
Peptide-drug Conjugate Information
General Information of This Peptide-drug Conjugate (PDC)
| PDC ID |
PDC_00500
|
|||||
|---|---|---|---|---|---|---|
| PDC Name |
RXRXRX - Nalidixic acid conjugate
|
|||||
| PDC Status |
Investigative
|
|||||
| Indication |
In total 1 Indication(s)
|
|||||
| Structure |
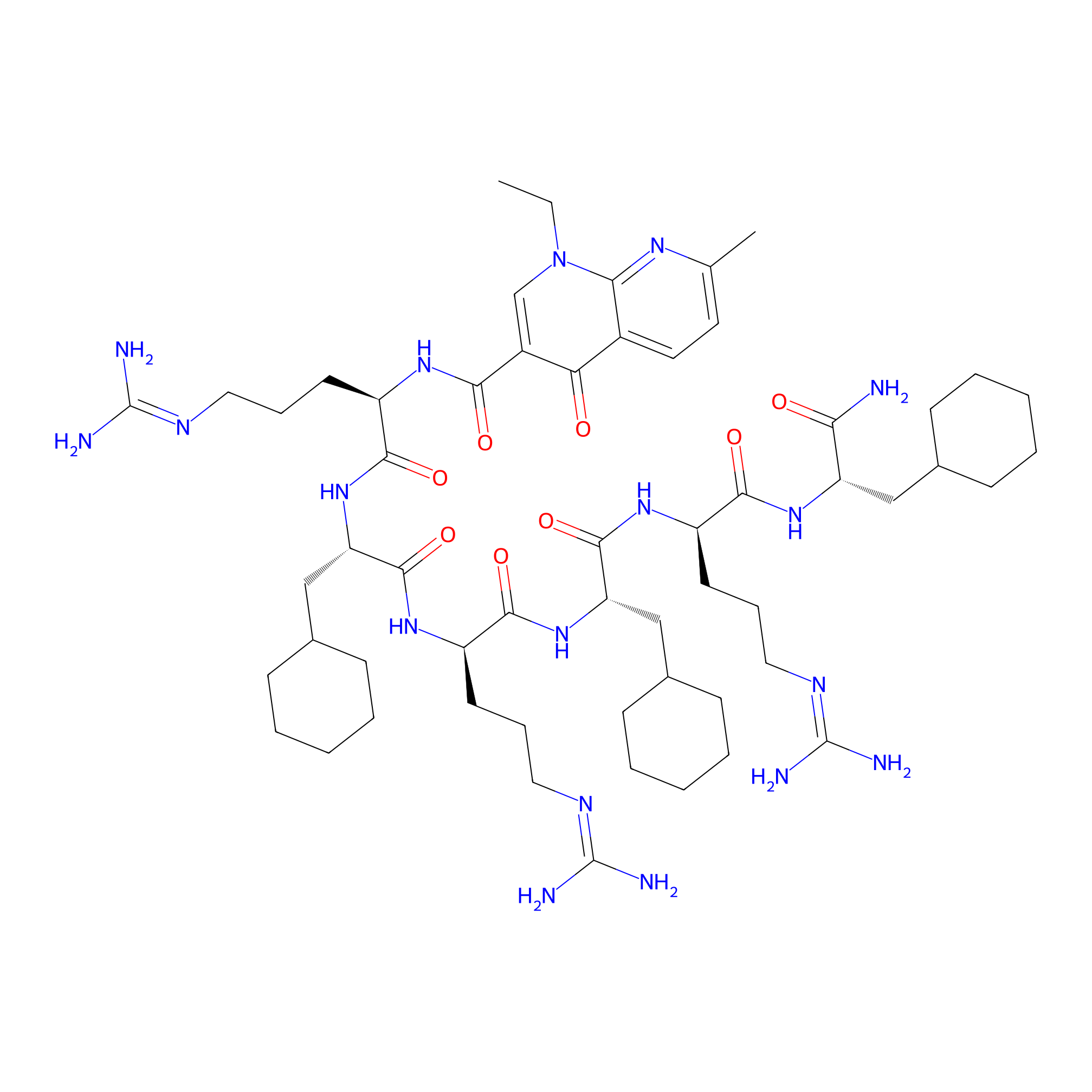
|
|||||
| Peptide Name |
RXRXRX
|
Peptide Info | ||||
| Receptor Name |
DNA gyrase subunit B (gyrB)
|
Receptor Info | ||||
| Drug Name |
Nalidixic Acid
|
Drug Info | ||||
| Linker Name |
Amide bond
|
Linker Info | ||||
| Formula |
C57H94N18O8
|
|||||
| #Ro5 Violations (Lipinski): 4 | Molecular Weight | 1159.497 | ||||
| Lipid-water partition coefficient (xlogp) | 0.80452 | |||||
| Hydrogen Bond Donor Count (hbonddonor) | 13 | |||||
| Hydrogen Bond Acceptor Count (hbondacc) | 13 | |||||
| Rotatable Bond Count (rotbonds) | 32 | |||||
Full List of Activity Data of This Peptide-drug Conjugate
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
6.0 ± 3.3 μM
|
|||
| Administration Time | 24 h | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
As expected, unconjugated NA showed limited activity toward MRSA and MSSA. Peptide vectors alone did not show significant toxicity to both MRSA and MSSA at all concentrations investigated. However, several of the peptide/drug conjugates, compounds 4 and 5 in particular, exhibited much lower IC50 and MIC values in both strains of the bacteria. A comparison of the hydrophobicities and molecular charges of the peptide/drug conjugates indicates that these two properties exert significant influence over the activities of these molecules. Hydrophobicity, measured by monitoring HPLC elution profiles, increased in the following order: 2 6 < 3 &tide; 4 < 5. The peptide conjugates that exhibited the highest potencies, compounds 4 and 5, had the highest levels of hydrophobicity. Compound 3, which had a similar level of hydrophobicity relative to these compounds, exhibited a lower level of toxicity, indicating that hydrophobicity alone does not control activity. This compound also carries a lower molecular charge, pointing to this parameter as another influencer of potency.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
7.0 ± 1.4 μM
|
|||
| Administration Time | 24 h | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
As expected, unconjugated NA showed limited activity toward MRSA and MSSA. Peptide vectors alone did not show significant toxicity to both MRSA and MSSA at all concentrations investigated. However, several of the peptide/drug conjugates, compounds 4 and 5 in particular, exhibited much lower IC50 and MIC values in both strains of the bacteria. A comparison of the hydrophobicities and molecular charges of the peptide/drug conjugates indicates that these two properties exert significant influence over the activities of these molecules. Hydrophobicity, measured by monitoring HPLC elution profiles, increased in the following order: 2 6 < 3 &tide; 4 < 5. The peptide conjugates that exhibited the highest potencies, compounds 4 and 5, had the highest levels of hydrophobicity. Compound 3, which had a similar level of hydrophobicity relative to these compounds, exhibited a lower level of toxicity, indicating that hydrophobicity alone does not control activity. This compound also carries a lower molecular charge, pointing to this parameter as another influencer of potency.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
25 μM
|
|||
| Administration Time | 2 h | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
As expected, unconjugated NA showed limited activity toward MRSA and MSSA. Peptide vectors alone did not show significant toxicity to both MRSA and MSSA at all concentrations investigated. However, several of the peptide/drug conjugates, compounds 4 and 5 in particular, exhibited much lower IC50 and MIC values in both strains of the bacteria. A comparison of the hydrophobicities and molecular charges of the peptide/drug conjugates indicates that these two properties exert significant influence over the activities of these molecules. Hydrophobicity, measured by monitoring HPLC elution profiles, increased in the following order: 2 6 < 3 &tide; 4 < 5. The peptide conjugates that exhibited the highest potencies, compounds 4 and 5, had the highest levels of hydrophobicity. Compound 3, which had a similar level of hydrophobicity relative to these compounds, exhibited a lower level of toxicity, indicating that hydrophobicity alone does not control activity. This compound also carries a lower molecular charge, pointing to this parameter as another influencer of potency.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus infection strain | 1280 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 80 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
To assess whether our most potent NA-peptide conjugates would possess a therapeutic window compatible with low levels of human toxicity, two types of human fibroblasts were treated with NA, compound 4, and the unconjugated peptide. The IC50 values were >10-fold higher for the fibroblasts versus the S. aureus strains tested. This trend indicates that the drug conjugates may possess a suitable therapeutic window for successful application as antibacterial agents.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human fibroblast strain 1 | Homo sapiens | ||
| Experiment 5 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 80 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
To assess whether our most potent NA-peptide conjugates would possess a therapeutic window compatible with low levels of human toxicity, two types of human fibroblasts were treated with NA, compound 4, and the unconjugated peptide. The IC50 values were >10-fold higher for the fibroblasts versus the S. aureus strains tested. This trend indicates that the drug conjugates may possess a suitable therapeutic window for successful application as antibacterial agents.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human fibroblast strain 2 | Homo sapiens | ||
| Experiment 6 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
18 μg/mL
|
|||
| Administration Time | 24 h | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
As expected, unconjugated NA showed limited activity toward MRSA and MSSA. Peptide vectors alone did not show significant toxicity to both MRSA and MSSA at all concentrations investigated. However, several of the peptide/drug conjugates, compounds 4 and 5 in particular, exhibited much lower IC50 and MIC values in both strains of the bacteria. A comparison of the hydrophobicities and molecular charges of the peptide/drug conjugates indicates that these two properties exert significant influence over the activities of these molecules. Hydrophobicity, measured by monitoring HPLC elution profiles, increased in the following order: 2 6 < 3 &tide; 4 < 5. The peptide conjugates that exhibited the highest potencies, compounds 4 and 5, had the highest levels of hydrophobicity. Compound 3, which had a similar level of hydrophobicity relative to these compounds, exhibited a lower level of toxicity, indicating that hydrophobicity alone does not control activity. This compound also carries a lower molecular charge, pointing to this parameter as another influencer of potency.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
18 μg/mL
|
|||
| Administration Time | 24 h | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
As expected, unconjugated NA showed limited activity toward MRSA and MSSA. Peptide vectors alone did not show significant toxicity to both MRSA and MSSA at all concentrations investigated. However, several of the peptide/drug conjugates, compounds 4 and 5 in particular, exhibited much lower IC50 and MIC values in both strains of the bacteria. A comparison of the hydrophobicities and molecular charges of the peptide/drug conjugates indicates that these two properties exert significant influence over the activities of these molecules. Hydrophobicity, measured by monitoring HPLC elution profiles, increased in the following order: 2 6 < 3 &tide; 4 < 5. The peptide conjugates that exhibited the highest potencies, compounds 4 and 5, had the highest levels of hydrophobicity. Compound 3, which had a similar level of hydrophobicity relative to these compounds, exhibited a lower level of toxicity, indicating that hydrophobicity alone does not control activity. This compound also carries a lower molecular charge, pointing to this parameter as another influencer of potency.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
References