE sensitivity. Tumor regression rates are comparable for small and large tumors, indicative of deep and rapid penetration throughout the tumor. BT8009 provides tumoricidal activity with several regimens, potentially allowing for clinical titration of dose and dose interval if required.
Click to Show/Hide
|
||||
| Description |
BT8009 demonstrated dose-related effects on tumor growth in CDX and PDX models over the range 1-3 mg/kg when administered qw. Full tumor regressions were routinely achieved in both models with 3 mg/kg, with no tumor regrowth in subsequent weeks off treatment. Stable disease was delivered by 2 mg/kg and at this dose, in the PDX model, tumor growth resumed after drug cessation. In both models animals from the vehicle treated group were treated with 3 mg/kg BT8009 when tumors reached approximately 800 mm3or 1,000 mm3. Profound tumor regression was rapidly initiated in response. In the majority of studies 3 mg/kg, qw, was adopted as the standard dosing regimen.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice triple-negative breast cancer cell-derived xenograft model. | ||||
| Half life period | 1-2 h | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
98%
|
|||
| Administration Time | 28 days | ||||
| Administration Dosage | 3 mg/kg | ||||
| MOA of PDC |
The cell adhesion molecule Nectin-4 shows elevated expression in multiple tumor types correlated with poor prognosis. Nectin-4-directed ADCs show efficacy in multiple xenograft tumor models. The ADC enfortumab vedotin (EV) delivers the antimitotic toxin MMAE to Nectin-4-expressing cells via internalization and cleavage of a valine-citrulline dipeptide linker component. Clinical validation of Nectin-4 as a target in urothelial cancer has been demonstrated with EV. In 2021, the FDA-approved EV for patients with locally advanced or metastatic urothelial cancer who had previously received a PD-1 or PD-L1 inhibitor and platinum-containing chemotherapy or were ineligible for cisplatin containing chemotherapy and had received one or more prior lines of therapy. Bicycle toxin conjugates (BTC) are structurally constrained bicyclic peptides conjugated through cleavable linkers to a toxin. Various, well-described linker toxin combinations can be incorporated into the BTC molecule (e.g., BT1718 and BT5528; refs.). They are of low molecular weight (approximately 4-4.5 kDa) and being chemically synthesized can be optimized for appropriate affinity, stability, and solubility relatively simply. Through intravenous administration, high systemic Cmax values can be attained, which, along with BTCs relatively small size, helps drive rapid diffusion into extra-vascular compartment, as reflected in a volume of distribution similar to extracellular fluid. We believe that delivery of a high number of BTCs, each carrying a reduced toxin load (peptide toxin ratio of 1:1) should improve tumor penetration and reduce the impact of the binding site barrier. BTCs show moderate clearance from the systemic vasculature, predominantly by the renal route. This overall profile marks them out from most ADCs and provides the possibility of enhanced clinical efficacy with a wider therapeutic index. BTCs are new therapeutic modality that shows a very different pharmacokinetic and structural profile to classic ADCs, whereas possessing robust tumoricidal properties. BTCs targeting MT1-MMP and EphA2 are currently in clinical trials. BT8009 is the most recent BTC to enter clinical trial. BT8009 is highly selective for Nectin-4 over other nectin family members and an extensive range of cell membrane expressed proteins. It shares the same cleavable linker and toxin combination as EV and can be cleaved by proteases in the TME, releasing cell penetrant MMAE that diffuses into tumor cells, or bystander stromal-supportive cells, evoking cell death, and tumor regression. It shows a robust and dose-dependent antitumor activity in multiple CDX and PDX models, representing lung, breast, bladder, head and neck cancers. Optimal tumor regression is associated with membrane expression of Nectin-4, in conjunction with MMAE sensitivity. Tumor regression rates are comparable for small and large tumors, indicative of deep and rapid penetration throughout the tumor. BT8009 provides tumoricidal activity with several regimens, potentially allowing for clinical titration of dose and dose interval if required.
Click to Show/Hide
|
||||
| Description |
BT8009 demonstrated dose-related effects on tumor growth in CDX and PDX models over the range 1-3 mg/kg when administered qw. Full tumor regressions were routinely achieved in both models with 3 mg/kg, with no tumor regrowth in subsequent weeks off treatment. Stable disease was delivered by 2 mg/kg and at this dose, in the PDX model, tumor growth resumed after drug cessation. In both models animals from the vehicle treated group were treated with 3 mg/kg BT8009 when tumors reached approximately 800 mm3or 1,000 mm3. Profound tumor regression was rapidly initiated in response. In the majority of studies 3 mg/kg, qw, was adopted as the standard dosing regimen.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice non-small cell lung cell-derived xenograft model. | ||||
| Half life period | 1-2 h | ||||
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
81%
|
|||
| Administration Time | 21 days | ||||
| Administration Dosage | 3 mg/kg qw | ||||
| MOA of PDC |
Nectin-4 is a cell adhesion molecule from the Nectin and Nectin-like family. It is a clinically validated tumor target and has been reported to be highly expressed in a wide range of solid tumors, including bladder, esophageal, pancreatic, and lung, but with limited distribution in healthy tissues.An antibody drug conjugate (PADCEV, enfortumab vedotin) that targets Nectin-4 was recently approved for the treatment of bladder cancer following the generation of positive data in a number of clinical studies. Herein we describe the discovery via phage display and subsequent chemical optimization of a Nectin-4 binding Bicycle and its incorporation into BT8009, a BTC that is currently under clinical evaluation. A detailed report of the pharmacologic properties of BT8009 has recently been described.In this, BT8009 shows potent efficacy in multiple tumor models, including patient-derived xenografts, across a variety of tumor indications and is well-tolerated in preclinical safety studies. In several models it demonstrated superior or equivalent efficacy to an analogue of the ADC PADCEV.
Click to Show/Hide
|
||||
| Description |
The efficacy of BT8009 was evaluated in a cell-derived xenograft (CDX) model using breast adenocarcinoma (MDA-MB-468) cells, which express Nectin-4.When tested at a dose of 3 mg/kg once weekly, significant antitumor activity was observed. At a dose of 3 mg/kg twice weekly or 5 mg/kg once weekly, potent efficacy was achieved with almost complete regression of the tumor after 18 days. Importantly, following cessation of dosing after 18 days, animals from the 5 mg/kg once weekly dosing group were monitored up to day 42, and no tumor regrowth was observed. Consistent animal body weights throughout the study indicate that BT8009 appeared to be well-tolerated at all doses tested. In additional studies reported elsewhere, BT8009 has shown preclinical efficacy in a wide range of CDX and PDX tumor types with full tumor regression seen in small and large tumors, where efficacy broadly correlates with Nectin-4 expression. Additionally, when BT8009 was codosed with an excess of an MMAE-free analogue, efficacy was attenuated, and a BTC incorporating a nonbinding Bicycle analogue showed a demonstrably lower rate and degree of tumor regression.
Click to Show/Hide
|
||||
| In Vivo Model | MDA-MB-468 cell-derived xenograft (CDX) model. | ||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-468 cell | CVCL_0419 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
93%
|
|||
| Administration Time | 21 days | ||||
| Administration Dosage | 3 mg/kg biw | ||||
| MOA of PDC |
Nectin-4 is a cell adhesion molecule from the Nectin and Nectin-like family. It is a clinically validated tumor target and has been reported to be highly expressed in a wide range of solid tumors, including bladder, esophageal, pancreatic, and lung, but with limited distribution in healthy tissues.An antibody drug conjugate (PADCEV, enfortumab vedotin) that targets Nectin-4 was recently approved for the treatment of bladder cancer following the generation of positive data in a number of clinical studies. Herein we describe the discovery via phage display and subsequent chemical optimization of a Nectin-4 binding Bicycle and its incorporation into BT8009, a BTC that is currently under clinical evaluation. A detailed report of the pharmacologic properties of BT8009 has recently been described.In this, BT8009 shows potent efficacy in multiple tumor models, including patient-derived xenografts, across a variety of tumor indications and is well-tolerated in preclinical safety studies. In several models it demonstrated superior or equivalent efficacy to an analogue of the ADC PADCEV.
Click to Show/Hide
|
||||
| Description |
The efficacy of BT8009 was evaluated in a cell-derived xenograft (CDX) model using breast adenocarcinoma (MDA-MB-468) cells, which express Nectin-4.When tested at a dose of 3 mg/kg once weekly, significant antitumor activity was observed. At a dose of 3 mg/kg twice weekly or 5 mg/kg once weekly, potent efficacy was achieved with almost complete regression of the tumor after 18 days. Importantly, following cessation of dosing after 18 days, animals from the 5 mg/kg once weekly dosing group were monitored up to day 42, and no tumor regrowth was observed. Consistent animal body weights throughout the study indicate that BT8009 appeared to be well-tolerated at all doses tested. In additional studies reported elsewhere, BT8009 has shown preclinical efficacy in a wide range of CDX and PDX tumor types with full tumor regression seen in small and large tumors, where efficacy broadly correlates with Nectin-4 expression. Additionally, when BT8009 was codosed with an excess of an MMAE-free analogue, efficacy was attenuated, and a BTC incorporating a nonbinding Bicycle analogue showed a demonstrably lower rate and degree of tumor regression.
Click to Show/Hide
|
||||
| In Vivo Model | MDA-MB-468 cell-derived xenograft (CDX) model. | ||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-468 cell | CVCL_0419 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
93%
|
|||
| Administration Time | 21 days | ||||
| Administration Dosage | 5 mg/kg qw | ||||
| MOA of PDC |
Nectin-4 is a cell adhesion molecule from the Nectin and Nectin-like family. It is a clinically validated tumor target and has been reported to be highly expressed in a wide range of solid tumors, including bladder, esophageal, pancreatic, and lung, but with limited distribution in healthy tissues.An antibody drug conjugate (PADCEV, enfortumab vedotin) that targets Nectin-4 was recently approved for the treatment of bladder cancer following the generation of positive data in a number of clinical studies. Herein we describe the discovery via phage display and subsequent chemical optimization of a Nectin-4 binding Bicycle and its incorporation into BT8009, a BTC that is currently under clinical evaluation. A detailed report of the pharmacologic properties of BT8009 has recently been described.In this, BT8009 shows potent efficacy in multiple tumor models, including patient-derived xenografts, across a variety of tumor indications and is well-tolerated in preclinical safety studies. In several models it demonstrated superior or equivalent efficacy to an analogue of the ADC PADCEV.
Click to Show/Hide
|
||||
| Description |
The efficacy of BT8009 was evaluated in a cell-derived xenograft (CDX) model using breast adenocarcinoma (MDA-MB-468) cells, which express Nectin-4.When tested at a dose of 3 mg/kg once weekly, significant antitumor activity was observed. At a dose of 3 mg/kg twice weekly or 5 mg/kg once weekly, potent efficacy was achieved with almost complete regression of the tumor after 18 days. Importantly, following cessation of dosing after 18 days, animals from the 5 mg/kg once weekly dosing group were monitored up to day 42, and no tumor regrowth was observed. Consistent animal body weights throughout the study indicate that BT8009 appeared to be well-tolerated at all doses tested. In additional studies reported elsewhere, BT8009 has shown preclinical efficacy in a wide range of CDX and PDX tumor types with full tumor regression seen in small and large tumors, where efficacy broadly correlates with Nectin-4 expression. Additionally, when BT8009 was codosed with an excess of an MMAE-free analogue, efficacy was attenuated, and a BTC incorporating a nonbinding Bicycle analogue showed a demonstrably lower rate and degree of tumor regression.
Click to Show/Hide
|
||||
| In Vivo Model | MDA-MB-468 cell-derived xenograft (CDX) model. | ||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-468 cell | CVCL_0419 | ||
BT5528 [Phase 1/2]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Fibrosarcoma | ||||
| Efficacy Data | Tumor volume |
1200 mm3
|
|||
| Administration Time | 20 day | ||||
| Administration Dosage | 0.167 mg/kg every week | ||||
| MOA of PDC |
Here we describe the development of BT5528, a bicyclic peptide (Bicycle) conjugated to the auristatin derivative maleimidocaproyl-monomethyl auristatin E to generate the Bicycle toxin conjugate BT5528. There are two potential mechanism of BT5528 bystander activity: extracellular linker cleavage and toxin penetration into neighboring cells, or receptor internalization and intracellular linker cleavage followed by release of cell penetrant toxin from lysed cells. The available data do not allow us to distinguish between these two mechanisms and it seems likely that BT5528 activity is mediated by toxin release following a combination of intracellular and extracellular linker cleavage.
Click to Show/Hide
|
||||
| Description |
BT5528 is efficacious in the PC3 xenograft model but control BTCs with noncleavable linkers and non-cell penetrant toxins lack comparable efficacy. A, The nonbinding BTC, BCY8245, is less active than BT5528 in the PC3 model (group mean ± SEM, n = 5) at both 0.5 and 0.0167 mg/kg dosing level (*, P < 0.05; **, P < 0.01; two-way ANOVA with Sidak's multiple comparisons test). B, Replacement of the cell penetrant toxin (MMAE: BT5528) with the non-cell penetrant toxin (MMAF: BCY10188) reduces activity in the PC3 model at 3, 1, and 0.33 mg/kg (group mean ± SEM; n = 5; *, P < 0.05; **, P < 0.01; two-way ANOVA with Sidak's multiple comparisons test). C, noncleavable linker chemistry abolishes activity in the HT1080 model; BCY6063, 10 mg/kg twice a week (group mean ± SEM, n = 3).
Click to Show/Hide
|
||||
| In Vivo Model | HT108 cells xenograft models in 6- to 8-week-old female balb/c nude or CB17-SCID mice. | ||||
| In Vitro Model | Fibrosarcoma | HT-1080 cell | CVCL_0317 | ||
| Half life period | 0.4 hour (Mouse); 0.3 hour (Rat); 0.6 hour (Nonhuman primate) | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Fibrosarcoma | ||||
| Efficacy Data | Tumor volume |
2200 mm3
|
|||
| Administration Time | 20 day | ||||
| Administration Dosage | 0.5 mg/kg every week | ||||
| MOA of PDC |
Here we describe the development of BT5528, a bicyclic peptide (Bicycle) conjugated to the auristatin derivative maleimidocaproyl-monomethyl auristatin E to generate the Bicycle toxin conjugate BT5528. There are two potential mechanism of BT5528 bystander activity: extracellular linker cleavage and toxin penetration into neighboring cells, or receptor internalization and intracellular linker cleavage followed by release of cell penetrant toxin from lysed cells. The available data do not allow us to distinguish between these two mechanisms and it seems likely that BT5528 activity is mediated by toxin release following a combination of intracellular and extracellular linker cleavage.
Click to Show/Hide
|
||||
| Description |
BT5528 is efficacious in the PC3 xenograft model but control BTCs with noncleavable linkers and non-cell penetrant toxins lack comparable efficacy. A, The nonbinding BTC, BCY8245, is less active than BT5528 in the PC3 model (group mean ± SEM, n = 5) at both 0.5 and 0.0167 mg/kg dosing level (*, P < 0.05; **, P < 0.01; two-way ANOVA with Sidak's multiple comparisons test). B, Replacement of the cell penetrant toxin (MMAE: BT5528) with the non-cell penetrant toxin (MMAF: BCY10188) reduces activity in the PC3 model at 3, 1, and 0.33 mg/kg (group mean ± SEM; n = 5; *, P < 0.05; **, P < 0.01; two-way ANOVA with Sidak's multiple comparisons test). C, noncleavable linker chemistry abolishes activity in the HT1080 model; BCY6063, 10 mg/kg twice a week (group mean ± SEM, n = 3).
Click to Show/Hide
|
||||
| In Vivo Model | HT108 cells xenograft models in 6- to 8-week-old female balb/c nude or CB17-SCID mice. | ||||
| In Vitro Model | Fibrosarcoma | HT-1080 cell | CVCL_0317 | ||
| Half life period | 0.4 hour (Mouse); 0.3 hour (Rat); 0.6 hour (Nonhuman primate) | ||||
TFα-peptide-duocarmycin conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Colon adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
402 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTS cell proliferation assay | ||||
| MOA of PDC |
The cancer phenotype is commonly associated with aberrant glycosylation patterns. One glycan that is directly linked to cancer is the Thomsen-Friedenreich antigen (TF or CD176). It is a disaccharide composed of a galactose β1-3 N-acetylgalactosamine, O-linked to a glycoprotein through serine or threonine residues and commonly written as Galβ1-3GalNAc--O-Ser/Thr. The TF is therapeutically attractive due to its cryptic nature in normal cells and exposure in embryonic and cancer cells. The expression of the TF has been demonstrated in 90% of primary human carcinomas, including in the lung, the breast, and the pancreas. Additionally, cancer initiating cells or cancer stem cells in the lung, liver, and colon express the TF. The peptide sequence HGRFILPWWYAFSPS (TF-peptide) is known to bind tightly to the TF (Kd = 1.2 M) and has been demonstrated to inhibit processes directly involved in TF accessibility.
Click to Show/Hide
|
||||
| Description |
All cell lines demonstrated sensitivity toward the DSA-PABA payload 5 in the nanomolar range. The cleaved peptide 6, and the benzyl protected control peptide 4, had no appreciable effect on cell proliferation (>100 uM), suggesting that any observed cytotoxic activity is due to the DSA warhead. Interestingly, PDC 7, without the cathepsin B cleavable sequence, also had no appreciable effect on cell proliferation (>100 uM), despite possessing the active DSA DNA alkylating moiety. Perhaps this PDC is either not being taken up into cells or not being broken down by proteases and peptidases in the cell. In both cases, the warhead may not be reaching the nucleus, the site of action of the duocarmycins. Cell lines that demonstrated appreciable levels of TF were sensitive to PDC 3. Interestingly, HCT116, which had relatively high TF expression and cathepsin B activity, demonstrated the greatest sensitivity to PDC 3, approaching similar potency to the payload 5. Excitingly, A549, H292, and 16HBE14o, which showed no detectable TF expression, appeared unaffected by PDC 3 up to 100 uM, suggesting that TF expression is required for the efficacy of PDC 3.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT 116 cell | CVCL_0291 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
526 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTS cell proliferation assay | ||||
| MOA of PDC |
As a payload for PDCs (and ADCs), analogues of the duocarmycins are attractive. Duocarmycin SA and yatakemycin rank among the most potent natural cytotoxins discovered. The cyclopropyl and prodrug seco forms are both naturally occurring and equipotent in most circumstances. Studies of the binding-driven bonding model of their interaction with DNA suggest that their utility will be enhanced when targeted to tumor cells. In fact, SYD985, an ADC that utilizes a peptide linker for a duocarmycin analogue to trastuzumab, has recently been progressed to phase III clinical trial.
Click to Show/Hide
|
||||
| Description |
All cell lines demonstrated sensitivity toward the DSA-PABA payload 5 in the nanomolar range. The cleaved peptide 6, and the benzyl protected control peptide 4, had no appreciable effect on cell proliferation (>100 uM), suggesting that any observed cytotoxic activity is due to the DSA warhead. Interestingly, PDC 7, without the cathepsin B cleavable sequence, also had no appreciable effect on cell proliferation (>100 uM), despite possessing the active DSA DNA alkylating moiety. Perhaps this PDC is either not being taken up into cells or not being broken down by proteases and peptidases in the cell. In both cases, the warhead may not be reaching the nucleus, the site of action of the duocarmycins. Cell lines that demonstrated appreciable levels of TF were sensitive to PDC 3. Interestingly, HCT116, which had relatively high TF expression and cathepsin B activity, demonstrated the greatest sensitivity to PDC 3, approaching similar potency to the payload 5. Excitingly, A549, H292, and 16HBE14o, which showed no detectable TF expression, appeared unaffected by PDC 3 up to 100 uM, suggesting that TF expression is required for the efficacy of PDC 3.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Invasive breast carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
864 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTS cell proliferation assay | ||||
| MOA of PDC |
The cancer phenotype is commonly associated with aberrant glycosylation patterns. One glycan that is directly linked to cancer is the Thomsen-Friedenreich antigen (TF or CD176). It is a disaccharide composed of a galactose β1-3 N-acetylgalactosamine, O-linked to a glycoprotein through serine or threonine residues and commonly written as Galβ1-3GalNAc--O-Ser/Thr. The TF is therapeutically attractive due to its cryptic nature in normal cells and exposure in embryonic and cancer cells. The expression of the TF has been demonstrated in 90% of primary human carcinomas, including in the lung, the breast, and the pancreas. Additionally, cancer initiating cells or cancer stem cells in the lung, liver, and colon express the TF. The peptide sequence HGRFILPWWYAFSPS (TFα-peptide) is known to bind tightly to the TF (Kd = 1.2 M) and has been demonstrated to inhibit processes directly involved in TF accessibility.
Click to Show/Hide
|
||||
| Description |
All cell lines demonstrated sensitivity toward the DSA-PABA payload 5 in the nanomolar range. The cleaved peptide 6, and the benzyl protected control peptide 4, had no appreciable effect on cell proliferation (>100 uM), suggesting that any observed cytotoxic activity is due to the DSA warhead. Interestingly, PDC 7, without the cathepsin B cleavable sequence, also had no appreciable effect on cell proliferation (>100 uM), despite possessing the active DSA DNA alkylating moiety. Perhaps this PDC is either not being taken up into cells or not being broken down by proteases and peptidases in the cell. In both cases, the warhead may not be reaching the nucleus, the site of action of the duocarmycins. Cell lines that demonstrated appreciable levels of TF were sensitive to PDC 3. Interestingly, HCT116, which had relatively high TF expression and cathepsin B activity, demonstrated the greatest sensitivity to PDC 3, approaching similar potency to the payload 5. Excitingly, A549, H292, and 16HBE14o, which showed no detectable TF expression, appeared unaffected by PDC 3 up to 100 uM, suggesting that TF expression is required for the efficacy of PDC 3.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Acute myeloid leukemia | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1831 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTS cell proliferation assay | ||||
| MOA of PDC |
The cancer phenotype is commonly associated with aberrant glycosylation patterns. One glycan that is directly linked to cancer is the Thomsen-Friedenreich antigen (TF or CD176). It is a disaccharide composed of a galactose β1-3 N-acetylgalactosamine, O-linked to a glycoprotein through serine or threonine residues and commonly written as Galβ1-3GalNAc--O-Ser/Thr. The TF is therapeutically attractive due to its cryptic nature in normal cells and exposure in embryonic and cancer cells. The expression of the TF has been demonstrated in 90% of primary human carcinomas, including in the lung, the breast, and the pancreas. Additionally, cancer initiating cells or cancer stem cells in the lung, liver, and colon express the TF. The peptide sequence HGRFILPWWYAFSPS (TF-peptide) is known to bind tightly to the TF (Kd = 1.2 M) and has been demonstrated to inhibit processes directly involved in TF accessibility.
Click to Show/Hide
|
||||
| Description |
All cell lines demonstrated sensitivity toward the DSA-PABA payload 5 in the nanomolar range. The cleaved peptide 6, and the benzyl protected control peptide 4, had no appreciable effect on cell proliferation (>100 uM), suggesting that any observed cytotoxic activity is due to the DSA warhead. Interestingly, PDC 7, without the cathepsin B cleavable sequence, also had no appreciable effect on cell proliferation (>100 uM), despite possessing the active DSA DNA alkylating moiety. Perhaps this PDC is either not being taken up into cells or not being broken down by proteases and peptidases in the cell. In both cases, the warhead may not be reaching the nucleus, the site of action of the duocarmycins. Cell lines that demonstrated appreciable levels of TF were sensitive to PDC 3. Interestingly, HCT116, which had relatively high TF expression and cathepsin B activity, demonstrated the greatest sensitivity to PDC 3, approaching similar potency to the payload 5. Excitingly, A549, H292, and 16HBE14o, which showed no detectable TF expression, appeared unaffected by PDC 3 up to 100 uM, suggesting that TF expression is required for the efficacy of PDC 3.
Click to Show/Hide
|
||||
| In Vitro Model | Acute myeloid leukemia | HL-60 cell | CVCL_0002 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Fibrosarcoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
8266 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTS cell proliferation assay | ||||
| MOA of PDC |
As a payload for PDCs (and ADCs), analogues of the duocarmycins are attractive. Duocarmycin SA and yatakemycin rank among the most potent natural cytotoxins discovered. The cyclopropyl and prodrug seco forms are both naturally occurring and equipotent in most circumstances. Studies of the binding-driven bonding model of their interaction with DNA suggest that their utility will be enhanced when targeted to tumor cells. In fact, SYD985, an ADC that utilizes a peptide linker for a duocarmycin analogue to trastuzumab, has recently been progressed to phase III clinical trial.
Click to Show/Hide
|
||||
| Description |
All cell lines demonstrated sensitivity toward the DSA-PABA payload 5 in the nanomolar range. The cleaved peptide 6, and the benzyl protected control peptide 4, had no appreciable effect on cell proliferation (>100 uM), suggesting that any observed cytotoxic activity is due to the DSA warhead. Interestingly, PDC 7, without the cathepsin B cleavable sequence, also had no appreciable effect on cell proliferation (>100 uM), despite possessing the active DSA DNA alkylating moiety. Perhaps this PDC is either not being taken up into cells or not being broken down by proteases and peptidases in the cell. In both cases, the warhead may not be reaching the nucleus, the site of action of the duocarmycins. Cell lines that demonstrated appreciable levels of TF were sensitive to PDC 3. Interestingly, HCT116, which had relatively high TF expression and cathepsin B activity, demonstrated the greatest sensitivity to PDC 3, approaching similar potency to the payload 5. Excitingly, A549, H292, and 16HBE14o, which showed no detectable TF expression, appeared unaffected by PDC 3 up to 100 uM, suggesting that TF expression is required for the efficacy of PDC 3.
Click to Show/Hide
|
||||
| In Vitro Model | Fibrosarcoma | HT-1080 cell | CVCL_0317 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Lung adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 100 µM | |||
| Administration Time | 72 h | ||||
| Evaluation Method | MTS cell proliferation assay | ||||
| MOA of PDC |
The cancer phenotype is commonly associated with aberrant glycosylation patterns. One glycan that is directly linked to cancer is the Thomsen-Friedenreich antigen (TF or CD176). It is a disaccharide composed of a galactose β1-3 N-acetylgalactosamine, O-linked to a glycoprotein through serine or threonine residues and commonly written as Galβ1-3GalNAc--O-Ser/Thr. The TF is therapeutically attractive due to its cryptic nature in normal cells and exposure in embryonic and cancer cells. The expression of the TF has been demonstrated in 90% of primary human carcinomas, including in the lung, the breast, and the pancreas. Additionally, cancer initiating cells or cancer stem cells in the lung, liver, and colon express the TF. The peptide sequence HGRFILPWWYAFSPS (TF-peptide) is known to bind tightly to the TF (Kd = 1.2 M) and has been demonstrated to inhibit processes directly involved in TF accessibility.
Click to Show/Hide
|
||||
| Description |
All cell lines demonstrated sensitivity toward the DSA-PABA payload 5 in the nanomolar range. The cleaved peptide 6, and the benzyl protected control peptide 4, had no appreciable effect on cell proliferation (>100 uM), suggesting that any observed cytotoxic activity is due to the DSA warhead. Interestingly, PDC 7, without the cathepsin B cleavable sequence, also had no appreciable effect on cell proliferation (>100 uM), despite possessing the active DSA DNA alkylating moiety. Perhaps this PDC is either not being taken up into cells or not being broken down by proteases and peptidases in the cell. In both cases, the warhead may not be reaching the nucleus, the site of action of the duocarmycins. Cell lines that demonstrated appreciable levels of TF were sensitive to PDC 3. Interestingly, HCT116, which had relatively high TF expression and cathepsin B activity, demonstrated the greatest sensitivity to PDC 3, approaching similar potency to the payload 5. Excitingly, A549, H292, and 16HBE14o, which showed no detectable TF expression, appeared unaffected by PDC 3 up to 100 uM, suggesting that TF expression is required for the efficacy of PDC 3.
Click to Show/Hide
|
||||
| In Vitro Model | Lung adenocarcinoma | A-549 cell | CVCL_0023 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Lung mucoepidermoid carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 100 µM | |||
| Administration Time | 72 h | ||||
| Evaluation Method | MTS cell proliferation assay | ||||
| MOA of PDC |
As a payload for PDCs (and ADCs), analogues of the duocarmycins are attractive. Duocarmycin SA and yatakemycin rank among the most potent natural cytotoxins discovered. The cyclopropyl and prodrug seco forms are both naturally occurring and equipotent in most circumstances. Studies of the binding-driven bonding model of their interaction with DNA suggest that their utility will be enhanced when targeted to tumor cells. In fact, SYD985, an ADC that utilizes a peptide linker for a duocarmycin analogue to trastuzumab, has recently been progressed to phase III clinical trial.
Click to Show/Hide
|
||||
| Description |
All cell lines demonstrated sensitivity toward the DSA-PABA payload 5 in the nanomolar range. The cleaved peptide 6, and the benzyl protected control peptide 4, had no appreciable effect on cell proliferation (>100 uM), suggesting that any observed cytotoxic activity is due to the DSA warhead. Interestingly, PDC 7, without the cathepsin B cleavable sequence, also had no appreciable effect on cell proliferation (>100 uM), despite possessing the active DSA DNA alkylating moiety. Perhaps this PDC is either not being taken up into cells or not being broken down by proteases and peptidases in the cell. In both cases, the warhead may not be reaching the nucleus, the site of action of the duocarmycins. Cell lines that demonstrated appreciable levels of TF were sensitive to PDC 3. Interestingly, HCT116, which had relatively high TF expression and cathepsin B activity, demonstrated the greatest sensitivity to PDC 3, approaching similar potency to the payload 5. Excitingly, A549, H292, and 16HBE14o, which showed no detectable TF expression, appeared unaffected by PDC 3 up to 100 uM, suggesting that TF expression is required for the efficacy of PDC 3.
Click to Show/Hide
|
||||
| In Vitro Model | Lung mucoepidermoid carcinoma | H292 cell | CVCL_0455 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Normal | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 100 µM | |||
| Administration Time | 72 h | ||||
| Evaluation Method | MTS cell proliferation assay | ||||
| MOA of PDC |
As a payload for PDCs (and ADCs), analogues of the duocarmycins are attractive. Duocarmycin SA and yatakemycin rank among the most potent natural cytotoxins discovered. The cyclopropyl and prodrug seco forms are both naturally occurring and equipotent in most circumstances. Studies of the binding-driven bonding model of their interaction with DNA suggest that their utility will be enhanced when targeted to tumor cells. In fact, SYD985, an ADC that utilizes a peptide linker for a duocarmycin analogue to trastuzumab, has recently been progressed to phase III clinical trial.
Click to Show/Hide
|
||||
| Description |
All cell lines demonstrated sensitivity toward the DSA-PABA payload 5 in the nanomolar range. The cleaved peptide 6, and the benzyl protected control peptide 4, had no appreciable effect on cell proliferation (>100 uM), suggesting that any observed cytotoxic activity is due to the DSA warhead. Interestingly, PDC 7, without the cathepsin B cleavable sequence, also had no appreciable effect on cell proliferation (>100 uM), despite possessing the active DSA DNA alkylating moiety. Perhaps this PDC is either not being taken up into cells or not being broken down by proteases and peptidases in the cell. In both cases, the warhead may not be reaching the nucleus, the site of action of the duocarmycins. Cell lines that demonstrated appreciable levels of TF were sensitive to PDC 3. Interestingly, HCT116, which had relatively high TF expression and cathepsin B activity, demonstrated the greatest sensitivity to PDC 3, approaching similar potency to the payload 5. Excitingly, A549, H292, and 16HBE14o, which showed no detectable TF expression, appeared unaffected by PDC 3 up to 100 uM, suggesting that TF expression is required for the efficacy of PDC 3.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | 16HBE14o- cell | CVCL_0112 | ||
GnRH-III-[2ΔHis-3D-Tic, 8Lys(glutaryl-Val-Ala-PABC-diamine-PTX)] conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
0.51 ± 0.11 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
0.67 ± 0.07 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
5.03 ± 1.91 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
6.44 ± 1.22 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
10.54 ± 2.01 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Normal human pituitary cell | Homo sapiens | ||
| Experiment 6 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
10.65 ± 1.82 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate cancer | Human prostate cancer cells | Homo sapiens | ||
| Experiment 7 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
14.88 ± 0.33 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate cancer | Human prostate cancer cells | Homo sapiens | ||
| Experiment 8 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
18.49 ± 2.72 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Normal human pituitary cell | Homo sapiens | ||
GnRH-III-[2ΔHis-3D-Tic,8Lys(glutaryl-Val-Cit-PABC-Dau)] conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4.24 ± 1.09 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
10.97 ± 1.23 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate cancer | Human prostate cancer cells | Homo sapiens | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
11.70 ± 0.42 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Normal human pituitary cell | Homo sapiens | ||
| Experiment 4 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 100 µM | |||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
TATE-VCit-Ama [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
8.7 ± 0.5 μM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTS assay | ||||
| MOA of PDC |
Towards these ends, we report on the design of a modular and user-friendly approach to enable bio-orthogonal conjugation of synthetic amanitins to three different linkers that are elaborated to three potent peptide-amanitin conjugates that differ by their mode of toxin release, as defined by the linker. Octreotate (TATE-N3, 2) was chosen as it is a circulation-stable octapeptide somatostatin analog that agonizes somatostatin receptors (Kd &tide; 0.4 nM for sstr2). Targeting sstr2 has been successfully exploited for drug delivery, cancer imaging, and radiotherapy owing to the high levels of sstr2 expression on neuroendocrine tumors (e.g. carcinoids, pancreatic islet cell tumors, thyroid carcinomas, and small lung cancer to name a few). While this is the first report of an octreotate-amanitin conjugate, herein, octreotate serves as an exemplar to highlight the potential for developing peptide-amanitin conjugates in targeted applications.
Click to Show/Hide
|
||||
| Description |
With bioconjugates 5-7 in hand, we evaluated their cytotoxicity by assaying cell viability on an sstr2-positive rat pancreatic cancer Ar42J cells, using free amatoxins as controls. The MTS cell viability assay showed that all three PDCs were effective at reducing the viability of Ar42J cells in the low nM range, providing calculated EC50 values of 4.2, and 2.3 nM for conjugates 6 and 7, respectively; these displayed up to 1000-fold enhancement in apparent cytotoxicity compared to free amatoxins 3 and 4, both of which gave calculated EC50 values of &tide;2 μM. This points to successful targeting and intracellular accumulation of toxin inside cells through an sstr2-mediated uptake inside the cells. Further evidence supporting target-mediated uptake is the micromolar toxicity of bioconjugates to the sstr2-negative CHO cell line that is otherwise sensitive to non-targeted amanitin. In comparison to -amanitin and synthetic amatoxins 3 and 4, bioconjugates 5-7 are 7- to 14-fold less toxic to CHO cells (control cell line), likely owing to impaired uptake of octreotate-amanitin conjugates that must enter via diffusion mediated processes or other non-specific import mechanisms that are currently unknown. In addition, a blocking study was run in the presence of free TATE-N32, which led to the expected reduction in apparent toxicity.
Click to Show/Hide
|
||||
| In Vitro Model | Digestive system neoplasms | AR42J (SSTR2+) cell | CVCL_0143 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half maximal effective concentration (EC50) |
2.3 ±0.8 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTS assay | ||||
| MOA of PDC |
Towards these ends, we report on the design of a modular and user-friendly approach to enable bio-orthogonal conjugation of synthetic amanitins to three different linkers that are elaborated to three potent peptide-amanitin conjugates that differ by their mode of toxin release, as defined by the linker. Octreotate (TATE-N3, 2) was chosen as it is a circulation-stable octapeptide somatostatin analog that agonizes somatostatin receptors (Kd &tide; 0.4 nM for sstr2). Targeting sstr2 has been successfully exploited for drug delivery, cancer imaging, and radiotherapy owing to the high levels of sstr2 expression on neuroendocrine tumors (e.g. carcinoids, pancreatic islet cell tumors, thyroid carcinomas, and small lung cancer to name a few). While this is the first report of an octreotate-amanitin conjugate, herein, octreotate serves as an exemplar to highlight the potential for developing peptide-amanitin conjugates in targeted applications.
Click to Show/Hide
|
||||
| Description |
With bioconjugates 5-7 in hand, we evaluated their cytotoxicity by assaying cell viability on an sstr2-positive rat pancreatic cancer Ar42J cells, using free amatoxins as controls. The MTS cell viability assay showed that all three PDCs were effective at reducing the viability of Ar42J cells in the low nM range, providing calculated EC50 values of 4.2, and 2.3 nM for conjugates 6 and 7, respectively; these displayed up to 1000-fold enhancement in apparent cytotoxicity compared to free amatoxins 3 and 4, both of which gave calculated EC50 values of &tide;2 μM. This points to successful targeting and intracellular accumulation of toxin inside cells through an sstr2-mediated uptake inside the cells. Further evidence supporting target-mediated uptake is the micromolar toxicity of bioconjugates to the sstr2-negative CHO cell line that is otherwise sensitive to non-targeted amanitin. In comparison to -amanitin and synthetic amatoxins 3 and 4, bioconjugates 5-7 are 7- to 14-fold less toxic to CHO cells (control cell line), likely owing to impaired uptake of octreotate-amanitin conjugates that must enter via diffusion mediated processes or other non-specific import mechanisms that are currently unknown. In addition, a blocking study was run in the presence of free TATE-N32, which led to the expected reduction in apparent toxicity.
Click to Show/Hide
|
||||
| In Vitro Model | Digestive system neoplasms | AR42J (SSTR2+) cell | CVCL_0143 | ||
GnRH-III-[2His-3Trp,8Lys(glutaryl-Val-Cit-PABC-Dau)] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
11.18 ± 0.38 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
13.66 ± 1.87 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate cancer | Human prostate cancer cells | Homo sapiens | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
16.12 ± 2.03 µM
|
|||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
Human pituitary and human prostate cancer tissues have been used to evaluate the binding affinities of the new GnRH-III-drug conjugates to GnRH-R. Therefore, increasing compound concentrations were applied and the displacement of radiolabeled [125I]-triptorelin from GnRH-Rs was detected. The obtained results were compared with the binding affinities of the oxime bond-linked GnRH-III-Dau conjugates (I, II). All compounds bind to the receptors with high affinities in the low nanomolar range, while GnRH unrelated peptides such as somatostatin or bombesin were not able to displace the radio-labelled triptorelin. However, in comparison to the GnRH-III-homing peptide (I), the self-immolative linker conjugate exhibited a 3- to 10-times reduced affinity to the GnRH receptors. Interestingly, most of the PTX-containing cleavable compounds possessed a slightly higher binding affinity than the corresponding Dau-equivalent, even if the targeting sequence and the cathepsin cleavage site remained the same.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Normal human pituitary cell | Homo sapiens | ||
| Experiment 4 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
85.57 ± 24.33 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | GraphPad prism assay | ||||
| MOA of PDC |
Drug delivery systems (DDS) are promising tools for targeted tumor therapy providing the selective delivery of cytotoxic drugs to malignant cells, while side-effects and systemic toxicity are reduced. In addition to monoclonal antibodies (mAb), peptide ligands with a high affinity for tumor-specific cell surface compartments (e.g., receptors) can be used as carriers for cytotoxic payloads, as they provide beneficial features such as good tissue penetration, low immunogenicity and structural simplicity which enables their cost-efficient production by chemical synthesi. Receptors for the human gonadotropin releasing hormone (GnRH-I, <EHWSYGLRPG-NH2, <E is pyroglutamic acid) were not only identified in the pituitary, but also in various reproductive system-related cancers, such as breast, prostate and ovarian cancers, as well as non-reproductive cancers, such as colon and lung cancer. Thus, GnRH-related peptides are promising homing devices to deliver cytotoxic drugs selectively to cancer cells. A natural isoform of GnRH-I is the sea lamprey analog GnRH-III. This weak GnRH agonist binds to GnRH receptors (GnRH-R) on cancer cells and induces, like GnRH-I, a direct antitumor activity on several cancer cell lines, but its gonadotropin releasing activity is 500-1000 times lower in vitro and in vivo. Due to the direct anticancer activity and the low endocrine effect, GnRH-III and its derivatives have been successfully used as homing devices in in vitro and in vivo experiments. Encouraged by these promising findings, we report on the synthesis and biochemical characterization of eight cleavable self-immolative linker containing GnRH-III-drug conjugates. Of particular interest was the comparison of (1) two GnRH-III targeting moieties (GnRH-III-[4Lys(Bu)] (I) and GnRH-III-[2His,3D-Tic,4Lys(Bu)] (II)), (2) two cathepsin B-cleavable dipeptidyl-PABC linkers (Val-Ala and Val-Cit) and (3) two traditional anticancer drugs with different modes of action (Dau and PTX). For a better comparison and to demonstrate the proof of concept, four corresponding non-cleavable GnRH-III-Dau and -PTX conjugates have been developed and analyzed. The 8Lys of the targeting peptide was used as the ligation site. In the case of the Dau conjugates, the amino group of the daunosamine sugar has been used for attachment to the linker, while in the case of PTX, the C2-OH group was exploited for this purpose. All synthesized GnRH-III-Dau and -PTX conjugates were studied for their anticancer activity on A2780 ovarian and Panc-1 pancreatic cancer cells. Furthermore, the release of the drug by lysosomal enzymes and the GnRH-R binding affinities of the SMDC were examined.
Click to Show/Hide
|
||||
| Description |
To investigate the anticancer activity of the GnRH-III drug conjugates, cell viability studies have been performed on A2780 ovarian cancer and Panc-1 pancreatic cancer cells. The GnRH-R expression of these cell lines was determined by Western blot studies. In the case of the A2780 cells, a distinct band at 38 kDa could be detected which corresponds to the full-length human GnRH-R. In contrast, the signal intensity of the 38 kDa band was much lower for Panc-1 pancreatic cancer cells being in line with our previous results. Thus, the antiproliferative activity of the GnRH-drug conjugates was studied on high-GnRH-R-expressing A2780 cells and low-GnRH-R-expressing Panc-1 cells. Since the release of free Dau and PTX can be assumed, both drugs were used as controls. The cells were treated for either 24 h (Dau conjugates) or six hours (PTX compounds), followed by additional incubation with fresh growth medium until 72 h after treatment initiation. The obtained results reveal, on the one hand, that the non-cleavable linker-containing conjugates possess a reduced anticancer activity in comparison to the cleavable conjugates and, on the other hand, that the activity of the all GnRH-III-drug conjugates was substantially reduced compared to the free drug. Moreover, all compounds displayed a lower biological activity on Panc-1 cells than on A2780. In the case of the cleavable GnRH-III-Dau conjugates, the IC50 values varied between 2.85-11.18 μM on A2780 cells, whereby the best activity was obtained for compound 13 (2.85 μM) which contained the cathepsin B-cleavage site Val-Ala and the GnRH-III-[2His-3D-Tic-4Lys(Bu)] peptide carrier. Apart from that, the IC50 values of the cleavable PTX conjugates on A2780 cells are in the same sub-micromolar range and vary between 0.51-0.77 μM, while the activity of these conjugates was approximately 10 times lower on Panc-1 cells (5.03-8.15 μM).
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
References