
Peptide-drug Conjugate Information
General Information of This Peptide-drug Conjugate (PDC)
| PDC ID |
PDC_02049
|
|||||
|---|---|---|---|---|---|---|
| PDC Name |
Boc-Asn-Phe-Pro-AZT
|
|||||
| PDC Status |
Investigative
|
|||||
| Indication |
In total 1 Indication(s)
|
|||||
| Structure |
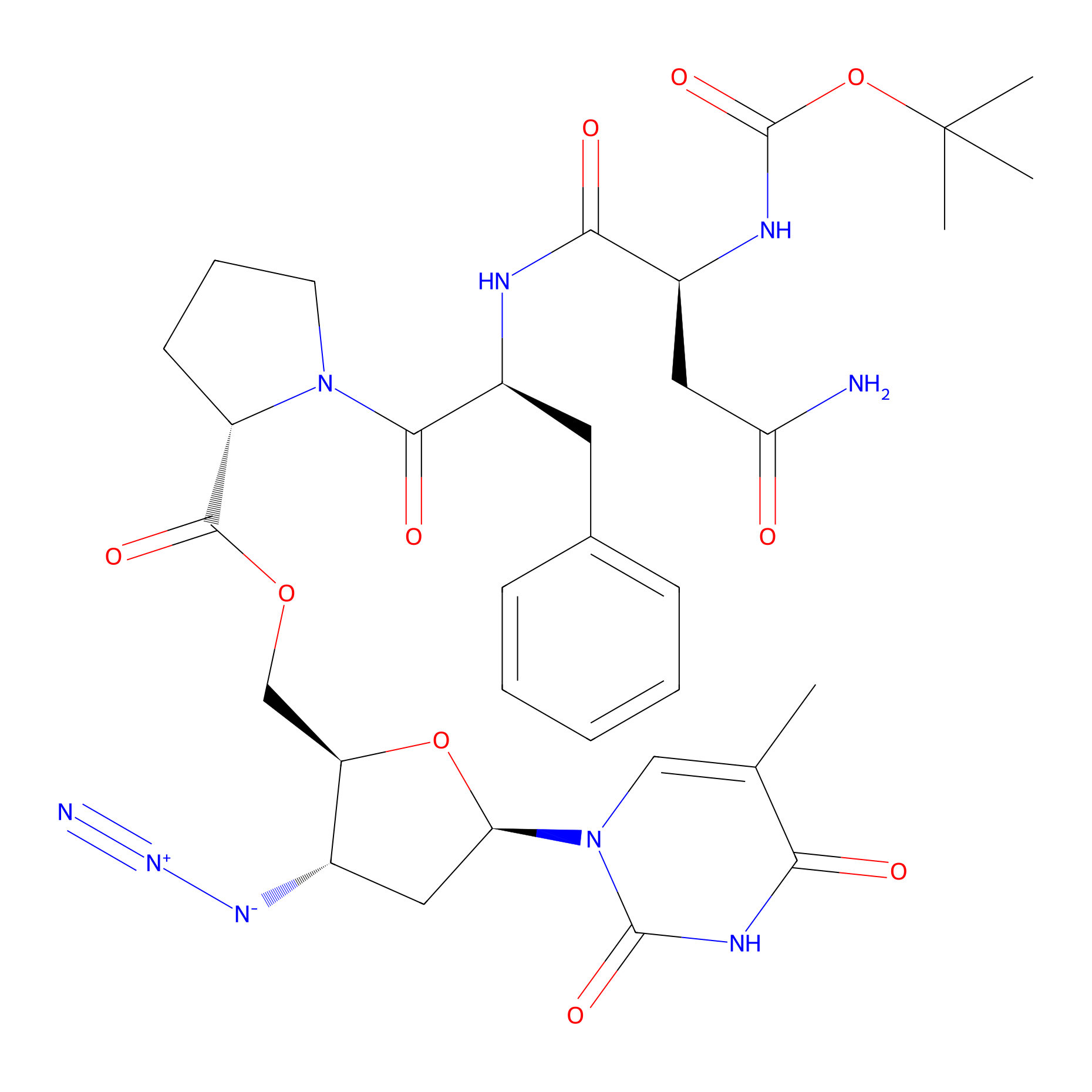
|
|||||
| Peptide Name |
Tripeptide 82
|
Peptide Info | ||||
| Receptor Name |
HIV-1 protease
|
Receptor Info | ||||
| Drug Name |
Zidovudine
|
Drug Info | ||||
| Linker Name |
Ester bond
|
Linker Info | ||||
| Formula |
C33H43N9O10
|
|||||
| #Ro5 Violations (Lipinski): 3 | Molecular Weight | 725.76 | ||||
| Lipid-water partition coefficient (xlogp) | 0.6733 | |||||
| Hydrogen Bond Donor Count (hbonddonor) | 4 | |||||
| Hydrogen Bond Acceptor Count (hbondacc) | 12 | |||||
| Rotatable Bond Count (rotbonds) | 13 | |||||
Full List of Activity Data of This Peptide-drug Conjugate
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Human immunodeficiency virus infection | ||||
| Efficacy Data | Half maximal cytotoxicity concentration (CC50) | > 100 μM | |||
| Administration Time | 5 days | ||||
| MOA of PDC |
We report on the synthesis of a series of peptideAZT (2A, 2B, 5A, 5B) and peptide-(AZT-MP) conjugates incorporating peptide sequences found in a nonapeptide known to be a HIV-PR substrate. We also selected the AZT-MP phosphoramidate peptide-conjugates containing an additional alanine residue directly linked to the phosphorus atom. Indeed, among the various aminoacid phosphoramidates of ddN analogues known so far, the alanine derivative was shown to be one of the most efficient ddN-MP delivery system. All these conjugates contain the scissile Phe-Pro motif. In the case of the AZT prodrugs, the AZT and peptide moieties were connected through an ester bond. To ensure a greater stability, particularly toward aminopeptidases, the N-terminal amino group of the peptide was masked by different protecting groups. In the case of the AZT-MP prodrugs, the various peptide moieties were connected via their N-terminal amino group to the AZT-phenoxy-phosphodiester group. We report also on (i) the stability of the peptide-AZT and peptide(AZT-MP) prodrugs when incubated in a physiological medium that does or does not contain 10% fetal calf serum, and in CEM cell lysates in order to mimic the behaviour of these compounds inside the cells, (ii) their ability to inhibit the HIV-PR activity and their susceptibility to be hydrolysed by PR, and (iii) their in vitro anti-HIV activities and cytotoxicities, which were evaluated in acutely-infected and uninfected MT4 and CEM cells, respectively. Moreover, their antiviral activities were also investigated in a thymidine kinase-deficient (TK- ) CEM cell line in order to gain further insight into their mechanism of action.
Click to Show/Hide
|
||||
| Description |
The in vitro anti-HIV activity and cytotoxicity of the peptide conjugates were determined both in infected and non-infected CEM-SS and MT-4 cells against HIV-1 (LAI and IIIB strain, respectively) according to published procedures. These assays were also performed on infected and non-infected TK- CEM cells.The data are collected in Tables 1 and 2 together along with data for AZT. Ester conjugates when incubated with the cells for 5 days were found to inhibit HIV replication at IC50 ranging from 6 to 140 nM in TK+ CEM-SS and from 5 to 530 nM in MT-4. Under these assay conditions, AZT inhibited HIV replication at IC50 of 14 nM (CEM-SS) and 16 nM (MT-4). As with their parent drug, none of the conjugates inhibited HIV replication in TK- CEM cells. Moreover, except for compound 5Bb, none of them was found to be cytotoxic on the three cell types. The antiviral activity level measured for these ester conjugates appeared to be correlated, to some extent, to their hydrolysis t1/2 value and hence, to the amount of AZT released during the 5 days of the experiment. Indeed, the prodrug series displaying the higher t1/2 value range, that is, the Ile-AZT series, correspond to those for which a lower HIV inhibition level was obtained. Conversely, the set of prodrug that gave a higher anti-HIV inhibition, that is, the Pro-AZT series, were those that were hydrolysed more rapidly. Moreover, the faster their hydrolysis, the closer their antiviral activity to that of free AZT. This is particularly relevant for the antiviral activities measured on MT-4 cells. However, it is noticeable that compounds Z-Phe-ProAZT (2Ac) and Qnc-Phe-Pro-Ile-AZT (2Bd), which are the most stable compounds of the series, constitute exception to this rule. Indeed, these derivatives exhibited, despite a quite high stability, a surprizingly high anti-HIV activity in CEM-SS and also in MT-4 cells for 2Ac.
Click to Show/Hide
|
||||
| In Vitro Model | HIV infection | HIV-1 LAI infected CEM/TK+ cell | CVCL_0207 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Human immunodeficiency virus infection | ||||
| Efficacy Data | Half maximal cytotoxicity concentration (CC50) | > 100 μM | |||
| Administration Time | 5 days | ||||
| MOA of PDC |
We report on the synthesis of a series of peptideAZT (2A, 2B, 5A, 5B) and peptide-(AZT-MP) conjugates incorporating peptide sequences found in a nonapeptide known to be a HIV-PR substrate. We also selected the AZT-MP phosphoramidate peptide-conjugates containing an additional alanine residue directly linked to the phosphorus atom. Indeed, among the various aminoacid phosphoramidates of ddN analogues known so far, the alanine derivative was shown to be one of the most efficient ddN-MP delivery system. All these conjugates contain the scissile Phe-Pro motif. In the case of the AZT prodrugs, the AZT and peptide moieties were connected through an ester bond. To ensure a greater stability, particularly toward aminopeptidases, the N-terminal amino group of the peptide was masked by different protecting groups. In the case of the AZT-MP prodrugs, the various peptide moieties were connected via their N-terminal amino group to the AZT-phenoxy-phosphodiester group. We report also on (i) the stability of the peptide-AZT and peptide(AZT-MP) prodrugs when incubated in a physiological medium that does or does not contain 10% fetal calf serum, and in CEM cell lysates in order to mimic the behaviour of these compounds inside the cells, (ii) their ability to inhibit the HIV-PR activity and their susceptibility to be hydrolysed by PR, and (iii) their in vitro anti-HIV activities and cytotoxicities, which were evaluated in acutely-infected and uninfected MT4 and CEM cells, respectively. Moreover, their antiviral activities were also investigated in a thymidine kinase-deficient (TK- ) CEM cell line in order to gain further insight into their mechanism of action.
Click to Show/Hide
|
||||
| Description |
The in vitro anti-HIV activity and cytotoxicity of the peptide conjugates were determined both in infected and non-infected CEM-SS and MT-4 cells against HIV-1 (LAI and IIIB strain, respectively) according to published procedures. These assays were also performed on infected and non-infected TK- CEM cells.The data are collected in Tables 1 and 2 together along with data for AZT. Ester conjugates when incubated with the cells for 5 days were found to inhibit HIV replication at IC50 ranging from 6 to 140 nM in TK+ CEM-SS and from 5 to 530 nM in MT-4. Under these assay conditions, AZT inhibited HIV replication at IC50 of 14 nM (CEM-SS) and 16 nM (MT-4). As with their parent drug, none of the conjugates inhibited HIV replication in TK- CEM cells. Moreover, except for compound 5Bb, none of them was found to be cytotoxic on the three cell types. The antiviral activity level measured for these ester conjugates appeared to be correlated, to some extent, to their hydrolysis t1/2 value and hence, to the amount of AZT released during the 5 days of the experiment. Indeed, the prodrug series displaying the higher t1/2 value range, that is, the Ile-AZT series, correspond to those for which a lower HIV inhibition level was obtained. Conversely, the set of prodrug that gave a higher anti-HIV inhibition, that is, the Pro-AZT series, were those that were hydrolysed more rapidly. Moreover, the faster their hydrolysis, the closer their antiviral activity to that of free AZT. This is particularly relevant for the antiviral activities measured on MT-4 cells. However, it is noticeable that compounds Z-Phe-ProAZT (2Ac) and Qnc-Phe-Pro-Ile-AZT (2Bd), which are the most stable compounds of the series, constitute exception to this rule. Indeed, these derivatives exhibited, despite a quite high stability, a surprizingly high anti-HIV activity in CEM-SS and also in MT-4 cells for 2Ac.
Click to Show/Hide
|
||||
| In Vitro Model | HIV infection | HIV-1 LAI infected CEM/TK- cell | CVCL_0207 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Human immunodeficiency virus infection | ||||
| Efficacy Data | Half maximal cytotoxicity concentration (CC50) | > 100 μM | |||
| Administration Time | 5 days | ||||
| MOA of PDC |
We report on the synthesis of a series of peptideAZT (2A, 2B, 5A, 5B) and peptide-(AZT-MP) conjugates incorporating peptide sequences found in a nonapeptide known to be a HIV-PR substrate. We also selected the AZT-MP phosphoramidate peptide-conjugates containing an additional alanine residue directly linked to the phosphorus atom. Indeed, among the various aminoacid phosphoramidates of ddN analogues known so far, the alanine derivative was shown to be one of the most efficient ddN-MP delivery system. All these conjugates contain the scissile Phe-Pro motif. In the case of the AZT prodrugs, the AZT and peptide moieties were connected through an ester bond. To ensure a greater stability, particularly toward aminopeptidases, the N-terminal amino group of the peptide was masked by different protecting groups. In the case of the AZT-MP prodrugs, the various peptide moieties were connected via their N-terminal amino group to the AZT-phenoxy-phosphodiester group. We report also on (i) the stability of the peptide-AZT and peptide(AZT-MP) prodrugs when incubated in a physiological medium that does or does not contain 10% fetal calf serum, and in CEM cell lysates in order to mimic the behaviour of these compounds inside the cells, (ii) their ability to inhibit the HIV-PR activity and their susceptibility to be hydrolysed by PR, and (iii) their in vitro anti-HIV activities and cytotoxicities, which were evaluated in acutely-infected and uninfected MT4 and CEM cells, respectively. Moreover, their antiviral activities were also investigated in a thymidine kinase-deficient (TK- ) CEM cell line in order to gain further insight into their mechanism of action.
Click to Show/Hide
|
||||
| Description |
The in vitro anti-HIV activity and cytotoxicity of the peptide conjugates were determined both in infected and non-infected CEM-SS and MT-4 cells against HIV-1 (LAI and IIIB strain, respectively) according to published procedures. These assays were also performed on infected and non-infected TK- CEM cells.The data are collected in Tables 1 and 2 together along with data for AZT. Ester conjugates when incubated with the cells for 5 days were found to inhibit HIV replication at IC50 ranging from 6 to 140 nM in TK+ CEM-SS and from 5 to 530 nM in MT-4. Under these assay conditions, AZT inhibited HIV replication at IC50 of 14 nM (CEM-SS) and 16 nM (MT-4). As with their parent drug, none of the conjugates inhibited HIV replication in TK- CEM cells. Moreover, except for compound 5Bb, none of them was found to be cytotoxic on the three cell types. The antiviral activity level measured for these ester conjugates appeared to be correlated, to some extent, to their hydrolysis t1/2 value and hence, to the amount of AZT released during the 5 days of the experiment. Indeed, the prodrug series displaying the higher t1/2 value range, that is, the Ile-AZT series, correspond to those for which a lower HIV inhibition level was obtained. Conversely, the set of prodrug that gave a higher anti-HIV inhibition, that is, the Pro-AZT series, were those that were hydrolysed more rapidly. Moreover, the faster their hydrolysis, the closer their antiviral activity to that of free AZT. This is particularly relevant for the antiviral activities measured on MT-4 cells. However, it is noticeable that compounds Z-Phe-ProAZT (2Ac) and Qnc-Phe-Pro-Ile-AZT (2Bd), which are the most stable compounds of the series, constitute exception to this rule. Indeed, these derivatives exhibited, despite a quite high stability, a surprizingly high anti-HIV activity in CEM-SS and also in MT-4 cells for 2Ac.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MT4/HIV-1 cell | CVCL_RW54 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Human immunodeficiency virus infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
35 nM
|
|||
| Administration Time | 5 days | ||||
| MOA of PDC |
We report on the synthesis of a series of peptideAZT (2A, 2B, 5A, 5B) and peptide-(AZT-MP) conjugates incorporating peptide sequences found in a nonapeptide known to be a HIV-PR substrate. We also selected the AZT-MP phosphoramidate peptide-conjugates containing an additional alanine residue directly linked to the phosphorus atom. Indeed, among the various aminoacid phosphoramidates of ddN analogues known so far, the alanine derivative was shown to be one of the most efficient ddN-MP delivery system. All these conjugates contain the scissile Phe-Pro motif. In the case of the AZT prodrugs, the AZT and peptide moieties were connected through an ester bond. To ensure a greater stability, particularly toward aminopeptidases, the N-terminal amino group of the peptide was masked by different protecting groups. In the case of the AZT-MP prodrugs, the various peptide moieties were connected via their N-terminal amino group to the AZT-phenoxy-phosphodiester group. We report also on (i) the stability of the peptide-AZT and peptide(AZT-MP) prodrugs when incubated in a physiological medium that does or does not contain 10% fetal calf serum, and in CEM cell lysates in order to mimic the behaviour of these compounds inside the cells, (ii) their ability to inhibit the HIV-PR activity and their susceptibility to be hydrolysed by PR, and (iii) their in vitro anti-HIV activities and cytotoxicities, which were evaluated in acutely-infected and uninfected MT4 and CEM cells, respectively. Moreover, their antiviral activities were also investigated in a thymidine kinase-deficient (TK- ) CEM cell line in order to gain further insight into their mechanism of action.
Click to Show/Hide
|
||||
| Description |
The in vitro anti-HIV activity and cytotoxicity of the peptide conjugates were determined both in infected and non-infected CEM-SS and MT-4 cells against HIV-1 (LAI and IIIB strain, respectively) according to published procedures. These assays were also performed on infected and non-infected TK- CEM cells.The data are collected in Tables 1 and 2 together along with data for AZT. Ester conjugates when incubated with the cells for 5 days were found to inhibit HIV replication at IC50 ranging from 6 to 140 nM in TK+ CEM-SS and from 5 to 530 nM in MT-4. Under these assay conditions, AZT inhibited HIV replication at IC50 of 14 nM (CEM-SS) and 16 nM (MT-4). As with their parent drug, none of the conjugates inhibited HIV replication in TK- CEM cells. Moreover, except for compound 5Bb, none of them was found to be cytotoxic on the three cell types. The antiviral activity level measured for these ester conjugates appeared to be correlated, to some extent, to their hydrolysis t1/2 value and hence, to the amount of AZT released during the 5 days of the experiment. Indeed, the prodrug series displaying the higher t1/2 value range, that is, the Ile-AZT series, correspond to those for which a lower HIV inhibition level was obtained. Conversely, the set of prodrug that gave a higher anti-HIV inhibition, that is, the Pro-AZT series, were those that were hydrolysed more rapidly. Moreover, the faster their hydrolysis, the closer their antiviral activity to that of free AZT. This is particularly relevant for the antiviral activities measured on MT-4 cells. However, it is noticeable that compounds Z-Phe-ProAZT (2Ac) and Qnc-Phe-Pro-Ile-AZT (2Bd), which are the most stable compounds of the series, constitute exception to this rule. Indeed, these derivatives exhibited, despite a quite high stability, a surprizingly high anti-HIV activity in CEM-SS and also in MT-4 cells for 2Ac.
Click to Show/Hide
|
||||
| In Vitro Model | HIV infection | HIV-1 LAI infected CEM/TK+ cell | CVCL_0207 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Human immunodeficiency virus infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
82 nM
|
|||
| Administration Time | 5 days | ||||
| MOA of PDC |
We report on the synthesis of a series of peptideAZT (2A, 2B, 5A, 5B) and peptide-(AZT-MP) conjugates incorporating peptide sequences found in a nonapeptide known to be a HIV-PR substrate. We also selected the AZT-MP phosphoramidate peptide-conjugates containing an additional alanine residue directly linked to the phosphorus atom. Indeed, among the various aminoacid phosphoramidates of ddN analogues known so far, the alanine derivative was shown to be one of the most efficient ddN-MP delivery system. All these conjugates contain the scissile Phe-Pro motif. In the case of the AZT prodrugs, the AZT and peptide moieties were connected through an ester bond. To ensure a greater stability, particularly toward aminopeptidases, the N-terminal amino group of the peptide was masked by different protecting groups. In the case of the AZT-MP prodrugs, the various peptide moieties were connected via their N-terminal amino group to the AZT-phenoxy-phosphodiester group. We report also on (i) the stability of the peptide-AZT and peptide(AZT-MP) prodrugs when incubated in a physiological medium that does or does not contain 10% fetal calf serum, and in CEM cell lysates in order to mimic the behaviour of these compounds inside the cells, (ii) their ability to inhibit the HIV-PR activity and their susceptibility to be hydrolysed by PR, and (iii) their in vitro anti-HIV activities and cytotoxicities, which were evaluated in acutely-infected and uninfected MT4 and CEM cells, respectively. Moreover, their antiviral activities were also investigated in a thymidine kinase-deficient (TK- ) CEM cell line in order to gain further insight into their mechanism of action.
Click to Show/Hide
|
||||
| Description |
The in vitro anti-HIV activity and cytotoxicity of the peptide conjugates were determined both in infected and non-infected CEM-SS and MT-4 cells against HIV-1 (LAI and IIIB strain, respectively) according to published procedures. These assays were also performed on infected and non-infected TK- CEM cells.The data are collected in Tables 1 and 2 together along with data for AZT. Ester conjugates when incubated with the cells for 5 days were found to inhibit HIV replication at IC50 ranging from 6 to 140 nM in TK+ CEM-SS and from 5 to 530 nM in MT-4. Under these assay conditions, AZT inhibited HIV replication at IC50 of 14 nM (CEM-SS) and 16 nM (MT-4). As with their parent drug, none of the conjugates inhibited HIV replication in TK- CEM cells. Moreover, except for compound 5Bb, none of them was found to be cytotoxic on the three cell types. The antiviral activity level measured for these ester conjugates appeared to be correlated, to some extent, to their hydrolysis t1/2 value and hence, to the amount of AZT released during the 5 days of the experiment. Indeed, the prodrug series displaying the higher t1/2 value range, that is, the Ile-AZT series, correspond to those for which a lower HIV inhibition level was obtained. Conversely, the set of prodrug that gave a higher anti-HIV inhibition, that is, the Pro-AZT series, were those that were hydrolysed more rapidly. Moreover, the faster their hydrolysis, the closer their antiviral activity to that of free AZT. This is particularly relevant for the antiviral activities measured on MT-4 cells. However, it is noticeable that compounds Z-Phe-ProAZT (2Ac) and Qnc-Phe-Pro-Ile-AZT (2Bd), which are the most stable compounds of the series, constitute exception to this rule. Indeed, these derivatives exhibited, despite a quite high stability, a surprizingly high anti-HIV activity in CEM-SS and also in MT-4 cells for 2Ac.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MT4/HIV-1 cell | CVCL_RW54 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Human immunodeficiency virus infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 10 | |||
| Administration Time | 5 days | ||||
| MOA of PDC |
We report on the synthesis of a series of peptideAZT (2A, 2B, 5A, 5B) and peptide-(AZT-MP) conjugates incorporating peptide sequences found in a nonapeptide known to be a HIV-PR substrate. We also selected the AZT-MP phosphoramidate peptide-conjugates containing an additional alanine residue directly linked to the phosphorus atom. Indeed, among the various aminoacid phosphoramidates of ddN analogues known so far, the alanine derivative was shown to be one of the most efficient ddN-MP delivery system. All these conjugates contain the scissile Phe-Pro motif. In the case of the AZT prodrugs, the AZT and peptide moieties were connected through an ester bond. To ensure a greater stability, particularly toward aminopeptidases, the N-terminal amino group of the peptide was masked by different protecting groups. In the case of the AZT-MP prodrugs, the various peptide moieties were connected via their N-terminal amino group to the AZT-phenoxy-phosphodiester group. We report also on (i) the stability of the peptide-AZT and peptide(AZT-MP) prodrugs when incubated in a physiological medium that does or does not contain 10% fetal calf serum, and in CEM cell lysates in order to mimic the behaviour of these compounds inside the cells, (ii) their ability to inhibit the HIV-PR activity and their susceptibility to be hydrolysed by PR, and (iii) their in vitro anti-HIV activities and cytotoxicities, which were evaluated in acutely-infected and uninfected MT4 and CEM cells, respectively. Moreover, their antiviral activities were also investigated in a thymidine kinase-deficient (TK- ) CEM cell line in order to gain further insight into their mechanism of action.
Click to Show/Hide
|
||||
| Description |
The in vitro anti-HIV activity and cytotoxicity of the peptide conjugates were determined both in infected and non-infected CEM-SS and MT-4 cells against HIV-1 (LAI and IIIB strain, respectively) according to published procedures. These assays were also performed on infected and non-infected TK- CEM cells.The data are collected in Tables 1 and 2 together along with data for AZT. Ester conjugates when incubated with the cells for 5 days were found to inhibit HIV replication at IC50 ranging from 6 to 140 nM in TK+ CEM-SS and from 5 to 530 nM in MT-4. Under these assay conditions, AZT inhibited HIV replication at IC50 of 14 nM (CEM-SS) and 16 nM (MT-4). As with their parent drug, none of the conjugates inhibited HIV replication in TK- CEM cells. Moreover, except for compound 5Bb, none of them was found to be cytotoxic on the three cell types. The antiviral activity level measured for these ester conjugates appeared to be correlated, to some extent, to their hydrolysis t1/2 value and hence, to the amount of AZT released during the 5 days of the experiment. Indeed, the prodrug series displaying the higher t1/2 value range, that is, the Ile-AZT series, correspond to those for which a lower HIV inhibition level was obtained. Conversely, the set of prodrug that gave a higher anti-HIV inhibition, that is, the Pro-AZT series, were those that were hydrolysed more rapidly. Moreover, the faster their hydrolysis, the closer their antiviral activity to that of free AZT. This is particularly relevant for the antiviral activities measured on MT-4 cells. However, it is noticeable that compounds Z-Phe-ProAZT (2Ac) and Qnc-Phe-Pro-Ile-AZT (2Bd), which are the most stable compounds of the series, constitute exception to this rule. Indeed, these derivatives exhibited, despite a quite high stability, a surprizingly high anti-HIV activity in CEM-SS and also in MT-4 cells for 2Ac.
Click to Show/Hide
|
||||
| In Vitro Model | HIV infection | HIV-1 LAI infected CEM/TK- cell | CVCL_0207 | ||
References