
Peptide Information
General Information of This Peptide
| Peptide ID |
PEP00091
|
|||||
|---|---|---|---|---|---|---|
| Peptide Name |
LRRYQWAVGHLNle-NH<sub>2</sub>
|
|||||
| Structure |
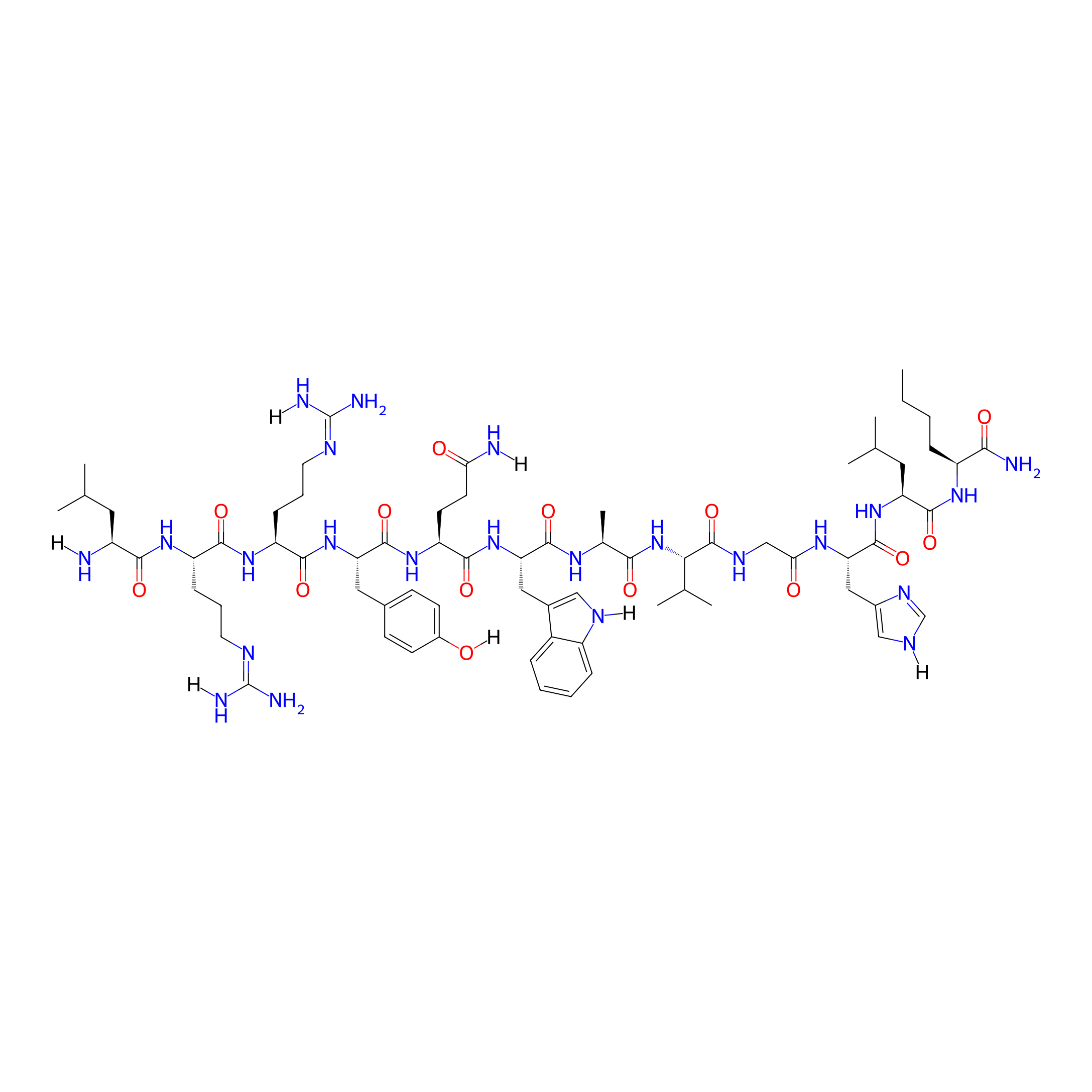
|
|||||
| Sequence |
LRRYQWAVGHLNle-NH2
|
|||||
| Peptide Type |
Linear
|
|||||
| Receptor Name |
Bombesin receptor
|
Receptor Info | ||||
| PDC Transmembrane Types | Cell targeting peptides (CTPs) | |||||
| Formula |
C71H111N23O14
|
|||||
| Isosmiles |
[H]NC(=O)CC[C@H](NC(=O)[C@H](Cc1ccc(O[H])cc1)NC(=O)[C@H](CCC/N=C(/N)N[H])NC(=O)[C@H](CCC/N=C(/N)N[H])NC(=O)[C@H](CC(C)C)N[H])C(=O)N[C@@H](Cc1cn([H])c2ccccc12)C(=O)N[C@@H](C)C(=O)N[C@H](C(=O)NCC(=O)N[C@@H](Cc1cn([H])cn1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCC)C(N)=O)C(C)C
|
|||||
| InChI |
InChI=1S/C71H111N23O14/c1-9-10-16-48(59(74)98)87-66(105)52(29-38(4)5)91-68(107)55(32-43-34-79-36-84-43)86-57(97)35-83-69(108)58(39(6)7)94-60(99)40(8)85-65(104)54(31-42-33-82-47-17-12-11-15-45(42)47)93-64(103)51(24-25-56(73)96)90-67(106)53(30-41-20-22-44(95)23-21-41)92-63(102)50(19-14-27-81-71(77)78)89-62(101)49(18-13-26-80-70(75)76)88-61(100)46(72)28-37(2)3/h11-12,15,17,20-23,33-34,36-40,46,48-55,58,82,95H,9-10,13-14,16,18-19,24-32,35,72H2,1-8H3,(H2,73,96)(H2,74,98)(H,79,84)(H,83,108)(H,85,104)(H,86,97)(H,87,105)(H,88,100)(H,89,101)(H,90,106)(H,91,107)(H,92,102)(H,93,103)(H,94,99)(H4,75,76,80)(H4,77,78,81)/t40-,46-,48-,49-,50-,51-,52-,53-,54-,55-,58-/m0/s1
|
|||||
| InChIKey |
HBMGEBVBKMLBSB-UTHWUPJASA-N
|
|||||
| Pharmaceutical Properties |
Molecule Weight
|
1510.816
|
Polar area
|
625.8
|
||
|
Complexity
|
1509.868085
|
xlogp Value
|
-3.2677
|
|||
|
Heavy Count
|
108
|
Rot Bonds
|
50
|
|||
|
Hbond acc
|
18
|
Hbond Donor
|
21
|
|||
Each Peptide-drug Conjugate Related to This Peptide
Full Information of The Activity Data of The PDC(s) Related to This Peptide
Dau=Aoa-LRRYQWAVGHLNle-NH2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Half Maximal Uptake Concentration (UC50) |
22.92 µM
|
|||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
Like GnRH and somatostatin, bombesin (BBN) is another example of peptide hor-556 mone which receptors are overexpressed in cancerous tissues. This 14-mer peptide (Glp-Gln-Arg-Leu-Gly-Asn-Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2), first discovered in the skin of the frog Bombina bombina, is associated with the gastrin-releasing peptide (GRP) as a mammalian counterpart. The bombesin receptor family comprises neuromedin receptor B (NMB-R, or BB1), gastrin-releasing peptide receptor (GRP-R, or BB2) and bombesin receptor subtype 3 (BRS-3, or BB3). Among them, GRP-R has been the most investigated so far and has been proven to be upregulated in breast, prostate, pancreas, small-cell lung cancers, among others, hence representing a suitable target for drug delivery to tumors. Many attempts have been made to modify and shorten the sequence of bombesin to tailor its stability, activity (agonist or antagonist), affinity and selectivity towards this receptor. Several research groups have encouraged the use of their optimized structures as putative drug delivery systems, but they were never directly compared. Moreover, the previous examples of conjugates between GRP-R ligands and anthracyclines display a labile ester bond that could cause the early release of the drug in vivo. Therefore, our group took the leap and produced conjugates based on the most promising bombesin analogs as homing devices attached to Dau via an oxime bond and two cathepsin B-cleavable linkers (LRRY or GFLG). Furthermore, a conjugate bearing a novel developed bombesin analog ([6D-Phe, 11-Ala, 13Sta, 14Nle]-BBN(7-14)) was synthesized. The use of oxime-linked Dau as a payload promoted the identification of three conjugates with improved cytostatic activity and cellular uptake by human prostate (PC-3) and breast cancer (MDA-MB-231 and MDA-MB-453) cell lines. The Dau=Aoa-Leu-OH active metabolite was readily released in all the cases in less than 30 min in rat liver lysosomal homogenate, but only L5 (Dau=Aoa-LRRY-[6D-Phe, 13Sta, 14Leu]-BBN(7-14), where Sta is statine) and L6 Dau=Aoa-LRRY-[6D-Phe, 11-Ala, 13Sta, 14Nle]-BBN(7-14) demonstrated a satisfactory stability in the mouse plasma. Therefore, only these two conjugates were further investigated in vivo. As mentioned above, their chronic toxicity was assessed in healthy mice by administering the conjugates doses of 5, 10 and 20 mg Dau content/kg body weight. The PDCs were not critically harmful at any dose, although a relatively high mouse weight loss (10-15%) was induced at the highest dose. The in vivo antitumor efficacy was studied in murine xenograft models bearing s.c.-inoculated PC-3 human prostatic adenocarcinoma. The conjugates were administered intraperitoneally every fifth day starting from day 9 after tumor inoculation, for a total of five treatments, at a dose of 10 mg Dau-content/kg body weight, and their tumor growth inhibition values were compared to those of the mice treated with the maximum tolerated dose of free Dau (1 mg/kg) and 0.9% saline solution (control group). On the last day of the experiment (day 33), L5 and L6 revealed reductions in the tumor size by 21.4% and 31.4% and tumor weights by 16.6% and 33.1% compared to those of the control, respectively. On the other hand, the Dau-treated group had to be terminated on day 26 because of severe toxicity, without showing a significant reduction in the tumor volume and weight. To better understand the long-term effect of the newly developed compounds, the tumor doubling time (DT) was calculated; both the PDCs significantly increased the DT compared to the treatment with free Dau.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4.38 ± 0.33 µM
|
|||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
Like GnRH and somatostatin, bombesin (BBN) is another example of peptide hor-556 mone which receptors are overexpressed in cancerous tissues. This 14-mer peptide (Glp-Gln-Arg-Leu-Gly-Asn-Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2), first discovered in the skin of the frog Bombina bombina, is associated with the gastrin-releasing peptide (GRP) as a mammalian counterpart. The bombesin receptor family comprises neuromedin receptor B (NMB-R, or BB1), gastrin-releasing peptide receptor (GRP-R, or BB2) and bombesin receptor subtype 3 (BRS-3, or BB3). Among them, GRP-R has been the most investigated so far and has been proven to be upregulated in breast, prostate, pancreas, small-cell lung cancers, among others, hence representing a suitable target for drug delivery to tumors. Many attempts have been made to modify and shorten the sequence of bombesin to tailor its stability, activity (agonist or antagonist), affinity and selectivity towards this receptor. Several research groups have encouraged the use of their optimized structures as putative drug delivery systems, but they were never directly compared. Moreover, the previous examples of conjugates between GRP-R ligands and anthracyclines display a labile ester bond that could cause the early release of the drug in vivo. Therefore, our group took the leap and produced conjugates based on the most promising bombesin analogs as homing devices attached to Dau via an oxime bond and two cathepsin B-cleavable linkers (LRRY or GFLG). Furthermore, a conjugate bearing a novel developed bombesin analog ([6D-Phe, 11-Ala, 13Sta, 14Nle]-BBN(7-14)) was synthesized. The use of oxime-linked Dau as a payload promoted the identification of three conjugates with improved cytostatic activity and cellular uptake by human prostate (PC-3) and breast cancer (MDA-MB-231 and MDA-MB-453) cell lines. The Dau=Aoa-Leu-OH active metabolite was readily released in all the cases in less than 30 min in rat liver lysosomal homogenate, but only L5 (Dau=Aoa-LRRY-[6D-Phe, 13Sta, 14Leu]-BBN(7-14), where Sta is statine) and L6 Dau=Aoa-LRRY-[6D-Phe, 11-Ala, 13Sta, 14Nle]-BBN(7-14) demonstrated a satisfactory stability in the mouse plasma. Therefore, only these two conjugates were further investigated in vivo. As mentioned above, their chronic toxicity was assessed in healthy mice by administering the conjugates doses of 5, 10 and 20 mg Dau content/kg body weight. The PDCs were not critically harmful at any dose, although a relatively high mouse weight loss (10-15%) was induced at the highest dose. The in vivo antitumor efficacy was studied in murine xenograft models bearing s.c.-inoculated PC-3 human prostatic adenocarcinoma. The conjugates were administered intraperitoneally every fifth day starting from day 9 after tumor inoculation, for a total of five treatments, at a dose of 10 mg Dau-content/kg body weight, and their tumor growth inhibition values were compared to those of the mice treated with the maximum tolerated dose of free Dau (1 mg/kg) and 0.9% saline solution (control group). On the last day of the experiment (day 33), L5 and L6 revealed reductions in the tumor size by 21.4% and 31.4% and tumor weights by 16.6% and 33.1% compared to those of the control, respectively. On the other hand, the Dau-treated group had to be terminated on day 26 because of severe toxicity, without showing a significant reduction in the tumor volume and weight. To better understand the long-term effect of the newly developed compounds, the tumor doubling time (DT) was calculated; both the PDCs significantly increased the DT compared to the treatment with free Dau.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
References