
Linker Information
General Information of This Linker
| Linker ID |
LIN00087
|
|||||
|---|---|---|---|---|---|---|
| Linker Name |
Amide bond
|
|||||
| Structure |
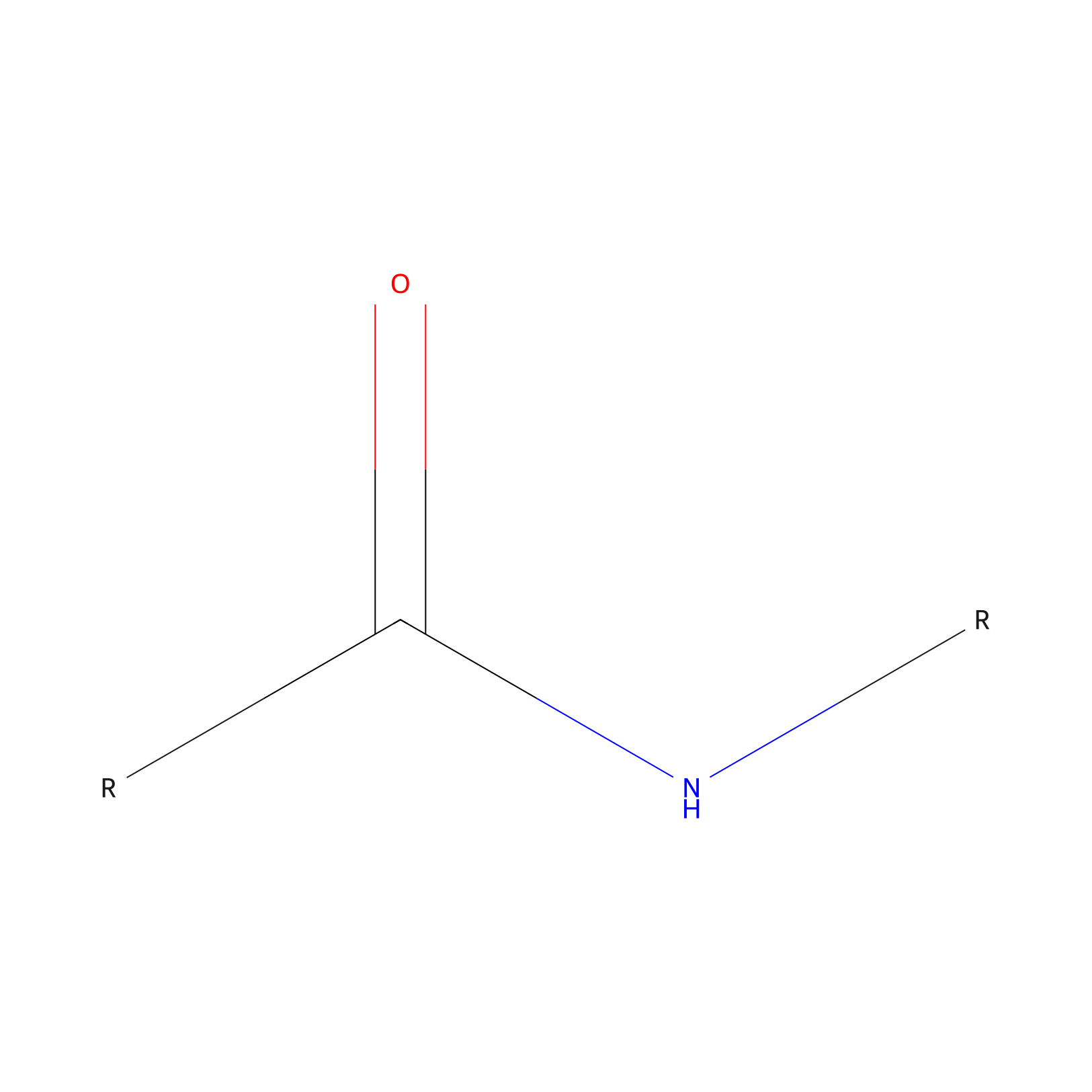
|
|||||
| Formula |
CH*2NO
|
|||||
| #Ro5 Violations (Lipinski): 0 | Molecular Weight (mw) | 43.025 | ||||
| Lipid-water partition coefficient (xlogp) | -0.2904 | |||||
| Hydrogen Bond Donor Count (hbonddonor) | 1 | |||||
| Hydrogen Bond Acceptor Count (hbondacc) | 1 | |||||
| Rotatable Bond Count (rotbonds) | 0 | |||||
| Canonical smiles |
*NC(*)=O
|
|||||
Each Peptide-drug Conjugate Related to This Linker
Full Information of The Activity Data of The PDC(s) Related to This Linker
177Lu-DOTATATE [Approved]
Identified from the Human Clinical Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Inoperable or metastatic paragangliomas | ||||
| Efficacy Data | Tumour inhibition rate |
85%
|
|||
| Patients Enrolled |
Patients with inoperable or metastatic paragangliomas and pheochromocytomas with baseline disease progression.
|
||||
| Administration Time | Four cycles | ||||
| Administration Dosage | 7.4 Gb per cycle | ||||
| Description |
In 20 patients with baseline disease progression, tumour control was observed in 17 (85%); the median progression-free survival was 91 months in patients with parasympathetic PGLs, 13 months in patients with sympathetic PGLs and 10 months in patients with metastatic PCCs.
|
||||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Inoperable or metastatic pheochromocytomas | ||||
| Efficacy Data | Tumour inhibition rate |
85%
|
|||
| Patients Enrolled |
Patients with inoperable or metastatic paragangliomas and pheochromocytomas with baseline disease progression.
|
||||
| Administration Time | Four cycles | ||||
| Administration Dosage | 7.4 Gb per cycle | ||||
| Description |
In 20 patients with baseline disease progression, tumour control was observed in 17 (85%); the median progression-free survival was 91 months in patients with parasympathetic PGLs, 13 months in patients with sympathetic PGLs and 10 months in patients with metastatic PCCs.
|
||||
| Experiment 3 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Atypical and anaplastic meningiomas | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
7%
|
|||
| Administration Dosage | 7.4 GBq (200 mCi) | ||||
| Evaluation Method | 111In-octreotide scan assay | ||||
| Description |
A transient increase in tumor volume was observed after the first cycle, which may have represented delayed treatment effect or pseudoprogression secondary to vasogenic edema. Subsequently, the tumor volume decreased by 7% and remained stable with minimal change in volume throughout the remainder of treatment. Over the 4 cycles (8 months) of treatment, the tumor volume increased only 16% during 177Lu-dotatate therapy in comparison to the rapid increase of 169% during the 8 months prior to 177Lu-dotatate initiation. However, the tumor resumed its previous rapid growth after therapy cessation.
Click to Show/Hide
|
||||
| In Vivo Model | A 62-year-old man presented with a history of atypical meningioma (World Health Organization grade II) and recurrent as anaplastic meningioma (World Health Organization grade III). | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Well-differentiated pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
7 mm reduction in tumor diameter
|
|||
| Patients Enrolled |
24 patients with morphologically WD-PanNETs who underwent PRRT preoperatively.
|
||||
| Administration Time | A median of 6 months (interquartile range [iqr] 6; 8 months) from the last cycle of prrt | ||||
| Evaluation Method | Stained with hematoxylin and eosin (H&E) & Formalin-fixed and paraffin-embedded (FFPE) assay | ||||
| Description |
In the neoadjuvant group, the average reduction in tumor diameter, before and after PRRT, assessed by computerized tomography (CT), was 7 mm. The average volume reduction showed a statistically significant correlation (p < 0.022) with the percentage of stroma after PRRT.
|
||||
| Experiment 5 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | SDHB positive metastatic paraganglioma | ||||
| Efficacy Data | TSH receptor antibody level | < 1 IU/L | |||
| Administration Time | 1 month | ||||
| Administration Dosage | 200mCi | ||||
| Evaluation Method | Immunoassay | ||||
| Description |
The patient was diagnosed with Hashimoto's thyrotoxicosis. However, the patient was asymptomatic. One month after the first dose of 200mCi 177Lu-DOTATATE, the patient noted fatigue and a 2.6 Kg weight gain. The TSH (73.04 μIU/ml), anti-TPO antibodies (>1,000 IU/ml), and anti-Tg antibodies (668 IU/ml) had substantially increased, with reductions in FT4 (0.3 ng/dl) and TT3 [54 ng/dl (87-169 ng/dl)]. Diagnostic gallium 68 - DOTATATE positron emission tomography-computed tomography performed prior to 177Lu-DOTATATE treatment revealed diffuse thyroid uptake. Post-therapy single-photon emission computed tomography also revealed diffuse uptake of 177Lu-DOTATATE in the thyroid gland. Levothyroxine therapy was initiated, and the patient's symptoms resolved.
Click to Show/Hide
|
||||
| In Vivo Model | A 29-year-old male with SDHB positive metastatic paraganglioma. | ||||
| Related Clinical Trial | |||||
| NCT Number | NCT03206060 | Clinical Status | Phase 2 | ||
| Clinical Description | Background:Pheochromocytoma and paraganglioma are rare tumors. They usually form inside and near the adrenal gland or in the neck region. Not all these tumors can be removed with surgery, and there are no good treatments if the disease has spread. Researchers think a new drug may be able to help.Objective:To learn the safety and tolerability of Lu-177-DOTATATE. Also, to see if it improves the length of time it takes for the cancer to return.Eligibility:Adults who have an inoperable tumor of the study cancer that can be detected with Ga-68-DOTATATE PET/CT imagingDesign:Participants will be screened with a medical history, physical exam, and blood tests.Eligible participants will be admitted to the NIH Clinical Center.Participants will get the study drug in an intravenous infusion. They will get 4 doses, given about 8 weeks apart.Between 4 and 24 hours after each study drug dose, participants will have scans taken. They will lie on their back on a scanner table.Participants will have vital signs taken. They will give blood and urine samples.During the study, participants will have other scans taken. Some scans will use a radioactive tracer.Participants will complete quality of life questionnaires.Participants will be contacted by phone 1-3 days after they leave the Clinical Center. They will then be followed every 3 to 6 months for 3 years or until their disease gets worse. | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | SDHB positive metastatic paraganglioma | ||||
| Efficacy Data | TSH receptor antibody level | ≤ 1.75 IU/L | |||
| Administration Time | 1 month | ||||
| Administration Dosage | 200mCi | ||||
| Description |
The patient was diagnosed with Hashimoto's thyrotoxicosis. However, the patient was asymptomatic. One month after the first dose of 200mCi 177Lu-DOTATATE, the patient noted fatigue and a 2.6 Kg weight gain. The TSH (73.04 μIU/ml), anti-TPO antibodies (>1,000 IU/ml), and anti-Tg antibodies (668 IU/ml) had substantially increased, with reductions in FT4 (0.3 ng/dl) and TT3 [54 ng/dl (87-169 ng/dl)]. Diagnostic gallium 68 - DOTATATE positron emission tomography-computed tomography performed prior to 177Lu-DOTATATE treatment revealed diffuse thyroid uptake. Post-therapy single-photon emission computed tomography also revealed diffuse uptake of 177Lu-DOTATATE in the thyroid gland. Levothyroxine therapy was initiated, and the patient's symptoms resolved.
Click to Show/Hide
|
||||
| In Vivo Model | A 29-year-old male with SDHB positive metastatic paraganglioma. | ||||
| Related Clinical Trial | |||||
| NCT Number | NCT03206060 | Clinical Status | Phase 2 | ||
| Clinical Description | Background:Pheochromocytoma and paraganglioma are rare tumors. They usually form inside and near the adrenal gland or in the neck region. Not all these tumors can be removed with surgery, and there are no good treatments if the disease has spread. Researchers think a new drug may be able to help.Objective:To learn the safety and tolerability of Lu-177-DOTATATE. Also, to see if it improves the length of time it takes for the cancer to return.Eligibility:Adults who have an inoperable tumor of the study cancer that can be detected with Ga-68-DOTATATE PET/CT imagingDesign:Participants will be screened with a medical history, physical exam, and blood tests.Eligible participants will be admitted to the NIH Clinical Center.Participants will get the study drug in an intravenous infusion. They will get 4 doses, given about 8 weeks apart.Between 4 and 24 hours after each study drug dose, participants will have scans taken. They will lie on their back on a scanner table.Participants will have vital signs taken. They will give blood and urine samples.During the study, participants will have other scans taken. Some scans will use a radioactive tracer.Participants will complete quality of life questionnaires.Participants will be contacted by phone 1-3 days after they leave the Clinical Center. They will then be followed every 3 to 6 months for 3 years or until their disease gets worse. | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Treatment-emergent adverse event |
0.40%
|
|||
| Patients Enrolled |
Patients with neuroendocrine tumours.
|
||||
| Description |
177Lu-DOTATATE was well tolerated with haematological, renal and hepatic treatment-related serious adverse events experienced by 9.7, 0.4 and 0.4% of patients respectively.
|
||||
| Experiment 8 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | SDHB positive metastatic paraganglioma | ||||
| Efficacy Data | Total triiodothyronine |
54 ng/dl
|
|||
| Administration Time | 1 month | ||||
| Administration Dosage | 200mCi | ||||
| Evaluation Method | Mass spectrometry assay | ||||
| Description |
The patient was diagnosed with Hashimoto's thyrotoxicosis. However, the patient was asymptomatic. One month after the first dose of 200mCi 177Lu-DOTATATE, the patient noted fatigue and a 2.6 Kg weight gain. The TSH (73.04 μIU/ml), anti-TPO antibodies (>1,000 IU/ml), and anti-Tg antibodies (668 IU/ml) had substantially increased, with reductions in FT4 (0.3 ng/dl) and TT3 [54 ng/dl (87-169 ng/dl)]. Diagnostic gallium 68 - DOTATATE positron emission tomography-computed tomography performed prior to 177Lu-DOTATATE treatment revealed diffuse thyroid uptake. Post-therapy single-photon emission computed tomography also revealed diffuse uptake of 177Lu-DOTATATE in the thyroid gland. Levothyroxine therapy was initiated, and the patient's symptoms resolved.
Click to Show/Hide
|
||||
| In Vivo Model | A 29-year-old male with SDHB positive metastatic paraganglioma. | ||||
| Related Clinical Trial | |||||
| NCT Number | NCT03206060 | Clinical Status | Phase 2 | ||
| Clinical Description | Background:Pheochromocytoma and paraganglioma are rare tumors. They usually form inside and near the adrenal gland or in the neck region. Not all these tumors can be removed with surgery, and there are no good treatments if the disease has spread. Researchers think a new drug may be able to help.Objective:To learn the safety and tolerability of Lu-177-DOTATATE. Also, to see if it improves the length of time it takes for the cancer to return.Eligibility:Adults who have an inoperable tumor of the study cancer that can be detected with Ga-68-DOTATATE PET/CT imagingDesign:Participants will be screened with a medical history, physical exam, and blood tests.Eligible participants will be admitted to the NIH Clinical Center.Participants will get the study drug in an intravenous infusion. They will get 4 doses, given about 8 weeks apart.Between 4 and 24 hours after each study drug dose, participants will have scans taken. They will lie on their back on a scanner table.Participants will have vital signs taken. They will give blood and urine samples.During the study, participants will have other scans taken. Some scans will use a radioactive tracer.Participants will complete quality of life questionnaires.Participants will be contacted by phone 1-3 days after they leave the Clinical Center. They will then be followed every 3 to 6 months for 3 years or until their disease gets worse. | ||||
| Experiment 9 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | SDHB positive metastatic paraganglioma | ||||
| Efficacy Data | Thyroid stimulating index | ≤ 1.3 | |||
| Administration Time | 1 month | ||||
| Administration Dosage | 200mCi | ||||
| Description |
The patient was diagnosed with Hashimoto's thyrotoxicosis. However, the patient was asymptomatic. One month after the first dose of 200mCi 177Lu-DOTATATE, the patient noted fatigue and a 2.6 Kg weight gain. The TSH (73.04 μIU/ml), anti-TPO antibodies (>1,000 IU/ml), and anti-Tg antibodies (668 IU/ml) had substantially increased, with reductions in FT4 (0.3 ng/dl) and TT3 [54 ng/dl (87-169 ng/dl)]. Diagnostic gallium 68 - DOTATATE positron emission tomography-computed tomography performed prior to 177Lu-DOTATATE treatment revealed diffuse thyroid uptake. Post-therapy single-photon emission computed tomography also revealed diffuse uptake of 177Lu-DOTATATE in the thyroid gland. Levothyroxine therapy was initiated, and the patient's symptoms resolved.
Click to Show/Hide
|
||||
| In Vivo Model | A 29-year-old male with SDHB positive metastatic paraganglioma. | ||||
| Related Clinical Trial | |||||
| NCT Number | NCT03206060 | Clinical Status | Phase 2 | ||
| Clinical Description | Background:Pheochromocytoma and paraganglioma are rare tumors. They usually form inside and near the adrenal gland or in the neck region. Not all these tumors can be removed with surgery, and there are no good treatments if the disease has spread. Researchers think a new drug may be able to help.Objective:To learn the safety and tolerability of Lu-177-DOTATATE. Also, to see if it improves the length of time it takes for the cancer to return.Eligibility:Adults who have an inoperable tumor of the study cancer that can be detected with Ga-68-DOTATATE PET/CT imagingDesign:Participants will be screened with a medical history, physical exam, and blood tests.Eligible participants will be admitted to the NIH Clinical Center.Participants will get the study drug in an intravenous infusion. They will get 4 doses, given about 8 weeks apart.Between 4 and 24 hours after each study drug dose, participants will have scans taken. They will lie on their back on a scanner table.Participants will have vital signs taken. They will give blood and urine samples.During the study, participants will have other scans taken. Some scans will use a radioactive tracer.Participants will complete quality of life questionnaires.Participants will be contacted by phone 1-3 days after they leave the Clinical Center. They will then be followed every 3 to 6 months for 3 years or until their disease gets worse. | ||||
| Experiment 10 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | SDHB positive metastatic paraganglioma | ||||
| Efficacy Data | Thyroid stimulating hormone levels |
73.04 µIU/ml
|
|||
| Administration Time | 1 month | ||||
| Administration Dosage | 200mCi | ||||
| Description |
The patient was diagnosed with Hashimoto's thyrotoxicosis. However, the patient was asymptomatic. One month after the first dose of 200mCi 177Lu-DOTATATE, the patient noted fatigue and a 2.6 Kg weight gain. The TSH (73.04 μIU/ml), anti-TPO antibodies (>1,000 IU/ml), and anti-Tg antibodies (668 IU/ml) had substantially increased, with reductions in FT4 (0.3 ng/dl) and TT3 [54 ng/dl (87-169 ng/dl)]. Diagnostic gallium 68 - DOTATATE positron emission tomography-computed tomography performed prior to 177Lu-DOTATATE treatment revealed diffuse thyroid uptake. Post-therapy single-photon emission computed tomography also revealed diffuse uptake of 177Lu-DOTATATE in the thyroid gland. Levothyroxine therapy was initiated, and the patient's symptoms resolved.
Click to Show/Hide
|
||||
| In Vivo Model | A 29-year-old male with SDHB positive metastatic paraganglioma. | ||||
| Related Clinical Trial | |||||
| NCT Number | NCT03206060 | Clinical Status | Phase 2 | ||
| Clinical Description | Background:Pheochromocytoma and paraganglioma are rare tumors. They usually form inside and near the adrenal gland or in the neck region. Not all these tumors can be removed with surgery, and there are no good treatments if the disease has spread. Researchers think a new drug may be able to help.Objective:To learn the safety and tolerability of Lu-177-DOTATATE. Also, to see if it improves the length of time it takes for the cancer to return.Eligibility:Adults who have an inoperable tumor of the study cancer that can be detected with Ga-68-DOTATATE PET/CT imagingDesign:Participants will be screened with a medical history, physical exam, and blood tests.Eligible participants will be admitted to the NIH Clinical Center.Participants will get the study drug in an intravenous infusion. They will get 4 doses, given about 8 weeks apart.Between 4 and 24 hours after each study drug dose, participants will have scans taken. They will lie on their back on a scanner table.Participants will have vital signs taken. They will give blood and urine samples.During the study, participants will have other scans taken. Some scans will use a radioactive tracer.Participants will complete quality of life questionnaires.Participants will be contacted by phone 1-3 days after they leave the Clinical Center. They will then be followed every 3 to 6 months for 3 years or until their disease gets worse. | ||||
| Experiment 11 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Thrombocytopenia and anemia |
16.67%
|
|||
| Patients Enrolled |
12 patients with pheocromocytomas (PCCs) and paragangliomas (PGLs) (1 PCC, 11PGL).
|
||||
| Administration Dosage | 7.4 GBq/cycle | ||||
| MOA of PDC |
We showed that PRRT based on 177Lu-DOTATATE and 90-YDOTATOC without radiosensitizers are efficacious therapeutic options (DCR of 83% and 76%, respectively) and can be considered in the multidisciplinary treatment of PCCs and PGLs as alternatives to I-131 MIBG and chemotherapy.
|
||||
| Description |
In detail, we report pooled data on 149 177Lu- and 64 90Y-treated patients (total: 213 patients). Most studies were retrospective. The largest one included 46 patients (34 treated with 177Lu-, 12 with 90Y- PRRTs). Median ages ranged from 32.5 to 60.4 years. The female gender was slightly predominant (83 women, 74 men, gender not specified in 3 studies). When reported, mutations of SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B) were the most frequent genetic alterations. According to the predominant retrospective and real practice nature of the selected studies, sites of metastases and previous treatments were heterogeneous, with bone the metastatic site most often involved, and surgery the most frequent previous therapy. Paramount descriptive information on PRRT doses per cycle, number of cycles, response criteria, best responses to treatments, and frequency of G3/G4 hematologic toxicities is reported in Table 2. The lowest applied dose was 3.4 GBq per cycle; the number of administered cycles was heterogeneous (at least 3 cycles in 7 studies). The most used response criteria were the RECIST 1.1. Only one study evaluated the response with SWOG criteria, which are more conservative with regard to the definition of PR (50% or greater reduction in the sum of the product of the perpendicular diameters of target lesions). The majority of the patients experienced PR or SD. Hematologic toxicity was manageable. Funnel plot of selected studies did not show publication bias. The MINORS and Newcastle-Ottawa Scale scoring for the selected articles are reported in Table 3.
Click to Show/Hide
|
||||
| Experiment 12 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Advanced paraganglioma | ||||
| Efficacy Data | Thrombocytopenia |
9%
|
|||
| Patients Enrolled |
A total of 201 patients with advanced paragangliomas.
|
||||
| MOA of PDC |
The clinical applications of radiolabeled SSAs and DOTA-SSA compounds in imaging and radionuclide therapy have been rapidly increasing over the past 2 decades and have been integrated into the current management guidelines of NETs. Several studies support the safety and clinical efficacy of FDA-approved radionuclide therapies including 177Lu-DOTATATE in advanced GEP NETs and 131I-MIBG in advanced paragangliomas and pheochromocytomas. The utility of SSTR-based imaging in PRRT response assessment and surveillance of NETs remains to be established.
Click to Show/Hide
|
||||
| Description |
Over the past decade, a small number of studies have evaluated the efficacy and safety of PRRT with 177Lu-DOTA-SSA in patients with metastatic or inoperable paragangliomas [66, 67]. A recent meta-analysis of 12 studies including a total of 201 patients with advanced paragangliomas showed an objective response rate of 25%, biochemical response rate of 64%, and disease control rate of 84% after treatment with PRRT (either 90Y- or 177Lu-based agents) [67]. The following minimal treatment-related adverse effects were observed: grade 3 or 4 lymphopenia, thrombocytopenia, neutropenia, and nephrotoxicity in 11%, 9%, 3%, and 4% of the patients, respectively.
Click to Show/Hide
|
||||
| Experiment 13 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Thrombocytopenia |
16.67%
|
|||
| Patients Enrolled |
30 patients with pheocromocytomas (PCCs) and paragangliomas (PGLs) (3 PCC, 27PGL).
|
||||
| Administration Time | 73% of patients received 4 cycles | ||||
| Administration Dosage | 7.4 GBq/cycle | ||||
| MOA of PDC |
We showed that PRRT based on 177Lu-DOTATATE and 90-YDOTATOC without radiosensitizers are efficacious therapeutic options (DCR of 83% and 76%, respectively) and can be considered in the multidisciplinary treatment of PCCs and PGLs as alternatives to I-131 MIBG and chemotherapy.
|
||||
| Description |
In detail, we report pooled data on 149 177Lu- and 64 90Y-treated patients (total: 213 patients). Most studies were retrospective. The largest one included 46 patients (34 treated with 177Lu-, 12 with 90Y- PRRTs). Median ages ranged from 32.5 to 60.4 years. The female gender was slightly predominant (83 women, 74 men, gender not specified in 3 studies). When reported, mutations of SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B) were the most frequent genetic alterations. According to the predominant retrospective and real practice nature of the selected studies, sites of metastases and previous treatments were heterogeneous, with bone the metastatic site most often involved, and surgery the most frequent previous therapy. Paramount descriptive information on PRRT doses per cycle, number of cycles, response criteria, best responses to treatments, and frequency of G3/G4 hematologic toxicities is reported in Table 2. The lowest applied dose was 3.4 GBq per cycle; the number of administered cycles was heterogeneous (at least 3 cycles in 7 studies). The most used response criteria were the RECIST 1.1. Only one study evaluated the response with SWOG criteria, which are more conservative with regard to the definition of PR (50% or greater reduction in the sum of the product of the perpendicular diameters of target lesions). The majority of the patients experienced PR or SD. Hematologic toxicity was manageable. Funnel plot of selected studies did not show publication bias. The MINORS and Newcastle-Ottawa Scale scoring for the selected articles are reported in Table 3.
Click to Show/Hide
|
||||
| Experiment 14 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Metastatic midgut neuroendocrine tumor | ||||
| Efficacy Data | Thrombocytopenia |
54 x 109 /L
|
|||
| Administration Dosage | 7.4 GBq | ||||
| Description |
Initial laboratory tests showed normocytic regenerative anemia (hemoglobin 85g/dL), thrombocytopenia (54 109/L), no signs of hemolysis, no schistocytes, prolonged prothrombin time (25.8seconds, 35%) and prolonged activated partial thromboplastin time (1.63), low fibrinogen (0.3g/L), elevated fibrin monomer (>400ug/mL) and elevated fibrin degradation products (>20ug/mL). Coagulation factors II (49%) and V (44%) were low; factors VII (97%) and X (81%) were normal. Liver function tests were normal with the exception of moderate elevation of gamma Glutamyl Transferase (γGT=100UI/L, N<38UI/L). C-reactive protein was <5mg/L. There was no sign of tumor lysis syndrome. The bone marrow aspirate and medullar karyotype were normal. Computed tomography showed stable metastatic disease, including a large stable necrotic liver metastasis.
Click to Show/Hide
|
||||
| In Vivo Model | 55-year-old woman with metastatic midgut neuroendocrine tumor. | ||||
| Experiment 15 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Pancreatic neuroendocrine tumor | ||||
| Efficacy Data | SUVmax decrease rate |
58% ± 30%
|
|||
| Administration Time | 3 injections at consecutive 2-month intervals | ||||
| Administration Dosage | 7.4 GBq | ||||
| Evaluation Method | SPECT/CT assay | ||||
| Description |
Subclavian nodes and hepatic and bone lesions could be easily detected on the whole-body SPECT images (A), and the absolute quantification of these lesions demonstrated gradual decreases in SUV max values (B), leading to a global mean decrease of 58% ± 30% and thus giving evidence of marked decreases in lesion dosimetry.
|
||||
| In Vivo Model | 70-year-old woman treated by 177Lu-DOTATATE injections for a metastatic recurrence of a pancreatic neuroendocrine tumor. | ||||
| Experiment 16 Reporting the Activity Data of This PDC | [10] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Survival rate |
83%
|
|||
| Patients Enrolled |
Patients with neuroendocrine tumor.
|
||||
| Administration Time | 24 months | ||||
| Experiment 17 Reporting the Activity Data of This PDC | [11] | ||||
| Indication | Head and neck paraganglioma | ||||
| Efficacy Data | Stable disease (SD) + Progressive disease (PD) | > 60% | |||
| Patients Enrolled |
7 patients with head and neck paraganglioma treated with PPRT between May 2014 and October 2016.
|
||||
| Administration Time | Underwent 3-5 cycles in 8- to 10-week intervals between may 2014 and october 2016 | ||||
| Administration Dosage | 7.2 ± 0.4 GBq | ||||
| Evaluation Method | PET/CT assay | ||||
| Description |
Overall results for PRRT in paraganglioma and pheochromocytoma are promising with response rates (SD and PR) of >60%.
|
||||
| Experiment 18 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
16.67%
|
|||
| Patients Enrolled |
12 patients with pheocromocytomas and paragangliomas.
|
||||
| Administration Time | At least 3 cycles | ||||
| Administration Dosage | 3.7-8.1 GBq/cycle | ||||
| MOA of PDC |
We showed that PRRT based on 177Lu-DOTATATE and 90-YDOTATOC without radiosensitizers are efficacious therapeutic options (DCR of 83% and 76%, respectively) and can be considered in the multidisciplinary treatment of PCCs and PGLs as alternatives to I-131 MIBG and chemotherapy.
|
||||
| Description |
There was no statistically significant heterogeneity among the 177Lu- and 90Y- PRRTs studies (I2 value = 0%, p = 0.68 and I2 value = 0%, p = 0.91, respectively). The pooled DCRs were 0.83 (95% CI: 0.75-0.88) and 0.76 (95% CI: 0.56-0.89) for 177Lu- and 90Y-PRRT treatments, respectively. The pooled DCR for PRRT agents was 0.81 (95% CI: 0.74-0.87). Although the present study is the largest description of DCR in PRRT therapy of PCCs and PGLs, given the small sample size, no formal attempts were made to compare efficacy between 177Lu- and 90Y-PRRT treatments. In detail, we report pooled data on 149 177Lu- and 64 90Y-treated patients (total: 213 patients). Most studies were retrospective. The largest one included 46 patients (34 treated with 177Lu-, 12 with 90Y- PRRTs). Median ages ranged from 32.5 to 60.4 years. The female gender was slightly predominant (83 women, 74 men, gender not specified in 3 studies). When reported, mutations of SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B) were the most frequent genetic alterations. According to the predominant retrospective and real practice nature of the selected studies, sites of metastases and previous treatments were heterogeneous, with bone the metastatic site most often involved, and surgery the most frequent previous therapy. Paramount descriptive information on PRRT doses per cycle, number of cycles, response criteria, best responses to treatments, and frequency of G3/G4 hematologic toxicities is reported in Table 2. The lowest applied dose was 3.4 GBq per cycle; the number of administered cycles was heterogeneous (at least 3 cycles in 7 studies). The most used response criteria were the RECIST 1.1. Only one study evaluated the response with SWOG criteria, which are more conservative with regard to the definition of PR (50% or greater reduction in the sum of the product of the perpendicular diameters of target lesions). The majority of the patients experienced PR or SD. Hematologic toxicity was manageable. Funnel plot of selected studies did not show publication bias. The MINORS and Newcastle-Ottawa Scale scoring for the selected articles are reported in Table 3.
Click to Show/Hide
|
||||
| Experiment 19 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | High-grade (WHO G3) neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
21.90%
|
|||
| Patients Enrolled |
Patients with neuroendocrine tumours including high-grade (WHO G3) neuroendocrine tumours.
|
||||
| Evaluation Method | RECIST criteria assay | ||||
| Description |
Among 160 patients whose responses to treatment could be evaluated according to the RECIST criteria, 28.1% (n=45) had a progressive disease, 21.9% (n=35) had a stable disease, 46.9% (n=75) had a partial response and 3.1% (n=5) had a complete response.
|
||||
| Experiment 20 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
24.20%
|
|||
| Patients Enrolled |
33 patients with neuroendocrine tumour.
|
||||
| Administration Time | 2 cycles | ||||
| Administration Dosage | 7.4 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 21 Reporting the Activity Data of This PDC | [14] | ||||
| Indication | Metastatic/Advanced neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
25.20%
|
|||
| Patients Enrolled |
468 patients with metastatic/advanced neuroendocrine tumor.
|
||||
| Administration Time | At least two cycles at 10-12 weeks interval | ||||
| Administration Dosage | 5.55 to 7.4 GBq per patient | ||||
| Description |
Long-term outcome (follow-up ranging from 4 to 97.6 months; median period:46 months following first 177Lu-DOTATATE PRRT) results showed, (i) on symptomatic response evaluation scale, complete response (CR) in 214 patients (45.7%), partial response (PR) in 108 (23.1%), stable disease (SD) in 118 (25.2%), progressive disease (PD) in 28 (6%).
Click to Show/Hide
|
||||
| Experiment 22 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Hindgut neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
31%
|
|||
| Patients Enrolled |
14 patients with hindgut neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 23 Reporting the Activity Data of This PDC | [16] | ||||
| Indication | Metastatic medullary thyroid carcinoma | ||||
| Efficacy Data | Stable disease (SD) |
33.33%
|
|||
| Patients Enrolled |
6 patients with medullary thyroid cancer (3 males and 3 females).
|
||||
| Administration Time | 3 cycles | ||||
| Administration Dosage | 5.7 GBq/cycle (range, 2.6-7.4 GBq) | ||||
| Evaluation Method | PET/CT assay | ||||
| MOA of PDC |
Radiolabeled somatostatin analogs are contributive for the management of recurrent MTC. 68Ga-DOTATAE PET-CT showed a relatively high detection rate in recurrent MTC. In addition, PRRT with 177Lu-DOTATATE was found to be a safe alternative therapeutic option for MTC.
|
||||
| Description |
Four of the 17 patients with positive SSTR-PET were scheduled for PRRT. In addition, 2 patients had positive 99mTc-octreotide scintigraphy results (Krenning score ≥ 2) and were scheduled for PRRT. Two of the 6 patients who underwent PRRT showed PR, 2 SD and 2 PD. Two patients died during the follow-up period. Median overall survival was 19 months (95% CI: 5.52-29.48). There were no cases of significant toxicity.
Click to Show/Hide
|
||||
| Experiment 24 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Stable disease (SD) |
35%
|
|||
| Patients Enrolled |
78 patients with pancreatic neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 25 Reporting the Activity Data of This PDC | [14] | ||||
| Indication | Metastatic/Advanced neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
38%
|
|||
| Patients Enrolled |
468 patients with metastatic/advanced neuroendocrine tumor.
|
||||
| Administration Time | At least two cycles at 10-12 weeks interval | ||||
| Administration Dosage | 5.55 to 7.4 GBq per patient | ||||
| Description |
Biochemical response evaluation showed CR in 52 (12%), PR in 172 (40%), SD in 161 (38%), and PD in 42 patients (10%).
|
||||
| Experiment 26 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
42.40%
|
|||
| Patients Enrolled |
33 patients with neuroendocrine tumour.
|
||||
| Administration Time | 2-4 cycles | ||||
| Administration Dosage | Mean administered activity during re-treatment: 17.7 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 27 Reporting the Activity Data of This PDC | [17] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
42.90%
|
|||
| Patients Enrolled |
7 patients with inoperable, progressive, metastatic, or locally advanced, somatostatin receptor-positive neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | 21.3-30.1 GBq total dose | ||||
| MOA of PDC |
In conclusion, for the first time in Korea, we have conducted a clinical trial of PRRT with SNU-KB-01, a NCA 177Lu-DOTATATE, in patients with SSTR-positive NET. Treatment with SNU-KB-01 was safe and resulted in control of disease in most of the patients with high SSTR expression. The recommended dose per cycle of SNU-KB-01 was determined to be 7.40 GBq for the phase 2 trial. Our results indicate SNU-KB-01 as a potentially safe and efficacious treatment option for NET patients and are expected to provide a wider choice of treatment opportunities for patients with various types of SSTR-positive tumors in Korea.
Click to Show/Hide
|
||||
| Description |
Among seven patients, the best objective response to SNU-KB-01 was the PR observed in three patients (42.9%). No CR was observed. Three patients (42.9%) had SD and one patient (14.3%) had PD. The ORR (CR+PR) was 42.9%, and the DCR (CR+PR+SD) was 85.7%.
|
||||
| Experiment 28 Reporting the Activity Data of This PDC | [18] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumor | ||||
| Efficacy Data | Stable disease (SD) |
43%
|
|||
| Patients Enrolled |
Patients treated with Lu-DOTATATE.
|
||||
| Administration Dosage | 100 mCi (3.7 GBq) | ||||
| Description |
The objective response rate of the total group of patients was 39%. Stable disease was reached in 43% of patients. Progression-free survival (PFS) and overall survival (OS) for all NET patients were 29 months [95% confidence interval (CI), 26-33 months] and 63 months (95% CI, 55-72 months). Long-term toxicity included acute leukemia in four patients (0.7%) and myelodysplastic syndrome in nine patients (1.5%).
Click to Show/Hide
|
||||
| Experiment 29 Reporting the Activity Data of This PDC | [19] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
43.55%
|
|||
| Patients Enrolled |
23 GEP neuroendocrine tumours patients.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | 7.4 GBq (200mCi) | ||||
| MOA of PDC |
From our initial experience, Lutathera has been well tolerated in patients with GEP-NET. Additional studies are needed to examine long-term clinical and patient-reported outcomes associated with treatment of GEP-NETs as well as financial considerations for hospitals embarking on a PRRT program. A multidisciplinary team and complete collaboration between hospital administration and clinical teams are required for successful implementation of a PRRT program.
Click to Show/Hide
|
||||
| Description |
PET/CT imaging were done to determine treatment responses 3 months post-PRRT treatment. Only patients who completed 4 cycles (n=23) were included. Five patients (21.7%) showed partial response, 10 patients (43.5 5%) showed stable disease, and 3 patients (13.0%) showed disease progression; the responses from 5 patients (21.7%) were unknown because no PET/CT scan was done.
Click to Show/Hide
|
||||
| Experiment 30 Reporting the Activity Data of This PDC | [20] | ||||
| Indication | SSR positive lung neuroendocrine tumor | ||||
| Efficacy Data | Stable disease (SD) |
44%
|
|||
| Patients Enrolled |
39 patients with neuroendocrine tumour with available <sup>68</sup>Ga-DOTATATE PET/CT.
|
||||
| Administration Time | 1-4 cycles | ||||
| Administration Dosage | Median cumulative activity of 27 GBq (range, 6-43 GBq) | ||||
| Evaluation Method | 68Ga-DOTATATE PET/CT assay | ||||
| MOA of PDC |
In patients with advanced progressive lung NET and satisfactory SSR expression, 177Lu-DOTATATE is effective and safe, with a high DCR and encouraging PFS and OS. Further prospective studies comparing 177Lu-DOTATATE with other systemic options are warranted.
|
||||
| Description |
Of 48 patients (median age, 63 y; 13 women), 43 (90%) had AC and 5 (10%) TC. Almost all patients (47, 98%) were treated due to progression. Most patients (40, 83%) received somatostatin analogs, and 10 patients (20%) had prior everolimus, chemotherapy, or both. All patients had high SSR expression (≥ modified Krenning score 3) on pretreatment 68Ga-DOTATATE PET/CT. Patients received a median 4 (range, 1-4) cycles of 177Lu-DOTATATE (33% with concurrent radiosensitizing chemotherapy) to a median cumulative activity of 27 GBq (range, 6-43GBq). At a median follow-up of 42 mo, the median PFS and OS were 23 mo (95% CI, 18-28 mo) and 59 mo (95% CI, 50-not reached [NR]), respectively. Of 40 patients with RECIST-measurable disease and 39 patients with available 68Ga-DOTATATE PET/CT, response categories were partial response, 20% (95% CI, 10%-35%) and 44% (95% CI, 30%-59%); stable disease, 68% (95% CI, 52%-80%) and 44% (95% CI, 30%-59%); and progressive disease, 12% (95% CI, 5%-27%) by both, respectively. There was a moderate concordance between response categories by RECIST and 68Ga-DOTATATE PET/CT, weighted of 0.51 (95% CI, 0.21-0.68).
Click to Show/Hide
|
||||
| Experiment 31 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Stable disease (SD) |
44.44%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Nineteen patients, all with progressive, heavily treated disease, were identified. Sites of tumor origin were: pancreas (74%), small bowel (11%), rectum (11%), and lung (5%); median Ki-67 was 32% (range 22-56). Thirteen patients (68%) completed all four 177Lu-DOTATATE cycles. Best response (N = 18 evaluable) was: 5/18 (28%) partial response, 8/18 (44%) stable disease, and 5/18 (28%) disease progression. Median PFS was 13.1 months (95% CI: 8.7-20.9).
Click to Show/Hide
|
||||
| Experiment 32 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
50%
|
|||
| Patients Enrolled |
12 patients with pheocromocytomas (PCCs) and paragangliomas (PGLs) (1 PCC, 11PGL).
|
||||
| Administration Dosage | 7.4 GBq/cycle | ||||
| MOA of PDC |
We showed that PRRT based on 177Lu-DOTATATE and 90-YDOTATOC without radiosensitizers are efficacious therapeutic options (DCR of 83% and 76%, respectively) and can be considered in the multidisciplinary treatment of PCCs and PGLs as alternatives to I-131 MIBG and chemotherapy.
|
||||
| Description |
There was no statistically significant heterogeneity among the 177Lu- and 90Y- PRRTs studies (I2 value = 0%, p = 0.68 and I2 value = 0%, p = 0.91, respectively). The pooled DCRs were 0.83 (95% CI: 0.75-0.88) and 0.76 (95% CI: 0.56-0.89) for 177Lu- and 90Y-PRRT treatments, respectively. The pooled DCR for PRRT agents was 0.81 (95% CI: 0.74-0.87). Although the present study is the largest description of DCR in PRRT therapy of PCCs and PGLs, given the small sample size, no formal attempts were made to compare efficacy between 177Lu- and 90Y-PRRT treatments. In detail, we report pooled data on 149 177Lu- and 64 90Y-treated patients (total: 213 patients). Most studies were retrospective. The largest one included 46 patients (34 treated with 177Lu-, 12 with 90Y- PRRTs). Median ages ranged from 32.5 to 60.4 years. The female gender was slightly predominant (83 women, 74 men, gender not specified in 3 studies). When reported, mutations of SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B) were the most frequent genetic alterations. According to the predominant retrospective and real practice nature of the selected studies, sites of metastases and previous treatments were heterogeneous, with bone the metastatic site most often involved, and surgery the most frequent previous therapy. Paramount descriptive information on PRRT doses per cycle, number of cycles, response criteria, best responses to treatments, and frequency of G3/G4 hematologic toxicities is reported in Table 2. The lowest applied dose was 3.4 GBq per cycle; the number of administered cycles was heterogeneous (at least 3 cycles in 7 studies). The most used response criteria were the RECIST 1.1. Only one study evaluated the response with SWOG criteria, which are more conservative with regard to the definition of PR (50% or greater reduction in the sum of the product of the perpendicular diameters of target lesions). The majority of the patients experienced PR or SD. Hematologic toxicity was manageable. Funnel plot of selected studies did not show publication bias. The MINORS and Newcastle-Ottawa Scale scoring for the selected articles are reported in Table 3.
Click to Show/Hide
|
||||
| Experiment 33 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Meningiomas | ||||
| Efficacy Data | Stable disease (SD) |
50%
|
|||
| Patients Enrolled |
20 patients with meningiomas.
|
||||
| Administration Time | Median 3 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
A few studies evaluated the efficacy of RLT in meningiomas, administrating 2-5 cycles of [90Y]Y-DOTATATE/-DOTATOC or [177Lu]Lu-DOTATATE/-DOTATOC. In a meta-analysis performed by Mirian et al., RLToffered as mono-therapy or in combination with other oncological treatmentsallowed a comprehensive DCR of 63% in refractory meningiomas. The 6-month PFS rates were 94%, 48%, and 0% for patients with WHO grade I, II, and III meningiomas, respectively, whereas the corresponding 1-year OS rates were 88%, 71%, and 52%, respectively. In a study by Seystahl et al. RLT, offered mainly with [177Lu]Lu-DOTATATE (85% of patients), obtained 6-month PFS rates of 100%, 57%, and 0% for grade I, II, and III refractory meningiomas, respectively. In a recent study on a small group of selected patients affected by WHO grade II refractory meningiomas, 6-month PFS was 85.7% and 1-year PFS was 66.7%. The treatment was safe and well tolerated.
Click to Show/Hide
|
||||
| Experiment 34 Reporting the Activity Data of This PDC | [23] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
50.20%
|
|||
| Patients Enrolled |
Patients with advanced neuroendocrine tumor.
|
||||
| Administration Dosage | Along with oral capecitabine therapy | ||||
| Description |
According to Response Evaluation Criteria in Solid Tumors 1.1 criteria, in group 1 patients, the response was partial response in 34% of the patients, stable disease in 50.2%, and progressive disease in 6.8% versus partial response in 6.3%, stable disease in 60.9%, and progressive disease in 26.5% among group 2 patients. The median overall survival (OS) and progression-free survival (PFS) was not reached in group 1 patients. The median OS and PFS in group 2 patients were 48 months.
Click to Show/Hide
|
||||
| Experiment 35 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
53.80%
|
|||
| Patients Enrolled |
13 Re-retreatment neuroendocrine tumour patients.
|
||||
| Administration Time | 2 cycles | ||||
| Administration Dosage | Median cumulative dose: 59.7 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 36 Reporting the Activity Data of This PDC | [24] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
55%
|
|||
| Patients Enrolled |
45 patients with advanced inoperable/metastatic neuroendocrine tumor (22 women, 23 men).
|
||||
| Administration Time | 2 to 7 cycles | ||||
| Administration Dosage | 27.0 GBq | ||||
| Evaluation Method | 68Ga-DOTATATE PET/CT assay | ||||
| Description |
Radiological response with the CT images of 68Ga-DOTANOC PET/CECT was assessed 8 to 12 weeks after the last treatment cycle. Three patients were lost to follow-up, and 2 other patients had nonmeasurable lesions on CT. Thus, according to RECIST 1.1, 40 patients had evaluable lesions, of which 12 patients (30%) had PR and 22 patients (55%) had SD, whereas disease progression was limited to 6 patients (15%). No case of CR was observed in our study cohort.
Click to Show/Hide
|
||||
| Experiment 37 Reporting the Activity Data of This PDC | [25] | ||||
| Indication | SDHB positive metastatic paraganglioma | ||||
| Efficacy Data | Stable disease (SD) |
55%
|
|||
| Patients Enrolled |
37 patients affected by neuroendocrine tumors.
|
||||
| Administration Time | Five cycles of 5.5 gbq each every 8 weeks | ||||
| Administration Dosage | Cumulative activity of 27.5 GBq | ||||
| Description |
Thirty-three of 37 patients were evaluable for response. Objective responses included partial response (PR) in 10 patients (30%), stable disease (SD) in 18 patients (55%), with a DCR of 85%. Median follow up was 38 months (range 4.6-51.1 months). Median PFS was 31.4 months (17.6-45.4) and mOS was not reached.
|
||||
| Experiment 38 Reporting the Activity Data of This PDC | [14] | ||||
| Indication | Metastatic/Advanced neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
57%
|
|||
| Patients Enrolled |
468 patients with metastatic/advanced neuroendocrine tumor.
|
||||
| Administration Time | At least two cycles at 10-12 weeks interval | ||||
| Administration Dosage | 5.55 to 7.4 GBq per patient | ||||
| Evaluation Method | PERCIST criteria assay | ||||
| Description |
Molecular imaging response (by PERCIST criteria) showed CR in 29 (6%), PR in 116 (25%), SD in 267 (57%) and PD in 56 (12%) patients.
|
||||
| Experiment 39 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
59.10%
|
|||
| Patients Enrolled |
Patients with neuroendocrine tumour (Retreatment).
|
||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 40 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
59.50%
|
|||
| Patients Enrolled |
168 patients with neuroendocrine tumour.
|
||||
| Administration Time | 2 cycles | ||||
| Administration Dosage | Median cumulative dose: 44.7 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 41 Reporting the Activity Data of This PDC | [14] | ||||
| Indication | Metastatic/Advanced neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
60%
|
|||
| Patients Enrolled |
468 patients with metastatic/advanced neuroendocrine tumor.
|
||||
| Administration Time | At least two cycles at 10-12 weeks interval | ||||
| Administration Dosage | 5.55 to 7.4 GBq per patient | ||||
| Description |
On RECIST 1.1 criteria, CR was observed in 14 patients (3%), PR in 126 patients (27%), SD in 282 patients (60%) and PD in 46 patients (10%).
|
||||
| Experiment 42 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
60%
|
|||
| Patients Enrolled |
5 patients with pheocromocytomas (PCCs) and paragangliomas (PGLs) (5 PGL, ).
|
||||
| Administration Time | 3 cycles | ||||
| Administration Dosage | 6.6-7.6 GBq/cycle | ||||
| MOA of PDC |
We showed that PRRT based on 177Lu-DOTATATE and 90-YDOTATOC without radiosensitizers are efficacious therapeutic options (DCR of 83% and 76%, respectively) and can be considered in the multidisciplinary treatment of PCCs and PGLs as alternatives to I-131 MIBG and chemotherapy.
|
||||
| Description |
There was no statistically significant heterogeneity among the 177Lu- and 90Y- PRRTs studies (I2 value = 0%, p = 0.68 and I2 value = 0%, p = 0.91, respectively). The pooled DCRs were 0.83 (95% CI: 0.75-0.88) and 0.76 (95% CI: 0.56-0.89) for 177Lu- and 90Y-PRRT treatments, respectively. The pooled DCR for PRRT agents was 0.81 (95% CI: 0.74-0.87). Although the present study is the largest description of DCR in PRRT therapy of PCCs and PGLs, given the small sample size, no formal attempts were made to compare efficacy between 177Lu- and 90Y-PRRT treatments. In detail, we report pooled data on 149 177Lu- and 64 90Y-treated patients (total: 213 patients). Most studies were retrospective. The largest one included 46 patients (34 treated with 177Lu-, 12 with 90Y- PRRTs). Median ages ranged from 32.5 to 60.4 years. The female gender was slightly predominant (83 women, 74 men, gender not specified in 3 studies). When reported, mutations of SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B) were the most frequent genetic alterations. According to the predominant retrospective and real practice nature of the selected studies, sites of metastases and previous treatments were heterogeneous, with bone the metastatic site most often involved, and surgery the most frequent previous therapy. Paramount descriptive information on PRRT doses per cycle, number of cycles, response criteria, best responses to treatments, and frequency of G3/G4 hematologic toxicities is reported in Table 2. The lowest applied dose was 3.4 GBq per cycle; the number of administered cycles was heterogeneous (at least 3 cycles in 7 studies). The most used response criteria were the RECIST 1.1. Only one study evaluated the response with SWOG criteria, which are more conservative with regard to the definition of PR (50% or greater reduction in the sum of the product of the perpendicular diameters of target lesions). The majority of the patients experienced PR or SD. Hematologic toxicity was manageable. Funnel plot of selected studies did not show publication bias. The MINORS and Newcastle-Ottawa Scale scoring for the selected articles are reported in Table 3.
Click to Show/Hide
|
||||
| Experiment 43 Reporting the Activity Data of This PDC | [23] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
60.90%
|
|||
| Patients Enrolled |
Patients with advanced neuroendocrine tumor.
|
||||
| Description |
According to Response Evaluation Criteria in Solid Tumors 1.1 criteria, in group 1 patients, the response was partial response in 34% of the patients, stable disease in 50.2%, and progressive disease in 6.8% versus partial response in 6.3%, stable disease in 60.9%, and progressive disease in 26.5% among group 2 patients. The median overall survival (OS) and progression-free survival (PFS) was not reached in group 1 patients. The median OS and PFS in group 2 patients were 48 months.
Click to Show/Hide
|
||||
| Experiment 44 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Bronchial neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
63%
|
|||
| Patients Enrolled |
21 patients with bronchial neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 45 Reporting the Activity Data of This PDC | [26] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumor | ||||
| Efficacy Data | Stable disease (SD) |
64%
|
|||
| Patients Enrolled |
Patients with advanced GEP-neuroendocrine tumour.
|
||||
| Administration Time | Four or five cycles | ||||
| Administration Dosage | 18.5 GBq/27.8 GBq | ||||
| Description |
Two patients (6%) achieved a partial response and 21 (64%) showed stable disease, giving a disease control rate (DCR) of 70%.
|
||||
| Experiment 46 Reporting the Activity Data of This PDC | [27] | ||||
| Indication | Metastatic neuroendocrine tumor | ||||
| Efficacy Data | Stable disease (SD) |
64%
|
|||
| Patients Enrolled |
Patients treated with Lu-DOTATATE.
|
||||
| Description |
At end-of-treatment radiological restaging, 13% of patients were found to have partial response and 64% to have stable disease; 23% of patients progressed through treatment.
|
||||
| Experiment 47 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
64%
|
|||
| Patients Enrolled |
395 patients with neuroendocrine tumour.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 48 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Unknown originb neuroendocrine tumor | ||||
| Efficacy Data | Stable disease (SD) |
65%
|
|||
| Patients Enrolled |
22 patients with unknown originb neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 49 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
66.67%
|
|||
| Patients Enrolled |
30 patients with pheocromocytomas (PCCs) and paragangliomas (PGLs) (3 PCC, 27PGL).
|
||||
| Administration Time | 73% of patients received 4 cycles | ||||
| Administration Dosage | 7.4 GBq/cycle | ||||
| MOA of PDC |
We showed that PRRT based on 177Lu-DOTATATE and 90-YDOTATOC without radiosensitizers are efficacious therapeutic options (DCR of 83% and 76%, respectively) and can be considered in the multidisciplinary treatment of PCCs and PGLs as alternatives to I-131 MIBG and chemotherapy.
|
||||
| Description |
There was no statistically significant heterogeneity among the 177Lu- and 90Y- PRRTs studies (I2 value = 0%, p = 0.68 and I2 value = 0%, p = 0.91, respectively). The pooled DCRs were 0.83 (95% CI: 0.75-0.88) and 0.76 (95% CI: 0.56-0.89) for 177Lu- and 90Y-PRRT treatments, respectively. The pooled DCR for PRRT agents was 0.81 (95% CI: 0.74-0.87). Although the present study is the largest description of DCR in PRRT therapy of PCCs and PGLs, given the small sample size, no formal attempts were made to compare efficacy between 177Lu- and 90Y-PRRT treatments. In detail, we report pooled data on 149 177Lu- and 64 90Y-treated patients (total: 213 patients). Most studies were retrospective. The largest one included 46 patients (34 treated with 177Lu-, 12 with 90Y- PRRTs). Median ages ranged from 32.5 to 60.4 years. The female gender was slightly predominant (83 women, 74 men, gender not specified in 3 studies). When reported, mutations of SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B) were the most frequent genetic alterations. According to the predominant retrospective and real practice nature of the selected studies, sites of metastases and previous treatments were heterogeneous, with bone the metastatic site most often involved, and surgery the most frequent previous therapy. Paramount descriptive information on PRRT doses per cycle, number of cycles, response criteria, best responses to treatments, and frequency of G3/G4 hematologic toxicities is reported in Table 2. The lowest applied dose was 3.4 GBq per cycle; the number of administered cycles was heterogeneous (at least 3 cycles in 7 studies). The most used response criteria were the RECIST 1.1. Only one study evaluated the response with SWOG criteria, which are more conservative with regard to the definition of PR (50% or greater reduction in the sum of the product of the perpendicular diameters of target lesions). The majority of the patients experienced PR or SD. Hematologic toxicity was manageable. Funnel plot of selected studies did not show publication bias. The MINORS and Newcastle-Ottawa Scale scoring for the selected articles are reported in Table 3.
Click to Show/Hide
|
||||
| Experiment 50 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
66.67%
|
|||
| Patients Enrolled |
9 patients with pheocromocytomas (PCCs) and paragangliomas (PGLs) (3 PCC , 6PGL).
|
||||
| Administration Time | 3.11 cycles | ||||
| Administration Dosage | 8.01 (7.4-8.4) GBq/cycle | ||||
| MOA of PDC |
We showed that PRRT based on 177Lu-DOTATATE and 90-YDOTATOC without radiosensitizers are efficacious therapeutic options (DCR of 83% and 76%, respectively) and can be considered in the multidisciplinary treatment of PCCs and PGLs as alternatives to I-131 MIBG and chemotherapy.
|
||||
| Description |
There was no statistically significant heterogeneity among the 177Lu- and 90Y- PRRTs studies (I2 value = 0%, p = 0.68 and I2 value = 0%, p = 0.91, respectively). The pooled DCRs were 0.83 (95% CI: 0.75-0.88) and 0.76 (95% CI: 0.56-0.89) for 177Lu- and 90Y-PRRT treatments, respectively. The pooled DCR for PRRT agents was 0.81 (95% CI: 0.74-0.87). Although the present study is the largest description of DCR in PRRT therapy of PCCs and PGLs, given the small sample size, no formal attempts were made to compare efficacy between 177Lu- and 90Y-PRRT treatments. In detail, we report pooled data on 149 177Lu- and 64 90Y-treated patients (total: 213 patients). Most studies were retrospective. The largest one included 46 patients (34 treated with 177Lu-, 12 with 90Y- PRRTs). Median ages ranged from 32.5 to 60.4 years. The female gender was slightly predominant (83 women, 74 men, gender not specified in 3 studies). When reported, mutations of SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B) were the most frequent genetic alterations. According to the predominant retrospective and real practice nature of the selected studies, sites of metastases and previous treatments were heterogeneous, with bone the metastatic site most often involved, and surgery the most frequent previous therapy. Paramount descriptive information on PRRT doses per cycle, number of cycles, response criteria, best responses to treatments, and frequency of G3/G4 hematologic toxicities is reported in Table 2. The lowest applied dose was 3.4 GBq per cycle; the number of administered cycles was heterogeneous (at least 3 cycles in 7 studies). The most used response criteria were the RECIST 1.1. Only one study evaluated the response with SWOG criteria, which are more conservative with regard to the definition of PR (50% or greater reduction in the sum of the product of the perpendicular diameters of target lesions). The majority of the patients experienced PR or SD. Hematologic toxicity was manageable. Funnel plot of selected studies did not show publication bias. The MINORS and Newcastle-Ottawa Scale scoring for the selected articles are reported in Table 3.
Click to Show/Hide
|
||||
| Experiment 51 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Inoperable or metastatic paragangliomas | ||||
| Efficacy Data | Stable disease (SD) |
67%
|
|||
| Patients Enrolled |
Patients with inoperable or metastatic paragangliomas and pheochromocytomas.
|
||||
| Administration Time | Four cycles | ||||
| Administration Dosage | 7.4 Gb per cycle | ||||
| Description |
Best tumour response was partial response in 7 (23%) and stable disease in 20 (67%)
|
||||
| Experiment 52 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Inoperable or metastatic pheochromocytomas | ||||
| Efficacy Data | Stable disease (SD) |
67%
|
|||
| Patients Enrolled |
Patients with inoperable or metastatic paragangliomas and pheochromocytomas.
|
||||
| Administration Time | Four cycles | ||||
| Administration Dosage | 7.4 Gb per cycle | ||||
| Description |
Best tumour response was partial response in 7 (23%) and stable disease in 20 (67%)
|
||||
| Experiment 53 Reporting the Activity Data of This PDC | [20] | ||||
| Indication | SSR positive lung neuroendocrine tumor | ||||
| Efficacy Data | Stable disease (SD) |
68%
|
|||
| Patients Enrolled |
40 patients with neuroendocrine tumour with RECIST-measurable disease.
|
||||
| Administration Time | 1-4 cycles | ||||
| Administration Dosage | Median cumulative activity of 27 GBq (range, 6-43 GBq) | ||||
| MOA of PDC |
In patients with advanced progressive lung NET and satisfactory SSR expression, 177Lu-DOTATATE is effective and safe, with a high DCR and encouraging PFS and OS. Further prospective studies comparing 177Lu-DOTATATE with other systemic options are warranted.
|
||||
| Description |
Of 48 patients (median age, 63 y; 13 women), 43 (90%) had AC and 5 (10%) TC. Almost all patients (47, 98%) were treated due to progression. Most patients (40, 83%) received somatostatin analogs, and 10 patients (20%) had prior everolimus, chemotherapy, or both. All patients had high SSR expression (≥ modified Krenning score 3) on pretreatment 68Ga-DOTATATE PET/CT. Patients received a median 4 (range, 1-4) cycles of 177Lu-DOTATATE (33% with concurrent radiosensitizing chemotherapy) to a median cumulative activity of 27 GBq (range, 6-43GBq). At a median follow-up of 42 mo, the median PFS and OS were 23 mo (95% CI, 18-28 mo) and 59 mo (95% CI, 50-not reached [NR]), respectively. Of 40 patients with RECIST-measurable disease and 39 patients with available 68Ga-DOTATATE PET/CT, response categories were partial response, 20% (95% CI, 10%-35%) and 44% (95% CI, 30%-59%); stable disease, 68% (95% CI, 52%-80%) and 44% (95% CI, 30%-59%); and progressive disease, 12% (95% CI, 5%-27%) by both, respectively. There was a moderate concordance between response categories by RECIST and 68Ga-DOTATATE PET/CT, weighted of 0.51 (95% CI, 0.21-0.68).
Click to Show/Hide
|
||||
| Experiment 54 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
68%
|
|||
| Patients Enrolled |
20 patients with others neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 55 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | PPGLa neuroendocrine tumor | ||||
| Efficacy Data | Stable disease (SD) |
70%
|
|||
| Patients Enrolled |
11 patients with PPGLa neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 56 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
73.33%
|
|||
| Patients Enrolled |
15 patients with pheocromocytomas (PCCs) and paragangliomas (PGLs) (5 PCC , 10PGL).
|
||||
| Administration Time | 4.13 cycles | ||||
| MOA of PDC |
We showed that PRRT based on 177Lu-DOTATATE and 90-YDOTATOC without radiosensitizers are efficacious therapeutic options (DCR of 83% and 76%, respectively) and can be considered in the multidisciplinary treatment of PCCs and PGLs as alternatives to I-131 MIBG and chemotherapy.
|
||||
| Description |
There was no statistically significant heterogeneity among the 177Lu- and 90Y- PRRTs studies (I2 value = 0%, p = 0.68 and I2 value = 0%, p = 0.91, respectively). The pooled DCRs were 0.83 (95% CI: 0.75-0.88) and 0.76 (95% CI: 0.56-0.89) for 177Lu- and 90Y-PRRT treatments, respectively. The pooled DCR for PRRT agents was 0.81 (95% CI: 0.74-0.87). Although the present study is the largest description of DCR in PRRT therapy of PCCs and PGLs, given the small sample size, no formal attempts were made to compare efficacy between 177Lu- and 90Y-PRRT treatments. In detail, we report pooled data on 149 177Lu- and 64 90Y-treated patients (total: 213 patients). Most studies were retrospective. The largest one included 46 patients (34 treated with 177Lu-, 12 with 90Y- PRRTs). Median ages ranged from 32.5 to 60.4 years. The female gender was slightly predominant (83 women, 74 men, gender not specified in 3 studies). When reported, mutations of SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B) were the most frequent genetic alterations. According to the predominant retrospective and real practice nature of the selected studies, sites of metastases and previous treatments were heterogeneous, with bone the metastatic site most often involved, and surgery the most frequent previous therapy. Paramount descriptive information on PRRT doses per cycle, number of cycles, response criteria, best responses to treatments, and frequency of G3/G4 hematologic toxicities is reported in Table 2. The lowest applied dose was 3.4 GBq per cycle; the number of administered cycles was heterogeneous (at least 3 cycles in 7 studies). The most used response criteria were the RECIST 1.1. Only one study evaluated the response with SWOG criteria, which are more conservative with regard to the definition of PR (50% or greater reduction in the sum of the product of the perpendicular diameters of target lesions). The majority of the patients experienced PR or SD. Hematologic toxicity was manageable. Funnel plot of selected studies did not show publication bias. The MINORS and Newcastle-Ottawa Scale scoring for the selected articles are reported in Table 3.
Click to Show/Hide
|
||||
| Experiment 57 Reporting the Activity Data of This PDC | [28] | ||||
| Indication | Phaeochromocytoma/Paraganglioma | ||||
| Efficacy Data | Stable disease (SD) |
73.50%
|
|||
| Patients Enrolled |
34 patients with SSTr2-positive progressive locally advanced or metastatic PPGLs.
|
||||
| Administration Time | 5 cycles | ||||
| Administration Dosage | 18.5/27.5 GBq | ||||
| MOA of PDC |
In a long-term follow-up of patients with mPPGL, PRRT demonstrated a tolerability and efficacy comparable to those obtained in GEP-NETs. Our results highlight the need for an appropriate cumulative administered dosage to achieve a very prolonged result and underline the importance of specific symptoms and presence of bone lesions as predictive factors. Given the substantial number of patients evaluated over time, the superior mOS of 177Lu-DOTATATE with respect to 90Y-DOTATOC indicates the former radiopharmaceutical as the better candidate for new prospective studies of single or associated therapies.
Click to Show/Hide
|
||||
| Description |
Both 90Y-DOTATOC and 177Lu-DOTATATE PRRT were well tolerated by patients without significant renal or bone marrow toxicity. The median follow-up was 73 months (range 5-146 months). The overall disease control rate (DCR) was 80% [95% confidence interval (CI) 68.9% to 91.9%] with a mean five cycles of therapy. However, 177Lu-DOTATATE patients showed a longer median overall survival (mOS) than those receiving 90Y-Dotatoc and a better DCR when higher dosages were administered, even if a direct comparison was not carried out. Syndromic patients had a poorer mOS. SDHx mutations did not interfere with treatment efficacy.
Click to Show/Hide
|
||||
| Experiment 58 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
73.53%
|
|||
| Patients Enrolled |
34 patients with pheocromocytomas and paragangliomas.
|
||||
| Administration Time | 5 cycles | ||||
| Administration Dosage | 3.7-5.5 GBq/cycle | ||||
| MOA of PDC |
We showed that PRRT based on 177Lu-DOTATATE and 90-YDOTATOC without radiosensitizers are efficacious therapeutic options (DCR of 83% and 76%, respectively) and can be considered in the multidisciplinary treatment of PCCs and PGLs as alternatives to I-131 MIBG and chemotherapy.
|
||||
| Description |
There was no statistically significant heterogeneity among the 177Lu- and 90Y- PRRTs studies (I2 value = 0%, p = 0.68 and I2 value = 0%, p = 0.91, respectively). The pooled DCRs were 0.83 (95% CI: 0.75-0.88) and 0.76 (95% CI: 0.56-0.89) for 177Lu- and 90Y-PRRT treatments, respectively. The pooled DCR for PRRT agents was 0.81 (95% CI: 0.74-0.87). Although the present study is the largest description of DCR in PRRT therapy of PCCs and PGLs, given the small sample size, no formal attempts were made to compare efficacy between 177Lu- and 90Y-PRRT treatments. In detail, we report pooled data on 149 177Lu- and 64 90Y-treated patients (total: 213 patients). Most studies were retrospective. The largest one included 46 patients (34 treated with 177Lu-, 12 with 90Y- PRRTs). Median ages ranged from 32.5 to 60.4 years. The female gender was slightly predominant (83 women, 74 men, gender not specified in 3 studies). When reported, mutations of SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B) were the most frequent genetic alterations. According to the predominant retrospective and real practice nature of the selected studies, sites of metastases and previous treatments were heterogeneous, with bone the metastatic site most often involved, and surgery the most frequent previous therapy. Paramount descriptive information on PRRT doses per cycle, number of cycles, response criteria, best responses to treatments, and frequency of G3/G4 hematologic toxicities is reported in Table 2. The lowest applied dose was 3.4 GBq per cycle; the number of administered cycles was heterogeneous (at least 3 cycles in 7 studies). The most used response criteria were the RECIST 1.1. Only one study evaluated the response with SWOG criteria, which are more conservative with regard to the definition of PR (50% or greater reduction in the sum of the product of the perpendicular diameters of target lesions). The majority of the patients experienced PR or SD. Hematologic toxicity was manageable. Funnel plot of selected studies did not show publication bias. The MINORS and Newcastle-Ottawa Scale scoring for the selected articles are reported in Table 3.
Click to Show/Hide
|
||||
| Experiment 59 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Midgut neuroendocrine tumor | ||||
| Efficacy Data | Stable disease (SD) |
76%
|
|||
| Patients Enrolled |
229 patients with midgut neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 60 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
76.90%
|
|||
| Patients Enrolled |
26 patients with neuroendocrine tumour.
|
||||
| Administration Time | 2-5 cycles | ||||
| Administration Dosage | Median activity for re-treatment: 16.5 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 61 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
78.72%
|
|||
| Patients Enrolled |
18 patients with neuroendocrine tumour.
|
||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 62 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
80%
|
|||
| Patients Enrolled |
5 patients with pheocromocytomas (PCCs) and paragangliomas (PGLs) (1PCC, 4PGL).
|
||||
| Administration Dosage | 7.4 GBq/cycle | ||||
| MOA of PDC |
We showed that PRRT based on 177Lu-DOTATATE and 90-YDOTATOC without radiosensitizers are efficacious therapeutic options (DCR of 83% and 76%, respectively) and can be considered in the multidisciplinary treatment of PCCs and PGLs as alternatives to I-131 MIBG and chemotherapy.
|
||||
| Description |
There was no statistically significant heterogeneity among the 177Lu- and 90Y- PRRTs studies (I2 value = 0%, p = 0.68 and I2 value = 0%, p = 0.91, respectively). The pooled DCRs were 0.83 (95% CI: 0.75-0.88) and 0.76 (95% CI: 0.56-0.89) for 177Lu- and 90Y-PRRT treatments, respectively. The pooled DCR for PRRT agents was 0.81 (95% CI: 0.74-0.87). Although the present study is the largest description of DCR in PRRT therapy of PCCs and PGLs, given the small sample size, no formal attempts were made to compare efficacy between 177Lu- and 90Y-PRRT treatments. In detail, we report pooled data on 149 177Lu- and 64 90Y-treated patients (total: 213 patients). Most studies were retrospective. The largest one included 46 patients (34 treated with 177Lu-, 12 with 90Y- PRRTs). Median ages ranged from 32.5 to 60.4 years. The female gender was slightly predominant (83 women, 74 men, gender not specified in 3 studies). When reported, mutations of SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B) were the most frequent genetic alterations. According to the predominant retrospective and real practice nature of the selected studies, sites of metastases and previous treatments were heterogeneous, with bone the metastatic site most often involved, and surgery the most frequent previous therapy. Paramount descriptive information on PRRT doses per cycle, number of cycles, response criteria, best responses to treatments, and frequency of G3/G4 hematologic toxicities is reported in Table 2. The lowest applied dose was 3.4 GBq per cycle; the number of administered cycles was heterogeneous (at least 3 cycles in 7 studies). The most used response criteria were the RECIST 1.1. Only one study evaluated the response with SWOG criteria, which are more conservative with regard to the definition of PR (50% or greater reduction in the sum of the product of the perpendicular diameters of target lesions). The majority of the patients experienced PR or SD. Hematologic toxicity was manageable. Funnel plot of selected studies did not show publication bias. The MINORS and Newcastle-Ottawa Scale scoring for the selected articles are reported in Table 3.
Click to Show/Hide
|
||||
| Experiment 63 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
81.30%
|
|||
| Patients Enrolled |
35 patients with neuroendocrine tumour.
|
||||
| Administration Time | 1-4 cycles | ||||
| Administration Dosage | Median cumulative activity 44 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 64 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
90.91%
|
|||
| Patients Enrolled |
22 patients with pheocromocytomas (PCCs) and paragangliomas (PGLs) (9 PCC , 13PGL).
|
||||
| Administration Time | 4.9 cycles | ||||
| Administration Dosage | 7.4 GBq/cycle | ||||
| MOA of PDC |
We showed that PRRT based on 177Lu-DOTATATE and 90-YDOTATOC without radiosensitizers are efficacious therapeutic options (DCR of 83% and 76%, respectively) and can be considered in the multidisciplinary treatment of PCCs and PGLs as alternatives to I-131 MIBG and chemotherapy.
|
||||
| Description |
There was no statistically significant heterogeneity among the 177Lu- and 90Y- PRRTs studies (I2 value = 0%, p = 0.68 and I2 value = 0%, p = 0.91, respectively). The pooled DCRs were 0.83 (95% CI: 0.75-0.88) and 0.76 (95% CI: 0.56-0.89) for 177Lu- and 90Y-PRRT treatments, respectively. The pooled DCR for PRRT agents was 0.81 (95% CI: 0.74-0.87). Although the present study is the largest description of DCR in PRRT therapy of PCCs and PGLs, given the small sample size, no formal attempts were made to compare efficacy between 177Lu- and 90Y-PRRT treatments. In detail, we report pooled data on 149 177Lu- and 64 90Y-treated patients (total: 213 patients). Most studies were retrospective. The largest one included 46 patients (34 treated with 177Lu-, 12 with 90Y- PRRTs). Median ages ranged from 32.5 to 60.4 years. The female gender was slightly predominant (83 women, 74 men, gender not specified in 3 studies). When reported, mutations of SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B) were the most frequent genetic alterations. According to the predominant retrospective and real practice nature of the selected studies, sites of metastases and previous treatments were heterogeneous, with bone the metastatic site most often involved, and surgery the most frequent previous therapy. Paramount descriptive information on PRRT doses per cycle, number of cycles, response criteria, best responses to treatments, and frequency of G3/G4 hematologic toxicities is reported in Table 2. The lowest applied dose was 3.4 GBq per cycle; the number of administered cycles was heterogeneous (at least 3 cycles in 7 studies). The most used response criteria were the RECIST 1.1. Only one study evaluated the response with SWOG criteria, which are more conservative with regard to the definition of PR (50% or greater reduction in the sum of the product of the perpendicular diameters of target lesions). The majority of the patients experienced PR or SD. Hematologic toxicity was manageable. Funnel plot of selected studies did not show publication bias. The MINORS and Newcastle-Ottawa Scale scoring for the selected articles are reported in Table 3.
Click to Show/Hide
|
||||
| Experiment 65 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Stable disease (SD) |
100%
|
|||
| Patients Enrolled |
5 patients with pheocromocytomas (PCCs) and paragangliomas (PGLs) (2 PCC, 3PGL).
|
||||
| Administration Dosage | 7.4 GBq/cycle | ||||
| MOA of PDC |
We showed that PRRT based on 177Lu-DOTATATE and 90-YDOTATOC without radiosensitizers are efficacious therapeutic options (DCR of 83% and 76%, respectively) and can be considered in the multidisciplinary treatment of PCCs and PGLs as alternatives to I-131 MIBG and chemotherapy.
|
||||
| Description |
There was no statistically significant heterogeneity among the 177Lu- and 90Y- PRRTs studies (I2 value = 0%, p = 0.68 and I2 value = 0%, p = 0.91, respectively). The pooled DCRs were 0.83 (95% CI: 0.75-0.88) and 0.76 (95% CI: 0.56-0.89) for 177Lu- and 90Y-PRRT treatments, respectively. The pooled DCR for PRRT agents was 0.81 (95% CI: 0.74-0.87). Although the present study is the largest description of DCR in PRRT therapy of PCCs and PGLs, given the small sample size, no formal attempts were made to compare efficacy between 177Lu- and 90Y-PRRT treatments. In detail, we report pooled data on 149 177Lu- and 64 90Y-treated patients (total: 213 patients). Most studies were retrospective. The largest one included 46 patients (34 treated with 177Lu-, 12 with 90Y- PRRTs). Median ages ranged from 32.5 to 60.4 years. The female gender was slightly predominant (83 women, 74 men, gender not specified in 3 studies). When reported, mutations of SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B) were the most frequent genetic alterations. According to the predominant retrospective and real practice nature of the selected studies, sites of metastases and previous treatments were heterogeneous, with bone the metastatic site most often involved, and surgery the most frequent previous therapy. Paramount descriptive information on PRRT doses per cycle, number of cycles, response criteria, best responses to treatments, and frequency of G3/G4 hematologic toxicities is reported in Table 2. The lowest applied dose was 3.4 GBq per cycle; the number of administered cycles was heterogeneous (at least 3 cycles in 7 studies). The most used response criteria were the RECIST 1.1. Only one study evaluated the response with SWOG criteria, which are more conservative with regard to the definition of PR (50% or greater reduction in the sum of the product of the perpendicular diameters of target lesions). The majority of the patients experienced PR or SD. Hematologic toxicity was manageable. Funnel plot of selected studies did not show publication bias. The MINORS and Newcastle-Ottawa Scale scoring for the selected articles are reported in Table 3.
Click to Show/Hide
|
||||
| Experiment 66 Reporting the Activity Data of This PDC | [29] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Serum troponin I decrease rate |
0.01 ng/ml
|
|||
| Patients Enrolled |
Patients with prostate cancer and neuroendocrine tumours referred for PRRT and RLT.
|
||||
| Description |
In all patients, the value of troponin I was in normal range. In all patients, the median values of serum troponin I before and after treatment were 0.2 ± 0.02 (range: 0.00-0.42) and 0.28 ± 0.02 (range: 0.00-0.46) ng/ml, respectively (p > 0.05). In the prostate cancer patients, the median values of serum troponin I before and after treatment were 0.26 ± 0.04 (0.04-0.42) and 0.30 ± 0.04 (0.00-0.41) ng/ml, respectively (p > 0.05). In the NET patients, the median values of serum troponin I before and after treatment were 0.18 ± 0.03 (0.00-0.42) and 0.17 ± 0.03 (0.00-0.46) ng/ml, respectively (p > 0.05).
Click to Show/Hide
|
||||
| Experiment 67 Reporting the Activity Data of This PDC | [30] | ||||
| Indication | Sinonasal neuroendocrine carcinomas | ||||
| Efficacy Data | Resolution rate |
100%
|
|||
| Administration Dosage | ˜7.4 GBq [200 mCi] | ||||
| Description |
On follow-up for a second PRRT cycle, there was a complete symptomatic response. Follow-up scans showed a significant decrease in the size of the sinonasal mass (˜1.9 0.8 cm vs. 7.0 4.6 5.0 cm at baseline), with a significant decrease in the size of the left cervical level II lymph node (1.5 1.1 cm vs. 2.2 1.3 cm at baseline) and complete resolution of the right hilar lymph node.
Click to Show/Hide
|
||||
| In Vivo Model | A 52-year-old man with SNC. | ||||
| Experiment 68 Reporting the Activity Data of This PDC | [31] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumour | ||||
| Efficacy Data | Relative risk of hematopoietic neoplasms |
2.7
|
|||
| Patients Enrolled |
Patients with gastroenteropancreatic neuroendocrine tumors.
|
||||
| Description |
The expected number of hematopoietic neoplasms based on The Netherlands Cancer Registry data was 3.0, resulting in a relative risk of 2.7 (95% confidence interval, 0.7-10.0).
|
||||
| Experiment 69 Reporting the Activity Data of This PDC | [32] | ||||
| Indication | Meningiomas | ||||
| Efficacy Data | Rate of Progression-free survival at 6 months |
0%
|
|||
| Patients Enrolled |
17 patients with WHO grade III meningiomas.
|
||||
| Administration Time | 4 cycles in 8-9 week intervals | ||||
| Administration Dosage | 3200-7400 MBq per cycle | ||||
| Evaluation Method | 68Ga-DOTATATE PET/CT assay | ||||
| MOA of PDC |
PRRT may be a promising treatment for patients with refractory meningiomas, particularly those with grade I and II meningiomas. The treatment is well tolerated and stabilizes or slows tumor progression for at least a few months.
|
||||
| Description |
PFS-6 according to WHO grade was analyzed for 72 patients. The 6-month PFS was 89.7% for WHO grade I meningiomas (n = 29); 57.1% for WHO grade II (n = 21) and 0 % for WHO grade III (n = 12). For all grades (n = 86), PFS-6 was 58.1%. PFS-6 for unknown grades was 100%. Based on the available data and following RANO criteria, the best radiological response obtained was stable disease.
Click to Show/Hide
|
||||
| Experiment 70 Reporting the Activity Data of This PDC | [32] | ||||
| Indication | Meningiomas | ||||
| Efficacy Data | Rate of Progression-free survival at 6 months |
57.10%
|
|||
| Patients Enrolled |
30 patients with WHO grade II meningiomas.
|
||||
| Administration Time | 4 cycles in 8-9 week intervals | ||||
| Administration Dosage | 3200-7400 MBq per cycle | ||||
| Evaluation Method | 68Ga-DOTATATE PET/CT assay | ||||
| MOA of PDC |
PRRT may be a promising treatment for patients with refractory meningiomas, particularly those with grade I and II meningiomas. The treatment is well tolerated and stabilizes or slows tumor progression for at least a few months.
|
||||
| Description |
PFS-6 according to WHO grade was analyzed for 72 patients. The 6-month PFS was 89.7% for WHO grade I meningiomas (n = 29); 57.1% for WHO grade II (n = 21) and 0 % for WHO grade III (n = 12). For all grades (n = 86), PFS-6 was 58.1%. PFS-6 for unknown grades was 100%. Based on the available data and following RANO criteria, the best radiological response obtained was stable disease.
Click to Show/Hide
|
||||
| Experiment 71 Reporting the Activity Data of This PDC | [32] | ||||
| Indication | Meningiomas | ||||
| Efficacy Data | Rate of Progression-free survival at 6 months |
89.70%
|
|||
| Patients Enrolled |
29 patients with WHO grade I meningiomas.
|
||||
| Administration Time | 4 cycles in 8-9 week intervals | ||||
| Administration Dosage | 3200-7400 MBq per cycle | ||||
| Evaluation Method | 68Ga-DOTATATE PET/CT assay | ||||
| MOA of PDC |
PRRT may be a promising treatment for patients with refractory meningiomas, particularly those with grade I and II meningiomas. The treatment is well tolerated and stabilizes or slows tumor progression for at least a few months.
|
||||
| Description |
PFS-6 according to WHO grade was analyzed for 72 patients. The 6-month PFS was 89.7% for WHO grade I meningiomas (n = 29); 57.1% for WHO grade II (n = 21) and 0 % for WHO grade III (n = 12). For all grades (n = 86), PFS-6 was 58.1%. PFS-6 for unknown grades was 100%. Based on the available data and following RANO criteria, the best radiological response obtained was stable disease.
Click to Show/Hide
|
||||
| Experiment 72 Reporting the Activity Data of This PDC | [32] | ||||
| Indication | Meningiomas | ||||
| Efficacy Data | Rate of Progression-free survival at 6 months |
100.00%
|
|||
| Patients Enrolled |
17 patients with unknown WHO grade meningiomas.
|
||||
| Administration Time | 4 cycles in 8-9 week intervals | ||||
| Administration Dosage | 3200-7400 MBq per cycle | ||||
| Evaluation Method | 68Ga-DOTATATE PET/CT assay | ||||
| MOA of PDC |
PRRT may be a promising treatment for patients with refractory meningiomas, particularly those with grade I and II meningiomas. The treatment is well tolerated and stabilizes or slows tumor progression for at least a few months.
|
||||
| Description |
PFS-6 according to WHO grade was analyzed for 72 patients. The 6-month PFS was 89.7% for WHO grade I meningiomas (n = 29); 57.1% for WHO grade II (n = 21) and 0 % for WHO grade III (n = 12). For all grades (n = 86), PFS-6 was 58.1%. PFS-6 for unknown grades was 100%. Based on the available data and following RANO criteria, the best radiological response obtained was stable disease.
Click to Show/Hide
|
||||
| Experiment 73 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Metastatic midgut neuroendocrine tumor | ||||
| Efficacy Data | Rate of low fibrinogen |
0.3 g/L
|
|||
| Administration Dosage | 7.4 GBq | ||||
| Description |
Initial laboratory tests showed normocytic regenerative anemia (hemoglobin 85g/dL), thrombocytopenia (54 109/L), no signs of hemolysis, no schistocytes, prolonged prothrombin time (25.8seconds, 35%) and prolonged activated partial thromboplastin time (1.63), low fibrinogen (0.3g/L), elevated fibrin monomer (>400ug/mL) and elevated fibrin degradation products (>20ug/mL). Coagulation factors II (49%) and V (44%) were low; factors VII (97%) and X (81%) were normal. Liver function tests were normal with the exception of moderate elevation of gamma Glutamyl Transferase (γGT=100UI/L, N<38UI/L). C-reactive protein was <5mg/L. There was no sign of tumor lysis syndrome. The bone marrow aspirate and medullar karyotype were normal. Computed tomography showed stable metastatic disease, including a large stable necrotic liver metastasis.
Click to Show/Hide
|
||||
| In Vivo Model | 55-year-old woman with metastatic midgut neuroendocrine tumor. | ||||
| Experiment 74 Reporting the Activity Data of This PDC | [18] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumor | ||||
| Efficacy Data | Rate of long-term toxicity |
0.70%
|
|||
| Patients Enrolled |
Patients treated with Lu-DOTATATE.
|
||||
| Administration Dosage | 100 mCi (3.7 GBq) | ||||
| Description |
The objective response rate of the total group of patients was 39%. Stable disease was reached in 43% of patients. Progression-free survival (PFS) and overall survival (OS) for all NET patients were 29 months [95% confidence interval (CI), 26-33 months] and 63 months (95% CI, 55-72 months). Long-term toxicity included acute leukemia in four patients (0.7%) and myelodysplastic syndrome in nine patients (1.5%).
Click to Show/Hide
|
||||
| Experiment 75 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Rate of liver toxicity |
10.49%
|
|||
| Patients Enrolled |
534 male patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (25.9-29.6 GBq) (800 mCi [700-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 76 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Rate of liver toxicity |
14.58%
|
|||
| Patients Enrolled |
439 female patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (22.2-29.6 GBq) (800 mCi [600-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 77 Reporting the Activity Data of This PDC | [34] | ||||
| Indication | Carcinoid heart valve disease | ||||
| Efficacy Data | Rate of death decrease |
46%
|
|||
| Patients Enrolled |
8 patients with histologically confirmed grade 1 or 2 NEN and echocardiographically proven carcinoid syndrome.
|
||||
| Administration Time | Four prrt infusions at 8-16 weeks intervals | ||||
| Administration Dosage | 7.4 GBq | ||||
| MOA of PDC |
Peptide receptor radionuclide therapy (PRRT) uses lutetium-177 oxodotreotide, a radio-labelled peptide with high affinity for somatostatin receptor subtype 2. It is indicated in well-differentiated NENs that are either metastatic or surgically unsuitable that have proven somatostatin receptor positivity on imaging. Safe administration protocol requires the concomitant infusion of one litre of intravenous peptides.
Click to Show/Hide
|
||||
| Description |
Patients with inoperable gastrointestinal NENs treated in the phase 3 trial of Lu-177-DOtatate for midgut neuroendocrine tumours study were found to have significantly improved survival with the use of lutetium-177 therapy versus somatostatin receptor analogues (SSRA) alone, with a reduced risk of death (46%) in the lutetium group.
|
||||
| Experiment 78 Reporting the Activity Data of This PDC | [35] | ||||
| Indication | Advanced medullary thyroid carcinoma | ||||
| Efficacy Data | Rate of calcitonin reduction |
60%
|
|||
| Patients Enrolled |
8 patients with advanced medullary thyroid cancer.
|
||||
| Administration Time | From days 0 to 14 of each prrt cycle | ||||
| Administration Dosage | 20.9 GBq (interquartile range 8.9-27.7 GBq) | ||||
| Description |
Biochemical response with reduction in serum calcitonin levels was observed in 3/5 (60%) patients. With the exception of grade 2 anaemia in one patient, no other significant toxicity was observed in this cohort.
|
||||
| Experiment 79 Reporting the Activity Data of This PDC | [36] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumor | ||||
| Efficacy Data | Radiation absorbed doses |
0.017 ± 0.016 mGy/MBq
|
|||
| Patients Enrolled |
Patients with non-resectable histologically confirmed gastroenteropancreatic neuroendocrine tumors with normal kidney function.
|
||||
| Administration Time | 14 days | ||||
| Administration Dosage | 6.4 GBq-7.6 GBq | ||||
| Description |
Radiation absorbed doses (mean ± standard deviation) were obtained as 0.29 ± 0.12 mGy/MBq for kidneys, 0.30 ± 0.18 mGy/MBq for liver, 0.63 ± 0.37 mGy/MBq for spleen, 0.019 ± 0.001 mGy/MBq for bone marrow and 3.85 ± 1.74 mGy/MBq for tumours in the case group and they were 0.31± 0.26, 0.24 ± 0.14, 0.64 ± 0.42, 0.017 ± 0.016, 5.6 ± 11.27 mGy/MBq in kidneys, liver, spleen, bone marrow and neuroendocrine tumour, respectively, in the control group.
Click to Show/Hide
|
||||
| Experiment 80 Reporting the Activity Data of This PDC | [36] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumor | ||||
| Efficacy Data | Radiation absorbed doses |
0.019 ± 0.001 mGy/MBq
|
|||
| Patients Enrolled |
Patients with non-resectable histologically confirmed gastroenteropancreatic neuroendocrine tumors with normal kidney function.
|
||||
| Administration Time | 14 days | ||||
| Administration Dosage | 600 mg m-2 bovine serum albumin twice a day | ||||
| Description |
Radiation absorbed doses (mean ± standard deviation) were obtained as 0.29 ± 0.12 mGy/MBq for kidneys, 0.30 ± 0.18 mGy/MBq for liver, 0.63 ± 0.37 mGy/MBq for spleen, 0.019 ± 0.001 mGy/MBq for bone marrow and 3.85 ± 1.74 mGy/MBq for tumours in the case group and they were 0.31± 0.26, 0.24 ± 0.14, 0.64 ± 0.42, 0.017 ± 0.016, 5.6 ± 11.27 mGy/MBq in kidneys, liver, spleen, bone marrow and neuroendocrine tumour, respectively, in the control group.
Click to Show/Hide
|
||||
| Experiment 81 Reporting the Activity Data of This PDC | [36] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumor | ||||
| Efficacy Data | Radiation absorbed doses |
0.24 ± 0.14 mGy/MBq
|
|||
| Patients Enrolled |
Patients with non-resectable histologically confirmed gastroenteropancreatic neuroendocrine tumors with normal kidney function.
|
||||
| Administration Time | 14 days | ||||
| Administration Dosage | 6.4 GBq-7.6 GBq | ||||
| Description |
Radiation absorbed doses (mean ± standard deviation) were obtained as 0.29 ± 0.12 mGy/MBq for kidneys, 0.30 ± 0.18 mGy/MBq for liver, 0.63 ± 0.37 mGy/MBq for spleen, 0.019 ± 0.001 mGy/MBq for bone marrow and 3.85 ± 1.74 mGy/MBq for tumours in the case group and they were 0.31± 0.26, 0.24 ± 0.14, 0.64 ± 0.42, 0.017 ± 0.016, 5.6 ± 11.27 mGy/MBq in kidneys, liver, spleen, bone marrow and neuroendocrine tumour, respectively, in the control group.
Click to Show/Hide
|
||||
| Experiment 82 Reporting the Activity Data of This PDC | [36] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumor | ||||
| Efficacy Data | Radiation absorbed doses |
0.29 ± 0.12 mGy/MBq
|
|||
| Patients Enrolled |
Patients with non-resectable histologically confirmed gastroenteropancreatic neuroendocrine tumors with normal kidney function.
|
||||
| Administration Time | 14 days | ||||
| Administration Dosage | 600 mg m-2 bovine serum albumin twice a day | ||||
| Description |
Radiation absorbed doses (mean ± standard deviation) were obtained as 0.29 ± 0.12 mGy/MBq for kidneys, 0.30 ± 0.18 mGy/MBq for liver, 0.63 ± 0.37 mGy/MBq for spleen, 0.019 ± 0.001 mGy/MBq for bone marrow and 3.85 ± 1.74 mGy/MBq for tumours in the case group and they were 0.31± 0.26, 0.24 ± 0.14, 0.64 ± 0.42, 0.017 ± 0.016, 5.6 ± 11.27 mGy/MBq in kidneys, liver, spleen, bone marrow and neuroendocrine tumour, respectively, in the control group.
Click to Show/Hide
|
||||
| Experiment 83 Reporting the Activity Data of This PDC | [36] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumor | ||||
| Efficacy Data | Radiation absorbed doses |
0.30 ± 0.18 mGy/MBq
|
|||
| Patients Enrolled |
Patients with non-resectable histologically confirmed gastroenteropancreatic neuroendocrine tumors with normal kidney function.
|
||||
| Administration Time | 14 days | ||||
| Administration Dosage | 600 mg m-2 bovine serum albumin twice a day | ||||
| Description |
Radiation absorbed doses (mean ± standard deviation) were obtained as 0.29 ± 0.12 mGy/MBq for kidneys, 0.30 ± 0.18 mGy/MBq for liver, 0.63 ± 0.37 mGy/MBq for spleen, 0.019 ± 0.001 mGy/MBq for bone marrow and 3.85 ± 1.74 mGy/MBq for tumours in the case group and they were 0.31± 0.26, 0.24 ± 0.14, 0.64 ± 0.42, 0.017 ± 0.016, 5.6 ± 11.27 mGy/MBq in kidneys, liver, spleen, bone marrow and neuroendocrine tumour, respectively, in the control group.
Click to Show/Hide
|
||||
| Experiment 84 Reporting the Activity Data of This PDC | [36] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumor | ||||
| Efficacy Data | Radiation absorbed doses |
0.31 ± 0.26 mGy/MBq
|
|||
| Patients Enrolled |
Patients with non-resectable histologically confirmed gastroenteropancreatic neuroendocrine tumors with normal kidney function.
|
||||
| Administration Time | 14 days | ||||
| Administration Dosage | 6.4 GBq-7.6 GBq | ||||
| Description |
Radiation absorbed doses (mean ± standard deviation) were obtained as 0.29 ± 0.12 mGy/MBq for kidneys, 0.30 ± 0.18 mGy/MBq for liver, 0.63 ± 0.37 mGy/MBq for spleen, 0.019 ± 0.001 mGy/MBq for bone marrow and 3.85 ± 1.74 mGy/MBq for tumours in the case group and they were 0.31± 0.26, 0.24 ± 0.14, 0.64 ± 0.42, 0.017 ± 0.016, 5.6 ± 11.27 mGy/MBq in kidneys, liver, spleen, bone marrow and neuroendocrine tumour, respectively, in the control group.
Click to Show/Hide
|
||||
| Experiment 85 Reporting the Activity Data of This PDC | [36] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumor | ||||
| Efficacy Data | Radiation absorbed doses |
0.63 ± 0.37 mGy/MBq
|
|||
| Patients Enrolled |
Patients with non-resectable histologically confirmed gastroenteropancreatic neuroendocrine tumors with normal kidney function.
|
||||
| Administration Time | 14 days | ||||
| Administration Dosage | 600 mg m-2 bovine serum albumin twice a day | ||||
| Description |
Radiation absorbed doses (mean ± standard deviation) were obtained as 0.29 ± 0.12 mGy/MBq for kidneys, 0.30 ± 0.18 mGy/MBq for liver, 0.63 ± 0.37 mGy/MBq for spleen, 0.019 ± 0.001 mGy/MBq for bone marrow and 3.85 ± 1.74 mGy/MBq for tumours in the case group and they were 0.31± 0.26, 0.24 ± 0.14, 0.64 ± 0.42, 0.017 ± 0.016, 5.6 ± 11.27 mGy/MBq in kidneys, liver, spleen, bone marrow and neuroendocrine tumour, respectively, in the control group.
Click to Show/Hide
|
||||
| Experiment 86 Reporting the Activity Data of This PDC | [36] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumor | ||||
| Efficacy Data | Radiation absorbed doses |
0.64 ± 0.42 mGy/MBq
|
|||
| Patients Enrolled |
Patients with non-resectable histologically confirmed gastroenteropancreatic neuroendocrine tumors with normal kidney function.
|
||||
| Administration Time | 14 days | ||||
| Administration Dosage | 6.4 GBq-7.6 GBq | ||||
| Description |
Radiation absorbed doses (mean ± standard deviation) were obtained as 0.29 ± 0.12 mGy/MBq for kidneys, 0.30 ± 0.18 mGy/MBq for liver, 0.63 ± 0.37 mGy/MBq for spleen, 0.019 ± 0.001 mGy/MBq for bone marrow and 3.85 ± 1.74 mGy/MBq for tumours in the case group and they were 0.31± 0.26, 0.24 ± 0.14, 0.64 ± 0.42, 0.017 ± 0.016, 5.6 ± 11.27 mGy/MBq in kidneys, liver, spleen, bone marrow and neuroendocrine tumour, respectively, in the control group.
Click to Show/Hide
|
||||
| Experiment 87 Reporting the Activity Data of This PDC | [36] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumor | ||||
| Efficacy Data | Radiation absorbed doses |
3.85 ± 1.74 mGy/MBq
|
|||
| Patients Enrolled |
Patients with non-resectable histologically confirmed gastroenteropancreatic neuroendocrine tumors with normal kidney function.
|
||||
| Administration Time | 14 days | ||||
| Administration Dosage | 600 mg m-2 bovine serum albumin twice a day | ||||
| Description |
Radiation absorbed doses (mean ± standard deviation) were obtained as 0.29 ± 0.12 mGy/MBq for kidneys, 0.30 ± 0.18 mGy/MBq for liver, 0.63 ± 0.37 mGy/MBq for spleen, 0.019 ± 0.001 mGy/MBq for bone marrow and 3.85 ± 1.74 mGy/MBq for tumours in the case group and they were 0.31± 0.26, 0.24 ± 0.14, 0.64 ± 0.42, 0.017 ± 0.016, 5.6 ± 11.27 mGy/MBq in kidneys, liver, spleen, bone marrow and neuroendocrine tumour, respectively, in the control group.
Click to Show/Hide
|
||||
| Experiment 88 Reporting the Activity Data of This PDC | [36] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumor | ||||
| Efficacy Data | Radiation absorbed doses |
5.6 ± 11.27 mGy/MBq
|
|||
| Patients Enrolled |
Patients with non-resectable histologically confirmed gastroenteropancreatic neuroendocrine tumors with normal kidney function.
|
||||
| Administration Time | 14 days | ||||
| Administration Dosage | 6.4 GBq-7.6 GBq | ||||
| Description |
Radiation absorbed doses (mean ± standard deviation) were obtained as 0.29 ± 0.12 mGy/MBq for kidneys, 0.30 ± 0.18 mGy/MBq for liver, 0.63 ± 0.37 mGy/MBq for spleen, 0.019 ± 0.001 mGy/MBq for bone marrow and 3.85 ± 1.74 mGy/MBq for tumours in the case group and they were 0.31± 0.26, 0.24 ± 0.14, 0.64 ± 0.42, 0.017 ± 0.016, 5.6 ± 11.27 mGy/MBq in kidneys, liver, spleen, bone marrow and neuroendocrine tumour, respectively, in the control group.
Click to Show/Hide
|
||||
| Experiment 89 Reporting the Activity Data of This PDC | [37] | ||||
| Indication | Neuroendocrine prostate cancer | ||||
| Efficacy Data | PSA decline rate |
24%
|
|||
| Patients Enrolled |
A patient with metastatic castration-resistant prostatic cancer.
|
||||
| Administration Time | 1 cycle | ||||
| Administration Dosage | 7.4 GBq | ||||
| Evaluation Method | 68Ga-DOTATATE PET/CT assay | ||||
| MOA of PDC |
Treatment with 177Lu-DOTATATE might be beneficial, but due to rarity of this entity, collaborative, multicentric studies are needed to assess the efficacy of this treatment modality. This case also highlights possible usefulness of PRLT even in cases with diffuse bone marrow involvement (not an indication of treatment in the phase III VISION trial).
Click to Show/Hide
|
||||
| Description |
In follow-up visits, he reported transient bone pain reduction, and interestingly, PSA levels dropped by 24% (from 326 ng/mL to 247.5 ng/mL). The patient was unable to receive the second cycle due to severe fatigue, which was concomitant with abrupt increase in PSA.
|
||||
| Experiment 90 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Metastatic midgut neuroendocrine tumor | ||||
| Efficacy Data | Prolonged prothrombin time |
25.8 seconds
|
|||
| Administration Dosage | 7.4 GBq | ||||
| Description |
Initial laboratory tests showed normocytic regenerative anemia (hemoglobin 85g/dL), thrombocytopenia (54 109/L), no signs of hemolysis, no schistocytes, prolonged prothrombin time (25.8seconds, 35%) and prolonged activated partial thromboplastin time (1.63), low fibrinogen (0.3g/L), elevated fibrin monomer (>400ug/mL) and elevated fibrin degradation products (>20ug/mL). Coagulation factors II (49%) and V (44%) were low; factors VII (97%) and X (81%) were normal. Liver function tests were normal with the exception of moderate elevation of gamma Glutamyl Transferase (γGT=100UI/L, N<38UI/L). C-reactive protein was <5mg/L. There was no sign of tumor lysis syndrome. The bone marrow aspirate and medullar karyotype were normal. Computed tomography showed stable metastatic disease, including a large stable necrotic liver metastasis.
Click to Show/Hide
|
||||
| In Vivo Model | 55-year-old woman with metastatic midgut neuroendocrine tumor. | ||||
| Experiment 91 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Metastatic midgut neuroendocrine tumor | ||||
| Efficacy Data | Prolonged activated partial thromboplastin time |
1.63 seconds
|
|||
| Administration Dosage | 7.4 GBq | ||||
| Description |
Initial laboratory tests showed normocytic regenerative anemia (hemoglobin 85g/dL), thrombocytopenia (54 109/L), no signs of hemolysis, no schistocytes, prolonged prothrombin time (25.8seconds, 35%) and prolonged activated partial thromboplastin time (1.63), low fibrinogen (0.3g/L), elevated fibrin monomer (>400ug/mL) and elevated fibrin degradation products (>20ug/mL). Coagulation factors II (49%) and V (44%) were low; factors VII (97%) and X (81%) were normal. Liver function tests were normal with the exception of moderate elevation of gamma Glutamyl Transferase (γGT=100UI/L, N<38UI/L). C-reactive protein was <5mg/L. There was no sign of tumor lysis syndrome. The bone marrow aspirate and medullar karyotype were normal. Computed tomography showed stable metastatic disease, including a large stable necrotic liver metastasis.
Click to Show/Hide
|
||||
| In Vivo Model | 55-year-old woman with metastatic midgut neuroendocrine tumor. | ||||
| Experiment 92 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Unknown originb neuroendocrine tumor | ||||
| Efficacy Data | Progressive Disease (PD) |
0%
|
|||
| Patients Enrolled |
22 patients with unknown originb neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 93 Reporting the Activity Data of This PDC | [14] | ||||
| Indication | Metastatic/Advanced neuroendocrine tumour | ||||
| Efficacy Data | Progressive Disease (PD) |
6%
|
|||
| Patients Enrolled |
468 patients with metastatic/advanced neuroendocrine tumor.
|
||||
| Administration Time | At least two cycles at 10-12 weeks interval | ||||
| Administration Dosage | 5.55 to 7.4 GBq per patient | ||||
| Description |
Long-term outcome (follow-up ranging from 4 to 97.6 months; median period:46 months following first 177Lu-DOTATATE PRRT) results showed, (i) on symptomatic response evaluation scale, complete response (CR) in 214 patients (45.7%), partial response (PR) in 108 (23.1%), stable disease (SD) in 118 (25.2%), progressive disease (PD) in 28 (6%).
Click to Show/Hide
|
||||
| Experiment 94 Reporting the Activity Data of This PDC | [23] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Progressive Disease (PD) |
6.80%
|
|||
| Patients Enrolled |
Patients with advanced neuroendocrine tumor.
|
||||
| Administration Dosage | Along with oral capecitabine therapy | ||||
| Description |
According to Response Evaluation Criteria in Solid Tumors 1.1 criteria, in group 1 patients, the response was partial response in 34% of the patients, stable disease in 50.2%, and progressive disease in 6.8% versus partial response in 6.3%, stable disease in 60.9%, and progressive disease in 26.5% among group 2 patients. The median overall survival (OS) and progression-free survival (PFS) was not reached in group 1 patients. The median OS and PFS in group 2 patients were 48 months.
Click to Show/Hide
|
||||
| Experiment 95 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Hindgut neuroendocrine tumour | ||||
| Efficacy Data | Progressive Disease (PD) |
7%
|
|||
| Patients Enrolled |
14 patients with hindgut neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 96 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Progressive Disease (PD) |
7.70%
|
|||
| Patients Enrolled |
13 Re-retreatment neuroendocrine tumour patients.
|
||||
| Administration Time | 2 cycles | ||||
| Administration Dosage | Median cumulative dose: 59.7 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 97 Reporting the Activity Data of This PDC | [14] | ||||
| Indication | Metastatic/Advanced neuroendocrine tumour | ||||
| Efficacy Data | Progressive Disease (PD) |
10%
|
|||
| Patients Enrolled |
468 patients with metastatic/advanced neuroendocrine tumor.
|
||||
| Administration Time | At least two cycles at 10-12 weeks interval | ||||
| Administration Dosage | 5.55 to 7.4 GBq per patient | ||||
| Description |
Biochemical response evaluation showed CR in 52 (12%), PR in 172 (40%), SD in 161 (38%), and PD in 42 patients (10%).
|
||||
| Experiment 98 Reporting the Activity Data of This PDC | [14] | ||||
| Indication | Metastatic/Advanced neuroendocrine tumour | ||||
| Efficacy Data | Progressive Disease (PD) |
10%
|
|||
| Patients Enrolled |
468 patients with metastatic/advanced neuroendocrine tumor.
|
||||
| Administration Time | At least two cycles at 10-12 weeks interval | ||||
| Administration Dosage | 5.55 to 7.4 GBq per patient | ||||
| Description |
On RECIST 1.1 criteria, CR was observed in 14 patients (3%), PR in 126 patients (27%), SD in 282 patients (60%) and PD in 46 patients (10%).
|
||||
| Experiment 99 Reporting the Activity Data of This PDC | [38] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Progressive Disease (PD) |
10%
|
|||
| Patients Enrolled |
100 neuroendocrine tumour patients.
|
||||
| Administration Time | 4 cycles every 10 weeks | ||||
| Administration Dosage | 7.4 GBq | ||||
| MOA of PDC |
The incidence of severe haematotoxicity was low with extensive screening and monitoring. The vast majority of patients (96/100) was not restricted in treatment continuation by haematotoxicity; therefore, our selection criteria appeared appropriate for safe PRRT treatment. Baseline parameters showed limited correlation with the degree of decline in haematological values.
Click to Show/Hide
|
||||
| Description |
Four cycles were completed by 86/100 of patients, 4/100 patients discontinued due to haematotoxicity, and 10/100 patients due to progressive disease. The treatment course was adjusted due to haematotoxicity in 24/100 patients, including postponed next cycle (n = 17), reduced administered activity (n = 13), and both adjustments (n = 10). The most observed haematotoxicity grade was grade 0-1 in 54/100 patients, grade 2 in 38/100 and grade 3-4 in 8/100. Significant differences in baseline leucocyte, neutrophil and platelet counts were observed between grade 0-1 and grade 2. However, the correlation between baseline and lowest observed values was poor to moderate. No differences between haematotoxicity grades and baseline parameters or somatostatin receptor positive tumour volume was observed.
Click to Show/Hide
|
||||
| Experiment 100 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Midgut neuroendocrine tumor | ||||
| Efficacy Data | Progressive Disease (PD) |
11%
|
|||
| Patients Enrolled |
229 patients with midgut neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 101 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Progressive Disease (PD) |
11%
|
|||
| Patients Enrolled |
20 patients with others neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 102 Reporting the Activity Data of This PDC | [14] | ||||
| Indication | Metastatic/Advanced neuroendocrine tumour | ||||
| Efficacy Data | Progressive Disease (PD) |
12%
|
|||
| Patients Enrolled |
468 patients with metastatic/advanced neuroendocrine tumor.
|
||||
| Administration Time | At least two cycles at 10-12 weeks interval | ||||
| Administration Dosage | 5.55 to 7.4 GBq per patient | ||||
| Evaluation Method | PERCIST criteria assay | ||||
| Description |
Molecular imaging response (by PERCIST criteria) showed CR in 29 (6%), PR in 116 (25%), SD in 267 (57%) and PD in 56 (12%) patients.
|
||||
| Experiment 103 Reporting the Activity Data of This PDC | [20] | ||||
| Indication | SSR positive lung neuroendocrine tumor | ||||
| Efficacy Data | Progressive Disease (PD) |
12%
|
|||
| Patients Enrolled |
40 patients with neuroendocrine tumour with RECIST-measurable disease.
|
||||
| Administration Time | 1-4 cycles | ||||
| Administration Dosage | Median cumulative activity of 27 GBq (range, 6-43 GBq) | ||||
| MOA of PDC |
In patients with advanced progressive lung NET and satisfactory SSR expression, 177Lu-DOTATATE is effective and safe, with a high DCR and encouraging PFS and OS. Further prospective studies comparing 177Lu-DOTATATE with other systemic options are warranted.
|
||||
| Description |
Of 48 patients (median age, 63 y; 13 women), 43 (90%) had AC and 5 (10%) TC. Almost all patients (47, 98%) were treated due to progression. Most patients (40, 83%) received somatostatin analogs, and 10 patients (20%) had prior everolimus, chemotherapy, or both. All patients had high SSR expression (≥ modified Krenning score 3) on pretreatment 68Ga-DOTATATE PET/CT. Patients received a median 4 (range, 1-4) cycles of 177Lu-DOTATATE (33% with concurrent radiosensitizing chemotherapy) to a median cumulative activity of 27 GBq (range, 6-43GBq). At a median follow-up of 42 mo, the median PFS and OS were 23 mo (95% CI, 18-28 mo) and 59 mo (95% CI, 50-not reached [NR]), respectively. Of 40 patients with RECIST-measurable disease and 39 patients with available 68Ga-DOTATATE PET/CT, response categories were partial response, 20% (95% CI, 10%-35%) and 44% (95% CI, 30%-59%); stable disease, 68% (95% CI, 52%-80%) and 44% (95% CI, 30%-59%); and progressive disease, 12% (95% CI, 5%-27%) by both, respectively. There was a moderate concordance between response categories by RECIST and 68Ga-DOTATATE PET/CT, weighted of 0.51 (95% CI, 0.21-0.68).
Click to Show/Hide
|
||||
| Experiment 104 Reporting the Activity Data of This PDC | [20] | ||||
| Indication | SSR positive lung neuroendocrine tumor | ||||
| Efficacy Data | Progressive Disease (PD) |
12%
|
|||
| Patients Enrolled |
39 patients with neuroendocrine tumour with available <sup>68</sup>Ga-DOTATATE PET/CT.
|
||||
| Administration Time | 1-4 cycles | ||||
| Administration Dosage | Median cumulative activity of 27 GBq (range, 6-43 GBq) | ||||
| Evaluation Method | 6AN1831:AN18988Ga-DOTATATE PET/CT assay | ||||
| MOA of PDC |
In patients with advanced progressive lung NET and satisfactory SSR expression, 177Lu-DOTATATE is effective and safe, with a high DCR and encouraging PFS and OS. Further prospective studies comparing 177Lu-DOTATATE with other systemic options are warranted.
|
||||
| Description |
Of 48 patients (median age, 63 y; 13 women), 43 (90%) had AC and 5 (10%) TC. Almost all patients (47, 98%) were treated due to progression. Most patients (40, 83%) received somatostatin analogs, and 10 patients (20%) had prior everolimus, chemotherapy, or both. All patients had high SSR expression (≥ modified Krenning score 3) on pretreatment 68Ga-DOTATATE PET/CT. Patients received a median 4 (range, 1-4) cycles of 177Lu-DOTATATE (33% with concurrent radiosensitizing chemotherapy) to a median cumulative activity of 27 GBq (range, 6-43GBq). At a median follow-up of 42 mo, the median PFS and OS were 23 mo (95% CI, 18-28 mo) and 59 mo (95% CI, 50-not reached [NR]), respectively. Of 40 patients with RECIST-measurable disease and 39 patients with available 68Ga-DOTATATE PET/CT, response categories were partial response, 20% (95% CI, 10%-35%) and 44% (95% CI, 30%-59%); stable disease, 68% (95% CI, 52%-80%) and 44% (95% CI, 30%-59%); and progressive disease, 12% (95% CI, 5%-27%) by both, respectively. There was a moderate concordance between response categories by RECIST and 68Ga-DOTATATE PET/CT, weighted of 0.51 (95% CI, 0.21-0.68).
Click to Show/Hide
|
||||
| Experiment 105 Reporting the Activity Data of This PDC | [19] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumour | ||||
| Efficacy Data | Progressive Disease (PD) |
13.00%
|
|||
| Patients Enrolled |
23 GEP neuroendocrine tumours patients.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | 7.4 GBq (200mCi) | ||||
| MOA of PDC |
From our initial experience, Lutathera has been well tolerated in patients with GEP-NET. Additional studies are needed to examine long-term clinical and patient-reported outcomes associated with treatment of GEP-NETs as well as financial considerations for hospitals embarking on a PRRT program. A multidisciplinary team and complete collaboration between hospital administration and clinical teams are required for successful implementation of a PRRT program.
Click to Show/Hide
|
||||
| Description |
PET/CT imaging were done to determine treatment responses 3 months post-PRRT treatment. Only patients who completed 4 cycles (n=23) were included. Five patients (21.7%) showed partial response, 10 patients (43.5 5%) showed stable disease, and 3 patients (13.0%) showed disease progression; the responses from 5 patients (21.7%) were unknown because no PET/CT scan was done.
Click to Show/Hide
|
||||
| Experiment 106 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Progressive Disease (PD) |
14%
|
|||
| Patients Enrolled |
395 patients with neuroendocrine tumour.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 107 Reporting the Activity Data of This PDC | [24] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Progressive Disease (PD) |
15%
|
|||
| Patients Enrolled |
45 patients with advanced inoperable/metastatic neuroendocrine tumor (22 women, 23 men).
|
||||
| Administration Time | 2 to 7 cycles | ||||
| Administration Dosage | 27.0 GBq | ||||
| Evaluation Method | 68Ga-DOTATATE PET/CT assay | ||||
| Description |
Radiological response with the CT images of 68Ga-DOTANOC PET/CECT was assessed 8 to 12 weeks after the last treatment cycle. Three patients were lost to follow-up, and 2 other patients had nonmeasurable lesions on CT. Thus, according to RECIST 1.1, 40 patients had evaluable lesions, of which 12 patients (30%) had PR and 22 patients (55%) had SD, whereas disease progression was limited to 6 patients (15%). No case of CR was observed in our study cohort.
Click to Show/Hide
|
||||
| Experiment 108 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Progressive Disease (PD) |
15.40%
|
|||
| Patients Enrolled |
26 patients with neuroendocrine tumour.
|
||||
| Administration Time | 2-5 cycles | ||||
| Administration Dosage | Median activity for re-treatment: 16.5 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 109 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Progressive Disease (PD) |
15.60%
|
|||
| Patients Enrolled |
35 patients with neuroendocrine tumour.
|
||||
| Administration Time | 1-4 cycles | ||||
| Administration Dosage | Median cumulative activity 44 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 110 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Progressive Disease (PD) |
19.60%
|
|||
| Patients Enrolled |
168 patients with neuroendocrine tumour.
|
||||
| Administration Time | 2 cycles | ||||
| Administration Dosage | Median cumulative dose: 44.7 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 111 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Progressive Disease (PD) |
22%
|
|||
| Patients Enrolled |
78 patients with pancreatic neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 112 Reporting the Activity Data of This PDC | [39] | ||||
| Indication | SSR positive lung neuroendocrine tumor | ||||
| Efficacy Data | Progressive Disease (PD) |
23.40%
|
|||
| Patients Enrolled |
Patients with somatostatin-expressing neuroendocrine tumors who underwent Lu-DOTATATE PRRT; with CRHE.
|
||||
| Description |
Following PRRT, 76.5% of patients with CRHE experienced benefit (partial response + stable disease), whereas 23.4% experienced progressive disease. Patients with CRHE showed more stable disease (P = 0.048) and less progressive disease (P = 0.046) following PRRT compared with no CRHE.
|
||||
| Experiment 113 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Progressive Disease (PD) |
25%
|
|||
| Patients Enrolled |
Patients with neuroendocrine tumour (Retreatment).
|
||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 114 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Bronchial neuroendocrine tumour | ||||
| Efficacy Data | Progressive Disease (PD) |
26%
|
|||
| Patients Enrolled |
21 patients with bronchial neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 115 Reporting the Activity Data of This PDC | [23] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Progressive Disease (PD) |
26.50%
|
|||
| Patients Enrolled |
Patients with advanced neuroendocrine tumor.
|
||||
| Description |
According to Response Evaluation Criteria in Solid Tumors 1.1 criteria, in group 1 patients, the response was partial response in 34% of the patients, stable disease in 50.2%, and progressive disease in 6.8% versus partial response in 6.3%, stable disease in 60.9%, and progressive disease in 26.5% among group 2 patients. The median overall survival (OS) and progression-free survival (PFS) was not reached in group 1 patients. The median OS and PFS in group 2 patients were 48 months.
Click to Show/Hide
|
||||
| Experiment 116 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Progressive Disease (PD) |
27.70%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Nineteen patients, all with progressive, heavily treated disease, were identified. Sites of tumor origin were: pancreas (74%), small bowel (11%), rectum (11%), and lung (5%); median Ki-67 was 32% (range 22-56). Thirteen patients (68%) completed all four 177Lu-DOTATATE cycles. Best response (N = 18 evaluable) was: 5/18 (28%) partial response, 8/18 (44%) stable disease, and 5/18 (28%) disease progression. Median PFS was 13.1 months (95% CI: 8.7-20.9).
Click to Show/Hide
|
||||
| Experiment 117 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | High-grade (WHO G3) neuroendocrine tumour | ||||
| Efficacy Data | Progressive Disease (PD) |
28.10%
|
|||
| Patients Enrolled |
Patients with neuroendocrine tumours including high-grade (WHO G3) neuroendocrine tumours.
|
||||
| Evaluation Method | RECIST criteria assay | ||||
| Description |
Among 160 patients whose responses to treatment could be evaluated according to the RECIST criteria, 28.1% (n=45) had a progressive disease, 21.9% (n=35) had a stable disease, 46.9% (n=75) had a partial response and 3.1% (n=5) had a complete response.
|
||||
| Experiment 118 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | PPGLa neuroendocrine tumor | ||||
| Efficacy Data | Progressive Disease (PD) |
30%
|
|||
| Patients Enrolled |
11 patients with PPGLa neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 119 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Progressive Disease (PD) |
33.30%
|
|||
| Patients Enrolled |
33 patients with neuroendocrine tumour.
|
||||
| Administration Time | 2-4 cycles | ||||
| Administration Dosage | Mean administered activity during re-treatment: 17.7 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 120 Reporting the Activity Data of This PDC | [16] | ||||
| Indication | Metastatic medullary thyroid carcinoma | ||||
| Efficacy Data | Progressive Disease (PD) |
33.33%
|
|||
| Patients Enrolled |
6 patients with medullary thyroid cancer (3 males and 3 females).
|
||||
| Administration Time | 3 cycles | ||||
| Administration Dosage | 5.7 GBq/cycle (range, 2.6-7.4 GBq) | ||||
| Evaluation Method | PET/CT assay | ||||
| MOA of PDC |
Radiolabeled somatostatin analogs are contributive for the management of recurrent MTC. 68Ga-DOTATAE PET-CT showed a relatively high detection rate in recurrent MTC. In addition, PRRT with 177Lu-DOTATATE was found to be a safe alternative therapeutic option for MTC.
|
||||
| Description |
Four of the 17 patients with positive SSTR-PET were scheduled for PRRT. In addition, 2 patients had positive 99mTc-octreotide scintigraphy results (Krenning score ≥ 2) and were scheduled for PRRT. Two of the 6 patients who underwent PRRT showed PR, 2 SD and 2 PD. Two patients died during the follow-up period. Median overall survival was 19 months (95% CI: 5.52-29.48). There were no cases of significant toxicity.
Click to Show/Hide
|
||||
| Experiment 121 Reporting the Activity Data of This PDC | [40] | ||||
| Indication | High-grade gliomas | ||||
| Efficacy Data | Progressive Disease (PD) |
37.50%
|
|||
| Patients Enrolled |
16 patients with high-grade gliomas (10 males and 6 females).
|
||||
| Administration Time | 1 to 4 cycles | ||||
| Administration Dosage | 3.7 to 29.6 GBq (median, 10.45 GBq) | ||||
| Evaluation Method | MRI assay | ||||
| Description |
In total, 16 subjects (10 males and 6 females) with a mean age of 55.68 ± 13.17 years (26-73 years) participated in the study. Of them, 8 patients were new HGG cases, and 8 patients had recurrent tumors. The participants' responses to treatments were complete remission in 12.5% of (n = 2), partial remission in 31.25% (n = 5), disease stability in 18.7% (n = 3), and disease progression in 37.5% (n = 6). In total, pretreatment and posttreatment Karnofsky Performance Score and Eastern Cooperative Oncology Group scores did not improved (P > 0.05). The patients were followed up from 1 month to 26 months (median, 3 months).
Click to Show/Hide
|
||||
| Experiment 122 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Progressive Disease (PD) |
51.50%
|
|||
| Patients Enrolled |
33 patients with neuroendocrine tumour.
|
||||
| Administration Time | 2 cycles | ||||
| Administration Dosage | 7.4 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 123 Reporting the Activity Data of This PDC | [28] | ||||
| Indication | Phaeochromocytoma/Paraganglioma | ||||
| Efficacy Data | Progressive Disease (PD) |
82%
|
|||
| Patients Enrolled |
34 patients with SSTr2-positive progressive locally advanced or metastatic PPGLs.
|
||||
| Administration Time | 5 cycles | ||||
| Administration Dosage | 18.5/27.5 GBq | ||||
| MOA of PDC |
In a long-term follow-up of patients with mPPGL, PRRT demonstrated a tolerability and efficacy comparable to those obtained in GEP-NETs. Our results highlight the need for an appropriate cumulative administered dosage to achieve a very prolonged result and underline the importance of specific symptoms and presence of bone lesions as predictive factors. Given the substantial number of patients evaluated over time, the superior mOS of 177Lu-DOTATATE with respect to 90Y-DOTATOC indicates the former radiopharmaceutical as the better candidate for new prospective studies of single or associated therapies.
Click to Show/Hide
|
||||
| Description |
Both 90Y-DOTATOC and 177Lu-DOTATATE PRRT were well tolerated by patients without significant renal or bone marrow toxicity. The median follow-up was 73 months (range 5-146 months). The overall disease control rate (DCR) was 80% [95% confidence interval (CI) 68.9% to 91.9%] with a mean five cycles of therapy. However, 177Lu-DOTATATE patients showed a longer median overall survival (mOS) than those receiving 90Y-Dotatoc and a better DCR when higher dosages were administered, even if a direct comparison was not carried out. Syndromic patients had a poorer mOS. SDHx mutations did not interfere with treatment efficacy.
Click to Show/Hide
|
||||
| Experiment 124 Reporting the Activity Data of This PDC | [41] | ||||
| Indication | Intracranial meningioma | ||||
| Efficacy Data | Progression-free survival at 6 months |
50%
|
|||
| Patients Enrolled |
14 patients with intracranial meningioma.
|
||||
| Administration Time | Every eight weeks for four cycles | ||||
| Administration Dosage | 7.4 GBq (200 mCi) per cycles | ||||
| Evaluation Method | 68Ga-DOTATATE PET-MRI assay | ||||
| MOA of PDC |
Treatment with 177Lu-DOTATATE was well tolerated. The predefined PFS-6 threshold was met in this interim analysis, thereby allowing this multicenter clinical trial to continue enrollment. 68Ga-DOTATATE PET may be a useful imaging biomarker to assess therapeutic outcome in patients with meningioma.
|
||||
| Description |
Fourteen patients (female = 11, male = 3) with progressive meningiomas (WHO 1 = 3, 2 = 10, 3 = 1) were enrolled. Median age was 63.1 (range 49.7-78) years. All patients previously underwent tumor resection and at least one course of radiation. Treatment with 177Lu-DOTATATE was well tolerated. Seven patients (50%) achieved PFS-6. Best radiographic response by modified Macdonald criteria was stable disease (SD) in all seven patients. A >25% reduction in 68Ga-DOTATATE uptake (PET) was observed in five meningiomas and two patients. In one lesion, this corresponded to >50% reduction in bidirectional tumor measurements (MRI).
Click to Show/Hide
|
||||
| Experiment 125 Reporting the Activity Data of This PDC | [42], [43] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Progression-free survival (PFS) |
65.20%
|
|||
| Patients Enrolled |
29 patients who had well-differentiated, metastatic midgut neuroendocrine tumor.
|
||||
| Administration Time | Every 8 weeks for a total of 4 cycles | ||||
| MOA of PDC |
177Lu-DOTATATE is the first FDA-approved PRRT and utilizes a somatostatin analogue (DOTATATE) covalently bound to the beta-minus emitting radioisotope 177Lu in order to provide targeted radiation directly to NET cells overexpressing SSTRs (primarily SSTR2).
|
||||
| Description |
NETTER-1 demonstrated that treatment with 177Lu-DOTATATE and octreotide resulted in a progression free survival (PFS) rate of 65.2% vs. 10.8% in the high-dose octreotide group.
|
||||
| Related Clinical Trial | |||||
| NCT Number | NCT01578239 | Clinical Status | Phase 3 | ||
| Clinical Description | This was a multicenter, stratified, open, randomized, comparator-controlled, parallel-group phase III study comparing treatment with Lutathera plus best supportive care (30 mg Octreotide LAR) to treatment with high dose (60 mg) Octreotide LAR in participants with metastasized or locally advanced, inoperable, somatostatin receptor positive, histologically proven midgut carcinoid tumours with progression despite LAR treatment. | ||||
| Experiment 126 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Progression-free survival (PFS) |
10-91 months
|
|||
| Patients Enrolled |
4 patients with paraganglioma.
|
||||
| Administration Time | 1-10 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The use of PRRT for patients with PPGLs who had high tumor SSTR expression has mainly been reported in retrospective small case series and the protocols varied in all these studies. So far, four meta-analyses (Taieb et al. and Satapathy et al. in 2019 as well as Su et al. and Marretta et al. in 2023) based on PRRT (177Lu/90Y-DOTAT-TATE/TOC) therapy have been reported. The reported number of pooled patients ranged from 179-330 without any complete response. The reported partial response ranged from 20-25% and stable disease from 59-70% giving an objective response rate range of 20-25% and disease control rate range of 8190%. The survival rates were reported by only 3 meta-analyses. The reported progression-free survival were 17-39 months in seven studies by Taieb et al., 10-91 months in 4 studies and a mean of 37.1 months in 2 studies by Satapathy et al., and a median of 31.8 months in 13 studies by Su et al. whereas overall survival were 49.6-68.0 months in 3 studies by Taieb et al., a mean of 54.5 months in 4 studies by Satapathy et al. and a median of 74.3 months in 11 studies by Su et al. Su et al. reports a pooled adverse events data in 274/330 patients which mentions a grade 3/4 hematotoxicity in 4.3% (grade 1-4, 22.3%) and grade 3/4 nephrotoxicity in 1.9% (grade 1-4, 1.9%), and myelodysplastic syndrome in 1 patient. Treatment discontinuation occurred in 4 patients due to thrombocytopenia (n=3) or nephrotoxicity (n=1).
Click to Show/Hide
|
||||
| Experiment 127 Reporting the Activity Data of This PDC | [45] | ||||
| Indication | Primary cardiac paragangliomas | ||||
| Efficacy Data | Progression-free survival (PFS) |
13 months
|
|||
| Patients Enrolled |
47 years old primary cardiac paragangliomas patient.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity of 40.7 GBq (1100 mCi) | ||||
| MOA of PDC |
Cardiac PGL is an exceedingly rare tumor and may lead to hemodynamic compromise. Surgical resection, if indicated, is technically challenging and frequently not feasible. This case demonstrates that personalized 177Lu-DOTATATE PRRT can be considered as a safe and effective palliative treatment for unresectable MIBG negative tumor which can substantially improve quality of life.
Click to Show/Hide
|
||||
| Description |
The patient was treated with personalized 177Lu-DOTATATE PRRT and showed complete symptomatic and partial anatomical responses, with a progression-free survival of 13 months.
|
||||
| Experiment 128 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Progression-free survival (PFS) |
15.4 months
|
|||
| Patients Enrolled |
18 patients with paraganglioma.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The efficacy and safety data of the interim analysis of an open-label, single-arm phase 2 study evaluating 177Lu-DOTATATE in paraganglioma patients conducted at the National Institutes of Health (NCT03206060) in 36 patients [(n=18, apparently sporadic cohort and n=18 in patients having germline variant in one of the genes encoding subunits of succinate dehydrogenase complex (SDHA=2, SDHB=15, SDHD=1)), SDHx cohort] after 177LuDOTATATE therapy with 7.4 GBq every 8 weeks for 4 cycles. The progression-free survival at 6 months since initiation of 177Lu-DOTATATE was 19.1 months (22.7 months in apparently sporadic cohort and 15.4 months in SDHx cohort) and overall survival was not reached in both cohorts (80). Per RECIST 1.1, overall, 86.1% patients showed partial response (13.9%) or stable disease (72.2%). In sporadic cohort; partial response was observed in 11.1% and stable disease in 88.9% patients whereas those in SDHx cohort were, 16.7% and 55.5%, respectively at the end of 6 months.
Click to Show/Hide
|
||||
| Experiment 129 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Progression-free survival (PFS) |
17-39 months
|
|||
| Patients Enrolled |
179 patients with paraganglioma.
|
||||
| Administration Time | 1-11 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The use of PRRT for patients with PPGLs who had high tumor SSTR expression has mainly been reported in retrospective small case series and the protocols varied in all these studies. So far, four meta-analyses (Taieb et al. and Satapathy et al. in 2019 as well as Su et al. and Marretta et al. in 2023) based on PRRT (177Lu/90Y-DOTAT-TATE/TOC) therapy have been reported. The reported number of pooled patients ranged from 179-330 without any complete response. The reported partial response ranged from 20-25% and stable disease from 59-70% giving an objective response rate range of 20-25% and disease control rate range of 8190%. The survival rates were reported by only 3 meta-analyses. The reported progression-free survival were 17-39 months in seven studies by Taieb et al., 10-91 months in 4 studies and a mean of 37.1 months in 2 studies by Satapathy et al., and a median of 31.8 months in 13 studies by Su et al. whereas overall survival were 49.6-68.0 months in 3 studies by Taieb et al., a mean of 54.5 months in 4 studies by Satapathy et al. and a median of 74.3 months in 11 studies by Su et al. Su et al. reports a pooled adverse events data in 274/330 patients which mentions a grade 3/4 hematotoxicity in 4.3% (grade 1-4, 22.3%) and grade 3/4 nephrotoxicity in 1.9% (grade 1-4, 1.9%), and myelodysplastic syndrome in 1 patient. Treatment discontinuation occurred in 4 patients due to thrombocytopenia (n=3) or nephrotoxicity (n=1).
Click to Show/Hide
|
||||
| Experiment 130 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Progression-free survival (PFS) |
19.1 months
|
|||
| Patients Enrolled |
36 patients with paraganglioma.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The efficacy and safety data of the interim analysis of an open-label, single-arm phase 2 study evaluating 177Lu-DOTATATE in paraganglioma patients conducted at the National Institutes of Health (NCT03206060) in 36 patients [(n=18, apparently sporadic cohort and n=18 in patients having germline variant in one of the genes encoding subunits of succinate dehydrogenase complex (SDHA=2, SDHB=15, SDHD=1)), SDHx cohort] after 177LuDOTATATE therapy with 7.4 GBq every 8 weeks for 4 cycles. The progression-free survival at 6 months since initiation of 177Lu-DOTATATE was 19.1 months (22.7 months in apparently sporadic cohort and 15.4 months in SDHx cohort) and overall survival was not reached in both cohorts (80). Per RECIST 1.1, overall, 86.1% patients showed partial response (13.9%) or stable disease (72.2%). In sporadic cohort; partial response was observed in 11.1% and stable disease in 88.9% patients whereas those in SDHx cohort were, 16.7% and 55.5%, respectively at the end of 6 months.
Click to Show/Hide
|
||||
| Experiment 131 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Progression-free survival (PFS) |
22.7 months
|
|||
| Patients Enrolled |
18 patients with paraganglioma.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The efficacy and safety data of the interim analysis of an open-label, single-arm phase 2 study evaluating 177Lu-DOTATATE in paraganglioma patients conducted at the National Institutes of Health (NCT03206060) in 36 patients [(n=18, apparently sporadic cohort and n=18 in patients having germline variant in one of the genes encoding subunits of succinate dehydrogenase complex (SDHA=2, SDHB=15, SDHD=1)), SDHx cohort] after 177LuDOTATATE therapy with 7.4 GBq every 8 weeks for 4 cycles. The progression-free survival at 6 months since initiation of 177Lu-DOTATATE was 19.1 months (22.7 months in apparently sporadic cohort and 15.4 months in SDHx cohort) and overall survival was not reached in both cohorts (80). Per RECIST 1.1, overall, 86.1% patients showed partial response (13.9%) or stable disease (72.2%). In sporadic cohort; partial response was observed in 11.1% and stable disease in 88.9% patients whereas those in SDHx cohort were, 16.7% and 55.5%, respectively at the end of 6 months.
Click to Show/Hide
|
||||
| Experiment 132 Reporting the Activity Data of This PDC | [46] | ||||
| Indication | Pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Progression-free survival (PFS) |
24 months
|
|||
| Patients Enrolled |
Patients with advanced Pan-neuroendocrine tumour, previously pretreated with one (67%) or several (33%) lines of chemotherapy.
|
||||
| Administration Time | 1-10 cycles with 6- to 8-week intervals | ||||
| Administration Dosage | 7.4 GBq | ||||
| Description |
The median follow-up was 34 months, the median PFS was 24 months, and the median OS was 42 months from start of PRRT.
|
||||
| Experiment 133 Reporting the Activity Data of This PDC | [18] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumor | ||||
| Efficacy Data | Progression-free survival (PFS) |
29 months
|
|||
| Patients Enrolled |
Patients treated with Lu-DOTATATE.
|
||||
| Administration Dosage | 100 mCi (3.7 GBq) | ||||
| Description |
The objective response rate of the total group of patients was 39%. Stable disease was reached in 43% of patients. Progression-free survival (PFS) and overall survival (OS) for all NET patients were 29 months [95% confidence interval (CI), 26-33 months] and 63 months (95% CI, 55-72 months). Long-term toxicity included acute leukemia in four patients (0.7%) and myelodysplastic syndrome in nine patients (1.5%).
Click to Show/Hide
|
||||
| Experiment 134 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Progression-free survival (PFS) |
31.8 months
|
|||
| Patients Enrolled |
330 patients with paraganglioma.
|
||||
| Administration Time | 1-10 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The use of PRRT for patients with PPGLs who had high tumor SSTR expression has mainly been reported in retrospective small case series and the protocols varied in all these studies. So far, four meta-analyses (Taieb et al. and Satapathy et al. in 2019 as well as Su et al. and Marretta et al. in 2023) based on PRRT (177Lu/90Y-DOTAT-TATE/TOC) therapy have been reported. The reported number of pooled patients ranged from 179-330 without any complete response. The reported partial response ranged from 20-25% and stable disease from 59-70% giving an objective response rate range of 20-25% and disease control rate range of 8190%. The survival rates were reported by only 3 meta-analyses. The reported progression-free survival were 17-39 months in seven studies by Taieb et al., 10-91 months in 4 studies and a mean of 37.1 months in 2 studies by Satapathy et al., and a median of 31.8 months in 13 studies by Su et al. whereas overall survival were 49.6-68.0 months in 3 studies by Taieb et al., a mean of 54.5 months in 4 studies by Satapathy et al. and a median of 74.3 months in 11 studies by Su et al. Su et al. reports a pooled adverse events data in 274/330 patients which mentions a grade 3/4 hematotoxicity in 4.3% (grade 1-4, 22.3%) and grade 3/4 nephrotoxicity in 1.9% (grade 1-4, 1.9%), and myelodysplastic syndrome in 1 patient. Treatment discontinuation occurred in 4 patients due to thrombocytopenia (n=3) or nephrotoxicity (n=1).
Click to Show/Hide
|
||||
| Experiment 135 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Progression-free survival (PFS) |
37.1 months
|
|||
| Patients Enrolled |
2 patients with paraganglioma.
|
||||
| Administration Time | 1-10 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The use of PRRT for patients with PPGLs who had high tumor SSTR expression has mainly been reported in retrospective small case series and the protocols varied in all these studies. So far, four meta-analyses (Taieb et al. and Satapathy et al. in 2019 as well as Su et al. and Marretta et al. in 2023) based on PRRT (177Lu/90Y-DOTAT-TATE/TOC) therapy have been reported. The reported number of pooled patients ranged from 179-330 without any complete response. The reported partial response ranged from 20-25% and stable disease from 59-70% giving an objective response rate range of 20-25% and disease control rate range of 8190%. The survival rates were reported by only 3 meta-analyses. The reported progression-free survival were 17-39 months in seven studies by Taieb et al., 10-91 months in 4 studies and a mean of 37.1 months in 2 studies by Satapathy et al., and a median of 31.8 months in 13 studies by Su et al. whereas overall survival were 49.6-68.0 months in 3 studies by Taieb et al., a mean of 54.5 months in 4 studies by Satapathy et al. and a median of 74.3 months in 11 studies by Su et al. Su et al. reports a pooled adverse events data in 274/330 patients which mentions a grade 3/4 hematotoxicity in 4.3% (grade 1-4, 22.3%) and grade 3/4 nephrotoxicity in 1.9% (grade 1-4, 1.9%), and myelodysplastic syndrome in 1 patient. Treatment discontinuation occurred in 4 patients due to thrombocytopenia (n=3) or nephrotoxicity (n=1).
Click to Show/Hide
|
||||
| Experiment 136 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Progression-free survival (PFS) |
38 months
|
|||
| Administration Time | Four doses every eight weeks | ||||
| Administration Dosage | 7.4 GBq (200 mCi) | ||||
| Description |
We present a 26-year-old man who started with pelvic pain and after a biopsy of a retro-rectal mass observed in a magnetic resonance was diagnosed with an advanced neuroendocrine tumour. After progression to lanreotide, everolimus and sunitinib, treatment with 177Lu-DOTATATE was initiated, achieving an excellent response with a progression free survival (PFS) of 38 months.
Click to Show/Hide
|
||||
| In Vivo Model | 26-year-old man diagnosed with an advanced neuroendocrine tumour. | ||||
| Experiment 137 Reporting the Activity Data of This PDC | [23] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Progression-free survival (PFS) |
48 months
|
|||
| Patients Enrolled |
Patients with advanced neuroendocrine tumor.
|
||||
| Description |
According to Response Evaluation Criteria in Solid Tumors 1.1 criteria, in group 1 patients, the response was partial response in 34% of the patients, stable disease in 50.2%, and progressive disease in 6.8% versus partial response in 6.3%, stable disease in 60.9%, and progressive disease in 26.5% among group 2 patients. The median overall survival (OS) and progression-free survival (PFS) was not reached in group 1 patients. The median OS and PFS in group 2 patients were 48 months.
Click to Show/Hide
|
||||
| Experiment 138 Reporting the Activity Data of This PDC | [27] | ||||
| Indication | Metastatic neuroendocrine tumor | ||||
| Efficacy Data | Progressed through treatment |
23%
|
|||
| Patients Enrolled |
Patients treated with Lu-DOTATATE.
|
||||
| Description |
At end-of-treatment radiological restaging, 13% of patients were found to have partial response and 64% to have stable disease; 23% of patients progressed through treatment.
|
||||
| Experiment 139 Reporting the Activity Data of This PDC | [45] | ||||
| Indication | Primary cardiac paragangliomas | ||||
| Efficacy Data | Platelets decrease rate |
137 x 109/L
|
|||
| Patients Enrolled |
47 years old primary cardiac paragangliomas patient.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity of 40.7 GBq (1100 mCi) | ||||
| MOA of PDC |
Cardiac PGL is an exceedingly rare tumor and may lead to hemodynamic compromise. Surgical resection, if indicated, is technically challenging and frequently not feasible. This case demonstrates that personalized 177Lu-DOTATATE PRRT can be considered as a safe and effective palliative treatment for unresectable MIBG negative tumor which can substantially improve quality of life.
Click to Show/Hide
|
||||
| Description |
At the end of the 4 cycles, fatigue, chest pain and dyspnea resolved, and episodes of tachyarrhythmia disappeared. The dosage of metoprolol and prazocin was considerably reduced, decreasing respectively from 125 mg to 50 mg and 2 mg to 1 mg TID. The chromogranin A decreased from 585 to 205 ng/mL and the serum norepinephrine decreased from 104 to 37 nmol/L after 4 cycles.
Click to Show/Hide
|
||||
| Experiment 140 Reporting the Activity Data of This PDC | [48] | ||||
| Indication | Functioning pancreatic neuroendocrine tumour | ||||
| Efficacy Data | Partial response (PR)/complete response (CP) |
59%
|
|||
| Patients Enrolled |
Patients with functioning pancreatic neuroendocrine tumors.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | 7.4 GBq/cycle | ||||
| Description |
Subacute hematological toxicity, grade 3 or 4 occurred in 4 patients (12%) and a hormonal crisis in 3 patients (9%). PRRT resulted in partial or complete response in 59% of patients and the disease control rate was 78% in patients with baseline progression. 71% of patients with uncontrolled symptoms had a reduction of symptoms and a more than 80% decrease of circulating hormone levels was measured during follow-up. After PRRT, median progression-free survival was 18.1 months (interquartile range: 3.3 to 35.7) with a concurrent increase in QOL.
Click to Show/Hide
|
||||
| Experiment 141 Reporting the Activity Data of This PDC | [39] | ||||
| Indication | SSR positive lung neuroendocrine tumor | ||||
| Efficacy Data | Partial response (PR) + stable disease (SD) |
76.50%
|
|||
| Patients Enrolled |
Patients with somatostatin-expressing neuroendocrine tumors who underwent Lu-DOTATATE PRRT; with CRHE.
|
||||
| Description |
Following PRRT, 76.5% of patients with CRHE experienced benefit (partial response + stable disease), whereas 23.4% experienced progressive disease. Patients with CRHE showed more stable disease (P = 0.048) and less progressive disease (P = 0.046) following PRRT compared with no CRHE.
|
||||
| Experiment 142 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | PPGLa neuroendocrine tumor | ||||
| Efficacy Data | Partial response (PR) |
0%
|
|||
| Patients Enrolled |
11 patients with PPGLa neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 143 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Partial response (PR) |
3.10%
|
|||
| Patients Enrolled |
35 patients with neuroendocrine tumour.
|
||||
| Administration Time | 1-4 cycles | ||||
| Administration Dosage | Median cumulative activity 44 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 144 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Partial response (PR) |
3.80%
|
|||
| Patients Enrolled |
26 patients with neuroendocrine tumour.
|
||||
| Administration Time | 2-5 cycles | ||||
| Administration Dosage | Median activity for re-treatment: 16.5 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 145 Reporting the Activity Data of This PDC | [26] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumor | ||||
| Efficacy Data | Partial response (PR) |
6%
|
|||
| Patients Enrolled |
Patients with advanced GEP-neuroendocrine tumour.
|
||||
| Administration Time | Four or five cycles | ||||
| Administration Dosage | 18.5 GBq/27.8 GBq | ||||
| Description |
Two patients (6%) achieved a partial response and 21 (64%) showed stable disease, giving a disease control rate (DCR) of 70%.
|
||||
| Experiment 146 Reporting the Activity Data of This PDC | [23] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Partial response (PR) |
6.30%
|
|||
| Patients Enrolled |
Patients with advanced neuroendocrine tumor.
|
||||
| Description |
According to Response Evaluation Criteria in Solid Tumors 1.1 criteria, in group 1 patients, the response was partial response in 34% of the patients, stable disease in 50.2%, and progressive disease in 6.8% versus partial response in 6.3%, stable disease in 60.9%, and progressive disease in 26.5% among group 2 patients. The median overall survival (OS) and progression-free survival (PFS) was not reached in group 1 patients. The median OS and PFS in group 2 patients were 48 months.
Click to Show/Hide
|
||||
| Experiment 147 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Partial response (PR) |
6.67%
|
|||
| Patients Enrolled |
15 patients with pheocromocytomas (PCCs) and paragangliomas (PGLs) (5 PCC , 10PGL).
|
||||
| Administration Time | 4.13 cycles | ||||
| MOA of PDC |
We showed that PRRT based on 177Lu-DOTATATE and 90-YDOTATOC without radiosensitizers are efficacious therapeutic options (DCR of 83% and 76%, respectively) and can be considered in the multidisciplinary treatment of PCCs and PGLs as alternatives to I-131 MIBG and chemotherapy.
|
||||
| Description |
There was no statistically significant heterogeneity among the 177Lu- and 90Y- PRRTs studies (I2 value = 0%, p = 0.68 and I2 value = 0%, p = 0.91, respectively). The pooled DCRs were 0.83 (95% CI: 0.75-0.88) and 0.76 (95% CI: 0.56-0.89) for 177Lu- and 90Y-PRRT treatments, respectively. The pooled DCR for PRRT agents was 0.81 (95% CI: 0.74-0.87). Although the present study is the largest description of DCR in PRRT therapy of PCCs and PGLs, given the small sample size, no formal attempts were made to compare efficacy between 177Lu- and 90Y-PRRT treatments. In detail, we report pooled data on 149 177Lu- and 64 90Y-treated patients (total: 213 patients). Most studies were retrospective. The largest one included 46 patients (34 treated with 177Lu-, 12 with 90Y- PRRTs). Median ages ranged from 32.5 to 60.4 years. The female gender was slightly predominant (83 women, 74 men, gender not specified in 3 studies). When reported, mutations of SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B) were the most frequent genetic alterations. According to the predominant retrospective and real practice nature of the selected studies, sites of metastases and previous treatments were heterogeneous, with bone the metastatic site most often involved, and surgery the most frequent previous therapy. Paramount descriptive information on PRRT doses per cycle, number of cycles, response criteria, best responses to treatments, and frequency of G3/G4 hematologic toxicities is reported in Table 2. The lowest applied dose was 3.4 GBq per cycle; the number of administered cycles was heterogeneous (at least 3 cycles in 7 studies). The most used response criteria were the RECIST 1.1. Only one study evaluated the response with SWOG criteria, which are more conservative with regard to the definition of PR (50% or greater reduction in the sum of the product of the perpendicular diameters of target lesions). The majority of the patients experienced PR or SD. Hematologic toxicity was manageable. Funnel plot of selected studies did not show publication bias. The MINORS and Newcastle-Ottawa Scale scoring for the selected articles are reported in Table 3.
Click to Show/Hide
|
||||
| Experiment 148 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Partial response (PR) |
8.82%
|
|||
| Patients Enrolled |
34 patients with pheocromocytomas and paragangliomas.
|
||||
| Administration Time | 5 cycles | ||||
| Administration Dosage | 3.7-5.5 GBq/cycle | ||||
| MOA of PDC |
We showed that PRRT based on 177Lu-DOTATATE and 90-YDOTATOC without radiosensitizers are efficacious therapeutic options (DCR of 83% and 76%, respectively) and can be considered in the multidisciplinary treatment of PCCs and PGLs as alternatives to I-131 MIBG and chemotherapy.
|
||||
| Description |
There was no statistically significant heterogeneity among the 177Lu- and 90Y- PRRTs studies (I2 value = 0%, p = 0.68 and I2 value = 0%, p = 0.91, respectively). The pooled DCRs were 0.83 (95% CI: 0.75-0.88) and 0.76 (95% CI: 0.56-0.89) for 177Lu- and 90Y-PRRT treatments, respectively. The pooled DCR for PRRT agents was 0.81 (95% CI: 0.74-0.87). Although the present study is the largest description of DCR in PRRT therapy of PCCs and PGLs, given the small sample size, no formal attempts were made to compare efficacy between 177Lu- and 90Y-PRRT treatments. In detail, we report pooled data on 149 177Lu- and 64 90Y-treated patients (total: 213 patients). Most studies were retrospective. The largest one included 46 patients (34 treated with 177Lu-, 12 with 90Y- PRRTs). Median ages ranged from 32.5 to 60.4 years. The female gender was slightly predominant (83 women, 74 men, gender not specified in 3 studies). When reported, mutations of SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B) were the most frequent genetic alterations. According to the predominant retrospective and real practice nature of the selected studies, sites of metastases and previous treatments were heterogeneous, with bone the metastatic site most often involved, and surgery the most frequent previous therapy. Paramount descriptive information on PRRT doses per cycle, number of cycles, response criteria, best responses to treatments, and frequency of G3/G4 hematologic toxicities is reported in Table 2. The lowest applied dose was 3.4 GBq per cycle; the number of administered cycles was heterogeneous (at least 3 cycles in 7 studies). The most used response criteria were the RECIST 1.1. Only one study evaluated the response with SWOG criteria, which are more conservative with regard to the definition of PR (50% or greater reduction in the sum of the product of the perpendicular diameters of target lesions). The majority of the patients experienced PR or SD. Hematologic toxicity was manageable. Funnel plot of selected studies did not show publication bias. The MINORS and Newcastle-Ottawa Scale scoring for the selected articles are reported in Table 3.
Click to Show/Hide
|
||||
| Experiment 149 Reporting the Activity Data of This PDC | [28] | ||||
| Indication | Phaeochromocytoma/Paraganglioma | ||||
| Efficacy Data | Partial response (PR) |
8.90%
|
|||
| Patients Enrolled |
34 patients with SSTr2-positive progressive locally advanced or metastatic PPGLs.
|
||||
| Administration Time | 5 cycles | ||||
| Administration Dosage | 18.5/27.5 GBq | ||||
| MOA of PDC |
In a long-term follow-up of patients with mPPGL, PRRT demonstrated a tolerability and efficacy comparable to those obtained in GEP-NETs. Our results highlight the need for an appropriate cumulative administered dosage to achieve a very prolonged result and underline the importance of specific symptoms and presence of bone lesions as predictive factors. Given the substantial number of patients evaluated over time, the superior mOS of 177Lu-DOTATATE with respect to 90Y-DOTATOC indicates the former radiopharmaceutical as the better candidate for new prospective studies of single or associated therapies.
Click to Show/Hide
|
||||
| Description |
Both 90Y-DOTATOC and 177Lu-DOTATATE PRRT were well tolerated by patients without significant renal or bone marrow toxicity. The median follow-up was 73 months (range 5-146 months). The overall disease control rate (DCR) was 80% [95% confidence interval (CI) 68.9% to 91.9%] with a mean five cycles of therapy. However, 177Lu-DOTATATE patients showed a longer median overall survival (mOS) than those receiving 90Y-Dotatoc and a better DCR when higher dosages were administered, even if a direct comparison was not carried out. Syndromic patients had a poorer mOS. SDHx mutations did not interfere with treatment efficacy.
Click to Show/Hide
|
||||
| Experiment 150 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Partial response (PR) |
9.09%
|
|||
| Patients Enrolled |
22 patients with pheocromocytomas (PCCs) and paragangliomas (PGLs) (9 PCC , 13PGL).
|
||||
| Administration Time | 4.9 cycles | ||||
| Administration Dosage | 7.4 GBq/cycle | ||||
| MOA of PDC |
We showed that PRRT based on 177Lu-DOTATATE and 90-YDOTATOC without radiosensitizers are efficacious therapeutic options (DCR of 83% and 76%, respectively) and can be considered in the multidisciplinary treatment of PCCs and PGLs as alternatives to I-131 MIBG and chemotherapy.
|
||||
| Description |
There was no statistically significant heterogeneity among the 177Lu- and 90Y- PRRTs studies (I2 value = 0%, p = 0.68 and I2 value = 0%, p = 0.91, respectively). The pooled DCRs were 0.83 (95% CI: 0.75-0.88) and 0.76 (95% CI: 0.56-0.89) for 177Lu- and 90Y-PRRT treatments, respectively. The pooled DCR for PRRT agents was 0.81 (95% CI: 0.74-0.87). Although the present study is the largest description of DCR in PRRT therapy of PCCs and PGLs, given the small sample size, no formal attempts were made to compare efficacy between 177Lu- and 90Y-PRRT treatments. In detail, we report pooled data on 149 177Lu- and 64 90Y-treated patients (total: 213 patients). Most studies were retrospective. The largest one included 46 patients (34 treated with 177Lu-, 12 with 90Y- PRRTs). Median ages ranged from 32.5 to 60.4 years. The female gender was slightly predominant (83 women, 74 men, gender not specified in 3 studies). When reported, mutations of SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B) were the most frequent genetic alterations. According to the predominant retrospective and real practice nature of the selected studies, sites of metastases and previous treatments were heterogeneous, with bone the metastatic site most often involved, and surgery the most frequent previous therapy. Paramount descriptive information on PRRT doses per cycle, number of cycles, response criteria, best responses to treatments, and frequency of G3/G4 hematologic toxicities is reported in Table 2. The lowest applied dose was 3.4 GBq per cycle; the number of administered cycles was heterogeneous (at least 3 cycles in 7 studies). The most used response criteria were the RECIST 1.1. Only one study evaluated the response with SWOG criteria, which are more conservative with regard to the definition of PR (50% or greater reduction in the sum of the product of the perpendicular diameters of target lesions). The majority of the patients experienced PR or SD. Hematologic toxicity was manageable. Funnel plot of selected studies did not show publication bias. The MINORS and Newcastle-Ottawa Scale scoring for the selected articles are reported in Table 3.
Click to Show/Hide
|
||||
| Experiment 151 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Bronchial neuroendocrine tumour | ||||
| Efficacy Data | Partial response (PR) |
11%
|
|||
| Patients Enrolled |
21 patients with bronchial neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 152 Reporting the Activity Data of This PDC | [27] | ||||
| Indication | Metastatic neuroendocrine tumor | ||||
| Efficacy Data | Partial response (PR) |
13%
|
|||
| Patients Enrolled |
Patients treated with Lu-DOTATATE.
|
||||
| Description |
At end-of-treatment radiological restaging, 13% of patients were found to have partial response and 64% to have stable disease; 23% of patients progressed through treatment.
|
||||
| Experiment 153 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Midgut neuroendocrine tumor | ||||
| Efficacy Data | Partial response (PR) |
13%
|
|||
| Patients Enrolled |
229 patients with midgut neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 154 Reporting the Activity Data of This PDC | [17] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Partial response (PR) |
14.30%
|
|||
| Patients Enrolled |
7 patients with inoperable, progressive, metastatic, or locally advanced, somatostatin receptor-positive neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | 21.3-30.1 GBq total dose | ||||
| MOA of PDC |
In conclusion, for the first time in Korea, we have conducted a clinical trial of PRRT with SNU-KB-01, a NCA 177Lu-DOTATATE, in patients with SSTR-positive NET. Treatment with SNU-KB-01 was safe and resulted in control of disease in most of the patients with high SSTR expression. The recommended dose per cycle of SNU-KB-01 was determined to be 7.40 GBq for the phase 2 trial. Our results indicate SNU-KB-01 as a potentially safe and efficacious treatment option for NET patients and are expected to provide a wider choice of treatment opportunities for patients with various types of SSTR-positive tumors in Korea.
Click to Show/Hide
|
||||
| Description |
Among seven patients, the best objective response to SNU-KB-01 was the PR observed in three patients (42.9%). No CR was observed. Three patients (42.9%) had SD and one patient (14.3%) had PD. The ORR (CR+PR) was 42.9%, and the DCR (CR+PR+SD) was 85.7%.
|
||||
| Experiment 155 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Partial response (PR) |
15.50%
|
|||
| Patients Enrolled |
168 patients with neuroendocrine tumour.
|
||||
| Administration Time | 2 cycles | ||||
| Administration Dosage | Median cumulative dose: 44.7 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 156 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Partial response (PR) |
15.90%
|
|||
| Patients Enrolled |
Patients with neuroendocrine tumour (Retreatment).
|
||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 157 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Partial response (PR) |
16.67%
|
|||
| Patients Enrolled |
12 patients with pheocromocytomas (PCCs) and paragangliomas (PGLs) (1 PCC, 11PGL).
|
||||
| Administration Dosage | 7.4 GBq/cycle | ||||
| MOA of PDC |
We showed that PRRT based on 177Lu-DOTATATE and 90-YDOTATOC without radiosensitizers are efficacious therapeutic options (DCR of 83% and 76%, respectively) and can be considered in the multidisciplinary treatment of PCCs and PGLs as alternatives to I-131 MIBG and chemotherapy.
|
||||
| Description |
There was no statistically significant heterogeneity among the 177Lu- and 90Y- PRRTs studies (I2 value = 0%, p = 0.68 and I2 value = 0%, p = 0.91, respectively). The pooled DCRs were 0.83 (95% CI: 0.75-0.88) and 0.76 (95% CI: 0.56-0.89) for 177Lu- and 90Y-PRRT treatments, respectively. The pooled DCR for PRRT agents was 0.81 (95% CI: 0.74-0.87). Although the present study is the largest description of DCR in PRRT therapy of PCCs and PGLs, given the small sample size, no formal attempts were made to compare efficacy between 177Lu- and 90Y-PRRT treatments. In detail, we report pooled data on 149 177Lu- and 64 90Y-treated patients (total: 213 patients). Most studies were retrospective. The largest one included 46 patients (34 treated with 177Lu-, 12 with 90Y- PRRTs). Median ages ranged from 32.5 to 60.4 years. The female gender was slightly predominant (83 women, 74 men, gender not specified in 3 studies). When reported, mutations of SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B) were the most frequent genetic alterations. According to the predominant retrospective and real practice nature of the selected studies, sites of metastases and previous treatments were heterogeneous, with bone the metastatic site most often involved, and surgery the most frequent previous therapy. Paramount descriptive information on PRRT doses per cycle, number of cycles, response criteria, best responses to treatments, and frequency of G3/G4 hematologic toxicities is reported in Table 2. The lowest applied dose was 3.4 GBq per cycle; the number of administered cycles was heterogeneous (at least 3 cycles in 7 studies). The most used response criteria were the RECIST 1.1. Only one study evaluated the response with SWOG criteria, which are more conservative with regard to the definition of PR (50% or greater reduction in the sum of the product of the perpendicular diameters of target lesions). The majority of the patients experienced PR or SD. Hematologic toxicity was manageable. Funnel plot of selected studies did not show publication bias. The MINORS and Newcastle-Ottawa Scale scoring for the selected articles are reported in Table 3.
Click to Show/Hide
|
||||
| Experiment 158 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Partial response (PR) |
18.20%
|
|||
| Patients Enrolled |
33 patients with neuroendocrine tumour.
|
||||
| Administration Time | 2 cycles | ||||
| Administration Dosage | 7.4 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 159 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Partial response (PR) |
18.20%
|
|||
| Patients Enrolled |
33 patients with neuroendocrine tumour.
|
||||
| Administration Time | 2-4 cycles | ||||
| Administration Dosage | Mean administered activity during re-treatment: 17.7 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 160 Reporting the Activity Data of This PDC | [20] | ||||
| Indication | SSR positive lung neuroendocrine tumor | ||||
| Efficacy Data | Partial response (PR) |
20%
|
|||
| Patients Enrolled |
40 patients with neuroendocrine tumour with RECIST-measurable disease.
|
||||
| Administration Time | 1-4 cycles | ||||
| Administration Dosage | Median cumulative activity of 27 GBq (range, 6-43 GBq) | ||||
| MOA of PDC |
In patients with advanced progressive lung NET and satisfactory SSR expression, 177Lu-DOTATATE is effective and safe, with a high DCR and encouraging PFS and OS. Further prospective studies comparing 177Lu-DOTATATE with other systemic options are warranted.
|
||||
| Description |
Of 48 patients (median age, 63 y; 13 women), 43 (90%) had AC and 5 (10%) TC. Almost all patients (47, 98%) were treated due to progression. Most patients (40, 83%) received somatostatin analogs, and 10 patients (20%) had prior everolimus, chemotherapy, or both. All patients had high SSR expression (≥ modified Krenning score 3) on pretreatment 68Ga-DOTATATE PET/CT. Patients received a median 4 (range, 1-4) cycles of 177Lu-DOTATATE (33% with concurrent radiosensitizing chemotherapy) to a median cumulative activity of 27 GBq (range, 6-43GBq). At a median follow-up of 42 mo, the median PFS and OS were 23 mo (95% CI, 18-28 mo) and 59 mo (95% CI, 50-not reached [NR]), respectively. Of 40 patients with RECIST-measurable disease and 39 patients with available 68Ga-DOTATATE PET/CT, response categories were partial response, 20% (95% CI, 10%-35%) and 44% (95% CI, 30%-59%); stable disease, 68% (95% CI, 52%-80%) and 44% (95% CI, 30%-59%); and progressive disease, 12% (95% CI, 5%-27%) by both, respectively. There was a moderate concordance between response categories by RECIST and 68Ga-DOTATATE PET/CT, weighted of 0.51 (95% CI, 0.21-0.68).
Click to Show/Hide
|
||||
| Experiment 161 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Partial response (PR) |
20%
|
|||
| Patients Enrolled |
5 patients with pheocromocytomas (PCCs) and paragangliomas (PGLs) (5 PGL, ).
|
||||
| Administration Time | 3 cycles | ||||
| Administration Dosage | 6.6-7.6 GBq/cycle | ||||
| MOA of PDC |
We showed that PRRT based on 177Lu-DOTATATE and 90-YDOTATOC without radiosensitizers are efficacious therapeutic options (DCR of 83% and 76%, respectively) and can be considered in the multidisciplinary treatment of PCCs and PGLs as alternatives to I-131 MIBG and chemotherapy.
|
||||
| Description |
There was no statistically significant heterogeneity among the 177Lu- and 90Y- PRRTs studies (I2 value = 0%, p = 0.68 and I2 value = 0%, p = 0.91, respectively). The pooled DCRs were 0.83 (95% CI: 0.75-0.88) and 0.76 (95% CI: 0.56-0.89) for 177Lu- and 90Y-PRRT treatments, respectively. The pooled DCR for PRRT agents was 0.81 (95% CI: 0.74-0.87). Although the present study is the largest description of DCR in PRRT therapy of PCCs and PGLs, given the small sample size, no formal attempts were made to compare efficacy between 177Lu- and 90Y-PRRT treatments. In detail, we report pooled data on 149 177Lu- and 64 90Y-treated patients (total: 213 patients). Most studies were retrospective. The largest one included 46 patients (34 treated with 177Lu-, 12 with 90Y- PRRTs). Median ages ranged from 32.5 to 60.4 years. The female gender was slightly predominant (83 women, 74 men, gender not specified in 3 studies). When reported, mutations of SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B) were the most frequent genetic alterations. According to the predominant retrospective and real practice nature of the selected studies, sites of metastases and previous treatments were heterogeneous, with bone the metastatic site most often involved, and surgery the most frequent previous therapy. Paramount descriptive information on PRRT doses per cycle, number of cycles, response criteria, best responses to treatments, and frequency of G3/G4 hematologic toxicities is reported in Table 2. The lowest applied dose was 3.4 GBq per cycle; the number of administered cycles was heterogeneous (at least 3 cycles in 7 studies). The most used response criteria were the RECIST 1.1. Only one study evaluated the response with SWOG criteria, which are more conservative with regard to the definition of PR (50% or greater reduction in the sum of the product of the perpendicular diameters of target lesions). The majority of the patients experienced PR or SD. Hematologic toxicity was manageable. Funnel plot of selected studies did not show publication bias. The MINORS and Newcastle-Ottawa Scale scoring for the selected articles are reported in Table 3.
Click to Show/Hide
|
||||
| Experiment 162 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Partial response (PR) |
21%
|
|||
| Patients Enrolled |
20 patients with others neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 163 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Partial response (PR) |
21.27%
|
|||
| Patients Enrolled |
18 patients with neuroendocrine tumour.
|
||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 164 Reporting the Activity Data of This PDC | [19] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumour | ||||
| Efficacy Data | Partial response (PR) |
21.70%
|
|||
| Patients Enrolled |
23 GEP neuroendocrine tumours patients.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | 7.4 GBq (200mCi) | ||||
| MOA of PDC |
From our initial experience, Lutathera has been well tolerated in patients with GEP-NET. Additional studies are needed to examine long-term clinical and patient-reported outcomes associated with treatment of GEP-NETs as well as financial considerations for hospitals embarking on a PRRT program. A multidisciplinary team and complete collaboration between hospital administration and clinical teams are required for successful implementation of a PRRT program.
Click to Show/Hide
|
||||
| Description |
PET/CT imaging were done to determine treatment responses 3 months post-PRRT treatment. Only patients who completed 4 cycles (n=23) were included. Five patients (21.7%) showed partial response, 10 patients (43.5 5%) showed stable disease, and 3 patients (13.0%) showed disease progression; the responses from 5 patients (21.7%) were unknown because no PET/CT scan was done.
Click to Show/Hide
|
||||
| Experiment 165 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Partial response (PR) |
22%
|
|||
| Patients Enrolled |
395 patients with neuroendocrine tumour.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 166 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Partial response (PR) |
22.22%
|
|||
| Patients Enrolled |
9 patients with pheocromocytomas (PCCs) and paragangliomas (PGLs) (3 PCC , 6PGL).
|
||||
| Administration Time | 3.11 cycles | ||||
| Administration Dosage | 8.01 (7.4-8.4) GBq/cycle | ||||
| MOA of PDC |
We showed that PRRT based on 177Lu-DOTATATE and 90-YDOTATOC without radiosensitizers are efficacious therapeutic options (DCR of 83% and 76%, respectively) and can be considered in the multidisciplinary treatment of PCCs and PGLs as alternatives to I-131 MIBG and chemotherapy.
|
||||
| Description |
There was no statistically significant heterogeneity among the 177Lu- and 90Y- PRRTs studies (I2 value = 0%, p = 0.68 and I2 value = 0%, p = 0.91, respectively). The pooled DCRs were 0.83 (95% CI: 0.75-0.88) and 0.76 (95% CI: 0.56-0.89) for 177Lu- and 90Y-PRRT treatments, respectively. The pooled DCR for PRRT agents was 0.81 (95% CI: 0.74-0.87). Although the present study is the largest description of DCR in PRRT therapy of PCCs and PGLs, given the small sample size, no formal attempts were made to compare efficacy between 177Lu- and 90Y-PRRT treatments. In detail, we report pooled data on 149 177Lu- and 64 90Y-treated patients (total: 213 patients). Most studies were retrospective. The largest one included 46 patients (34 treated with 177Lu-, 12 with 90Y- PRRTs). Median ages ranged from 32.5 to 60.4 years. The female gender was slightly predominant (83 women, 74 men, gender not specified in 3 studies). When reported, mutations of SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B) were the most frequent genetic alterations. According to the predominant retrospective and real practice nature of the selected studies, sites of metastases and previous treatments were heterogeneous, with bone the metastatic site most often involved, and surgery the most frequent previous therapy. Paramount descriptive information on PRRT doses per cycle, number of cycles, response criteria, best responses to treatments, and frequency of G3/G4 hematologic toxicities is reported in Table 2. The lowest applied dose was 3.4 GBq per cycle; the number of administered cycles was heterogeneous (at least 3 cycles in 7 studies). The most used response criteria were the RECIST 1.1. Only one study evaluated the response with SWOG criteria, which are more conservative with regard to the definition of PR (50% or greater reduction in the sum of the product of the perpendicular diameters of target lesions). The majority of the patients experienced PR or SD. Hematologic toxicity was manageable. Funnel plot of selected studies did not show publication bias. The MINORS and Newcastle-Ottawa Scale scoring for the selected articles are reported in Table 3.
Click to Show/Hide
|
||||
| Experiment 167 Reporting the Activity Data of This PDC | [14] | ||||
| Indication | Metastatic/Advanced neuroendocrine tumour | ||||
| Efficacy Data | Partial response (PR) |
23.10%
|
|||
| Patients Enrolled |
468 patients with metastatic/advanced neuroendocrine tumor.
|
||||
| Administration Time | At least two cycles at 10-12 weeks interval | ||||
| Administration Dosage | 5.55 to 7.4 GBq per patient | ||||
| Description |
Long-term outcome (follow-up ranging from 4 to 97.6 months; median period:46 months following first 177Lu-DOTATATE PRRT) results showed, (i) on symptomatic response evaluation scale, complete response (CR) in 214 patients (45.7%), partial response (PR) in 108 (23.1%), stable disease (SD) in 118 (25.2%), progressive disease (PD) in 28 (6%).
Click to Show/Hide
|
||||
| Experiment 168 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Partial response (PR) |
23.33%
|
|||
| Patients Enrolled |
30 patients with pheocromocytomas (PCCs) and paragangliomas (PGLs) (3 PCC, 27PGL).
|
||||
| Administration Time | 73% of patients received 4 cycles | ||||
| Administration Dosage | 7.4 GBq/cycle | ||||
| MOA of PDC |
We showed that PRRT based on 177Lu-DOTATATE and 90-YDOTATOC without radiosensitizers are efficacious therapeutic options (DCR of 83% and 76%, respectively) and can be considered in the multidisciplinary treatment of PCCs and PGLs as alternatives to I-131 MIBG and chemotherapy.
|
||||
| Description |
There was no statistically significant heterogeneity among the 177Lu- and 90Y- PRRTs studies (I2 value = 0%, p = 0.68 and I2 value = 0%, p = 0.91, respectively). The pooled DCRs were 0.83 (95% CI: 0.75-0.88) and 0.76 (95% CI: 0.56-0.89) for 177Lu- and 90Y-PRRT treatments, respectively. The pooled DCR for PRRT agents was 0.81 (95% CI: 0.74-0.87). Although the present study is the largest description of DCR in PRRT therapy of PCCs and PGLs, given the small sample size, no formal attempts were made to compare efficacy between 177Lu- and 90Y-PRRT treatments. In detail, we report pooled data on 149 177Lu- and 64 90Y-treated patients (total: 213 patients). Most studies were retrospective. The largest one included 46 patients (34 treated with 177Lu-, 12 with 90Y- PRRTs). Median ages ranged from 32.5 to 60.4 years. The female gender was slightly predominant (83 women, 74 men, gender not specified in 3 studies). When reported, mutations of SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B) were the most frequent genetic alterations. According to the predominant retrospective and real practice nature of the selected studies, sites of metastases and previous treatments were heterogeneous, with bone the metastatic site most often involved, and surgery the most frequent previous therapy. Paramount descriptive information on PRRT doses per cycle, number of cycles, response criteria, best responses to treatments, and frequency of G3/G4 hematologic toxicities is reported in Table 2. The lowest applied dose was 3.4 GBq per cycle; the number of administered cycles was heterogeneous (at least 3 cycles in 7 studies). The most used response criteria were the RECIST 1.1. Only one study evaluated the response with SWOG criteria, which are more conservative with regard to the definition of PR (50% or greater reduction in the sum of the product of the perpendicular diameters of target lesions). The majority of the patients experienced PR or SD. Hematologic toxicity was manageable. Funnel plot of selected studies did not show publication bias. The MINORS and Newcastle-Ottawa Scale scoring for the selected articles are reported in Table 3.
Click to Show/Hide
|
||||
| Experiment 169 Reporting the Activity Data of This PDC | [14] | ||||
| Indication | Metastatic/Advanced neuroendocrine tumour | ||||
| Efficacy Data | Partial response (PR) |
25%
|
|||
| Patients Enrolled |
468 patients with metastatic/advanced neuroendocrine tumor.
|
||||
| Administration Time | At least two cycles at 10-12 weeks interval | ||||
| Administration Dosage | 5.55 to 7.4 GBq per patient | ||||
| Evaluation Method | PERCIST criteria assay | ||||
| Description |
Molecular imaging response (by PERCIST criteria) showed CR in 29 (6%), PR in 116 (25%), SD in 267 (57%) and PD in 56 (12%) patients.
|
||||
| Experiment 170 Reporting the Activity Data of This PDC | [14] | ||||
| Indication | Metastatic/Advanced neuroendocrine tumour | ||||
| Efficacy Data | Partial response (PR) |
27%
|
|||
| Patients Enrolled |
468 patients with metastatic/advanced neuroendocrine tumor.
|
||||
| Administration Time | At least two cycles at 10-12 weeks interval | ||||
| Administration Dosage | 5.55 to 7.4 GBq per patient | ||||
| Description |
On RECIST 1.1 criteria, CR was observed in 14 patients (3%), PR in 126 patients (27%), SD in 282 patients (60%) and PD in 46 patients (10%).
|
||||
| Experiment 171 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Partial response (PR) |
27.78%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Nineteen patients, all with progressive, heavily treated disease, were identified. Sites of tumor origin were: pancreas (74%), small bowel (11%), rectum (11%), and lung (5%); median Ki-67 was 32% (range 22-56). Thirteen patients (68%) completed all four 177Lu-DOTATATE cycles. Best response (N = 18 evaluable) was: 5/18 (28%) partial response, 8/18 (44%) stable disease, and 5/18 (28%) disease progression. Median PFS was 13.1 months (95% CI: 8.7-20.9).
Click to Show/Hide
|
||||
| Experiment 172 Reporting the Activity Data of This PDC | [24] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Partial response (PR) |
30%
|
|||
| Patients Enrolled |
45 patients with advanced inoperable/metastatic neuroendocrine tumor (22 women, 23 men).
|
||||
| Administration Time | 2 to 7 cycles | ||||
| Administration Dosage | 27.0 GBq | ||||
| Evaluation Method | 68Ga-DOTATATE PET/CT assay | ||||
| Description |
Radiological response with the CT images of 68Ga-DOTANOC PET/CECT was assessed 8 to 12 weeks after the last treatment cycle. Three patients were lost to follow-up, and 2 other patients had nonmeasurable lesions on CT. Thus, according to RECIST 1.1, 40 patients had evaluable lesions, of which 12 patients (30%) had PR and 22 patients (55%) had SD, whereas disease progression was limited to 6 patients (15%). No case of CR was observed in our study cohort.
Click to Show/Hide
|
||||
| Experiment 173 Reporting the Activity Data of This PDC | [25] | ||||
| Indication | SDHB positive metastatic paraganglioma | ||||
| Efficacy Data | Partial response (PR) |
30%
|
|||
| Patients Enrolled |
37 patients affected by neuroendocrine tumors.
|
||||
| Administration Time | Five cycles of 5.5 gbq each every 8 weeks | ||||
| Administration Dosage | Cumulative activity of 27.5 GBq | ||||
| Description |
Thirty-three of 37 patients were evaluable for response. Objective responses included partial response (PR) in 10 patients (30%), stable disease (SD) in 18 patients (55%), with a DCR of 85%. Median follow up was 38 months (range 4.6-51.1 months). Median PFS was 31.4 months (17.6-45.4) and mOS was not reached.
|
||||
| Experiment 174 Reporting the Activity Data of This PDC | [40] | ||||
| Indication | High-grade gliomas | ||||
| Efficacy Data | Partial response (PR) |
31.25%
|
|||
| Patients Enrolled |
16 patients with high-grade gliomas (10 males and 6 females).
|
||||
| Administration Time | 1 to 4 cycles | ||||
| Administration Dosage | 3.7 to 29.6 GBq (median, 10.45 GBq) | ||||
| Evaluation Method | MRI assay | ||||
| Description |
In total, 16 subjects (10 males and 6 females) with a mean age of 55.68 ± 13.17 years (26-73 years) participated in the study. Of them, 8 patients were new HGG cases, and 8 patients had recurrent tumors. The participants' responses to treatments were complete remission in 12.5% of (n = 2), partial remission in 31.25% (n = 5), disease stability in 18.7% (n = 3), and disease progression in 37.5% (n = 6). In total, pretreatment and posttreatment Karnofsky Performance Score and Eastern Cooperative Oncology Group scores did not improved (P > 0.05). The patients were followed up from 1 month to 26 months (median, 3 months).
Click to Show/Hide
|
||||
| Experiment 175 Reporting the Activity Data of This PDC | [16] | ||||
| Indication | Metastatic medullary thyroid carcinoma | ||||
| Efficacy Data | Partial response (PR) |
33.33%
|
|||
| Patients Enrolled |
6 patients with medullary thyroid cancer (3 males and 3 females).
|
||||
| Administration Time | 3 cycles | ||||
| Administration Dosage | 5.7 GBq/cycle (range, 2.6-7.4 GBq) | ||||
| Evaluation Method | PET/CT assay | ||||
| MOA of PDC |
Radiolabeled somatostatin analogs are contributive for the management of recurrent MTC. 68Ga-DOTATAE PET-CT showed a relatively high detection rate in recurrent MTC. In addition, PRRT with 177Lu-DOTATATE was found to be a safe alternative therapeutic option for MTC.
|
||||
| Description |
Four of the 17 patients with positive SSTR-PET were scheduled for PRRT. In addition, 2 patients had positive 99mTc-octreotide scintigraphy results (Krenning score ≥ 2) and were scheduled for PRRT. Two of the 6 patients who underwent PRRT showed PR, 2 SD and 2 PD. Two patients died during the follow-up period. Median overall survival was 19 months (95% CI: 5.52-29.48). There were no cases of significant toxicity.
Click to Show/Hide
|
||||
| Experiment 176 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Partial response (PR) |
33.33%
|
|||
| Patients Enrolled |
12 patients with pheocromocytomas and paragangliomas.
|
||||
| Administration Time | At least 3 cycles | ||||
| Administration Dosage | 3.7-8.1 GBq/cycle | ||||
| MOA of PDC |
We showed that PRRT based on 177Lu-DOTATATE and 90-YDOTATOC without radiosensitizers are efficacious therapeutic options (DCR of 83% and 76%, respectively) and can be considered in the multidisciplinary treatment of PCCs and PGLs as alternatives to I-131 MIBG and chemotherapy.
|
||||
| Description |
There was no statistically significant heterogeneity among the 177Lu- and 90Y- PRRTs studies (I2 value = 0%, p = 0.68 and I2 value = 0%, p = 0.91, respectively). The pooled DCRs were 0.83 (95% CI: 0.75-0.88) and 0.76 (95% CI: 0.56-0.89) for 177Lu- and 90Y-PRRT treatments, respectively. The pooled DCR for PRRT agents was 0.81 (95% CI: 0.74-0.87). Although the present study is the largest description of DCR in PRRT therapy of PCCs and PGLs, given the small sample size, no formal attempts were made to compare efficacy between 177Lu- and 90Y-PRRT treatments. In detail, we report pooled data on 149 177Lu- and 64 90Y-treated patients (total: 213 patients). Most studies were retrospective. The largest one included 46 patients (34 treated with 177Lu-, 12 with 90Y- PRRTs). Median ages ranged from 32.5 to 60.4 years. The female gender was slightly predominant (83 women, 74 men, gender not specified in 3 studies). When reported, mutations of SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B) were the most frequent genetic alterations. According to the predominant retrospective and real practice nature of the selected studies, sites of metastases and previous treatments were heterogeneous, with bone the metastatic site most often involved, and surgery the most frequent previous therapy. Paramount descriptive information on PRRT doses per cycle, number of cycles, response criteria, best responses to treatments, and frequency of G3/G4 hematologic toxicities is reported in Table 2. The lowest applied dose was 3.4 GBq per cycle; the number of administered cycles was heterogeneous (at least 3 cycles in 7 studies). The most used response criteria were the RECIST 1.1. Only one study evaluated the response with SWOG criteria, which are more conservative with regard to the definition of PR (50% or greater reduction in the sum of the product of the perpendicular diameters of target lesions). The majority of the patients experienced PR or SD. Hematologic toxicity was manageable. Funnel plot of selected studies did not show publication bias. The MINORS and Newcastle-Ottawa Scale scoring for the selected articles are reported in Table 3.
Click to Show/Hide
|
||||
| Experiment 177 Reporting the Activity Data of This PDC | [23] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Partial response (PR) |
34%
|
|||
| Patients Enrolled |
Patients with advanced neuroendocrine tumor.
|
||||
| Administration Dosage | Along with oral capecitabine therapy | ||||
| Description |
According to Response Evaluation Criteria in Solid Tumors 1.1 criteria, in group 1 patients, the response was partial response in 34% of the patients, stable disease in 50.2%, and progressive disease in 6.8% versus partial response in 6.3%, stable disease in 60.9%, and progressive disease in 26.5% among group 2 patients. The median overall survival (OS) and progression-free survival (PFS) was not reached in group 1 patients. The median OS and PFS in group 2 patients were 48 months.
Click to Show/Hide
|
||||
| Experiment 178 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Unknown originb neuroendocrine tumor | ||||
| Efficacy Data | Partial response (PR) |
35%
|
|||
| Patients Enrolled |
22 patients with unknown originb neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 179 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Partial response (PR) |
38.50%
|
|||
| Patients Enrolled |
13 Re-retreatment neuroendocrine tumour patients.
|
||||
| Administration Time | 2 cycles | ||||
| Administration Dosage | Median cumulative dose: 59.7 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 180 Reporting the Activity Data of This PDC | [14] | ||||
| Indication | Metastatic/Advanced neuroendocrine tumour | ||||
| Efficacy Data | Partial response (PR) |
40%
|
|||
| Patients Enrolled |
468 patients with metastatic/advanced neuroendocrine tumor.
|
||||
| Administration Time | At least two cycles at 10-12 weeks interval | ||||
| Administration Dosage | 5.55 to 7.4 GBq per patient | ||||
| Description |
Biochemical response evaluation showed CR in 52 (12%), PR in 172 (40%), SD in 161 (38%), and PD in 42 patients (10%).
|
||||
| Experiment 181 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Partial response (PR) |
43%
|
|||
| Patients Enrolled |
78 patients with pancreatic neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 182 Reporting the Activity Data of This PDC | [20] | ||||
| Indication | SSR positive lung neuroendocrine tumor | ||||
| Efficacy Data | Partial response (PR) |
44%
|
|||
| Patients Enrolled |
39 patients with neuroendocrine tumour with available <sup>68</sup>Ga-DOTATATE PET/CT.
|
||||
| Administration Time | 1-4 cycles | ||||
| Administration Dosage | Median cumulative activity of 27 GBq (range, 6-43 GBq) | ||||
| Evaluation Method | 68Ga-DOTATATE PET/CT assay | ||||
| MOA of PDC |
In patients with advanced progressive lung NET and satisfactory SSR expression, 177Lu-DOTATATE is effective and safe, with a high DCR and encouraging PFS and OS. Further prospective studies comparing 177Lu-DOTATATE with other systemic options are warranted.
|
||||
| Description |
Of 48 patients (median age, 63 y; 13 women), 43 (90%) had AC and 5 (10%) TC. Almost all patients (47, 98%) were treated due to progression. Most patients (40, 83%) received somatostatin analogs, and 10 patients (20%) had prior everolimus, chemotherapy, or both. All patients had high SSR expression (≥ modified Krenning score 3) on pretreatment 68Ga-DOTATATE PET/CT. Patients received a median 4 (range, 1-4) cycles of 177Lu-DOTATATE (33% with concurrent radiosensitizing chemotherapy) to a median cumulative activity of 27 GBq (range, 6-43GBq). At a median follow-up of 42 mo, the median PFS and OS were 23 mo (95% CI, 18-28 mo) and 59 mo (95% CI, 50-not reached [NR]), respectively. Of 40 patients with RECIST-measurable disease and 39 patients with available 68Ga-DOTATATE PET/CT, response categories were partial response, 20% (95% CI, 10%-35%) and 44% (95% CI, 30%-59%); stable disease, 68% (95% CI, 52%-80%) and 44% (95% CI, 30%-59%); and progressive disease, 12% (95% CI, 5%-27%) by both, respectively. There was a moderate concordance between response categories by RECIST and 68Ga-DOTATATE PET/CT, weighted of 0.51 (95% CI, 0.21-0.68).
Click to Show/Hide
|
||||
| Experiment 183 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | High-grade (WHO G3) neuroendocrine tumour | ||||
| Efficacy Data | Partial response (PR) |
46.90%
|
|||
| Patients Enrolled |
Patients with neuroendocrine tumours including high-grade (WHO G3) neuroendocrine tumours.
|
||||
| Evaluation Method | RECIST criteria assay | ||||
| Description |
Among 160 patients whose responses to treatment could be evaluated according to the RECIST criteria, 28.1% (n=45) had a progressive disease, 21.9% (n=35) had a stable disease, 46.9% (n=75) had a partial response and 3.1% (n=5) had a complete response.
|
||||
| Experiment 184 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Hindgut neuroendocrine tumour | ||||
| Efficacy Data | Partial response (PR) |
62%
|
|||
| Patients Enrolled |
14 patients with hindgut neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 185 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | High-grade (WHO G3) neuroendocrine tumour | ||||
| Efficacy Data | Overall survival (OS) |
38.4 months
|
|||
| Patients Enrolled |
Patients with WHO grades III.
|
||||
| Description |
The disease control rates in patients with WHO grades I, II and III were 74, 73 and 60%, respectively, and the OS rates were 61.9, 52.2 and 38.4 months, respectively.
|
||||
| Experiment 186 Reporting the Activity Data of This PDC | [46] | ||||
| Indication | Pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Overall survival (OS) |
42 months
|
|||
| Patients Enrolled |
Patients with advanced Pan-neuroendocrine tumour, previously pretreated with one (67%) or several (33%) lines of chemotherapy.
|
||||
| Administration Time | 1-10 cycles with 6- to 8-week intervals | ||||
| Administration Dosage | 7.4 GBq | ||||
| Description |
The median follow-up was 34 months, the median PFS was 24 months, and the median OS was 42 months from start of PRRT.
|
||||
| Experiment 187 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Overall survival (OS) |
49.6-68.0 months
|
|||
| Patients Enrolled |
179 patients with paraganglioma.
|
||||
| Administration Time | 1-11 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The use of PRRT for patients with PPGLs who had high tumor SSTR expression has mainly been reported in retrospective small case series and the protocols varied in all these studies. So far, four meta-analyses (Taieb et al. and Satapathy et al. in 2019 as well as Su et al. and Marretta et al. in 2023) based on PRRT (177Lu/90Y-DOTAT-TATE/TOC) therapy have been reported. The reported number of pooled patients ranged from 179-330 without any complete response. The reported partial response ranged from 20-25% and stable disease from 59-70% giving an objective response rate range of 20-25% and disease control rate range of 8190%. The survival rates were reported by only 3 meta-analyses. The reported progression-free survival were 17-39 months in seven studies by Taieb et al., 10-91 months in 4 studies and a mean of 37.1 months in 2 studies by Satapathy et al., and a median of 31.8 months in 13 studies by Su et al. whereas overall survival were 49.6-68.0 months in 3 studies by Taieb et al., a mean of 54.5 months in 4 studies by Satapathy et al. and a median of 74.3 months in 11 studies by Su et al. Su et al. reports a pooled adverse events data in 274/330 patients which mentions a grade 3/4 hematotoxicity in 4.3% (grade 1-4, 22.3%) and grade 3/4 nephrotoxicity in 1.9% (grade 1-4, 1.9%), and myelodysplastic syndrome in 1 patient. Treatment discontinuation occurred in 4 patients due to thrombocytopenia (n=3) or nephrotoxicity (n=1).
Click to Show/Hide
|
||||
| Experiment 188 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | High-grade (WHO G3) neuroendocrine tumour | ||||
| Efficacy Data | Overall survival (OS) |
52.2 months
|
|||
| Patients Enrolled |
Patients with WHO grades II.
|
||||
| Description |
The disease control rates in patients with WHO grades I, II and III were 74, 73 and 60%, respectively, and the OS rates were 61.9, 52.2 and 38.4 months, respectively.
|
||||
| Experiment 189 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Overall survival (OS) |
54.5 months
|
|||
| Patients Enrolled |
201 patients with paraganglioma.
|
||||
| Administration Time | 1-10 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The use of PRRT for patients with PPGLs who had high tumor SSTR expression has mainly been reported in retrospective small case series and the protocols varied in all these studies. So far, four meta-analyses (Taieb et al. and Satapathy et al. in 2019 as well as Su et al. and Marretta et al. in 2023) based on PRRT (177Lu/90Y-DOTAT-TATE/TOC) therapy have been reported. The reported number of pooled patients ranged from 179-330 without any complete response. The reported partial response ranged from 20-25% and stable disease from 59-70% giving an objective response rate range of 20-25% and disease control rate range of 8190%. The survival rates were reported by only 3 meta-analyses. The reported progression-free survival were 17-39 months in seven studies by Taieb et al., 10-91 months in 4 studies and a mean of 37.1 months in 2 studies by Satapathy et al., and a median of 31.8 months in 13 studies by Su et al. whereas overall survival were 49.6-68.0 months in 3 studies by Taieb et al., a mean of 54.5 months in 4 studies by Satapathy et al. and a median of 74.3 months in 11 studies by Su et al. Su et al. reports a pooled adverse events data in 274/330 patients which mentions a grade 3/4 hematotoxicity in 4.3% (grade 1-4, 22.3%) and grade 3/4 nephrotoxicity in 1.9% (grade 1-4, 1.9%), and myelodysplastic syndrome in 1 patient. Treatment discontinuation occurred in 4 patients due to thrombocytopenia (n=3) or nephrotoxicity (n=1).
Click to Show/Hide
|
||||
| Experiment 190 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | High-grade (WHO G3) neuroendocrine tumour | ||||
| Efficacy Data | Overall survival (OS) |
61.9 months
|
|||
| Patients Enrolled |
Patients with WHO grades I.
|
||||
| Description |
The disease control rates in patients with WHO grades I, II and III were 74, 73 and 60%, respectively, and the OS rates were 61.9, 52.2 and 38.4 months, respectively.
|
||||
| Experiment 191 Reporting the Activity Data of This PDC | [18] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumor | ||||
| Efficacy Data | Overall survival (OS) |
63 months
|
|||
| Patients Enrolled |
Patients treated with Lu-DOTATATE.
|
||||
| Administration Dosage | 100 mCi (3.7 GBq) | ||||
| Description |
The objective response rate of the total group of patients was 39%. Stable disease was reached in 43% of patients. Progression-free survival (PFS) and overall survival (OS) for all NET patients were 29 months [95% confidence interval (CI), 26-33 months] and 63 months (95% CI, 55-72 months). Long-term toxicity included acute leukemia in four patients (0.7%) and myelodysplastic syndrome in nine patients (1.5%).
Click to Show/Hide
|
||||
| Experiment 192 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Overall survival (OS) |
74.3 months
|
|||
| Patients Enrolled |
330 patients with paraganglioma.
|
||||
| Administration Time | 1-10 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The use of PRRT for patients with PPGLs who had high tumor SSTR expression has mainly been reported in retrospective small case series and the protocols varied in all these studies. So far, four meta-analyses (Taieb et al. and Satapathy et al. in 2019 as well as Su et al. and Marretta et al. in 2023) based on PRRT (177Lu/90Y-DOTAT-TATE/TOC) therapy have been reported. The reported number of pooled patients ranged from 179-330 without any complete response. The reported partial response ranged from 20-25% and stable disease from 59-70% giving an objective response rate range of 20-25% and disease control rate range of 8190%. The survival rates were reported by only 3 meta-analyses. The reported progression-free survival were 17-39 months in seven studies by Taieb et al., 10-91 months in 4 studies and a mean of 37.1 months in 2 studies by Satapathy et al., and a median of 31.8 months in 13 studies by Su et al. whereas overall survival were 49.6-68.0 months in 3 studies by Taieb et al., a mean of 54.5 months in 4 studies by Satapathy et al. and a median of 74.3 months in 11 studies by Su et al. Su et al. reports a pooled adverse events data in 274/330 patients which mentions a grade 3/4 hematotoxicity in 4.3% (grade 1-4, 22.3%) and grade 3/4 nephrotoxicity in 1.9% (grade 1-4, 1.9%), and myelodysplastic syndrome in 1 patient. Treatment discontinuation occurred in 4 patients due to thrombocytopenia (n=3) or nephrotoxicity (n=1).
Click to Show/Hide
|
||||
| Experiment 193 Reporting the Activity Data of This PDC | [49] | ||||
| Indication | Bronchial and gastroenteropancreatic neuroendocrine tumour | ||||
| Efficacy Data | Overall survival (OS) |
80.8 months
|
|||
| Patients Enrolled |
Patients with bronchial and gastroenteropancreatic neuroendocrine tumours.
|
||||
| Administration Time | Over two cycles | ||||
| Administration Dosage | 14.8 GBq | ||||
| Description |
Combined overall survival (OS) after I-PRRT plus R-PRRT and RR-PRRT was 80.8 months (95% CI 66.0-95.6).
|
||||
| Experiment 194 Reporting the Activity Data of This PDC | [50] | ||||
| Indication | Differentiated thyroid cancer | ||||
| Efficacy Data | Objective response rate (ORR) |
10.50%
|
|||
| Patients Enrolled |
157 patients with RR-DTC treated with PPRT.
|
||||
| MOA of PDC |
Findings of the study revealed that in the absence of the established treatment for the patients with RR-DTC and metastatic MTC, PRRT could be effective with few adverse events.
|
||||
| Description |
Among 2284 related papers, 41 papers met the inclusion criteria. A total of 157 patients with RR-DTC were treated with PPRT. Biochemical and objective responses (partial and complete) were observed in 25.3 and 10.5% of patients, respectively. Among 220 patients with metastatic MTC, biochemical and objective responses were observed in 37.2 and 10.6% of the patients, respectively. Forty-six deaths were reported in 95 patients with advanced RR-DTC. In addition, 63 deaths were observed in 144 patients with metastatic MTC. Major side effects were reported in 124 patients treated with 90Y -based agent. In the patients treated with 177Lu-DOTA-TATE and 111In-Octreotide, mild and transient hematologic or renal complications were reported.
Click to Show/Hide
|
||||
| Experiment 195 Reporting the Activity Data of This PDC | [50] | ||||
| Indication | Medullary thyroid carcinoma | ||||
| Efficacy Data | Objective response rate (ORR) |
10.60%
|
|||
| Patients Enrolled |
220 patients with metastatic medullary thyroid cancer.
|
||||
| MOA of PDC |
Findings of the study revealed that in the absence of the established treatment for the patients with RR-DTC and metastatic MTC, PRRT could be effective with few adverse events.
|
||||
| Description |
Among 2284 related papers, 41 papers met the inclusion criteria. A total of 157 patients with RR-DTC were treated with PPRT. Biochemical and objective responses (partial and complete) were observed in 25.3 and 10.5% of patients, respectively. Among 220 patients with metastatic MTC, biochemical and objective responses were observed in 37.2 and 10.6% of the patients, respectively. Forty-six deaths were reported in 95 patients with advanced RR-DTC. In addition, 63 deaths were observed in 144 patients with metastatic MTC. Major side effects were reported in 124 patients treated with 90Y -based agent. In the patients treated with 177Lu-DOTA-TATE and 111In-Octreotide, mild and transient hematologic or renal complications were reported.
Click to Show/Hide
|
||||
| Experiment 196 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Objective response rate (ORR) |
11.10%
|
|||
| Patients Enrolled |
18 patients with paraganglioma.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The efficacy and safety data of the interim analysis of an open-label, single-arm phase 2 study evaluating 177Lu-DOTATATE in paraganglioma patients conducted at the National Institutes of Health (NCT03206060) in 36 patients [(n=18, apparently sporadic cohort and n=18 in patients having germline variant in one of the genes encoding subunits of succinate dehydrogenase complex (SDHA=2, SDHB=15, SDHD=1)), SDHx cohort] after 177LuDOTATATE therapy with 7.4 GBq every 8 weeks for 4 cycles. The progression-free survival at 6 months since initiation of 177Lu-DOTATATE was 19.1 months (22.7 months in apparently sporadic cohort and 15.4 months in SDHx cohort) and overall survival was not reached in both cohorts (80). Per RECIST 1.1, overall, 86.1% patients showed partial response (13.9%) or stable disease (72.2%). In sporadic cohort; partial response was observed in 11.1% and stable disease in 88.9% patients whereas those in SDHx cohort were, 16.7% and 55.5%, respectively at the end of 6 months.
Click to Show/Hide
|
||||
| Experiment 197 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Objective response rate (ORR) |
13.90%
|
|||
| Patients Enrolled |
36 patients with paraganglioma.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The efficacy and safety data of the interim analysis of an open-label, single-arm phase 2 study evaluating 177Lu-DOTATATE in paraganglioma patients conducted at the National Institutes of Health (NCT03206060) in 36 patients [(n=18, apparently sporadic cohort and n=18 in patients having germline variant in one of the genes encoding subunits of succinate dehydrogenase complex (SDHA=2, SDHB=15, SDHD=1)), SDHx cohort] after 177LuDOTATATE therapy with 7.4 GBq every 8 weeks for 4 cycles. The progression-free survival at 6 months since initiation of 177Lu-DOTATATE was 19.1 months (22.7 months in apparently sporadic cohort and 15.4 months in SDHx cohort) and overall survival was not reached in both cohorts (80). Per RECIST 1.1, overall, 86.1% patients showed partial response (13.9%) or stable disease (72.2%). In sporadic cohort; partial response was observed in 11.1% and stable disease in 88.9% patients whereas those in SDHx cohort were, 16.7% and 55.5%, respectively at the end of 6 months.
Click to Show/Hide
|
||||
| Experiment 198 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Objective response rate (ORR) |
16.70%
|
|||
| Patients Enrolled |
18 patients with paraganglioma.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The efficacy and safety data of the interim analysis of an open-label, single-arm phase 2 study evaluating 177Lu-DOTATATE in paraganglioma patients conducted at the National Institutes of Health (NCT03206060) in 36 patients [(n=18, apparently sporadic cohort and n=18 in patients having germline variant in one of the genes encoding subunits of succinate dehydrogenase complex (SDHA=2, SDHB=15, SDHD=1)), SDHx cohort] after 177LuDOTATATE therapy with 7.4 GBq every 8 weeks for 4 cycles. The progression-free survival at 6 months since initiation of 177Lu-DOTATATE was 19.1 months (22.7 months in apparently sporadic cohort and 15.4 months in SDHx cohort) and overall survival was not reached in both cohorts (80). Per RECIST 1.1, overall, 86.1% patients showed partial response (13.9%) or stable disease (72.2%). In sporadic cohort; partial response was observed in 11.1% and stable disease in 88.9% patients whereas those in SDHx cohort were, 16.7% and 55.5%, respectively at the end of 6 months.
Click to Show/Hide
|
||||
| Experiment 199 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Phaeochromocytoma | ||||
| Efficacy Data | Objective response rate (ORR) |
18%
|
|||
| Patients Enrolled |
12 patients with pheocromocytomas and paragangliomas.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
As a consequence, we saw a rising interest in a potential radiolabeled-SST agonist theranostic approach for PPGLs, with several spontaneous studies reporting of the treatment of metastatic or inoperable tumors with Lutathera and other similar radiocompounds, with promising preliminary results. The first RLT experience in PPGLs was reported by van Essen et al. who treated a heterogeneous cohort of patients, including 12 PGLs. Despite the authors reporting lower response rates than those obtained in GEP-NET patients, ORR and DCR were 18% and 73%, respectively. These results are consistent with other reports in the literature. Kong et al. treated 20 PPGLs (8 PHEOs and 12 PGLs) with [117Lu]Lu-DOTATATE, 14 due to uncontrolled secondary hypertension and 6 to radiological PD. A DCR equal to 86% was reached, including five patients with ORR (36%). Of note, 62% of symptomatic patients required a dose reduction of antihypertensive medications, and 57% reported a subjective therapeutic benefit in terms of tumor-related symptoms. Comprehensive PFS was 39 months, including two patients with early recurrence (2 and 5 months post-RLT, respectively) and one patient with remarkable tumor downsizing that allowed a second-step curative liver surgery. Of note, the patient was still disease-free at the time of the study. The results of the largest cohort of PPGLs treated with RLT were reported by Severi et al., who treated 46 patients reaching a comprehensive DCR of 80%. Interestingly, 34 patients treated with [177Lu]Lu-DOTATATE obtained a longer median OS in comparison to those treated with [90Y]Y-DOTATOC (143 vs. 92 months, respectively). According to the authors, this result may be related both to DOTATATEs higher affinity for SSTR2 (which is the type most overexpressed in PPGLs) and to the longer half-life of 177Lu and, consequently, prolonged residence time within the tumor lesions. Moreover, this study reports that sympathetic functioning PPGLs were associated to a shorter median PFS compared to non-functioning PPGLs. Prado-Wohlwend et al. treated nine patients with [177Lu]Lu-DOTATATE and eight with [131I]MIBG, obtaining a PFS of 29 and 18.5 months and a DCR of 88.8% and 62.5%, respectively, for each therapy. Despite the low number of patients involved, a trend for a longer PFS was found for adrenal primary PPGLs treated with [177Lu]Lu-DOTATATE. Nevertheless, the authors suggest performing both [68Ga]Ga-DOTA-SSA PET/CT and [123I]MIBG SPECT/CT in each patient in order to offer a personalized theranostic approach based on the highest uptake intensity in one of the two imaging scans. This is consistent with the results by Jaiswal et al. who found that a baseline SUVmax > 21 at [68Ga]Ga-DOTA-SSA PET/CT is a very strong predictor of response to [177Lu]Lu-DOTATATE (p < 0.0001).
Click to Show/Hide
|
||||
| Experiment 200 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Paraganglioma | ||||
| Efficacy Data | Objective response rate (ORR) |
18%
|
|||
| Patients Enrolled |
12 patients with pheocromocytomas and paragangliomas.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
As a consequence, we saw a rising interest in a potential radiolabeled-SST agonist theranostic approach for PPGLs, with several spontaneous studies reporting of the treatment of metastatic or inoperable tumors with Lutathera and other similar radiocompounds, with promising preliminary results. The first RLT experience in PPGLs was reported by van Essen et al. who treated a heterogeneous cohort of patients, including 12 PGLs. Despite the authors reporting lower response rates than those obtained in GEP-NET patients, ORR and DCR were 18% and 73%, respectively. These results are consistent with other reports in the literature. Kong et al. treated 20 PPGLs (8 PHEOs and 12 PGLs) with [117Lu]Lu-DOTATATE, 14 due to uncontrolled secondary hypertension and 6 to radiological PD. A DCR equal to 86% was reached, including five patients with ORR (36%). Of note, 62% of symptomatic patients required a dose reduction of antihypertensive medications, and 57% reported a subjective therapeutic benefit in terms of tumor-related symptoms. Comprehensive PFS was 39 months, including two patients with early recurrence (2 and 5 months post-RLT, respectively) and one patient with remarkable tumor downsizing that allowed a second-step curative liver surgery. Of note, the patient was still disease-free at the time of the study. The results of the largest cohort of PPGLs treated with RLT were reported by Severi et al., who treated 46 patients reaching a comprehensive DCR of 80%. Interestingly, 34 patients treated with [177Lu]Lu-DOTATATE obtained a longer median OS in comparison to those treated with [90Y]Y-DOTATOC (143 vs. 92 months, respectively). According to the authors, this result may be related both to DOTATATEs higher affinity for SSTR2 (which is the type most overexpressed in PPGLs) and to the longer half-life of 177Lu and, consequently, prolonged residence time within the tumor lesions. Moreover, this study reports that sympathetic functioning PPGLs were associated to a shorter median PFS compared to non-functioning PPGLs. Prado-Wohlwend et al. treated nine patients with [177Lu]Lu-DOTATATE and eight with [131I]MIBG, obtaining a PFS of 29 and 18.5 months and a DCR of 88.8% and 62.5%, respectively, for each therapy. Despite the low number of patients involved, a trend for a longer PFS was found for adrenal primary PPGLs treated with [177Lu]Lu-DOTATATE. Nevertheless, the authors suggest performing both [68Ga]Ga-DOTA-SSA PET/CT and [123I]MIBG SPECT/CT in each patient in order to offer a personalized theranostic approach based on the highest uptake intensity in one of the two imaging scans. This is consistent with the results by Jaiswal et al. who found that a baseline SUVmax > 21 at [68Ga]Ga-DOTA-SSA PET/CT is a very strong predictor of response to [177Lu]Lu-DOTATATE (p < 0.0001).
Click to Show/Hide
|
||||
| Experiment 201 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Objective response rate (ORR) |
20%
|
|||
| Patients Enrolled |
330 patients with paraganglioma.
|
||||
| Administration Time | 1-10 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The use of PRRT for patients with PPGLs who had high tumor SSTR expression has mainly been reported in retrospective small case series and the protocols varied in all these studies. So far, four meta-analyses (Taieb et al. and Satapathy et al. in 2019 as well as Su et al. and Marretta et al. in 2023) based on PRRT (177Lu/90Y-DOTAT-TATE/TOC) therapy have been reported. The reported number of pooled patients ranged from 179-330 without any complete response. The reported partial response ranged from 20-25% and stable disease from 59-70% giving an objective response rate range of 20-25% and disease control rate range of 8190%. The survival rates were reported by only 3 meta-analyses. The reported progression-free survival were 17-39 months in seven studies by Taieb et al., 10-91 months in 4 studies and a mean of 37.1 months in 2 studies by Satapathy et al., and a median of 31.8 months in 13 studies by Su et al. whereas overall survival were 49.6-68.0 months in 3 studies by Taieb et al., a mean of 54.5 months in 4 studies by Satapathy et al. and a median of 74.3 months in 11 studies by Su et al. Su et al. reports a pooled adverse events data in 274/330 patients which mentions a grade 3/4 hematotoxicity in 4.3% (grade 1-4, 22.3%) and grade 3/4 nephrotoxicity in 1.9% (grade 1-4, 1.9%), and myelodysplastic syndrome in 1 patient. Treatment discontinuation occurred in 4 patients due to thrombocytopenia (n=3) or nephrotoxicity (n=1).
Click to Show/Hide
|
||||
| Experiment 202 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Advanced paraganglioma | ||||
| Efficacy Data | Objective response rate (ORR) |
25%
|
|||
| Patients Enrolled |
A total of 201 patients with advanced paragangliomas.
|
||||
| MOA of PDC |
The clinical applications of radiolabeled SSAs and DOTA-SSA compounds in imaging and radionuclide therapy have been rapidly increasing over the past 2 decades and have been integrated into the current management guidelines of NETs. Several studies support the safety and clinical efficacy of FDA-approved radionuclide therapies including 177Lu-DOTATATE in advanced GEP NETs and 131I-MIBG in advanced paragangliomas and pheochromocytomas. The utility of SSTR-based imaging in PRRT response assessment and surveillance of NETs remains to be established.
Click to Show/Hide
|
||||
| Description |
Over the past decade, a small number of studies have evaluated the efficacy and safety of PRRT with 177Lu-DOTA-SSA in patients with metastatic or inoperable paragangliomas [66, 67]. A recent meta-analysis of 12 studies including a total of 201 patients with advanced paragangliomas showed an objective response rate of 25%, biochemical response rate of 64%, and disease control rate of 84% after treatment with PRRT (either 90Y- or 177Lu-based agents) [67]. The following minimal treatment-related adverse effects were observed: grade 3 or 4 lymphopenia, thrombocytopenia, neutropenia, and nephrotoxicity in 11%, 9%, 3%, and 4% of the patients, respectively.
Click to Show/Hide
|
||||
| Experiment 203 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Objective response rate (ORR) |
25%
|
|||
| Patients Enrolled |
201 patients with paraganglioma.
|
||||
| Administration Time | 1-10 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The use of PRRT for patients with PPGLs who had high tumor SSTR expression has mainly been reported in retrospective small case series and the protocols varied in all these studies. So far, four meta-analyses (Taieb et al. and Satapathy et al. in 2019 as well as Su et al. and Marretta et al. in 2023) based on PRRT (177Lu/90Y-DOTAT-TATE/TOC) therapy have been reported. The reported number of pooled patients ranged from 179-330 without any complete response. The reported partial response ranged from 20-25% and stable disease from 59-70% giving an objective response rate range of 20-25% and disease control rate range of 8190%. The survival rates were reported by only 3 meta-analyses. The reported progression-free survival were 17-39 months in seven studies by Taieb et al., 10-91 months in 4 studies and a mean of 37.1 months in 2 studies by Satapathy et al., and a median of 31.8 months in 13 studies by Su et al. whereas overall survival were 49.6-68.0 months in 3 studies by Taieb et al., a mean of 54.5 months in 4 studies by Satapathy et al. and a median of 74.3 months in 11 studies by Su et al. Su et al. reports a pooled adverse events data in 274/330 patients which mentions a grade 3/4 hematotoxicity in 4.3% (grade 1-4, 22.3%) and grade 3/4 nephrotoxicity in 1.9% (grade 1-4, 1.9%), and myelodysplastic syndrome in 1 patient. Treatment discontinuation occurred in 4 patients due to thrombocytopenia (n=3) or nephrotoxicity (n=1).
Click to Show/Hide
|
||||
| Experiment 204 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Bronchial carcinoids | ||||
| Efficacy Data | Objective response rate (ORR) |
29.20%
|
|||
| Patients Enrolled |
114 patients with bronchial carcinoids.
|
||||
| Administration Time | 4-6 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
Treatment of bronchial carcinoids is not simple and requires a multidisciplinary approach. Surgery remains the mainstay of treatment for local disease, while in advanced disease, management includes chemotherapy, cold somatostatin analogues, immunotherapy, everolimus, and others target therapies. RLT is an option for selected patients with advanced or metastatic BCs overexpressing SSTR in progression to cold SSAs therapy. Although the experience of [177Lu]Lu-DOTATATE is more limited in BCs than GEP-NET, off-label use can be considered with promising results. A prospective phase II trial in 34 patients with stage IV BCs treated with cumulative activity of 18.5-27.8 GBq in four or five cycles of [177Lu]Lu-DOTATATE documented a DCR of 80% (6% complete response, 27% partial response, and 47% stable disease) and a median PFS of 20 months. In this study, negative prognostic factors were AC histology, tumor thyroid transcription factor-1 (TTF-1) expression, and a positive [18F]FDG PET imaging. Comparable results (median PFS of 17 months) were reported in a group of patients with diffuse extrahepatic metastases treated with RLT after several lines of therapies. Higher activities were administered in a study by van Essen et al., who treated nine patients with metastatic BCs, delivering a cumulative dose of 22.2-29.6 GBq of [177Lu]Lu-DOTATATE, demonstrating an overall response rate comparable to that of other GEP-NET (50% vs. 47%, respectively for BCs and GEP-NET). Moreover, tumor regression was reported in 66.6% of patients, without any outcome discrepancies between TCs and ACs. Comparable median OS and response rate between BCs and others GEP-NETs were reported also in another recent retrospective study. Likewise, the efficacy of RLT in BCs was demonstrated by Brabander et al. in a study involving 443 patients affected by midgut, bronchial, and CUP-NETs treated with 7.4 GBq of [177Lu]Lu-DOTATATE every 8 weeks. The authors found that objective response rate (ORR) for BCs was 30%, and an additional 30% of patients had a stable disease. Nevertheless, median OS for BCs was 52 months versus 71 months for pancreatic-NET. Long term results were analyzed by Mariniello et al. in 114 patients with advanced BCs treated with different RLT protocols. They documented a median PFS and OS of 28.0 and 58.8 months, respectively, while morphological responses were observed in 26.5% of patients. [177Lu]Lu-DOTATATE monotherapy protocol resulted in the highest 5-year OS (61.4%), despite tandem protocol ([90Y]Y-DOTATOC + [177Lu]Lu-DOTATATE) provided the highest response rates (38.1%). Best outcome was reached in patients who underwent RLT in early stage of disease, suggesting that this treatment should also be considered for newly diagnosed unresectable BCs. Moreover, despite most patients having only mild and transient adverse events, patients with hematologic toxicity showed worse survival outcomes.
Click to Show/Hide
|
||||
| Experiment 205 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Bronchial carcinoids | ||||
| Efficacy Data | Objective response rate (ORR) |
33%
|
|||
| Patients Enrolled |
48 patients with bronchial carcinoids.
|
||||
| Administration Time | Median 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
Treatment of bronchial carcinoids is not simple and requires a multidisciplinary approach. Surgery remains the mainstay of treatment for local disease, while in advanced disease, management includes chemotherapy, cold somatostatin analogues, immunotherapy, everolimus, and others target therapies. RLT is an option for selected patients with advanced or metastatic BCs overexpressing SSTR in progression to cold SSAs therapy. Although the experience of [177Lu]Lu-DOTATATE is more limited in BCs than GEP-NET, off-label use can be considered with promising results. A prospective phase II trial in 34 patients with stage IV BCs treated with cumulative activity of 18.5-27.8 GBq in four or five cycles of [177Lu]Lu-DOTATATE documented a DCR of 80% (6% complete response, 27% partial response, and 47% stable disease) and a median PFS of 20 months. In this study, negative prognostic factors were AC histology, tumor thyroid transcription factor-1 (TTF-1) expression, and a positive [18F]FDG PET imaging. Comparable results (median PFS of 17 months) were reported in a group of patients with diffuse extrahepatic metastases treated with RLT after several lines of therapies. Higher activities were administered in a study by van Essen et al., who treated nine patients with metastatic BCs, delivering a cumulative dose of 22.2-29.6 GBq of [177Lu]Lu-DOTATATE, demonstrating an overall response rate comparable to that of other GEP-NET (50% vs. 47%, respectively for BCs and GEP-NET). Moreover, tumor regression was reported in 66.6% of patients, without any outcome discrepancies between TCs and ACs. Comparable median OS and response rate between BCs and others GEP-NETs were reported also in another recent retrospective study. Likewise, the efficacy of RLT in BCs was demonstrated by Brabander et al. in a study involving 443 patients affected by midgut, bronchial, and CUP-NETs treated with 7.4 GBq of [177Lu]Lu-DOTATATE every 8 weeks. The authors found that objective response rate (ORR) for BCs was 30%, and an additional 30% of patients had a stable disease. Nevertheless, median OS for BCs was 52 months versus 71 months for pancreatic-NET. Long term results were analyzed by Mariniello et al. in 114 patients with advanced BCs treated with different RLT protocols. They documented a median PFS and OS of 28.0 and 58.8 months, respectively, while morphological responses were observed in 26.5% of patients. [177Lu]Lu-DOTATATE monotherapy protocol resulted in the highest 5-year OS (61.4%), despite tandem protocol ([90Y]Y-DOTATOC + [177Lu]Lu-DOTATATE) provided the highest response rates (38.1%). Best outcome was reached in patients who underwent RLT in early stage of disease, suggesting that this treatment should also be considered for newly diagnosed unresectable BCs. Moreover, despite most patients having only mild and transient adverse events, patients with hematologic toxicity showed worse survival outcomes.
Click to Show/Hide
|
||||
| Experiment 206 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Objective response rate (ORR) |
35%
|
|||
| Patients Enrolled |
82 patients with CUP-neuroendocrine tumour.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
Unfortunately, the literature lacks specific trials assessing RLT efficacy and safety on CUP-NETs, and the only available literature evidence is provided by mixed trials also including a few patients with SSTR-positive CUP-NETs. In a large cohort of patients treated with [177Lu]Lu-DOTATATE, Brabander et al.reported of 82 CUP-NET patients. Overall, DCR was reached in 78% of patients and median PFS and OS were 29 and 53 months, respectively. These results show that RLT may be effective in CUP-NETs patients, with a response rate intermediate between that obtained in GEP-NETswho showed longer median OS (60 vs. 53 months, respectively)and BCswho showed shorter median PFS (20 vs. 29 months, respectively). These results are consistent with those reported by Demirci et al. who treated 19 CUP-NETs, with an overall DCR of 84.2% and mean PFS and OS of 40 and 48 months, respectively. Once again, RLT outcomes in CUP-NETs were intermediate between those in GEP-NETs and BCs. In a large mixed cohort, Baum et al. treated 151 CUP-NETs (mostly with [177Lu]Lu-DOTATATE, although a small percentage of patients was treated with [90Y]Y-DOTATATE/DOTATOC in the study), obtaining median PFS and OS of 13 and 46 months, respectively. CUP-NETStogether with BCswere associated with a significantly shorter PFS at multivariate analysis if compared to pancreatic NETs despite showing a longer OS (50 vs. 43 months, respectively). Future prospective trials assessing efficacy and safety of RLT in CUP-NETs are desirable.
Click to Show/Hide
|
||||
| Experiment 207 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Phaeochromocytoma | ||||
| Efficacy Data | Objective response rate (ORR) |
36%
|
|||
| Patients Enrolled |
20 patients with phaeochromocytoma and paraganglioma.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
As a consequence, we saw a rising interest in a potential radiolabeled-SST agonist theranostic approach for PPGLs, with several spontaneous studies reporting of the treatment of metastatic or inoperable tumors with Lutathera and other similar radiocompounds, with promising preliminary results. The first RLT experience in PPGLs was reported by van Essen et al. who treated a heterogeneous cohort of patients, including 12 PGLs. Despite the authors reporting lower response rates than those obtained in GEP-NET patients, ORR and DCR were 18% and 73%, respectively. These results are consistent with other reports in the literature. Kong et al. treated 20 PPGLs (8 PHEOs and 12 PGLs) with [117Lu]Lu-DOTATATE, 14 due to uncontrolled secondary hypertension and 6 to radiological PD. A DCR equal to 86% was reached, including five patients with ORR (36%). Of note, 62% of symptomatic patients required a dose reduction of antihypertensive medications, and 57% reported a subjective therapeutic benefit in terms of tumor-related symptoms. Comprehensive PFS was 39 months, including two patients with early recurrence (2 and 5 months post-RLT, respectively) and one patient with remarkable tumor downsizing that allowed a second-step curative liver surgery. Of note, the patient was still disease-free at the time of the study. The results of the largest cohort of PPGLs treated with RLT were reported by Severi et al., who treated 46 patients reaching a comprehensive DCR of 80%. Interestingly, 34 patients treated with [177Lu]Lu-DOTATATE obtained a longer median OS in comparison to those treated with [90Y]Y-DOTATOC (143 vs. 92 months, respectively). According to the authors, this result may be related both to DOTATATEs higher affinity for SSTR2 (which is the type most overexpressed in PPGLs) and to the longer half-life of 177Lu and, consequently, prolonged residence time within the tumor lesions. Moreover, this study reports that sympathetic functioning PPGLs were associated to a shorter median PFS compared to non-functioning PPGLs. Prado-Wohlwend et al. treated nine patients with [177Lu]Lu-DOTATATE and eight with [131I]MIBG, obtaining a PFS of 29 and 18.5 months and a DCR of 88.8% and 62.5%, respectively, for each therapy. Despite the low number of patients involved, a trend for a longer PFS was found for adrenal primary PPGLs treated with [177Lu]Lu-DOTATATE. Nevertheless, the authors suggest performing both [68Ga]Ga-DOTA-SSA PET/CT and [123I]MIBG SPECT/CT in each patient in order to offer a personalized theranostic approach based on the highest uptake intensity in one of the two imaging scans. This is consistent with the results by Jaiswal et al. who found that a baseline SUVmax > 21 at [68Ga]Ga-DOTA-SSA PET/CT is a very strong predictor of response to [177Lu]Lu-DOTATATE (p < 0.0001).
Click to Show/Hide
|
||||
| Experiment 208 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Paraganglioma | ||||
| Efficacy Data | Objective response rate (ORR) |
36%
|
|||
| Patients Enrolled |
20 patients with phaeochromocytoma and paraganglioma.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
As a consequence, we saw a rising interest in a potential radiolabeled-SST agonist theranostic approach for PPGLs, with several spontaneous studies reporting of the treatment of metastatic or inoperable tumors with Lutathera and other similar radiocompounds, with promising preliminary results. The first RLT experience in PPGLs was reported by van Essen et al. who treated a heterogeneous cohort of patients, including 12 PGLs. Despite the authors reporting lower response rates than those obtained in GEP-NET patients, ORR and DCR were 18% and 73%, respectively. These results are consistent with other reports in the literature. Kong et al. treated 20 PPGLs (8 PHEOs and 12 PGLs) with [117Lu]Lu-DOTATATE, 14 due to uncontrolled secondary hypertension and 6 to radiological PD. A DCR equal to 86% was reached, including five patients with ORR (36%). Of note, 62% of symptomatic patients required a dose reduction of antihypertensive medications, and 57% reported a subjective therapeutic benefit in terms of tumor-related symptoms. Comprehensive PFS was 39 months, including two patients with early recurrence (2 and 5 months post-RLT, respectively) and one patient with remarkable tumor downsizing that allowed a second-step curative liver surgery. Of note, the patient was still disease-free at the time of the study. The results of the largest cohort of PPGLs treated with RLT were reported by Severi et al., who treated 46 patients reaching a comprehensive DCR of 80%. Interestingly, 34 patients treated with [177Lu]Lu-DOTATATE obtained a longer median OS in comparison to those treated with [90Y]Y-DOTATOC (143 vs. 92 months, respectively). According to the authors, this result may be related both to DOTATATEs higher affinity for SSTR2 (which is the type most overexpressed in PPGLs) and to the longer half-life of 177Lu and, consequently, prolonged residence time within the tumor lesions. Moreover, this study reports that sympathetic functioning PPGLs were associated to a shorter median PFS compared to non-functioning PPGLs. Prado-Wohlwend et al. treated nine patients with [177Lu]Lu-DOTATATE and eight with [131I]MIBG, obtaining a PFS of 29 and 18.5 months and a DCR of 88.8% and 62.5%, respectively, for each therapy. Despite the low number of patients involved, a trend for a longer PFS was found for adrenal primary PPGLs treated with [177Lu]Lu-DOTATATE. Nevertheless, the authors suggest performing both [68Ga]Ga-DOTA-SSA PET/CT and [123I]MIBG SPECT/CT in each patient in order to offer a personalized theranostic approach based on the highest uptake intensity in one of the two imaging scans. This is consistent with the results by Jaiswal et al. who found that a baseline SUVmax > 21 at [68Ga]Ga-DOTA-SSA PET/CT is a very strong predictor of response to [177Lu]Lu-DOTATATE (p < 0.0001).
Click to Show/Hide
|
||||
| Experiment 209 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Objective response rate (ORR) |
36.80%
|
|||
| Patients Enrolled |
19 patients with CUP-neuroendocrine tumor.
|
||||
| Administration Time | Median 6 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
Unfortunately, the literature lacks specific trials assessing RLT efficacy and safety on CUP-NETs, and the only available literature evidence is provided by mixed trials also including a few patients with SSTR-positive CUP-NETs. In a large cohort of patients treated with [177Lu]Lu-DOTATATE, Brabander et al.reported of 82 CUP-NET patients. Overall, DCR was reached in 78% of patients and median PFS and OS were 29 and 53 months, respectively. These results show that RLT may be effective in CUP-NETs patients, with a response rate intermediate between that obtained in GEP-NETswho showed longer median OS (60 vs. 53 months, respectively)and BCswho showed shorter median PFS (20 vs. 29 months, respectively). These results are consistent with those reported by Demirci et al. who treated 19 CUP-NETs, with an overall DCR of 84.2% and mean PFS and OS of 40 and 48 months, respectively. Once again, RLT outcomes in CUP-NETs were intermediate between those in GEP-NETs and BCs. In a large mixed cohort, Baum et al. treated 151 CUP-NETs (mostly with [177Lu]Lu-DOTATATE, although a small percentage of patients was treated with [90Y]Y-DOTATATE/DOTATOC in the study), obtaining median PFS and OS of 13 and 46 months, respectively. CUP-NETStogether with BCswere associated with a significantly shorter PFS at multivariate analysis if compared to pancreatic NETs despite showing a longer OS (50 vs. 43 months, respectively). Future prospective trials assessing efficacy and safety of RLT in CUP-NETs are desirable.
Click to Show/Hide
|
||||
| Experiment 210 Reporting the Activity Data of This PDC | [18] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumor | ||||
| Efficacy Data | Objective response rate (ORR) |
39%
|
|||
| Patients Enrolled |
Patients treated with Lu-DOTATATE.
|
||||
| Administration Dosage | 100 mCi (3.7 GBq) | ||||
| Description |
The objective response rate of the total group of patients was 39%. Stable disease was reached in 43% of patients. Progression-free survival (PFS) and overall survival (OS) for all NET patients were 29 months [95% confidence interval (CI), 26-33 months] and 63 months (95% CI, 55-72 months). Long-term toxicity included acute leukemia in four patients (0.7%) and myelodysplastic syndrome in nine patients (1.5%).
Click to Show/Hide
|
||||
| Experiment 211 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Bronchial carcinoids | ||||
| Efficacy Data | Objective response rate (ORR) |
39%
|
|||
| Patients Enrolled |
443 patients with bronchial carcinoids.
|
||||
| Administration Dosage | Cumulative dose: 27.8-29.6 GBq | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
Treatment of bronchial carcinoids is not simple and requires a multidisciplinary approach. Surgery remains the mainstay of treatment for local disease, while in advanced disease, management includes chemotherapy, cold somatostatin analogues, immunotherapy, everolimus, and others target therapies. RLT is an option for selected patients with advanced or metastatic BCs overexpressing SSTR in progression to cold SSAs therapy. Although the experience of [177Lu]Lu-DOTATATE is more limited in BCs than GEP-NET, off-label use can be considered with promising results. A prospective phase II trial in 34 patients with stage IV BCs treated with cumulative activity of 18.5-27.8 GBq in four or five cycles of [177Lu]Lu-DOTATATE documented a DCR of 80% (6% complete response, 27% partial response, and 47% stable disease) and a median PFS of 20 months. In this study, negative prognostic factors were AC histology, tumor thyroid transcription factor-1 (TTF-1) expression, and a positive [18F]FDG PET imaging. Comparable results (median PFS of 17 months) were reported in a group of patients with diffuse extrahepatic metastases treated with RLT after several lines of therapies. Higher activities were administered in a study by van Essen et al., who treated nine patients with metastatic BCs, delivering a cumulative dose of 22.2-29.6 GBq of [177Lu]Lu-DOTATATE, demonstrating an overall response rate comparable to that of other GEP-NET (50% vs. 47%, respectively for BCs and GEP-NET). Moreover, tumor regression was reported in 66.6% of patients, without any outcome discrepancies between TCs and ACs. Comparable median OS and response rate between BCs and others GEP-NETs were reported also in another recent retrospective study. Likewise, the efficacy of RLT in BCs was demonstrated by Brabander et al. in a study involving 443 patients affected by midgut, bronchial, and CUP-NETs treated with 7.4 GBq of [177Lu]Lu-DOTATATE every 8 weeks. The authors found that objective response rate (ORR) for BCs was 30%, and an additional 30% of patients had a stable disease. Nevertheless, median OS for BCs was 52 months versus 71 months for pancreatic-NET. Long term results were analyzed by Mariniello et al. in 114 patients with advanced BCs treated with different RLT protocols. They documented a median PFS and OS of 28.0 and 58.8 months, respectively, while morphological responses were observed in 26.5% of patients. [177Lu]Lu-DOTATATE monotherapy protocol resulted in the highest 5-year OS (61.4%), despite tandem protocol ([90Y]Y-DOTATOC + [177Lu]Lu-DOTATATE) provided the highest response rates (38.1%). Best outcome was reached in patients who underwent RLT in early stage of disease, suggesting that this treatment should also be considered for newly diagnosed unresectable BCs. Moreover, despite most patients having only mild and transient adverse events, patients with hematologic toxicity showed worse survival outcomes.
Click to Show/Hide
|
||||
| Experiment 212 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Meningiomas | ||||
| Efficacy Data | Objective response rate (ORR) |
40%
|
|||
| Patients Enrolled |
15 patients with meningiomas.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
A few studies evaluated the efficacy of RLT in meningiomas, administrating 2-5 cycles of [90Y]Y-DOTATATE/-DOTATOC or [177Lu]Lu-DOTATATE/-DOTATOC. In a meta-analysis performed by Mirian et al., RLToffered as mono-therapy or in combination with other oncological treatmentsallowed a comprehensive DCR of 63% in refractory meningiomas. The 6-month PFS rates were 94%, 48%, and 0% for patients with WHO grade I, II, and III meningiomas, respectively, whereas the corresponding 1-year OS rates were 88%, 71%, and 52%, respectively. In a study by Seystahl et al. RLT, offered mainly with [177Lu]Lu-DOTATATE (85% of patients), obtained 6-month PFS rates of 100%, 57%, and 0% for grade I, II, and III refractory meningiomas, respectively. In a recent study on a small group of selected patients affected by WHO grade II refractory meningiomas, 6-month PFS was 85.7% and 1-year PFS was 66.7%. The treatment was safe and well tolerated.
Click to Show/Hide
|
||||
| Experiment 213 Reporting the Activity Data of This PDC | [17] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Objective response rate (ORR) |
42.90%
|
|||
| Patients Enrolled |
7 patients with inoperable, progressive, metastatic, or locally advanced, somatostatin receptor-positive neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | 21.3-30.1 GBq total dose | ||||
| MOA of PDC |
In conclusion, for the first time in Korea, we have conducted a clinical trial of PRRT with SNU-KB-01, a NCA 177Lu-DOTATATE, in patients with SSTR-positive NET. Treatment with SNU-KB-01 was safe and resulted in control of disease in most of the patients with high SSTR expression. The recommended dose per cycle of SNU-KB-01 was determined to be 7.40 GBq for the phase 2 trial. Our results indicate SNU-KB-01 as a potentially safe and efficacious treatment option for NET patients and are expected to provide a wider choice of treatment opportunities for patients with various types of SSTR-positive tumors in Korea.
Click to Show/Hide
|
||||
| Description |
Grade 3 thrombocytopenia occurred in one patient, but no other grade 3 or 4 major hematologic or renal toxicity was observed. The best objective response to SNU-KB-01 was partial response. Overall response rate was 42.9% and disease control rate was 85.7%.
|
||||
| Experiment 214 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Bronchial carcinoids | ||||
| Efficacy Data | Objective response rate (ORR) |
56%
|
|||
| Patients Enrolled |
9 patients with bronchial carcinoids.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
Treatment of bronchial carcinoids is not simple and requires a multidisciplinary approach. Surgery remains the mainstay of treatment for local disease, while in advanced disease, management includes chemotherapy, cold somatostatin analogues, immunotherapy, everolimus, and others target therapies. RLT is an option for selected patients with advanced or metastatic BCs overexpressing SSTR in progression to cold SSAs therapy. Although the experience of [177Lu]Lu-DOTATATE is more limited in BCs than GEP-NET, off-label use can be considered with promising results. A prospective phase II trial in 34 patients with stage IV BCs treated with cumulative activity of 18.5-27.8 GBq in four or five cycles of [177Lu]Lu-DOTATATE documented a DCR of 80% (6% complete response, 27% partial response, and 47% stable disease) and a median PFS of 20 months. In this study, negative prognostic factors were AC histology, tumor thyroid transcription factor-1 (TTF-1) expression, and a positive [18F]FDG PET imaging. Comparable results (median PFS of 17 months) were reported in a group of patients with diffuse extrahepatic metastases treated with RLT after several lines of therapies. Higher activities were administered in a study by van Essen et al., who treated nine patients with metastatic BCs, delivering a cumulative dose of 22.2-29.6 GBq of [177Lu]Lu-DOTATATE, demonstrating an overall response rate comparable to that of other GEP-NET (50% vs. 47%, respectively for BCs and GEP-NET). Moreover, tumor regression was reported in 66.6% of patients, without any outcome discrepancies between TCs and ACs. Comparable median OS and response rate between BCs and others GEP-NETs were reported also in another recent retrospective study. Likewise, the efficacy of RLT in BCs was demonstrated by Brabander et al. in a study involving 443 patients affected by midgut, bronchial, and CUP-NETs treated with 7.4 GBq of [177Lu]Lu-DOTATATE every 8 weeks. The authors found that objective response rate (ORR) for BCs was 30%, and an additional 30% of patients had a stable disease. Nevertheless, median OS for BCs was 52 months versus 71 months for pancreatic-NET. Long term results were analyzed by Mariniello et al. in 114 patients with advanced BCs treated with different RLT protocols. They documented a median PFS and OS of 28.0 and 58.8 months, respectively, while morphological responses were observed in 26.5% of patients. [177Lu]Lu-DOTATATE monotherapy protocol resulted in the highest 5-year OS (61.4%), despite tandem protocol ([90Y]Y-DOTATOC + [177Lu]Lu-DOTATATE) provided the highest response rates (38.1%). Best outcome was reached in patients who underwent RLT in early stage of disease, suggesting that this treatment should also be considered for newly diagnosed unresectable BCs. Moreover, despite most patients having only mild and transient adverse events, patients with hematologic toxicity showed worse survival outcomes.
Click to Show/Hide
|
||||
| Experiment 215 Reporting the Activity Data of This PDC | [14] | ||||
| Indication | Metastatic/Advanced neuroendocrine tumour | ||||
| Efficacy Data | Objective response rate (ORR) |
87.90%
|
|||
| Patients Enrolled |
29 patients with neuroendocrine tumour with progressive disease at the initiation point of PRRT.
|
||||
| Description |
On a separate sub-analysis of 322 NET patients with progressive disease at the initiation point of PRRT, overall response rates (CR + PR + SD) were 93.5%, 88.5%, 89.1 and 87.9% on symptomatic, biochemical, RECIST 1.1 and PERCIST criteria and PFS and OS at 7 years 68.3% and 79.2%, respectively.
|
||||
| Experiment 216 Reporting the Activity Data of This PDC | [14] | ||||
| Indication | Metastatic/Advanced neuroendocrine tumour | ||||
| Efficacy Data | Objective response rate (ORR) |
88.50%
|
|||
| Patients Enrolled |
29 patients with neuroendocrine tumour with progressive disease at the initiation point of PRRT.
|
||||
| Description |
On a separate sub-analysis of 322 NET patients with progressive disease at the initiation point of PRRT, overall response rates (CR + PR + SD) were 93.5%, 88.5%, 89.1 and 87.9% on symptomatic, biochemical, RECIST 1.1 and PERCIST criteria and PFS and OS at 7 years 68.3% and 79.2%, respectively.
|
||||
| Experiment 217 Reporting the Activity Data of This PDC | [14] | ||||
| Indication | Metastatic/Advanced neuroendocrine tumour | ||||
| Efficacy Data | Objective response rate (ORR) |
89.10%
|
|||
| Patients Enrolled |
29 patients with neuroendocrine tumour with progressive disease at the initiation point of PRRT.
|
||||
| Description |
On a separate sub-analysis of 322 NET patients with progressive disease at the initiation point of PRRT, overall response rates (CR + PR + SD) were 93.5%, 88.5%, 89.1 and 87.9% on symptomatic, biochemical, RECIST 1.1 and PERCIST criteria and PFS and OS at 7 years 68.3% and 79.2%, respectively.
|
||||
| Experiment 218 Reporting the Activity Data of This PDC | [14] | ||||
| Indication | Metastatic/Advanced neuroendocrine tumour | ||||
| Efficacy Data | Objective response rate (ORR) |
93.50%
|
|||
| Patients Enrolled |
29 patients with neuroendocrine tumour with progressive disease at the initiation point of PRRT.
|
||||
| Description |
On a separate sub-analysis of 322 NET patients with progressive disease at the initiation point of PRRT, overall response rates (CR + PR + SD) were 93.5%, 88.5%, 89.1 and 87.9% on symptomatic, biochemical, RECIST 1.1 and PERCIST criteria and PFS and OS at 7 years 68.3% and 79.2%, respectively.
|
||||
| Experiment 219 Reporting the Activity Data of This PDC | [31] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumour | ||||
| Efficacy Data | Number of hematopoietic neoplasms |
3
|
|||
| Patients Enrolled |
Patients with gastroenteropancreatic neuroendocrine tumors.
|
||||
| Description |
The expected number of hematopoietic neoplasms based on The Netherlands Cancer Registry data was 3.0, resulting in a relative risk of 2.7 (95% confidence interval, 0.7-10.0).
|
||||
| Experiment 220 Reporting the Activity Data of This PDC | [45] | ||||
| Indication | Primary cardiac paragangliomas | ||||
| Efficacy Data | Neutrophils decrease rate |
4 x 109/L
|
|||
| Patients Enrolled |
47 years old primary cardiac paragangliomas patient.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity of 40.7 GBq (1100 mCi) | ||||
| MOA of PDC |
Cardiac PGL is an exceedingly rare tumor and may lead to hemodynamic compromise. Surgical resection, if indicated, is technically challenging and frequently not feasible. This case demonstrates that personalized 177Lu-DOTATATE PRRT can be considered as a safe and effective palliative treatment for unresectable MIBG negative tumor which can substantially improve quality of life.
Click to Show/Hide
|
||||
| Description |
At the end of the 4 cycles, fatigue, chest pain and dyspnea resolved, and episodes of tachyarrhythmia disappeared. The dosage of metoprolol and prazocin was considerably reduced, decreasing respectively from 125 mg to 50 mg and 2 mg to 1 mg TID. The chromogranin A decreased from 585 to 205 ng/mL and the serum norepinephrine decreased from 104 to 37 nmol/L after 4 cycles.
Click to Show/Hide
|
||||
| Experiment 221 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Advanced paraganglioma | ||||
| Efficacy Data | Neutropenia |
3%
|
|||
| Patients Enrolled |
A total of 201 patients with advanced paragangliomas.
|
||||
| MOA of PDC |
The clinical applications of radiolabeled SSAs and DOTA-SSA compounds in imaging and radionuclide therapy have been rapidly increasing over the past 2 decades and have been integrated into the current management guidelines of NETs. Several studies support the safety and clinical efficacy of FDA-approved radionuclide therapies including 177Lu-DOTATATE in advanced GEP NETs and 131I-MIBG in advanced paragangliomas and pheochromocytomas. The utility of SSTR-based imaging in PRRT response assessment and surveillance of NETs remains to be established.
Click to Show/Hide
|
||||
| Description |
Over the past decade, a small number of studies have evaluated the efficacy and safety of PRRT with 177Lu-DOTA-SSA in patients with metastatic or inoperable paragangliomas [66, 67]. A recent meta-analysis of 12 studies including a total of 201 patients with advanced paragangliomas showed an objective response rate of 25%, biochemical response rate of 64%, and disease control rate of 84% after treatment with PRRT (either 90Y- or 177Lu-based agents) [67]. The following minimal treatment-related adverse effects were observed: grade 3 or 4 lymphopenia, thrombocytopenia, neutropenia, and nephrotoxicity in 11%, 9%, 3%, and 4% of the patients, respectively.
Click to Show/Hide
|
||||
| Experiment 222 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Advanced paraganglioma | ||||
| Efficacy Data | Nephrotoxicity |
4%
|
|||
| Patients Enrolled |
A total of 201 patients with advanced paragangliomas.
|
||||
| MOA of PDC |
The clinical applications of radiolabeled SSAs and DOTA-SSA compounds in imaging and radionuclide therapy have been rapidly increasing over the past 2 decades and have been integrated into the current management guidelines of NETs. Several studies support the safety and clinical efficacy of FDA-approved radionuclide therapies including 177Lu-DOTATATE in advanced GEP NETs and 131I-MIBG in advanced paragangliomas and pheochromocytomas. The utility of SSTR-based imaging in PRRT response assessment and surveillance of NETs remains to be established.
Click to Show/Hide
|
||||
| Description |
Over the past decade, a small number of studies have evaluated the efficacy and safety of PRRT with 177Lu-DOTA-SSA in patients with metastatic or inoperable paragangliomas [66, 67]. A recent meta-analysis of 12 studies including a total of 201 patients with advanced paragangliomas showed an objective response rate of 25%, biochemical response rate of 64%, and disease control rate of 84% after treatment with PRRT (either 90Y- or 177Lu-based agents) [67]. The following minimal treatment-related adverse effects were observed: grade 3 or 4 lymphopenia, thrombocytopenia, neutropenia, and nephrotoxicity in 11%, 9%, 3%, and 4% of the patients, respectively.
Click to Show/Hide
|
||||
| Experiment 223 Reporting the Activity Data of This PDC | [18] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumor | ||||
| Efficacy Data | Myelodysplastic syndrome |
1.50%
|
|||
| Patients Enrolled |
Patients treated with Lu-DOTATATE.
|
||||
| Administration Dosage | 100 mCi (3.7 GBq) | ||||
| Description |
The objective response rate of the total group of patients was 39%. Stable disease was reached in 43% of patients. Progression-free survival (PFS) and overall survival (OS) for all NET patients were 29 months [95% confidence interval (CI), 26-33 months] and 63 months (95% CI, 55-72 months). Long-term toxicity included acute leukemia in four patients (0.7%) and myelodysplastic syndrome in nine patients (1.5%).
Click to Show/Hide
|
||||
| Experiment 224 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Minor response (MR) |
3%
|
|||
| Patients Enrolled |
33 patients with neuroendocrine tumour.
|
||||
| Administration Time | 2-4 cycles | ||||
| Administration Dosage | Mean administered activity during re-treatment: 17.7 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 225 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Minor response (MR) |
3.80%
|
|||
| Patients Enrolled |
26 patients with neuroendocrine tumour.
|
||||
| Administration Time | 2-5 cycles | ||||
| Administration Dosage | Median activity for re-treatment: 16.5 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 226 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Minor response (MR) |
6.10%
|
|||
| Patients Enrolled |
33 patients with neuroendocrine tumour.
|
||||
| Administration Time | 2 cycles | ||||
| Administration Dosage | 7.4 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 227 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Well-differentiated pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Median stroma percentage |
40%
|
|||
| Patients Enrolled |
24 patients with morphologically WD-PanNETs who underwent PRRT preoperatively.
|
||||
| Administration Time | A median of 6 months (interquartile range [iqr] 6; 8 months) from the last cycle of prrt | ||||
| Evaluation Method | Stained with hematoxylin and eosin (H&E) & Formalin-fixed and paraffin-embedded (FFPE) assay | ||||
| Description |
In the PRRT group, the median stroma percentage was 40% compared to 20% in the control group (p < 0.0001).
|
||||
| Experiment 228 Reporting the Activity Data of This PDC | [51] | ||||
| Indication | Negative progressive/Symptomatic locally advanced/Metastatic paragangliomas | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
63%
|
|||
| Patients Enrolled |
9 patients with paragangliomas.
|
||||
| Administration Time | ≥ 4cycles | ||||
| Administration Dosage | > 22.2 GBq) | ||||
| Experiment 229 Reporting the Activity Data of This PDC | [14] | ||||
| Indication | Metastatic/Advanced neuroendocrine tumour | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
71.10%
|
|||
| Patients Enrolled |
468 patients with metastatic/advanced neuroendocrine tumor.
|
||||
| Administration Time | At least two cycles at 10-12 weeks interval | ||||
| Administration Dosage | 5.55 to 7.4 GBq per patient | ||||
| Description |
The median PFS and OS were not reached at a median follow-up of 46 months. Observed PFS and OS at 7 years were 71.1% 95% CI (62.4-79.7%) and 79.4% 95% CI (71.4-86.9%) respectively.
|
||||
| Experiment 230 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Meningiomas | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
2.1 months
|
|||
| Patients Enrolled |
Patients with WHO grade III meningiomas.
|
||||
| Administration Time | Median 3 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
A few studies evaluated the efficacy of RLT in meningiomas, administrating 2-5 cycles of [90Y]Y-DOTATATE/-DOTATOC or [177Lu]Lu-DOTATATE/-DOTATOC. In a meta-analysis performed by Mirian et al., RLToffered as mono-therapy or in combination with other oncological treatmentsallowed a comprehensive DCR of 63% in refractory meningiomas. The 6-month PFS rates were 94%, 48%, and 0% for patients with WHO grade I, II, and III meningiomas, respectively, whereas the corresponding 1-year OS rates were 88%, 71%, and 52%, respectively. In a study by Seystahl et al. RLT, offered mainly with [177Lu]Lu-DOTATATE (85% of patients), obtained 6-month PFS rates of 100%, 57%, and 0% for grade I, II, and III refractory meningiomas, respectively. In a recent study on a small group of selected patients affected by WHO grade II refractory meningiomas, 6-month PFS was 85.7% and 1-year PFS was 66.7%. The treatment was safe and well tolerated.
Click to Show/Hide
|
||||
| Experiment 231 Reporting the Activity Data of This PDC | [52] | ||||
| Indication | Progressive well-differentiated neuroendocrine tumour | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
3.06 months
|
|||
| Patients Enrolled |
122 patients with progressive well-differentiated neuroendocrine tumour (small intestinal, pancreatic, colon, gastric, lung, thymic and unknown primaries).
|
||||
| Administration Time | 1-2 doses of prrt | ||||
| Administration Dosage | 7.4/3.7 GBq | ||||
| Description |
Among patients who received 3-4 doses of PRRT, patients with a CS ≤ 4 experienced a median PFS of not reached (NR) (95% CI 18 - NR) while patients with a CS > 4 experienced a median PFS of 16.92 months (95% CI 13.21 - NR). Among patients who received zero doses of PRRT, patients with a CS ≤ 4 experienced a median PFS of 9.82 months (95% CI 5.78 - 18.6) while patients with a CS > 4 experienced a median PFS of 9.35 months (95% CI 2.43 - 18.1). Among patients who received 1-2 doses of PRRT, patients with a CS ≤ 4 experienced a median PFS of 6.83 months (95% CI NR - NR) while patients with a CS > 4 experienced a median PFS of 3.06 months (95% CI.49 - 8.51) (Log rank-test p <.0001).
Click to Show/Hide
|
||||
| Experiment 232 Reporting the Activity Data of This PDC | [53] | ||||
| Indication | Well-differentiated neuroendocrine tumor | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
3.06 months
|
|||
| Patients Enrolled |
26 patients with WD neuroendocrine tumour with a clinical score (CS) greater than 4 points.
|
||||
| Administration Time | 1 or 2 doses | ||||
| MOA of PDC |
Increases in CS were associated with worsening PFS in the validation cohort, validating findings from the original cohort. These findings suggest that the CS, to our knowledge, represents the first clinical metric to estimate anticipated benefit from PRRT for patients with WD NETs and may be a clinical tool for patients being considered for PRRT.
|
||||
| Description |
Among patients who received 1 or 2 doses of PRRT, patients with a CS less than or equal to 4 points experienced a median PFS of 6.83 months (95% CI, 4.37 months to NR) whereas patients with a CS greater than 4 points experienced a median PFS of 3.06 months (95% CI 1.25-7.16 months.
|
||||
| Experiment 233 Reporting the Activity Data of This PDC | [54] | ||||
| Indication | Primary cardiac paragangliomas | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
3.2 months
|
|||
| Patients Enrolled |
7 patients with biopsy-proven neuroendocrine tumour.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | 200 mCi (7.4 GBq) | ||||
| Evaluation Method | 68Ga-DOTATATE PET/CT assay | ||||
| Description |
The median time from the last cycle to PD was 3.2 months (range, 1.1-4.6 months). The median progression-free survival was 7.7 months (95% confidence interval, 5.7-9.8 months).
|
||||
| Experiment 234 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
6 months
|
|||
| Patients Enrolled |
35 patients with neuroendocrine tumour.
|
||||
| Administration Time | 1-4 cycles | ||||
| Administration Dosage | Median cumulative activity 44 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 235 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Meningiomas | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
6 months
|
|||
| Patients Enrolled |
8 patients with WHO grade II meningiomas.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
A few studies evaluated the efficacy of RLT in meningiomas, administrating 2-5 cycles of [90Y]Y-DOTATATE/-DOTATOC or [177Lu]Lu-DOTATATE/-DOTATOC. In a meta-analysis performed by Mirian et al., RLToffered as mono-therapy or in combination with other oncological treatmentsallowed a comprehensive DCR of 63% in refractory meningiomas. The 6-month PFS rates were 94%, 48%, and 0% for patients with WHO grade I, II, and III meningiomas, respectively, whereas the corresponding 1-year OS rates were 88%, 71%, and 52%, respectively. In a study by Seystahl et al. RLT, offered mainly with [177Lu]Lu-DOTATATE (85% of patients), obtained 6-month PFS rates of 100%, 57%, and 0% for grade I, II, and III refractory meningiomas, respectively. In a recent study on a small group of selected patients affected by WHO grade II refractory meningiomas, 6-month PFS was 85.7% and 1-year PFS was 66.7%. The treatment was safe and well tolerated.
Click to Show/Hide
|
||||
| Experiment 236 Reporting the Activity Data of This PDC | [52] | ||||
| Indication | Progressive well-differentiated neuroendocrine tumour | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
6.83 months
|
|||
| Patients Enrolled |
122 patients with progressive well-differentiated neuroendocrine tumour (small intestinal, pancreatic, colon, gastric, lung, thymic and unknown primaries).
|
||||
| Administration Time | 1-2 doses of prrt | ||||
| Administration Dosage | 7.4/3.7 GBq | ||||
| Description |
Among patients who received 3-4 doses of PRRT, patients with a CS ≤ 4 experienced a median PFS of not reached (NR) (95% CI 18 - NR) while patients with a CS > 4 experienced a median PFS of 16.92 months (95% CI 13.21 - NR). Among patients who received zero doses of PRRT, patients with a CS ≤ 4 experienced a median PFS of 9.82 months (95% CI 5.78 - 18.6) while patients with a CS > 4 experienced a median PFS of 9.35 months (95% CI 2.43 - 18.1). Among patients who received 1-2 doses of PRRT, patients with a CS ≤ 4 experienced a median PFS of 6.83 months (95% CI NR - NR) while patients with a CS > 4 experienced a median PFS of 3.06 months (95% CI.49 - 8.51) (Log rank-test p <.0001).
Click to Show/Hide
|
||||
| Experiment 237 Reporting the Activity Data of This PDC | [53] | ||||
| Indication | Well-differentiated neuroendocrine tumor | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
6.83 months
|
|||
| Patients Enrolled |
26 patients with WD neuroendocrine tumour with a clinical score (CS) less than or equal to 4 points.
|
||||
| Administration Time | 1 or 2 doses | ||||
| MOA of PDC |
Increases in CS were associated with worsening PFS in the validation cohort, validating findings from the original cohort. These findings suggest that the CS, to our knowledge, represents the first clinical metric to estimate anticipated benefit from PRRT for patients with WD NETs and may be a clinical tool for patients being considered for PRRT.
|
||||
| Description |
Among patients who received 1 or 2 doses of PRRT, patients with a CS less than or equal to 4 points experienced a median PFS of 6.83 months (95% CI, 4.37 months to NR) whereas patients with a CS greater than 4 points experienced a median PFS of 3.06 months (95% CI 1.25-7.16 months.
|
||||
| Experiment 238 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Meningiomas | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
7.6 months
|
|||
| Patients Enrolled |
Patients with WHO grade II meningiomas.
|
||||
| Administration Time | Median 3 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
A few studies evaluated the efficacy of RLT in meningiomas, administrating 2-5 cycles of [90Y]Y-DOTATATE/-DOTATOC or [177Lu]Lu-DOTATATE/-DOTATOC. In a meta-analysis performed by Mirian et al., RLToffered as mono-therapy or in combination with other oncological treatmentsallowed a comprehensive DCR of 63% in refractory meningiomas. The 6-month PFS rates were 94%, 48%, and 0% for patients with WHO grade I, II, and III meningiomas, respectively, whereas the corresponding 1-year OS rates were 88%, 71%, and 52%, respectively. In a study by Seystahl et al. RLT, offered mainly with [177Lu]Lu-DOTATATE (85% of patients), obtained 6-month PFS rates of 100%, 57%, and 0% for grade I, II, and III refractory meningiomas, respectively. In a recent study on a small group of selected patients affected by WHO grade II refractory meningiomas, 6-month PFS was 85.7% and 1-year PFS was 66.7%. The treatment was safe and well tolerated.
Click to Show/Hide
|
||||
| Experiment 239 Reporting the Activity Data of This PDC | [54] | ||||
| Indication | Primary cardiac paragangliomas | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
7.7 months
|
|||
| Patients Enrolled |
7 patients with biopsy-proven neuroendocrine tumour.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | 200 mCi (7.4 GBq) | ||||
| Evaluation Method | 68Ga-DOTATATE PET/CT assay | ||||
| Description |
The median time from the last cycle to PD was 3.2 months (range, 1.1-4.6 months). The median progression-free survival was 7.7 months (95% confidence interval, 5.7-9.8 months).
|
||||
| Experiment 240 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Meningiomas | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
7.8 months
|
|||
| Patients Enrolled |
15 patients with meningiomas.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
A few studies evaluated the efficacy of RLT in meningiomas, administrating 2-5 cycles of [90Y]Y-DOTATATE/-DOTATOC or [177Lu]Lu-DOTATATE/-DOTATOC. In a meta-analysis performed by Mirian et al., RLToffered as mono-therapy or in combination with other oncological treatmentsallowed a comprehensive DCR of 63% in refractory meningiomas. The 6-month PFS rates were 94%, 48%, and 0% for patients with WHO grade I, II, and III meningiomas, respectively, whereas the corresponding 1-year OS rates were 88%, 71%, and 52%, respectively. In a study by Seystahl et al. RLT, offered mainly with [177Lu]Lu-DOTATATE (85% of patients), obtained 6-month PFS rates of 100%, 57%, and 0% for grade I, II, and III refractory meningiomas, respectively. In a recent study on a small group of selected patients affected by WHO grade II refractory meningiomas, 6-month PFS was 85.7% and 1-year PFS was 66.7%. The treatment was safe and well tolerated.
Click to Show/Hide
|
||||
| Experiment 241 Reporting the Activity Data of This PDC | [52] | ||||
| Indication | Progressive well-differentiated neuroendocrine tumour | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
9.35 months
|
|||
| Patients Enrolled |
122 patients with progressive well-differentiated neuroendocrine tumour (small intestinal, pancreatic, colon, gastric, lung, thymic and unknown primaries).
|
||||
| Administration Time | Zero doses of prrt | ||||
| Administration Dosage | 7.4 /3.7 GBq ( GBq) | ||||
| Description |
Among patients who received 3-4 doses of PRRT, patients with a CS ≤ 4 experienced a median PFS of not reached (NR) (95% CI 18 - NR) while patients with a CS > 4 experienced a median PFS of 16.92 months (95% CI 13.21 - NR). Among patients who received zero doses of PRRT, patients with a CS ≤ 4 experienced a median PFS of 9.82 months (95% CI 5.78 - 18.6) while patients with a CS > 4 experienced a median PFS of 9.35 months (95% CI 2.43 - 18.1). Among patients who received 1-2 doses of PRRT, patients with a CS ≤ 4 experienced a median PFS of 6.83 months (95% CI NR - NR) while patients with a CS > 4 experienced a median PFS of 3.06 months (95% CI.49 - 8.51) (Log rank-test p <.0001).
Click to Show/Hide
|
||||
| Experiment 242 Reporting the Activity Data of This PDC | [52] | ||||
| Indication | Progressive well-differentiated neuroendocrine tumour | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
9.82 months
|
|||
| Patients Enrolled |
122 patients with progressive well-differentiated neuroendocrine tumour (small intestinal, pancreatic, colon, gastric, lung, thymic and unknown primaries).
|
||||
| Administration Time | Zero doses of prrt | ||||
| Administration Dosage | 7.4 /3.7 GBq ( GBq) | ||||
| Description |
Among patients who received 3-4 doses of PRRT, patients with a CS ≤ 4 experienced a median PFS of not reached (NR) (95% CI 18 - NR) while patients with a CS > 4 experienced a median PFS of 16.92 months (95% CI 13.21 - NR). Among patients who received zero doses of PRRT, patients with a CS ≤ 4 experienced a median PFS of 9.82 months (95% CI 5.78 - 18.6) while patients with a CS > 4 experienced a median PFS of 9.35 months (95% CI 2.43 - 18.1). Among patients who received 1-2 doses of PRRT, patients with a CS ≤ 4 experienced a median PFS of 6.83 months (95% CI NR - NR) while patients with a CS > 4 experienced a median PFS of 3.06 months (95% CI.49 - 8.51) (Log rank-test p <.0001).
Click to Show/Hide
|
||||
| Experiment 243 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Inoperable or metastatic paragangliomas | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
10 months
|
|||
| Patients Enrolled |
Patients with inoperable or metastatic paragangliomas and pheochromocytomas with metastatic pheochromocytomas.
|
||||
| Administration Time | Four cycles | ||||
| Administration Dosage | 7.4 Gb per cycle | ||||
| Description |
In 20 patients with baseline disease progression, tumour control was observed in 17 (85%); the median progression-free survival was 91 months in patients with parasympathetic PGLs, 13 months in patients with sympathetic PGLs and 10 months in patients with metastatic PCCs.
|
||||
| Experiment 244 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Inoperable or metastatic pheochromocytomas | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
10 months
|
|||
| Patients Enrolled |
Patients with inoperable or metastatic paragangliomas and pheochromocytomas with metastatic pheochromocytomas.
|
||||
| Administration Time | Four cycles | ||||
| Administration Dosage | 7.4 Gb per cycle | ||||
| Description |
In 20 patients with baseline disease progression, tumour control was observed in 17 (85%); the median progression-free survival was 91 months in patients with parasympathetic PGLs, 13 months in patients with sympathetic PGLs and 10 months in patients with metastatic PCCs.
|
||||
| Experiment 245 Reporting the Activity Data of This PDC | [53] | ||||
| Indication | Well-differentiated neuroendocrine tumor | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
12.55 months
|
|||
| Patients Enrolled |
82 patients with WD neuroendocrine tumour with a clinical score (CS) greater than 4 points.
|
||||
| Administration Time | 0 doses | ||||
| MOA of PDC |
Increases in CS were associated with worsening PFS in the validation cohort, validating findings from the original cohort. These findings suggest that the CS, to our knowledge, represents the first clinical metric to estimate anticipated benefit from PRRT for patients with WD NETs and may be a clinical tool for patients being considered for PRRT.
|
||||
| Description |
Among patients who received 0 doses of PRRT, patients with a CS less than or equal to 4 points experienced a median PFS of 23.52 months (95% CI, 16.76-26.94 months) whereas patients with a CS greater than 4 points experienced a median PFS of 12.55 months (95% CI, 4.99-14.95).
|
||||
| Experiment 246 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Inoperable or metastatic paragangliomas | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
13 months
|
|||
| Patients Enrolled |
Patients with inoperable or metastatic paragangliomas and pheochromocytomas with sympathetic paragangliomas.
|
||||
| Administration Time | Four cycles | ||||
| Administration Dosage | 7.4 Gb per cycle | ||||
| Description |
In 20 patients with baseline disease progression, tumour control was observed in 17 (85%); the median progression-free survival was 91 months in patients with parasympathetic PGLs, 13 months in patients with sympathetic PGLs and 10 months in patients with metastatic PCCs.
|
||||
| Experiment 247 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Inoperable or metastatic pheochromocytomas | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
13 months
|
|||
| Patients Enrolled |
Patients with inoperable or metastatic paragangliomas and pheochromocytomas with sympathetic paragangliomas.
|
||||
| Administration Time | Four cycles | ||||
| Administration Dosage | 7.4 Gb per cycle | ||||
| Description |
In 20 patients with baseline disease progression, tumour control was observed in 17 (85%); the median progression-free survival was 91 months in patients with parasympathetic PGLs, 13 months in patients with sympathetic PGLs and 10 months in patients with metastatic PCCs.
|
||||
| Experiment 248 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
13 months
|
|||
| Patients Enrolled |
33 patients with neuroendocrine tumour.
|
||||
| Administration Time | 2-4 cycles | ||||
| Administration Dosage | Mean administered activity during re-treatment: 17.7 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 249 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
13 months
|
|||
| Patients Enrolled |
1048 patients with CUP-neuroendocrine tumour.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
Unfortunately, the literature lacks specific trials assessing RLT efficacy and safety on CUP-NETs, and the only available literature evidence is provided by mixed trials also including a few patients with SSTR-positive CUP-NETs. In a large cohort of patients treated with [177Lu]Lu-DOTATATE, Brabander et al.reported of 82 CUP-NET patients. Overall, DCR was reached in 78% of patients and median PFS and OS were 29 and 53 months, respectively. These results show that RLT may be effective in CUP-NETs patients, with a response rate intermediate between that obtained in GEP-NETswho showed longer median OS (60 vs. 53 months, respectively)and BCswho showed shorter median PFS (20 vs. 29 months, respectively). These results are consistent with those reported by Demirci et al. who treated 19 CUP-NETs, with an overall DCR of 84.2% and mean PFS and OS of 40 and 48 months, respectively. Once again, RLT outcomes in CUP-NETs were intermediate between those in GEP-NETs and BCs. In a large mixed cohort, Baum et al. treated 151 CUP-NETs (mostly with [177Lu]Lu-DOTATATE, although a small percentage of patients was treated with [90Y]Y-DOTATATE/DOTATOC in the study), obtaining median PFS and OS of 13 and 46 months, respectively. CUP-NETStogether with BCswere associated with a significantly shorter PFS at multivariate analysis if compared to pancreatic NETs despite showing a longer OS (50 vs. 43 months, respectively). Future prospective trials assessing efficacy and safety of RLT in CUP-NETs are desirable.
Click to Show/Hide
|
||||
| Experiment 250 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
13.1 months
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Nineteen patients, all with progressive, heavily treated disease, were identified. Sites of tumor origin were: pancreas (74%), small bowel (11%), rectum (11%), and lung (5%); median Ki-67 was 32% (range 22-56). Thirteen patients (68%) completed all four 177Lu-DOTATATE cycles. Best response (N = 18 evaluable) was: 5/18 (28%) partial response, 8/18 (44%) stable disease, and 5/18 (28%) disease progression. Median PFS was 13.1 months (95% CI: 8.7-20.9).
Click to Show/Hide
|
||||
| Experiment 251 Reporting the Activity Data of This PDC | [49] | ||||
| Indication | Bronchial and gastroenteropancreatic neuroendocrine tumour | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
14.2 months
|
|||
| Patients Enrolled |
Patients with bronchial and gastroenteropancreatic neuroendocrine tumours.
|
||||
| Administration Time | Over two cycles | ||||
| Administration Dosage | 14.8 GBq | ||||
| Description |
Median PFS was 14.6 months (95% CI 12.4-16.9) following R-PRRT and 14.2 months (95% CI 9.8-18.5) following RR-PRRT.
|
||||
| Experiment 252 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
14.2 months
|
|||
| Patients Enrolled |
13 Re-retreatment neuroendocrine tumour patients.
|
||||
| Administration Time | 2 cycles | ||||
| Administration Dosage | Median cumulative dose: 59.7 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 253 Reporting the Activity Data of This PDC | [49] | ||||
| Indication | Bronchial and gastroenteropancreatic neuroendocrine tumour | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
14.6 months
|
|||
| Patients Enrolled |
Patients with bronchial and gastroenteropancreatic neuroendocrine tumours.
|
||||
| Administration Time | Over two cycles | ||||
| Administration Dosage | 14.8 GBq | ||||
| Description |
Median PFS was 14.6 months (95% CI 12.4-16.9) following R-PRRT and 14.2 months (95% CI 9.8-18.5) following RR-PRRT.
|
||||
| Experiment 254 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
14.6 months
|
|||
| Patients Enrolled |
168 patients with neuroendocrine tumour.
|
||||
| Administration Time | 2 cycles | ||||
| Administration Dosage | Median cumulative dose: 44.7 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 255 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Atypical bronchial carcinoids | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
15.7 months
|
|||
| Patients Enrolled |
34 patients with bronchial carcinoids.
|
||||
| Administration Time | 4-5 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
Treatment of bronchial carcinoids is not simple and requires a multidisciplinary approach. Surgery remains the mainstay of treatment for local disease, while in advanced disease, management includes chemotherapy, cold somatostatin analogues, immunotherapy, everolimus, and others target therapies. RLT is an option for selected patients with advanced or metastatic BCs overexpressing SSTR in progression to cold SSAs therapy. Although the experience of [177Lu]Lu-DOTATATE is more limited in BCs than GEP-NET, off-label use can be considered with promising results. A prospective phase II trial in 34 patients with stage IV BCs treated with cumulative activity of 18.5-27.8 GBq in four or five cycles of [177Lu]Lu-DOTATATE documented a DCR of 80% (6% complete response, 27% partial response, and 47% stable disease) and a median PFS of 20 months. In this study, negative prognostic factors were AC histology, tumor thyroid transcription factor-1 (TTF-1) expression, and a positive [18F]FDG PET imaging. Comparable results (median PFS of 17 months) were reported in a group of patients with diffuse extrahepatic metastases treated with RLT after several lines of therapies. Higher activities were administered in a study by van Essen et al., who treated nine patients with metastatic BCs, delivering a cumulative dose of 22.2-29.6 GBq of [177Lu]Lu-DOTATATE, demonstrating an overall response rate comparable to that of other GEP-NET (50% vs. 47%, respectively for BCs and GEP-NET). Moreover, tumor regression was reported in 66.6% of patients, without any outcome discrepancies between TCs and ACs. Comparable median OS and response rate between BCs and others GEP-NETs were reported also in another recent retrospective study. Likewise, the efficacy of RLT in BCs was demonstrated by Brabander et al. in a study involving 443 patients affected by midgut, bronchial, and CUP-NETs treated with 7.4 GBq of [177Lu]Lu-DOTATATE every 8 weeks. The authors found that objective response rate (ORR) for BCs was 30%, and an additional 30% of patients had a stable disease. Nevertheless, median OS for BCs was 52 months versus 71 months for pancreatic-NET. Long term results were analyzed by Mariniello et al. in 114 patients with advanced BCs treated with different RLT protocols. They documented a median PFS and OS of 28.0 and 58.8 months, respectively, while morphological responses were observed in 26.5% of patients. [177Lu]Lu-DOTATATE monotherapy protocol resulted in the highest 5-year OS (61.4%), despite tandem protocol ([90Y]Y-DOTATOC + [177Lu]Lu-DOTATATE) provided the highest response rates (38.1%). Best outcome was reached in patients who underwent RLT in early stage of disease, suggesting that this treatment should also be considered for newly diagnosed unresectable BCs. Moreover, despite most patients having only mild and transient adverse events, patients with hematologic toxicity showed worse survival outcomes.
Click to Show/Hide
|
||||
| Experiment 256 Reporting the Activity Data of This PDC | [52] | ||||
| Indication | Progressive well-differentiated neuroendocrine tumour | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
16.92 months
|
|||
| Patients Enrolled |
122 patients with progressive well-differentiated neuroendocrine tumour (small intestinal, pancreatic, colon, gastric, lung, thymic and unknown primaries).
|
||||
| Administration Time | 3-4 doses of prrt | ||||
| Administration Dosage | 7.4/3.7 GBq | ||||
| Description |
Among patients who received 3-4 doses of PRRT, patients with a CS ≤ 4 experienced a median PFS of not reached (NR) (95% CI 18 - NR) while patients with a CS > 4 experienced a median PFS of 16.92 months (95% CI 13.21 - NR). Among patients who received zero doses of PRRT, patients with a CS ≤ 4 experienced a median PFS of 9.82 months (95% CI 5.78 - 18.6) while patients with a CS > 4 experienced a median PFS of 9.35 months (95% CI 2.43 - 18.1). Among patients who received 1-2 doses of PRRT, patients with a CS ≤ 4 experienced a median PFS of 6.83 months (95% CI NR - NR) while patients with a CS > 4 experienced a median PFS of 3.06 months (95% CI.49 - 8.51) (Log rank-test p <.0001).
Click to Show/Hide
|
||||
| Experiment 257 Reporting the Activity Data of This PDC | [53] | ||||
| Indication | Well-differentiated neuroendocrine tumor | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
16.92 months
|
|||
| Patients Enrolled |
140 patients who has well-differentiated neuroendocrine tumor with a clinical score (CS) greater than 4 points.
|
||||
| Administration Time | 3 or 4 doses | ||||
| MOA of PDC |
Increases in CS were associated with worsening PFS in the validation cohort, validating findings from the original cohort. These findings suggest that the CS, to our knowledge, represents the first clinical metric to estimate anticipated benefit from PRRT for patients with WD NETs and may be a clinical tool for patients being considered for PRRT.
|
||||
| Description |
Among patients who received 3 or 4 doses of PRRT, patients with a CS less than or equal to 4 points experienced a median PFS of not reached (NR) (95% CI, NR-NR) whereas patients with a CS greater than 4 points experienced a median PFS of 16.92 months (95% CI, 13.50-24.74 months).
|
||||
| Experiment 258 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
17 months
|
|||
| Patients Enrolled |
33 patients with neuroendocrine tumour.
|
||||
| Administration Time | 2 cycles | ||||
| Administration Dosage | 7.4 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 259 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
17.5 months
|
|||
| Patients Enrolled |
18 patients with neuroendocrine tumour.
|
||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 260 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
17.5 months
|
|||
| Patients Enrolled |
Patients with neuroendocrine tumour (Retreatment).
|
||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 261 Reporting the Activity Data of This PDC | [48] | ||||
| Indication | Functioning pancreatic neuroendocrine tumour | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
18.1
|
|||
| Patients Enrolled |
Patients with functioning pancreatic neuroendocrine tumors.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | 7.4 GBq/cycle | ||||
| Description |
Subacute hematological toxicity, grade 3 or 4 occurred in 4 patients (12%) and a hormonal crisis in 3 patients (9%). PRRT resulted in partial or complete response in 59% of patients and the disease control rate was 78% in patients with baseline progression. 71% of patients with uncontrolled symptoms had a reduction of symptoms and a more than 80% decrease of circulating hormone levels was measured during follow-up. After PRRT, median progression-free survival was 18.1 months (interquartile range: 3.3 to 35.7) with a concurrent increase in QOL.
Click to Show/Hide
|
||||
| Experiment 262 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
18.9 months
|
|||
| Patients Enrolled |
15 patients with neuroendocrine tumour.
|
||||
| Administration Time | 3-6 cycles | ||||
| Administration Dosage | Median cumulative activity: 63.9 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 263 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Typical bronchial carcinoids | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
20.1 months
|
|||
| Patients Enrolled |
34 patients with bronchial carcinoids.
|
||||
| Administration Time | 4-5 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
Treatment of bronchial carcinoids is not simple and requires a multidisciplinary approach. Surgery remains the mainstay of treatment for local disease, while in advanced disease, management includes chemotherapy, cold somatostatin analogues, immunotherapy, everolimus, and others target therapies. RLT is an option for selected patients with advanced or metastatic BCs overexpressing SSTR in progression to cold SSAs therapy. Although the experience of [177Lu]Lu-DOTATATE is more limited in BCs than GEP-NET, off-label use can be considered with promising results. A prospective phase II trial in 34 patients with stage IV BCs treated with cumulative activity of 18.5-27.8 GBq in four or five cycles of [177Lu]Lu-DOTATATE documented a DCR of 80% (6% complete response, 27% partial response, and 47% stable disease) and a median PFS of 20 months. In this study, negative prognostic factors were AC histology, tumor thyroid transcription factor-1 (TTF-1) expression, and a positive [18F]FDG PET imaging. Comparable results (median PFS of 17 months) were reported in a group of patients with diffuse extrahepatic metastases treated with RLT after several lines of therapies. Higher activities were administered in a study by van Essen et al., who treated nine patients with metastatic BCs, delivering a cumulative dose of 22.2-29.6 GBq of [177Lu]Lu-DOTATATE, demonstrating an overall response rate comparable to that of other GEP-NET (50% vs. 47%, respectively for BCs and GEP-NET). Moreover, tumor regression was reported in 66.6% of patients, without any outcome discrepancies between TCs and ACs. Comparable median OS and response rate between BCs and others GEP-NETs were reported also in another recent retrospective study. Likewise, the efficacy of RLT in BCs was demonstrated by Brabander et al. in a study involving 443 patients affected by midgut, bronchial, and CUP-NETs treated with 7.4 GBq of [177Lu]Lu-DOTATATE every 8 weeks. The authors found that objective response rate (ORR) for BCs was 30%, and an additional 30% of patients had a stable disease. Nevertheless, median OS for BCs was 52 months versus 71 months for pancreatic-NET. Long term results were analyzed by Mariniello et al. in 114 patients with advanced BCs treated with different RLT protocols. They documented a median PFS and OS of 28.0 and 58.8 months, respectively, while morphological responses were observed in 26.5% of patients. [177Lu]Lu-DOTATATE monotherapy protocol resulted in the highest 5-year OS (61.4%), despite tandem protocol ([90Y]Y-DOTATOC + [177Lu]Lu-DOTATATE) provided the highest response rates (38.1%). Best outcome was reached in patients who underwent RLT in early stage of disease, suggesting that this treatment should also be considered for newly diagnosed unresectable BCs. Moreover, despite most patients having only mild and transient adverse events, patients with hematologic toxicity showed worse survival outcomes.
Click to Show/Hide
|
||||
| Experiment 264 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
22 months
|
|||
| Patients Enrolled |
26 patients with neuroendocrine tumour.
|
||||
| Administration Time | 2-5 cycles | ||||
| Administration Dosage | Median activity for re-treatment: 16.5 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 265 Reporting the Activity Data of This PDC | [26] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumor | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
23 months
|
|||
| Patients Enrolled |
Patients with advanced GEP-neuroendocrine tumour.
|
||||
| Administration Time | Four or five cycles | ||||
| Administration Dosage | 18.5 GBq/27.8 GBq | ||||
| Description |
The median progression-free survival (PFS) was 23 months (95% CI 14.9-31.0 months) and the median overall survival was 52.9 months (95% CI 17.1-68.9 months).
|
||||
| Experiment 266 Reporting the Activity Data of This PDC | [20] | ||||
| Indication | SSR positive lung neuroendocrine tumor | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
23 months
|
|||
| Patients Enrolled |
48 patients with neuroendocrine tumour.
|
||||
| Administration Time | 1-4 cycles | ||||
| Administration Dosage | Median cumulative activity of 27 GBq (range, 6-43 GBq) | ||||
| MOA of PDC |
In patients with advanced progressive lung NET and satisfactory SSR expression, 177Lu-DOTATATE is effective and safe, with a high DCR and encouraging PFS and OS. Further prospective studies comparing 177Lu-DOTATATE with other systemic options are warranted.
|
||||
| Description |
Of 48 patients (median age, 63 y; 13 women), 43 (90%) had AC and 5 (10%) TC. Almost all patients (47, 98%) were treated due to progression. Most patients (40, 83%) received somatostatin analogs, and 10 patients (20%) had prior everolimus, chemotherapy, or both. All patients had high SSR expression (≥ modified Krenning score 3) on pretreatment 68Ga-DOTATATE PET/CT. Patients received a median 4 (range, 1-4) cycles of 177Lu-DOTATATE (33% with concurrent radiosensitizing chemotherapy) to a median cumulative activity of 27 GBq (range, 6-43GBq). At a median follow-up of 42 mo, the median PFS and OS were 23 mo (95% CI, 18-28 mo) and 59 mo (95% CI, 50-not reached [NR]), respectively. Of 40 patients with RECIST-measurable disease and 39 patients with available 68Ga-DOTATATE PET/CT, response categories were partial response, 20% (95% CI, 10%-35%) and 44% (95% CI, 30%-59%); stable disease, 68% (95% CI, 52%-80%) and 44% (95% CI, 30%-59%); and progressive disease, 12% (95% CI, 5%-27%) by both, respectively. There was a moderate concordance between response categories by RECIST and 68Ga-DOTATATE PET/CT, weighted of 0.51 (95% CI, 0.21-0.68).
Click to Show/Hide
|
||||
| Experiment 267 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | PPGLa neuroendocrine tumor | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
23 months
|
|||
| Patients Enrolled |
11 patients with PPGLa neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 268 Reporting the Activity Data of This PDC | [53] | ||||
| Indication | Well-differentiated neuroendocrine tumor | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
23.52 months
|
|||
| Patients Enrolled |
82 patients with WD neuroendocrine tumour with a clinical score (CS) less than or equal to 4 points.
|
||||
| Administration Time | 0 doses | ||||
| MOA of PDC |
Increases in CS were associated with worsening PFS in the validation cohort, validating findings from the original cohort. These findings suggest that the CS, to our knowledge, represents the first clinical metric to estimate anticipated benefit from PRRT for patients with WD NETs and may be a clinical tool for patients being considered for PRRT.
|
||||
| Description |
Among patients who received 0 doses of PRRT, patients with a CS less than or equal to 4 points experienced a median PFS of 23.52 months (95% CI, 16.76-26.94 months) whereas patients with a CS greater than 4 points experienced a median PFS of 12.55 months (95% CI, 4.99-14.95).
|
||||
| Experiment 269 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
26 months
|
|||
| Patients Enrolled |
78 patients with pancreatic neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 270 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Bronchial carcinoids | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
28 months
|
|||
| Patients Enrolled |
114 patients with bronchial carcinoids.
|
||||
| Administration Time | 4-6 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
Treatment of bronchial carcinoids is not simple and requires a multidisciplinary approach. Surgery remains the mainstay of treatment for local disease, while in advanced disease, management includes chemotherapy, cold somatostatin analogues, immunotherapy, everolimus, and others target therapies. RLT is an option for selected patients with advanced or metastatic BCs overexpressing SSTR in progression to cold SSAs therapy. Although the experience of [177Lu]Lu-DOTATATE is more limited in BCs than GEP-NET, off-label use can be considered with promising results. A prospective phase II trial in 34 patients with stage IV BCs treated with cumulative activity of 18.5-27.8 GBq in four or five cycles of [177Lu]Lu-DOTATATE documented a DCR of 80% (6% complete response, 27% partial response, and 47% stable disease) and a median PFS of 20 months. In this study, negative prognostic factors were AC histology, tumor thyroid transcription factor-1 (TTF-1) expression, and a positive [18F]FDG PET imaging. Comparable results (median PFS of 17 months) were reported in a group of patients with diffuse extrahepatic metastases treated with RLT after several lines of therapies. Higher activities were administered in a study by van Essen et al., who treated nine patients with metastatic BCs, delivering a cumulative dose of 22.2-29.6 GBq of [177Lu]Lu-DOTATATE, demonstrating an overall response rate comparable to that of other GEP-NET (50% vs. 47%, respectively for BCs and GEP-NET). Moreover, tumor regression was reported in 66.6% of patients, without any outcome discrepancies between TCs and ACs. Comparable median OS and response rate between BCs and others GEP-NETs were reported also in another recent retrospective study. Likewise, the efficacy of RLT in BCs was demonstrated by Brabander et al. in a study involving 443 patients affected by midgut, bronchial, and CUP-NETs treated with 7.4 GBq of [177Lu]Lu-DOTATATE every 8 weeks. The authors found that objective response rate (ORR) for BCs was 30%, and an additional 30% of patients had a stable disease. Nevertheless, median OS for BCs was 52 months versus 71 months for pancreatic-NET. Long term results were analyzed by Mariniello et al. in 114 patients with advanced BCs treated with different RLT protocols. They documented a median PFS and OS of 28.0 and 58.8 months, respectively, while morphological responses were observed in 26.5% of patients. [177Lu]Lu-DOTATATE monotherapy protocol resulted in the highest 5-year OS (61.4%), despite tandem protocol ([90Y]Y-DOTATOC + [177Lu]Lu-DOTATATE) provided the highest response rates (38.1%). Best outcome was reached in patients who underwent RLT in early stage of disease, suggesting that this treatment should also be considered for newly diagnosed unresectable BCs. Moreover, despite most patients having only mild and transient adverse events, patients with hematologic toxicity showed worse survival outcomes.
Click to Show/Hide
|
||||
| Experiment 271 Reporting the Activity Data of This PDC | [55] | ||||
| Indication | Midgut neuroendocrine tumour | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
28.5 months
|
|||
| Patients Enrolled |
Patients with midgut neuroendocrine tumour treated with <sup>177</sup>Lu-Dotatate.
|
||||
| Description |
The approval of 177Lu-Dotatate was based on the findings of the phase III NETTER-1 trial, which showed that for patients with midgut (jejunum, ileum, appendix and proximal colon) NETs, the use of 177Lu-Dotatate (plus octreotide long-acting repeatable [LAR] 30 mg for symptom control) led to a significant gain in both progression-free survival (PFS), overall survival (OS) and quality of life relative to BSC involving octreotide LAR 60 mg Findings from the pivotal phase III NETTER-1 trial were supported by data from the ERASMUS study, in which midgut-NET patients treated with 177Lu-Dotatate had a median (95% CI) PFS and OS of 28.5 (23.9-33.3) months and 54.9 (47.5-63.2) months, respectively.
Click to Show/Hide
|
||||
| Experiment 272 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Bronchial neuroendocrine tumour | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
29 months
|
|||
| Patients Enrolled |
21 patients with bronchial neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 273 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Bronchial carcinoids | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
29 months
|
|||
| Patients Enrolled |
443 patients with bronchial carcinoids.
|
||||
| Administration Dosage | Cumulative dose: 27.8-29.6 GBq | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
Treatment of bronchial carcinoids is not simple and requires a multidisciplinary approach. Surgery remains the mainstay of treatment for local disease, while in advanced disease, management includes chemotherapy, cold somatostatin analogues, immunotherapy, everolimus, and others target therapies. RLT is an option for selected patients with advanced or metastatic BCs overexpressing SSTR in progression to cold SSAs therapy. Although the experience of [177Lu]Lu-DOTATATE is more limited in BCs than GEP-NET, off-label use can be considered with promising results. A prospective phase II trial in 34 patients with stage IV BCs treated with cumulative activity of 18.5-27.8 GBq in four or five cycles of [177Lu]Lu-DOTATATE documented a DCR of 80% (6% complete response, 27% partial response, and 47% stable disease) and a median PFS of 20 months. In this study, negative prognostic factors were AC histology, tumor thyroid transcription factor-1 (TTF-1) expression, and a positive [18F]FDG PET imaging. Comparable results (median PFS of 17 months) were reported in a group of patients with diffuse extrahepatic metastases treated with RLT after several lines of therapies. Higher activities were administered in a study by van Essen et al., who treated nine patients with metastatic BCs, delivering a cumulative dose of 22.2-29.6 GBq of [177Lu]Lu-DOTATATE, demonstrating an overall response rate comparable to that of other GEP-NET (50% vs. 47%, respectively for BCs and GEP-NET). Moreover, tumor regression was reported in 66.6% of patients, without any outcome discrepancies between TCs and ACs. Comparable median OS and response rate between BCs and others GEP-NETs were reported also in another recent retrospective study. Likewise, the efficacy of RLT in BCs was demonstrated by Brabander et al. in a study involving 443 patients affected by midgut, bronchial, and CUP-NETs treated with 7.4 GBq of [177Lu]Lu-DOTATATE every 8 weeks. The authors found that objective response rate (ORR) for BCs was 30%, and an additional 30% of patients had a stable disease. Nevertheless, median OS for BCs was 52 months versus 71 months for pancreatic-NET. Long term results were analyzed by Mariniello et al. in 114 patients with advanced BCs treated with different RLT protocols. They documented a median PFS and OS of 28.0 and 58.8 months, respectively, while morphological responses were observed in 26.5% of patients. [177Lu]Lu-DOTATATE monotherapy protocol resulted in the highest 5-year OS (61.4%), despite tandem protocol ([90Y]Y-DOTATOC + [177Lu]Lu-DOTATATE) provided the highest response rates (38.1%). Best outcome was reached in patients who underwent RLT in early stage of disease, suggesting that this treatment should also be considered for newly diagnosed unresectable BCs. Moreover, despite most patients having only mild and transient adverse events, patients with hematologic toxicity showed worse survival outcomes.
Click to Show/Hide
|
||||
| Experiment 274 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Phaeochromocytoma | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
29 months
|
|||
| Patients Enrolled |
9 patients with Pheochromocytomas (PPGL) and paragangliomas (PHEO).
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
As a consequence, we saw a rising interest in a potential radiolabeled-SST agonist theranostic approach for PPGLs, with several spontaneous studies reporting of the treatment of metastatic or inoperable tumors with Lutathera and other similar radiocompounds, with promising preliminary results. The first RLT experience in PPGLs was reported by van Essen et al. who treated a heterogeneous cohort of patients, including 12 PGLs. Despite the authors reporting lower response rates than those obtained in GEP-NET patients, ORR and DCR were 18% and 73%, respectively. These results are consistent with other reports in the literature. Kong et al. treated 20 PPGLs (8 PHEOs and 12 PGLs) with [117Lu]Lu-DOTATATE, 14 due to uncontrolled secondary hypertension and 6 to radiological PD. A DCR equal to 86% was reached, including five patients with ORR (36%). Of note, 62% of symptomatic patients required a dose reduction of antihypertensive medications, and 57% reported a subjective therapeutic benefit in terms of tumor-related symptoms. Comprehensive PFS was 39 months, including two patients with early recurrence (2 and 5 months post-RLT, respectively) and one patient with remarkable tumor downsizing that allowed a second-step curative liver surgery. Of note, the patient was still disease-free at the time of the study. The results of the largest cohort of PPGLs treated with RLT were reported by Severi et al., who treated 46 patients reaching a comprehensive DCR of 80%. Interestingly, 34 patients treated with [177Lu]Lu-DOTATATE obtained a longer median OS in comparison to those treated with [90Y]Y-DOTATOC (143 vs. 92 months, respectively). According to the authors, this result may be related both to DOTATATEs higher affinity for SSTR2 (which is the type most overexpressed in PPGLs) and to the longer half-life of 177Lu and, consequently, prolonged residence time within the tumor lesions. Moreover, this study reports that sympathetic functioning PPGLs were associated to a shorter median PFS compared to non-functioning PPGLs. Prado-Wohlwend et al. treated nine patients with [177Lu]Lu-DOTATATE and eight with [131I]MIBG, obtaining a PFS of 29 and 18.5 months and a DCR of 88.8% and 62.5%, respectively, for each therapy. Despite the low number of patients involved, a trend for a longer PFS was found for adrenal primary PPGLs treated with [177Lu]Lu-DOTATATE. Nevertheless, the authors suggest performing both [68Ga]Ga-DOTA-SSA PET/CT and [123I]MIBG SPECT/CT in each patient in order to offer a personalized theranostic approach based on the highest uptake intensity in one of the two imaging scans. This is consistent with the results by Jaiswal et al. who found that a baseline SUVmax > 21 at [68Ga]Ga-DOTA-SSA PET/CT is a very strong predictor of response to [177Lu]Lu-DOTATATE (p < 0.0001).
Click to Show/Hide
|
||||
| Experiment 275 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Paraganglioma | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
29 months
|
|||
| Patients Enrolled |
9 patients with Pheochromocytomas (PPGL) and paragangliomas (PHEO).
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
As a consequence, we saw a rising interest in a potential radiolabeled-SST agonist theranostic approach for PPGLs, with several spontaneous studies reporting of the treatment of metastatic or inoperable tumors with Lutathera and other similar radiocompounds, with promising preliminary results. The first RLT experience in PPGLs was reported by van Essen et al. who treated a heterogeneous cohort of patients, including 12 PGLs. Despite the authors reporting lower response rates than those obtained in GEP-NET patients, ORR and DCR were 18% and 73%, respectively. These results are consistent with other reports in the literature. Kong et al. treated 20 PPGLs (8 PHEOs and 12 PGLs) with [117Lu]Lu-DOTATATE, 14 due to uncontrolled secondary hypertension and 6 to radiological PD. A DCR equal to 86% was reached, including five patients with ORR (36%). Of note, 62% of symptomatic patients required a dose reduction of antihypertensive medications, and 57% reported a subjective therapeutic benefit in terms of tumor-related symptoms. Comprehensive PFS was 39 months, including two patients with early recurrence (2 and 5 months post-RLT, respectively) and one patient with remarkable tumor downsizing that allowed a second-step curative liver surgery. Of note, the patient was still disease-free at the time of the study. The results of the largest cohort of PPGLs treated with RLT were reported by Severi et al., who treated 46 patients reaching a comprehensive DCR of 80%. Interestingly, 34 patients treated with [177Lu]Lu-DOTATATE obtained a longer median OS in comparison to those treated with [90Y]Y-DOTATOC (143 vs. 92 months, respectively). According to the authors, this result may be related both to DOTATATEs higher affinity for SSTR2 (which is the type most overexpressed in PPGLs) and to the longer half-life of 177Lu and, consequently, prolonged residence time within the tumor lesions. Moreover, this study reports that sympathetic functioning PPGLs were associated to a shorter median PFS compared to non-functioning PPGLs. Prado-Wohlwend et al. treated nine patients with [177Lu]Lu-DOTATATE and eight with [131I]MIBG, obtaining a PFS of 29 and 18.5 months and a DCR of 88.8% and 62.5%, respectively, for each therapy. Despite the low number of patients involved, a trend for a longer PFS was found for adrenal primary PPGLs treated with [177Lu]Lu-DOTATATE. Nevertheless, the authors suggest performing both [68Ga]Ga-DOTA-SSA PET/CT and [123I]MIBG SPECT/CT in each patient in order to offer a personalized theranostic approach based on the highest uptake intensity in one of the two imaging scans. This is consistent with the results by Jaiswal et al. who found that a baseline SUVmax > 21 at [68Ga]Ga-DOTA-SSA PET/CT is a very strong predictor of response to [177Lu]Lu-DOTATATE (p < 0.0001).
Click to Show/Hide
|
||||
| Experiment 276 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
29 months
|
|||
| Patients Enrolled |
82 patients with CUP-neuroendocrine tumour.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
Unfortunately, the literature lacks specific trials assessing RLT efficacy and safety on CUP-NETs, and the only available literature evidence is provided by mixed trials also including a few patients with SSTR-positive CUP-NETs. In a large cohort of patients treated with [177Lu]Lu-DOTATATE, Brabander et al.reported of 82 CUP-NET patients. Overall, DCR was reached in 78% of patients and median PFS and OS were 29 and 53 months, respectively. These results show that RLT may be effective in CUP-NETs patients, with a response rate intermediate between that obtained in GEP-NETswho showed longer median OS (60 vs. 53 months, respectively)and BCswho showed shorter median PFS (20 vs. 29 months, respectively). These results are consistent with those reported by Demirci et al. who treated 19 CUP-NETs, with an overall DCR of 84.2% and mean PFS and OS of 40 and 48 months, respectively. Once again, RLT outcomes in CUP-NETs were intermediate between those in GEP-NETs and BCs. In a large mixed cohort, Baum et al. treated 151 CUP-NETs (mostly with [177Lu]Lu-DOTATATE, although a small percentage of patients was treated with [90Y]Y-DOTATATE/DOTATOC in the study), obtaining median PFS and OS of 13 and 46 months, respectively. CUP-NETStogether with BCswere associated with a significantly shorter PFS at multivariate analysis if compared to pancreatic NETs despite showing a longer OS (50 vs. 43 months, respectively). Future prospective trials assessing efficacy and safety of RLT in CUP-NETs are desirable.
Click to Show/Hide
|
||||
| Experiment 277 Reporting the Activity Data of This PDC | [56] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
30 month
|
|||
| Patients Enrolled |
42 patients with neuroendocrine tumour.
|
||||
| Administration Time | 2-5 cycles at 8-12-week intervals | ||||
| Administration Dosage | Median cumulative activity of 29.6 GBq | ||||
| MOA of PDC |
A higher baseline PLR was shown to be associated with a negative outcome on PFS after 177Lu-DOTATATE therapy and is a promising marker for future larger studies.
|
||||
| Experiment 278 Reporting the Activity Data of This PDC | [25] | ||||
| Indication | SDHB positive metastatic paraganglioma | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
31.4 months
|
|||
| Patients Enrolled |
37 patients affected by neuroendocrine tumors.
|
||||
| Administration Time | Five cycles of 5.5 gbq each every 8 weeks | ||||
| Administration Dosage | Cumulative activity of 27.5 GBq | ||||
| Description |
Thirty-three of 37 patients were evaluable for response. Objective responses included partial response (PR) in 10 patients (30%), stable disease (SD) in 18 patients (55%), with a DCR of 85%. Median follow up was 38 months (range 4.6-51.1 months). Median PFS was 31.4 months (17.6-45.4) and mOS was not reached.
|
||||
| Experiment 279 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Meningiomas | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
32.2 months
|
|||
| Patients Enrolled |
Patients with WHO grade I meningiomas.
|
||||
| Administration Time | Median 3 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
A few studies evaluated the efficacy of RLT in meningiomas, administrating 2-5 cycles of [90Y]Y-DOTATATE/-DOTATOC or [177Lu]Lu-DOTATATE/-DOTATOC. In a meta-analysis performed by Mirian et al., RLToffered as mono-therapy or in combination with other oncological treatmentsallowed a comprehensive DCR of 63% in refractory meningiomas. The 6-month PFS rates were 94%, 48%, and 0% for patients with WHO grade I, II, and III meningiomas, respectively, whereas the corresponding 1-year OS rates were 88%, 71%, and 52%, respectively. In a study by Seystahl et al. RLT, offered mainly with [177Lu]Lu-DOTATATE (85% of patients), obtained 6-month PFS rates of 100%, 57%, and 0% for grade I, II, and III refractory meningiomas, respectively. In a recent study on a small group of selected patients affected by WHO grade II refractory meningiomas, 6-month PFS was 85.7% and 1-year PFS was 66.7%. The treatment was safe and well tolerated.
Click to Show/Hide
|
||||
| Experiment 280 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
33 months
|
|||
| Patients Enrolled |
395 patients with neuroendocrine tumour.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 281 Reporting the Activity Data of This PDC | [57] | ||||
| Indication | Advanced paraganglioma | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
34.1 months
|
|||
| Patients Enrolled |
47 patients with neuroendocrine tumour.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | 5.5 GBq (150 mCi) | ||||
| MOA of PDC |
High burden of skeletal and hepatic metastases, non-GEP-NETs, chromogranin A of 4 ULN, unusual metastatic sites, pre-existing and interim ascites are predictors of poor outcomes in patients treated with 177Lu-Dotatate. These common indicators can be used for the risk stratification and identification of patients most likely to benefit from PRRT.
Click to Show/Hide
|
||||
| Description |
Median follow-up was 63.1 months with a median progression-free survival (PFS) of 34.1 months. However, median overall survival (OS) was not reached at the time of analysis.
|
||||
| Experiment 282 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Unknown originb neuroendocrine tumor | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
35 months
|
|||
| Patients Enrolled |
22 patients with unknown originb neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 283 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Midgut neuroendocrine tumor | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
36 months
|
|||
| Patients Enrolled |
229 patients with midgut neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 284 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | High-grade (WHO G3) neuroendocrine tumour | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
36.4 months
|
|||
| Patients Enrolled |
Patients with neuroendocrine tumours including high-grade (WHO G3) neuroendocrine tumours.
|
||||
| Evaluation Method | Kaplan-Meier estimated assay | ||||
| Description |
The Kaplan-Meier estimated median PFS was 36.4 months, mean PFS was 38 months and the mean OS was 55 months.
|
||||
| Experiment 285 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Phaeochromocytoma | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
39 months
|
|||
| Patients Enrolled |
20 patients with phaeochromocytoma and paraganglioma.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
As a consequence, we saw a rising interest in a potential radiolabeled-SST agonist theranostic approach for PPGLs, with several spontaneous studies reporting of the treatment of metastatic or inoperable tumors with Lutathera and other similar radiocompounds, with promising preliminary results. The first RLT experience in PPGLs was reported by van Essen et al. who treated a heterogeneous cohort of patients, including 12 PGLs. Despite the authors reporting lower response rates than those obtained in GEP-NET patients, ORR and DCR were 18% and 73%, respectively. These results are consistent with other reports in the literature. Kong et al. treated 20 PPGLs (8 PHEOs and 12 PGLs) with [117Lu]Lu-DOTATATE, 14 due to uncontrolled secondary hypertension and 6 to radiological PD. A DCR equal to 86% was reached, including five patients with ORR (36%). Of note, 62% of symptomatic patients required a dose reduction of antihypertensive medications, and 57% reported a subjective therapeutic benefit in terms of tumor-related symptoms. Comprehensive PFS was 39 months, including two patients with early recurrence (2 and 5 months post-RLT, respectively) and one patient with remarkable tumor downsizing that allowed a second-step curative liver surgery. Of note, the patient was still disease-free at the time of the study. The results of the largest cohort of PPGLs treated with RLT were reported by Severi et al., who treated 46 patients reaching a comprehensive DCR of 80%. Interestingly, 34 patients treated with [177Lu]Lu-DOTATATE obtained a longer median OS in comparison to those treated with [90Y]Y-DOTATOC (143 vs. 92 months, respectively). According to the authors, this result may be related both to DOTATATEs higher affinity for SSTR2 (which is the type most overexpressed in PPGLs) and to the longer half-life of 177Lu and, consequently, prolonged residence time within the tumor lesions. Moreover, this study reports that sympathetic functioning PPGLs were associated to a shorter median PFS compared to non-functioning PPGLs. Prado-Wohlwend et al. treated nine patients with [177Lu]Lu-DOTATATE and eight with [131I]MIBG, obtaining a PFS of 29 and 18.5 months and a DCR of 88.8% and 62.5%, respectively, for each therapy. Despite the low number of patients involved, a trend for a longer PFS was found for adrenal primary PPGLs treated with [177Lu]Lu-DOTATATE. Nevertheless, the authors suggest performing both [68Ga]Ga-DOTA-SSA PET/CT and [123I]MIBG SPECT/CT in each patient in order to offer a personalized theranostic approach based on the highest uptake intensity in one of the two imaging scans. This is consistent with the results by Jaiswal et al. who found that a baseline SUVmax > 21 at [68Ga]Ga-DOTA-SSA PET/CT is a very strong predictor of response to [177Lu]Lu-DOTATATE (p < 0.0001).
Click to Show/Hide
|
||||
| Experiment 286 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Paraganglioma | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
39 months
|
|||
| Patients Enrolled |
20 patients with phaeochromocytoma and paraganglioma.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
As a consequence, we saw a rising interest in a potential radiolabeled-SST agonist theranostic approach for PPGLs, with several spontaneous studies reporting of the treatment of metastatic or inoperable tumors with Lutathera and other similar radiocompounds, with promising preliminary results. The first RLT experience in PPGLs was reported by van Essen et al. who treated a heterogeneous cohort of patients, including 12 PGLs. Despite the authors reporting lower response rates than those obtained in GEP-NET patients, ORR and DCR were 18% and 73%, respectively. These results are consistent with other reports in the literature. Kong et al. treated 20 PPGLs (8 PHEOs and 12 PGLs) with [117Lu]Lu-DOTATATE, 14 due to uncontrolled secondary hypertension and 6 to radiological PD. A DCR equal to 86% was reached, including five patients with ORR (36%). Of note, 62% of symptomatic patients required a dose reduction of antihypertensive medications, and 57% reported a subjective therapeutic benefit in terms of tumor-related symptoms. Comprehensive PFS was 39 months, including two patients with early recurrence (2 and 5 months post-RLT, respectively) and one patient with remarkable tumor downsizing that allowed a second-step curative liver surgery. Of note, the patient was still disease-free at the time of the study. The results of the largest cohort of PPGLs treated with RLT were reported by Severi et al., who treated 46 patients reaching a comprehensive DCR of 80%. Interestingly, 34 patients treated with [177Lu]Lu-DOTATATE obtained a longer median OS in comparison to those treated with [90Y]Y-DOTATOC (143 vs. 92 months, respectively). According to the authors, this result may be related both to DOTATATEs higher affinity for SSTR2 (which is the type most overexpressed in PPGLs) and to the longer half-life of 177Lu and, consequently, prolonged residence time within the tumor lesions. Moreover, this study reports that sympathetic functioning PPGLs were associated to a shorter median PFS compared to non-functioning PPGLs. Prado-Wohlwend et al. treated nine patients with [177Lu]Lu-DOTATATE and eight with [131I]MIBG, obtaining a PFS of 29 and 18.5 months and a DCR of 88.8% and 62.5%, respectively, for each therapy. Despite the low number of patients involved, a trend for a longer PFS was found for adrenal primary PPGLs treated with [177Lu]Lu-DOTATATE. Nevertheless, the authors suggest performing both [68Ga]Ga-DOTA-SSA PET/CT and [123I]MIBG SPECT/CT in each patient in order to offer a personalized theranostic approach based on the highest uptake intensity in one of the two imaging scans. This is consistent with the results by Jaiswal et al. who found that a baseline SUVmax > 21 at [68Ga]Ga-DOTA-SSA PET/CT is a very strong predictor of response to [177Lu]Lu-DOTATATE (p < 0.0001).
Click to Show/Hide
|
||||
| Experiment 287 Reporting the Activity Data of This PDC | [10] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
40 months
|
|||
| Patients Enrolled |
Patients with neuroendocrine tumor.
|
||||
| Experiment 288 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
40.9 months
|
|||
| Patients Enrolled |
19 patients with CUP-neuroendocrine tumor.
|
||||
| Administration Time | Median 6 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
Unfortunately, the literature lacks specific trials assessing RLT efficacy and safety on CUP-NETs, and the only available literature evidence is provided by mixed trials also including a few patients with SSTR-positive CUP-NETs. In a large cohort of patients treated with [177Lu]Lu-DOTATATE, Brabander et al.reported of 82 CUP-NET patients. Overall, DCR was reached in 78% of patients and median PFS and OS were 29 and 53 months, respectively. These results show that RLT may be effective in CUP-NETs patients, with a response rate intermediate between that obtained in GEP-NETswho showed longer median OS (60 vs. 53 months, respectively)and BCswho showed shorter median PFS (20 vs. 29 months, respectively). These results are consistent with those reported by Demirci et al. who treated 19 CUP-NETs, with an overall DCR of 84.2% and mean PFS and OS of 40 and 48 months, respectively. Once again, RLT outcomes in CUP-NETs were intermediate between those in GEP-NETs and BCs. In a large mixed cohort, Baum et al. treated 151 CUP-NETs (mostly with [177Lu]Lu-DOTATATE, although a small percentage of patients was treated with [90Y]Y-DOTATATE/DOTATOC in the study), obtaining median PFS and OS of 13 and 46 months, respectively. CUP-NETStogether with BCswere associated with a significantly shorter PFS at multivariate analysis if compared to pancreatic NETs despite showing a longer OS (50 vs. 43 months, respectively). Future prospective trials assessing efficacy and safety of RLT in CUP-NETs are desirable.
Click to Show/Hide
|
||||
| Experiment 289 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Hindgut neuroendocrine tumour | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
41 months
|
|||
| Patients Enrolled |
14 patients with hindgut neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 290 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
41 months
|
|||
| Patients Enrolled |
20 patients with others neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 291 Reporting the Activity Data of This PDC | [58] | ||||
| Indication | Small bowel neuroendocrine tumor | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
42.7 months
|
|||
| Patients Enrolled |
Patients with consecutive patients with small bowel neuroendocrine tumour.
|
||||
| Administration Time | Every eight to 12 weeks | ||||
| Administration Dosage | 7.4 GBq (200 mCi) | ||||
| Evaluation Method | 68Ga-DOTATATE PET/CT assay | ||||
| MOA of PDC |
Surgery combined with 177Lu PRRT is safe and provides favourable PFS and OS in selected patients with advanced SBNEN. Liquid biopsy (NETest) has the potential to accurately delineate disease status.
|
||||
| Description |
A surgical cohort of 39 SBNEN patients met eligibility criteria. Thirty-two patients underwent ileal resection and 7 right hemicolectomy. The mean number of 177Lu PRRT cycles was 4. Mortality was nil. Surgical morbidity was 10.3%. Transient grade 1/2 toxicity occurred in 41% (PRRT). NETest scores (n=9 patients) decreased in 100% following treatment and correlated with diminished tumour volume and disease stabilization following surgery and PRRT. Median follow-up: 78 months. Median PFS and OS: 42.7 and 110 months, respectively. Progression-free survival at 1-, 3-, and 5-years was 79.4%, 57.1% and 40.5%, respectively. Overall survival at 1-, 3-, and 5-years was 97.4%, 97.4%, and 94.1%, respectively
Click to Show/Hide
|
||||
| Experiment 292 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Inoperable or metastatic paragangliomas | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
91 months
|
|||
| Patients Enrolled |
Patients with inoperable or metastatic paragangliomas and pheochromocytomas with parasympathetic paragangliomas.
|
||||
| Administration Time | Four cycles | ||||
| Administration Dosage | 7.4 Gb per cycle | ||||
| Description |
In 20 patients with baseline disease progression, tumour control was observed in 17 (85%); the median progression-free survival was 91 months in patients with parasympathetic PGLs, 13 months in patients with sympathetic PGLs and 10 months in patients with metastatic PCCs.
|
||||
| Experiment 293 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Inoperable or metastatic pheochromocytomas | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
91 months
|
|||
| Patients Enrolled |
Patients with inoperable or metastatic paragangliomas and pheochromocytomas with parasympathetic paragangliomas.
|
||||
| Administration Time | Four cycles | ||||
| Administration Dosage | 7.4 Gb per cycle | ||||
| Description |
In 20 patients with baseline disease progression, tumour control was observed in 17 (85%); the median progression-free survival was 91 months in patients with parasympathetic PGLs, 13 months in patients with sympathetic PGLs and 10 months in patients with metastatic PCCs.
|
||||
| Experiment 294 Reporting the Activity Data of This PDC | [43], [59] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Median overall survival (mOS) |
48.0 months
|
|||
| Patients Enrolled |
Patients were 18 years and older with locally advanced or metastatic, well differentiated, somatostatin receptor-positive midgut neuroendocrine tumours (Karnofsky performance status score 60) and disease progression on fixed-dose long-acting octreotide.
|
||||
| Administration Time | Every 8 weeks for a total of 4 cycles | ||||
| MOA of PDC |
177Lu-DOTATATE is the first FDA-approved PRRT and utilizes a somatostatin analogue (DOTATATE) covalently bound to the beta-minus emitting radioisotope 177Lu in order to provide targeted radiation directly to NET cells overexpressing SSTRs (primarily SSTR2).
|
||||
| Description |
In 2021, Strosberg et al. provided updated survival results from the NETTER-1 trial, which showed no statistically significant difference in median overall survival in the 177Lu-DOTATATE group and octreotide group vs. the control group (48.0 months vs. 36.3 months, respectively, p = 0.30), a result that was likely impacted by the high rate of crossover (36%) in the investigational arm of the study.
Click to Show/Hide
|
||||
| Related Clinical Trial | |||||
| NCT Number | NCT01578239 | Clinical Status | Phase 3 | ||
| Clinical Description | This was a multicenter, stratified, open, randomized, comparator-controlled, parallel-group phase III study comparing treatment with Lutathera plus best supportive care (30 mg Octreotide LAR) to treatment with high dose (60 mg) Octreotide LAR in participants with metastasized or locally advanced, inoperable, somatostatin receptor positive, histologically proven midgut carcinoid tumours with progression despite LAR treatment. | ||||
| Experiment 295 Reporting the Activity Data of This PDC | [51] | ||||
| Indication | Negative progressive/Symptomatic locally advanced/Metastatic paragangliomas | ||||
| Efficacy Data | Median overall survival (mOS) |
65%
|
|||
| Patients Enrolled |
9 patients with paragangliomas.
|
||||
| Administration Time | ≥ 4cycles | ||||
| Administration Dosage | > 22.2 GBq) | ||||
| Experiment 296 Reporting the Activity Data of This PDC | [14] | ||||
| Indication | Metastatic/Advanced neuroendocrine tumour | ||||
| Efficacy Data | Median overall survival (mOS) |
79.40%
|
|||
| Patients Enrolled |
468 patients with metastatic/advanced neuroendocrine tumor.
|
||||
| Administration Time | At least two cycles at 10-12 weeks interval | ||||
| Administration Dosage | 5.55 to 7.4 GBq per patient | ||||
| Description |
The median PFS and OS were not reached at a median follow-up of 46 months. Observed PFS and OS at 7 years were 71.1% 95% CI (62.4-79.7%) and 79.4% 95% CI (71.4-86.9%) respectively.
|
||||
| Experiment 297 Reporting the Activity Data of This PDC | [53] | ||||
| Indication | Well-differentiated neuroendocrine tumor | ||||
| Efficacy Data | Median overall survival (mOS) |
4.53 months
|
|||
| Patients Enrolled |
26 patients with WD neuroendocrine tumour with a clinical score (CS) greater than 4 points.
|
||||
| Administration Time | 1 or 2 doses | ||||
| MOA of PDC |
Increases in CS were associated with worsening PFS in the validation cohort, validating findings from the original cohort. These findings suggest that the CS, to our knowledge, represents the first clinical metric to estimate anticipated benefit from PRRT for patients with WD NETs and may be a clinical tool for patients being considered for PRRT.
|
||||
| Description |
Among patients who received 1 to 2 doses of PRRT, patients with CS less than or equal to 4 points experienced a median OS of 7.98 months (95% CI, 4.37 months to NR) whereas patients with a CS greater than 4 points experienced a median OS of 4.53 months (95% CI, 1.35 months to NR).
|
||||
| Experiment 298 Reporting the Activity Data of This PDC | [53] | ||||
| Indication | Well-differentiated neuroendocrine tumor | ||||
| Efficacy Data | Median overall survival (mOS) |
7.98 months
|
|||
| Patients Enrolled |
26 patients with WD neuroendocrine tumour with a clinical score (CS) less than or equal to 4 points.
|
||||
| Administration Time | 1 or 2 doses | ||||
| MOA of PDC |
Increases in CS were associated with worsening PFS in the validation cohort, validating findings from the original cohort. These findings suggest that the CS, to our knowledge, represents the first clinical metric to estimate anticipated benefit from PRRT for patients with WD NETs and may be a clinical tool for patients being considered for PRRT.
|
||||
| Description |
Among patients who received 1 to 2 doses of PRRT, patients with CS less than or equal to 4 points experienced a median OS of 7.98 months (95% CI, 4.37 months to NR) whereas patients with a CS greater than 4 points experienced a median OS of 4.53 months (95% CI, 1.35 months to NR).
|
||||
| Experiment 299 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Meningiomas | ||||
| Efficacy Data | Median overall survival (mOS) |
13.6 months
|
|||
| Patients Enrolled |
15 patients with meningiomas.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
A few studies evaluated the efficacy of RLT in meningiomas, administrating 2-5 cycles of [90Y]Y-DOTATATE/-DOTATOC or [177Lu]Lu-DOTATATE/-DOTATOC. In a meta-analysis performed by Mirian et al., RLToffered as mono-therapy or in combination with other oncological treatmentsallowed a comprehensive DCR of 63% in refractory meningiomas. The 6-month PFS rates were 94%, 48%, and 0% for patients with WHO grade I, II, and III meningiomas, respectively, whereas the corresponding 1-year OS rates were 88%, 71%, and 52%, respectively. In a study by Seystahl et al. RLT, offered mainly with [177Lu]Lu-DOTATATE (85% of patients), obtained 6-month PFS rates of 100%, 57%, and 0% for grade I, II, and III refractory meningiomas, respectively. In a recent study on a small group of selected patients affected by WHO grade II refractory meningiomas, 6-month PFS was 85.7% and 1-year PFS was 66.7%. The treatment was safe and well tolerated.
Click to Show/Hide
|
||||
| Experiment 300 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Meningiomas | ||||
| Efficacy Data | Median overall survival (mOS) |
17.2 months
|
|||
| Patients Enrolled |
20 patients with WHO grade III meningiomas.
|
||||
| Administration Time | Median 3 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
A few studies evaluated the efficacy of RLT in meningiomas, administrating 2-5 cycles of [90Y]Y-DOTATATE/-DOTATOC or [177Lu]Lu-DOTATATE/-DOTATOC. In a meta-analysis performed by Mirian et al., RLToffered as mono-therapy or in combination with other oncological treatmentsallowed a comprehensive DCR of 63% in refractory meningiomas. The 6-month PFS rates were 94%, 48%, and 0% for patients with WHO grade I, II, and III meningiomas, respectively, whereas the corresponding 1-year OS rates were 88%, 71%, and 52%, respectively. In a study by Seystahl et al. RLT, offered mainly with [177Lu]Lu-DOTATATE (85% of patients), obtained 6-month PFS rates of 100%, 57%, and 0% for grade I, II, and III refractory meningiomas, respectively. In a recent study on a small group of selected patients affected by WHO grade II refractory meningiomas, 6-month PFS was 85.7% and 1-year PFS was 66.7%. The treatment was safe and well tolerated.
Click to Show/Hide
|
||||
| Experiment 301 Reporting the Activity Data of This PDC | [16] | ||||
| Indication | Metastatic medullary thyroid carcinoma | ||||
| Efficacy Data | Median overall survival (mOS) |
19 months
|
|||
| Patients Enrolled |
6 patients with medullary thyroid cancer (3 males and 3 females).
|
||||
| Administration Time | 3 cycles | ||||
| Administration Dosage | 5.7 GBq/cycle (range, 2.6-7.4 GBq) | ||||
| Evaluation Method | PET/CT assay | ||||
| MOA of PDC |
Radiolabeled somatostatin analogs are contributive for the management of recurrent MTC. 68Ga-DOTATAE PET-CT showed a relatively high detection rate in recurrent MTC. In addition, PRRT with 177Lu-DOTATATE was found to be a safe alternative therapeutic option for MTC.
|
||||
| Description |
Four of the 17 patients with positive SSTR-PET were scheduled for PRRT. In addition, 2 patients had positive 99mTc-octreotide scintigraphy results (Krenning score ≥ 2) and were scheduled for PRRT. Two of the 6 patients who underwent PRRT showed PR, 2 SD and 2 PD. Two patients died during the follow-up period. Median overall survival was 19 months (95% CI: 5.52-29.48). There were no cases of significant toxicity.
Click to Show/Hide
|
||||
| Experiment 302 Reporting the Activity Data of This PDC | [53] | ||||
| Indication | Well-differentiated neuroendocrine tumor | ||||
| Efficacy Data | Median overall survival (mOS) |
27.47 months
|
|||
| Patients Enrolled |
82 patients with WD neuroendocrine tumour with a clinical score (CS) greater than 4 points.
|
||||
| Administration Time | 0 doses | ||||
| MOA of PDC |
Increases in CS were associated with worsening PFS in the validation cohort, validating findings from the original cohort. These findings suggest that the CS, to our knowledge, represents the first clinical metric to estimate anticipated benefit from PRRT for patients with WD NETs and may be a clinical tool for patients being considered for PRRT.
|
||||
| Description |
Among patients who received 0 doses of PRRT, patients with a CS less than or equal to 4 points experienced a median OS of NR (95% CI, NR-NR) whereas patients with a CS greater than 4 points experienced a median OS of 27.47 months (95% CI, 10.35 points to NR).
|
||||
| Experiment 303 Reporting the Activity Data of This PDC | [27] | ||||
| Indication | Metastatic neuroendocrine tumor | ||||
| Efficacy Data | Median overall survival (mOS) |
28 months
|
|||
| Patients Enrolled |
Patients treated with Lu-DOTATATE.
|
||||
| Description |
The overall estimated median time to progression from the start of treatment was 28 months.
|
||||
| Experiment 304 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Phaeochromocytoma | ||||
| Efficacy Data | Median overall survival (mOS) |
28 months
|
|||
| Patients Enrolled |
20 patients with phaeochromocytoma and paraganglioma.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
As a consequence, we saw a rising interest in a potential radiolabeled-SST agonist theranostic approach for PPGLs, with several spontaneous studies reporting of the treatment of metastatic or inoperable tumors with Lutathera and other similar radiocompounds, with promising preliminary results. The first RLT experience in PPGLs was reported by van Essen et al. who treated a heterogeneous cohort of patients, including 12 PGLs. Despite the authors reporting lower response rates than those obtained in GEP-NET patients, ORR and DCR were 18% and 73%, respectively. These results are consistent with other reports in the literature. Kong et al. treated 20 PPGLs (8 PHEOs and 12 PGLs) with [117Lu]Lu-DOTATATE, 14 due to uncontrolled secondary hypertension and 6 to radiological PD. A DCR equal to 86% was reached, including five patients with ORR (36%). Of note, 62% of symptomatic patients required a dose reduction of antihypertensive medications, and 57% reported a subjective therapeutic benefit in terms of tumor-related symptoms. Comprehensive PFS was 39 months, including two patients with early recurrence (2 and 5 months post-RLT, respectively) and one patient with remarkable tumor downsizing that allowed a second-step curative liver surgery. Of note, the patient was still disease-free at the time of the study. The results of the largest cohort of PPGLs treated with RLT were reported by Severi et al., who treated 46 patients reaching a comprehensive DCR of 80%. Interestingly, 34 patients treated with [177Lu]Lu-DOTATATE obtained a longer median OS in comparison to those treated with [90Y]Y-DOTATOC (143 vs. 92 months, respectively). According to the authors, this result may be related both to DOTATATEs higher affinity for SSTR2 (which is the type most overexpressed in PPGLs) and to the longer half-life of 177Lu and, consequently, prolonged residence time within the tumor lesions. Moreover, this study reports that sympathetic functioning PPGLs were associated to a shorter median PFS compared to non-functioning PPGLs. Prado-Wohlwend et al. treated nine patients with [177Lu]Lu-DOTATATE and eight with [131I]MIBG, obtaining a PFS of 29 and 18.5 months and a DCR of 88.8% and 62.5%, respectively, for each therapy. Despite the low number of patients involved, a trend for a longer PFS was found for adrenal primary PPGLs treated with [177Lu]Lu-DOTATATE. Nevertheless, the authors suggest performing both [68Ga]Ga-DOTA-SSA PET/CT and [123I]MIBG SPECT/CT in each patient in order to offer a personalized theranostic approach based on the highest uptake intensity in one of the two imaging scans. This is consistent with the results by Jaiswal et al. who found that a baseline SUVmax > 21 at [68Ga]Ga-DOTA-SSA PET/CT is a very strong predictor of response to [177Lu]Lu-DOTATATE (p < 0.0001).
Click to Show/Hide
|
||||
| Experiment 305 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Paraganglioma | ||||
| Efficacy Data | Median overall survival (mOS) |
28 months
|
|||
| Patients Enrolled |
20 patients with phaeochromocytoma and paraganglioma.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
As a consequence, we saw a rising interest in a potential radiolabeled-SST agonist theranostic approach for PPGLs, with several spontaneous studies reporting of the treatment of metastatic or inoperable tumors with Lutathera and other similar radiocompounds, with promising preliminary results. The first RLT experience in PPGLs was reported by van Essen et al. who treated a heterogeneous cohort of patients, including 12 PGLs. Despite the authors reporting lower response rates than those obtained in GEP-NET patients, ORR and DCR were 18% and 73%, respectively. These results are consistent with other reports in the literature. Kong et al. treated 20 PPGLs (8 PHEOs and 12 PGLs) with [117Lu]Lu-DOTATATE, 14 due to uncontrolled secondary hypertension and 6 to radiological PD. A DCR equal to 86% was reached, including five patients with ORR (36%). Of note, 62% of symptomatic patients required a dose reduction of antihypertensive medications, and 57% reported a subjective therapeutic benefit in terms of tumor-related symptoms. Comprehensive PFS was 39 months, including two patients with early recurrence (2 and 5 months post-RLT, respectively) and one patient with remarkable tumor downsizing that allowed a second-step curative liver surgery. Of note, the patient was still disease-free at the time of the study. The results of the largest cohort of PPGLs treated with RLT were reported by Severi et al., who treated 46 patients reaching a comprehensive DCR of 80%. Interestingly, 34 patients treated with [177Lu]Lu-DOTATATE obtained a longer median OS in comparison to those treated with [90Y]Y-DOTATOC (143 vs. 92 months, respectively). According to the authors, this result may be related both to DOTATATEs higher affinity for SSTR2 (which is the type most overexpressed in PPGLs) and to the longer half-life of 177Lu and, consequently, prolonged residence time within the tumor lesions. Moreover, this study reports that sympathetic functioning PPGLs were associated to a shorter median PFS compared to non-functioning PPGLs. Prado-Wohlwend et al. treated nine patients with [177Lu]Lu-DOTATATE and eight with [131I]MIBG, obtaining a PFS of 29 and 18.5 months and a DCR of 88.8% and 62.5%, respectively, for each therapy. Despite the low number of patients involved, a trend for a longer PFS was found for adrenal primary PPGLs treated with [177Lu]Lu-DOTATATE. Nevertheless, the authors suggest performing both [68Ga]Ga-DOTA-SSA PET/CT and [123I]MIBG SPECT/CT in each patient in order to offer a personalized theranostic approach based on the highest uptake intensity in one of the two imaging scans. This is consistent with the results by Jaiswal et al. who found that a baseline SUVmax > 21 at [68Ga]Ga-DOTA-SSA PET/CT is a very strong predictor of response to [177Lu]Lu-DOTATATE (p < 0.0001).
Click to Show/Hide
|
||||
| Experiment 306 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Median overall survival (mOS) |
34.2 months
|
|||
| Patients Enrolled |
Patients with neuroendocrine tumours who failed exclusion criteria.
|
||||
| Description |
Estimated median overall survival was significantly longer for patients who met selection criteria compared with those who did not (50.7 vs 34.2 months) (P = 0.018).
|
||||
| Experiment 307 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Unknown originb neuroendocrine tumor | ||||
| Efficacy Data | Median overall survival (mOS) |
39 months
|
|||
| Patients Enrolled |
22 patients with unknown originb neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 308 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Median overall survival (mOS) |
39 months
|
|||
| Patients Enrolled |
20 patients with others neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 309 Reporting the Activity Data of This PDC | [60] | ||||
| Indication | Metastatic/Advanced pulmonary neuroendocrine tumor | ||||
| Efficacy Data | Median overall survival (mOS) |
40 months
|
|||
| Patients Enrolled |
Patients treated with Lu-DOTATATE.
|
||||
| Administration Time | 12-16 weeks' intervals [range, 1-5 cycles; average, 4 cycles] | ||||
| Administration Dosage | 150 mCi [5.55 GBq]/cycle | ||||
| Description |
The observed annual OS rates were as follows: 1 year: 94.7% (95% CI, 68.1%-99.2%), 2 years: 66% (95% CI, 35.5%-84.5%), 3 years: 57.7% (95% CI, 28-78.9%), and 4 years: 38.5% (95% CI, 8.1%-69.5%); median OS was 40 months with 39% (95% CI, 13.1%-64.8%).
|
||||
| Experiment 310 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | PPGLa neuroendocrine tumor | ||||
| Efficacy Data | Median overall survival (mOS) |
44 months
|
|||
| Patients Enrolled |
11 patients with PPGLa neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 311 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Median overall survival (mOS) |
46 months
|
|||
| Patients Enrolled |
395 patients with neuroendocrine tumour.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 312 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Midgut neuroendocrine tumor | ||||
| Efficacy Data | Median overall survival (mOS) |
47 months
|
|||
| Patients Enrolled |
229 patients with midgut neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 313 Reporting the Activity Data of This PDC | [23] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Median overall survival (mOS) |
48 months
|
|||
| Patients Enrolled |
Patients with advanced neuroendocrine tumor.
|
||||
| Description |
According to Response Evaluation Criteria in Solid Tumors 1.1 criteria, in group 1 patients, the response was partial response in 34% of the patients, stable disease in 50.2%, and progressive disease in 6.8% versus partial response in 6.3%, stable disease in 60.9%, and progressive disease in 26.5% among group 2 patients. The median overall survival (OS) and progression-free survival (PFS) was not reached in group 1 patients. The median OS and PFS in group 2 patients were 48 months.
Click to Show/Hide
|
||||
| Experiment 314 Reporting the Activity Data of This PDC | [59] | ||||
| Indication | Midgut neuroendocrine tumor | ||||
| Efficacy Data | Median overall survival (mOS) |
48.0 months
|
|||
| Patients Enrolled |
231 NETTER-1 patients.
|
||||
| Administration Time | 4 cycles every 8 weeks | ||||
| Administration Dosage | 7.4 GBq (200 mCi) | ||||
| MOA of PDC |
177Lu-Dotatate treatment did not significantly improve median overall survival versus high-dose long-acting octreotide. Despite final overall survival not reaching statistical significance, the 117 month difference in median overall survival with 177Lu-Dotatate treatment versus high-dose long-acting octreotide alone might be considered clinically relevant. No new safety signals were reported during long-term follow-up.
Click to Show/Hide
|
||||
| Description |
The secondary endpoint of overall survival was not met: median overall survival was 480 months (95% CI 374-552) in the 177Lu-Dotatate group and 363 months (259-517) in the control group (HR 084 [95% CI 060-117]; two-sided p=030).
|
||||
| Experiment 315 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Median overall survival (mOS) |
48.3 months
|
|||
| Patients Enrolled |
19 patients with CUP-neuroendocrine tumor.
|
||||
| Administration Time | Median 6 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
Unfortunately, the literature lacks specific trials assessing RLT efficacy and safety on CUP-NETs, and the only available literature evidence is provided by mixed trials also including a few patients with SSTR-positive CUP-NETs. In a large cohort of patients treated with [177Lu]Lu-DOTATATE, Brabander et al.reported of 82 CUP-NET patients. Overall, DCR was reached in 78% of patients and median PFS and OS were 29 and 53 months, respectively. These results show that RLT may be effective in CUP-NETs patients, with a response rate intermediate between that obtained in GEP-NETswho showed longer median OS (60 vs. 53 months, respectively)and BCswho showed shorter median PFS (20 vs. 29 months, respectively). These results are consistent with those reported by Demirci et al. who treated 19 CUP-NETs, with an overall DCR of 84.2% and mean PFS and OS of 40 and 48 months, respectively. Once again, RLT outcomes in CUP-NETs were intermediate between those in GEP-NETs and BCs. In a large mixed cohort, Baum et al. treated 151 CUP-NETs (mostly with [177Lu]Lu-DOTATATE, although a small percentage of patients was treated with [90Y]Y-DOTATATE/DOTATOC in the study), obtaining median PFS and OS of 13 and 46 months, respectively. CUP-NETStogether with BCswere associated with a significantly shorter PFS at multivariate analysis if compared to pancreatic NETs despite showing a longer OS (50 vs. 43 months, respectively). Future prospective trials assessing efficacy and safety of RLT in CUP-NETs are desirable.
Click to Show/Hide
|
||||
| Experiment 316 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Bronchial carcinoids | ||||
| Efficacy Data | Median overall survival (mOS) |
48.6 months
|
|||
| Patients Enrolled |
34 patients with bronchial carcinoids.
|
||||
| Administration Time | 4-5 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
Treatment of bronchial carcinoids is not simple and requires a multidisciplinary approach. Surgery remains the mainstay of treatment for local disease, while in advanced disease, management includes chemotherapy, cold somatostatin analogues, immunotherapy, everolimus, and others target therapies. RLT is an option for selected patients with advanced or metastatic BCs overexpressing SSTR in progression to cold SSAs therapy. Although the experience of [177Lu]Lu-DOTATATE is more limited in BCs than GEP-NET, off-label use can be considered with promising results. A prospective phase II trial in 34 patients with stage IV BCs treated with cumulative activity of 18.5-27.8 GBq in four or five cycles of [177Lu]Lu-DOTATATE documented a DCR of 80% (6% complete response, 27% partial response, and 47% stable disease) and a median PFS of 20 months. In this study, negative prognostic factors were AC histology, tumor thyroid transcription factor-1 (TTF-1) expression, and a positive [18F]FDG PET imaging. Comparable results (median PFS of 17 months) were reported in a group of patients with diffuse extrahepatic metastases treated with RLT after several lines of therapies. Higher activities were administered in a study by van Essen et al., who treated nine patients with metastatic BCs, delivering a cumulative dose of 22.2-29.6 GBq of [177Lu]Lu-DOTATATE, demonstrating an overall response rate comparable to that of other GEP-NET (50% vs. 47%, respectively for BCs and GEP-NET). Moreover, tumor regression was reported in 66.6% of patients, without any outcome discrepancies between TCs and ACs. Comparable median OS and response rate between BCs and others GEP-NETs were reported also in another recent retrospective study. Likewise, the efficacy of RLT in BCs was demonstrated by Brabander et al. in a study involving 443 patients affected by midgut, bronchial, and CUP-NETs treated with 7.4 GBq of [177Lu]Lu-DOTATATE every 8 weeks. The authors found that objective response rate (ORR) for BCs was 30%, and an additional 30% of patients had a stable disease. Nevertheless, median OS for BCs was 52 months versus 71 months for pancreatic-NET. Long term results were analyzed by Mariniello et al. in 114 patients with advanced BCs treated with different RLT protocols. They documented a median PFS and OS of 28.0 and 58.8 months, respectively, while morphological responses were observed in 26.5% of patients. [177Lu]Lu-DOTATATE monotherapy protocol resulted in the highest 5-year OS (61.4%), despite tandem protocol ([90Y]Y-DOTATOC + [177Lu]Lu-DOTATATE) provided the highest response rates (38.1%). Best outcome was reached in patients who underwent RLT in early stage of disease, suggesting that this treatment should also be considered for newly diagnosed unresectable BCs. Moreover, despite most patients having only mild and transient adverse events, patients with hematologic toxicity showed worse survival outcomes.
Click to Show/Hide
|
||||
| Experiment 317 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Bronchial carcinoids | ||||
| Efficacy Data | Median overall survival (mOS) |
49 months
|
|||
| Patients Enrolled |
48 patients with bronchial carcinoids.
|
||||
| Administration Time | Median 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
Treatment of bronchial carcinoids is not simple and requires a multidisciplinary approach. Surgery remains the mainstay of treatment for local disease, while in advanced disease, management includes chemotherapy, cold somatostatin analogues, immunotherapy, everolimus, and others target therapies. RLT is an option for selected patients with advanced or metastatic BCs overexpressing SSTR in progression to cold SSAs therapy. Although the experience of [177Lu]Lu-DOTATATE is more limited in BCs than GEP-NET, off-label use can be considered with promising results. A prospective phase II trial in 34 patients with stage IV BCs treated with cumulative activity of 18.5-27.8 GBq in four or five cycles of [177Lu]Lu-DOTATATE documented a DCR of 80% (6% complete response, 27% partial response, and 47% stable disease) and a median PFS of 20 months. In this study, negative prognostic factors were AC histology, tumor thyroid transcription factor-1 (TTF-1) expression, and a positive [18F]FDG PET imaging. Comparable results (median PFS of 17 months) were reported in a group of patients with diffuse extrahepatic metastases treated with RLT after several lines of therapies. Higher activities were administered in a study by van Essen et al., who treated nine patients with metastatic BCs, delivering a cumulative dose of 22.2-29.6 GBq of [177Lu]Lu-DOTATATE, demonstrating an overall response rate comparable to that of other GEP-NET (50% vs. 47%, respectively for BCs and GEP-NET). Moreover, tumor regression was reported in 66.6% of patients, without any outcome discrepancies between TCs and ACs. Comparable median OS and response rate between BCs and others GEP-NETs were reported also in another recent retrospective study. Likewise, the efficacy of RLT in BCs was demonstrated by Brabander et al. in a study involving 443 patients affected by midgut, bronchial, and CUP-NETs treated with 7.4 GBq of [177Lu]Lu-DOTATATE every 8 weeks. The authors found that objective response rate (ORR) for BCs was 30%, and an additional 30% of patients had a stable disease. Nevertheless, median OS for BCs was 52 months versus 71 months for pancreatic-NET. Long term results were analyzed by Mariniello et al. in 114 patients with advanced BCs treated with different RLT protocols. They documented a median PFS and OS of 28.0 and 58.8 months, respectively, while morphological responses were observed in 26.5% of patients. [177Lu]Lu-DOTATATE monotherapy protocol resulted in the highest 5-year OS (61.4%), despite tandem protocol ([90Y]Y-DOTATOC + [177Lu]Lu-DOTATATE) provided the highest response rates (38.1%). Best outcome was reached in patients who underwent RLT in early stage of disease, suggesting that this treatment should also be considered for newly diagnosed unresectable BCs. Moreover, despite most patients having only mild and transient adverse events, patients with hematologic toxicity showed worse survival outcomes.
Click to Show/Hide
|
||||
| Experiment 318 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Hindgut neuroendocrine tumour | ||||
| Efficacy Data | Median overall survival (mOS) |
50 months
|
|||
| Patients Enrolled |
14 patients with hindgut neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 319 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Median overall survival (mOS) |
50 months
|
|||
| Patients Enrolled |
78 patients with pancreatic neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 320 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Median overall survival (mOS) |
50.7 months
|
|||
| Patients Enrolled |
Patients with neuroendocrine tumours who met selection criteria.
|
||||
| Description |
Estimated median overall survival was significantly longer for patients who met selection criteria compared with those who did not (50.7 vs 34.2 months) (P = 0.018).
|
||||
| Experiment 321 Reporting the Activity Data of This PDC | [26] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumor | ||||
| Efficacy Data | Median overall survival (mOS) |
52.9 months
|
|||
| Patients Enrolled |
Patients with advanced GEP-neuroendocrine tumour.
|
||||
| Administration Time | Four or five cycles | ||||
| Administration Dosage | 18.5 GBq/27.8 GBq | ||||
| Description |
The median progression-free survival (PFS) was 23 months (95% CI 14.9-31.0 months) and the median overall survival was 52.9 months (95% CI 17.1-68.9 months).
|
||||
| Experiment 322 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Median overall survival (mOS) |
53 months
|
|||
| Patients Enrolled |
82 patients with CUP-neuroendocrine tumour.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
Unfortunately, the literature lacks specific trials assessing RLT efficacy and safety on CUP-NETs, and the only available literature evidence is provided by mixed trials also including a few patients with SSTR-positive CUP-NETs. In a large cohort of patients treated with [177Lu]Lu-DOTATATE, Brabander et al.reported of 82 CUP-NET patients. Overall, DCR was reached in 78% of patients and median PFS and OS were 29 and 53 months, respectively. These results show that RLT may be effective in CUP-NETs patients, with a response rate intermediate between that obtained in GEP-NETswho showed longer median OS (60 vs. 53 months, respectively)and BCswho showed shorter median PFS (20 vs. 29 months, respectively). These results are consistent with those reported by Demirci et al. who treated 19 CUP-NETs, with an overall DCR of 84.2% and mean PFS and OS of 40 and 48 months, respectively. Once again, RLT outcomes in CUP-NETs were intermediate between those in GEP-NETs and BCs. In a large mixed cohort, Baum et al. treated 151 CUP-NETs (mostly with [177Lu]Lu-DOTATATE, although a small percentage of patients was treated with [90Y]Y-DOTATATE/DOTATOC in the study), obtaining median PFS and OS of 13 and 46 months, respectively. CUP-NETStogether with BCswere associated with a significantly shorter PFS at multivariate analysis if compared to pancreatic NETs despite showing a longer OS (50 vs. 43 months, respectively). Future prospective trials assessing efficacy and safety of RLT in CUP-NETs are desirable.
Click to Show/Hide
|
||||
| Experiment 323 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Median overall survival (mOS) |
53 months
|
|||
| Patients Enrolled |
1048 patients with CUP-neuroendocrine tumour.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
Unfortunately, the literature lacks specific trials assessing RLT efficacy and safety on CUP-NETs, and the only available literature evidence is provided by mixed trials also including a few patients with SSTR-positive CUP-NETs. In a large cohort of patients treated with [177Lu]Lu-DOTATATE, Brabander et al.reported of 82 CUP-NET patients. Overall, DCR was reached in 78% of patients and median PFS and OS were 29 and 53 months, respectively. These results show that RLT may be effective in CUP-NETs patients, with a response rate intermediate between that obtained in GEP-NETswho showed longer median OS (60 vs. 53 months, respectively)and BCswho showed shorter median PFS (20 vs. 29 months, respectively). These results are consistent with those reported by Demirci et al. who treated 19 CUP-NETs, with an overall DCR of 84.2% and mean PFS and OS of 40 and 48 months, respectively. Once again, RLT outcomes in CUP-NETs were intermediate between those in GEP-NETs and BCs. In a large mixed cohort, Baum et al. treated 151 CUP-NETs (mostly with [177Lu]Lu-DOTATATE, although a small percentage of patients was treated with [90Y]Y-DOTATATE/DOTATOC in the study), obtaining median PFS and OS of 13 and 46 months, respectively. CUP-NETStogether with BCswere associated with a significantly shorter PFS at multivariate analysis if compared to pancreatic NETs despite showing a longer OS (50 vs. 43 months, respectively). Future prospective trials assessing efficacy and safety of RLT in CUP-NETs are desirable.
Click to Show/Hide
|
||||
| Experiment 324 Reporting the Activity Data of This PDC | [55] | ||||
| Indication | Midgut neuroendocrine tumour | ||||
| Efficacy Data | Median overall survival (mOS) |
54.9 months
|
|||
| Patients Enrolled |
Patients with midgut neuroendocrine tumour treated with <sup>177</sup>Lu-Dotatate.
|
||||
| Description |
The approval of 177Lu-Dotatate was based on the findings of the phase III NETTER-1 trial, which showed that for patients with midgut (jejunum, ileum, appendix and proximal colon) NETs, the use of 177Lu-Dotatate (plus octreotide long-acting repeatable [LAR] 30 mg for symptom control) led to a significant gain in both progression-free survival (PFS), overall survival (OS) and quality of life relative to BSC involving octreotide LAR 60 mg Findings from the pivotal phase III NETTER-1 trial were supported by data from the ERASMUS study, in which midgut-NET patients treated with 177Lu-Dotatate had a median (95% CI) PFS and OS of 28.5 (23.9-33.3) months and 54.9 (47.5-63.2) months, respectively.
Click to Show/Hide
|
||||
| Experiment 325 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Bronchial carcinoids | ||||
| Efficacy Data | Median overall survival (mOS) |
58.8 months
|
|||
| Patients Enrolled |
114 patients with bronchial carcinoids.
|
||||
| Administration Time | 4-6 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
Treatment of bronchial carcinoids is not simple and requires a multidisciplinary approach. Surgery remains the mainstay of treatment for local disease, while in advanced disease, management includes chemotherapy, cold somatostatin analogues, immunotherapy, everolimus, and others target therapies. RLT is an option for selected patients with advanced or metastatic BCs overexpressing SSTR in progression to cold SSAs therapy. Although the experience of [177Lu]Lu-DOTATATE is more limited in BCs than GEP-NET, off-label use can be considered with promising results. A prospective phase II trial in 34 patients with stage IV BCs treated with cumulative activity of 18.5-27.8 GBq in four or five cycles of [177Lu]Lu-DOTATATE documented a DCR of 80% (6% complete response, 27% partial response, and 47% stable disease) and a median PFS of 20 months. In this study, negative prognostic factors were AC histology, tumor thyroid transcription factor-1 (TTF-1) expression, and a positive [18F]FDG PET imaging. Comparable results (median PFS of 17 months) were reported in a group of patients with diffuse extrahepatic metastases treated with RLT after several lines of therapies. Higher activities were administered in a study by van Essen et al., who treated nine patients with metastatic BCs, delivering a cumulative dose of 22.2-29.6 GBq of [177Lu]Lu-DOTATATE, demonstrating an overall response rate comparable to that of other GEP-NET (50% vs. 47%, respectively for BCs and GEP-NET). Moreover, tumor regression was reported in 66.6% of patients, without any outcome discrepancies between TCs and ACs. Comparable median OS and response rate between BCs and others GEP-NETs were reported also in another recent retrospective study. Likewise, the efficacy of RLT in BCs was demonstrated by Brabander et al. in a study involving 443 patients affected by midgut, bronchial, and CUP-NETs treated with 7.4 GBq of [177Lu]Lu-DOTATATE every 8 weeks. The authors found that objective response rate (ORR) for BCs was 30%, and an additional 30% of patients had a stable disease. Nevertheless, median OS for BCs was 52 months versus 71 months for pancreatic-NET. Long term results were analyzed by Mariniello et al. in 114 patients with advanced BCs treated with different RLT protocols. They documented a median PFS and OS of 28.0 and 58.8 months, respectively, while morphological responses were observed in 26.5% of patients. [177Lu]Lu-DOTATATE monotherapy protocol resulted in the highest 5-year OS (61.4%), despite tandem protocol ([90Y]Y-DOTATOC + [177Lu]Lu-DOTATATE) provided the highest response rates (38.1%). Best outcome was reached in patients who underwent RLT in early stage of disease, suggesting that this treatment should also be considered for newly diagnosed unresectable BCs. Moreover, despite most patients having only mild and transient adverse events, patients with hematologic toxicity showed worse survival outcomes.
Click to Show/Hide
|
||||
| Experiment 326 Reporting the Activity Data of This PDC | [20] | ||||
| Indication | SSR positive lung neuroendocrine tumor | ||||
| Efficacy Data | Median overall survival (mOS) |
59 months
|
|||
| Patients Enrolled |
48 patients with neuroendocrine tumour.
|
||||
| Administration Time | 1-4 cycles | ||||
| Administration Dosage | Median cumulative activity of 27 GBq (range, 6-43 GBq) | ||||
| MOA of PDC |
In patients with advanced progressive lung NET and satisfactory SSR expression, 177Lu-DOTATATE is effective and safe, with a high DCR and encouraging PFS and OS. Further prospective studies comparing 177Lu-DOTATATE with other systemic options are warranted.
|
||||
| Description |
Of 48 patients (median age, 63 y; 13 women), 43 (90%) had AC and 5 (10%) TC. Almost all patients (47, 98%) were treated due to progression. Most patients (40, 83%) received somatostatin analogs, and 10 patients (20%) had prior everolimus, chemotherapy, or both. All patients had high SSR expression (≥ modified Krenning score 3) on pretreatment 68Ga-DOTATATE PET/CT. Patients received a median 4 (range, 1-4) cycles of 177Lu-DOTATATE (33% with concurrent radiosensitizing chemotherapy) to a median cumulative activity of 27 GBq (range, 6-43GBq). At a median follow-up of 42 mo, the median PFS and OS were 23 mo (95% CI, 18-28 mo) and 59 mo (95% CI, 50-not reached [NR]), respectively. Of 40 patients with RECIST-measurable disease and 39 patients with available 68Ga-DOTATATE PET/CT, response categories were partial response, 20% (95% CI, 10%-35%) and 44% (95% CI, 30%-59%); stable disease, 68% (95% CI, 52%-80%) and 44% (95% CI, 30%-59%); and progressive disease, 12% (95% CI, 5%-27%) by both, respectively. There was a moderate concordance between response categories by RECIST and 68Ga-DOTATATE PET/CT, weighted of 0.51 (95% CI, 0.21-0.68).
Click to Show/Hide
|
||||
| Experiment 327 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Bronchial neuroendocrine tumour | ||||
| Efficacy Data | Median overall survival (mOS) |
59 months
|
|||
| Patients Enrolled |
21 patients with bronchial neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| Evaluation Method | Computed tomography (CT) or magnetic resonance imaging (MRI) assay | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Of the 395 patients, 280 (71%) completed all four cycles, 28 patients (7%) completed three cycles, 54 patients (14%) completed two cycles and 33 patients (8%) completed one cycle. The estimated median PFS for the entire cohort (n = 395) was 33 months (95% CI, 29-37) with 227 (57%) of patients having PD by the end of the study (Figure 1). Mean Follow Up time was 41 months (95% CI, 37-45). The estimated median OS for the entire cohort (n = 395) was 46 months (95% CI, 48-56). The number of patients who had died by the end of the study was 192 (49%). The median OS in patients completing only one or two cycles (n = 87) was 13 months compared to 48 months for patients who had completed three or four cycles (n = 308). Reasons for not completing four cycles were radiological progression in 40 patients, toxicity in seven patients (2 due to extreme fatigue, 5 due to bone marrow toxicity), clinical deterioration in 31 patients, death in 26 patients and other reasons, including patient choice and loss to follow up, in 11 patients. The median OS for patients who had been treated with four cycles of 177Lu-DOTATATE and were retreated with a further two cycles following PD (n = 26) was 59 months. Patients who had early PD (i.e., on the first response assessment cross-sectional study) following 177Lu-DOTATATE had statistically significant worse OS; p-value <.001 (median OS 33 months).
Click to Show/Hide
|
||||
| Experiment 328 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Bronchial carcinoids | ||||
| Efficacy Data | Median overall survival (mOS) |
63 months
|
|||
| Patients Enrolled |
443 patients with bronchial carcinoids.
|
||||
| Administration Dosage | Cumulative dose: 27.8-29.6 GBq | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
Treatment of bronchial carcinoids is not simple and requires a multidisciplinary approach. Surgery remains the mainstay of treatment for local disease, while in advanced disease, management includes chemotherapy, cold somatostatin analogues, immunotherapy, everolimus, and others target therapies. RLT is an option for selected patients with advanced or metastatic BCs overexpressing SSTR in progression to cold SSAs therapy. Although the experience of [177Lu]Lu-DOTATATE is more limited in BCs than GEP-NET, off-label use can be considered with promising results. A prospective phase II trial in 34 patients with stage IV BCs treated with cumulative activity of 18.5-27.8 GBq in four or five cycles of [177Lu]Lu-DOTATATE documented a DCR of 80% (6% complete response, 27% partial response, and 47% stable disease) and a median PFS of 20 months. In this study, negative prognostic factors were AC histology, tumor thyroid transcription factor-1 (TTF-1) expression, and a positive [18F]FDG PET imaging. Comparable results (median PFS of 17 months) were reported in a group of patients with diffuse extrahepatic metastases treated with RLT after several lines of therapies. Higher activities were administered in a study by van Essen et al., who treated nine patients with metastatic BCs, delivering a cumulative dose of 22.2-29.6 GBq of [177Lu]Lu-DOTATATE, demonstrating an overall response rate comparable to that of other GEP-NET (50% vs. 47%, respectively for BCs and GEP-NET). Moreover, tumor regression was reported in 66.6% of patients, without any outcome discrepancies between TCs and ACs. Comparable median OS and response rate between BCs and others GEP-NETs were reported also in another recent retrospective study. Likewise, the efficacy of RLT in BCs was demonstrated by Brabander et al. in a study involving 443 patients affected by midgut, bronchial, and CUP-NETs treated with 7.4 GBq of [177Lu]Lu-DOTATATE every 8 weeks. The authors found that objective response rate (ORR) for BCs was 30%, and an additional 30% of patients had a stable disease. Nevertheless, median OS for BCs was 52 months versus 71 months for pancreatic-NET. Long term results were analyzed by Mariniello et al. in 114 patients with advanced BCs treated with different RLT protocols. They documented a median PFS and OS of 28.0 and 58.8 months, respectively, while morphological responses were observed in 26.5% of patients. [177Lu]Lu-DOTATATE monotherapy protocol resulted in the highest 5-year OS (61.4%), despite tandem protocol ([90Y]Y-DOTATOC + [177Lu]Lu-DOTATATE) provided the highest response rates (38.1%). Best outcome was reached in patients who underwent RLT in early stage of disease, suggesting that this treatment should also be considered for newly diagnosed unresectable BCs. Moreover, despite most patients having only mild and transient adverse events, patients with hematologic toxicity showed worse survival outcomes.
Click to Show/Hide
|
||||
| Experiment 329 Reporting the Activity Data of This PDC | [58] | ||||
| Indication | Small bowel neuroendocrine tumor | ||||
| Efficacy Data | Median overall survival (mOS) |
110 months
|
|||
| Patients Enrolled |
Patients with consecutive patients with small bowel neuroendocrine tumour.
|
||||
| Administration Time | Every eight to 12 weeks | ||||
| Administration Dosage | 7.4 GBq (200 mCi) | ||||
| Evaluation Method | 68Ga-DOTATATE PET/CT assay | ||||
| MOA of PDC |
Surgery combined with 177Lu PRRT is safe and provides favourable PFS and OS in selected patients with advanced SBNEN. Liquid biopsy (NETest) has the potential to accurately delineate disease status.
|
||||
| Description |
A surgical cohort of 39 SBNEN patients met eligibility criteria. Thirty-two patients underwent ileal resection and 7 right hemicolectomy. The mean number of 177Lu PRRT cycles was 4. Mortality was nil. Surgical morbidity was 10.3%. Transient grade 1/2 toxicity occurred in 41% (PRRT). NETest scores (n=9 patients) decreased in 100% following treatment and correlated with diminished tumour volume and disease stabilization following surgery and PRRT. Median follow-up: 78 months. Median PFS and OS: 42.7 and 110 months, respectively. Progression-free survival at 1-, 3-, and 5-years was 79.4%, 57.1% and 40.5%, respectively. Overall survival at 1-, 3-, and 5-years was 97.4%, 97.4%, and 94.1%, respectively
Click to Show/Hide
|
||||
| Experiment 330 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Phaeochromocytoma | ||||
| Efficacy Data | Median overall survival (mOS) |
142.6 months
|
|||
| Patients Enrolled |
46 patients with Pheochromocytomas (PPGL) and paragangliomas (PHEO).
|
||||
| Administration Time | 4-5 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
As a consequence, we saw a rising interest in a potential radiolabeled-SST agonist theranostic approach for PPGLs, with several spontaneous studies reporting of the treatment of metastatic or inoperable tumors with Lutathera and other similar radiocompounds, with promising preliminary results. The first RLT experience in PPGLs was reported by van Essen et al. who treated a heterogeneous cohort of patients, including 12 PGLs. Despite the authors reporting lower response rates than those obtained in GEP-NET patients, ORR and DCR were 18% and 73%, respectively. These results are consistent with other reports in the literature. Kong et al. treated 20 PPGLs (8 PHEOs and 12 PGLs) with [117Lu]Lu-DOTATATE, 14 due to uncontrolled secondary hypertension and 6 to radiological PD. A DCR equal to 86% was reached, including five patients with ORR (36%). Of note, 62% of symptomatic patients required a dose reduction of antihypertensive medications, and 57% reported a subjective therapeutic benefit in terms of tumor-related symptoms. Comprehensive PFS was 39 months, including two patients with early recurrence (2 and 5 months post-RLT, respectively) and one patient with remarkable tumor downsizing that allowed a second-step curative liver surgery. Of note, the patient was still disease-free at the time of the study. The results of the largest cohort of PPGLs treated with RLT were reported by Severi et al., who treated 46 patients reaching a comprehensive DCR of 80%. Interestingly, 34 patients treated with [177Lu]Lu-DOTATATE obtained a longer median OS in comparison to those treated with [90Y]Y-DOTATOC (143 vs. 92 months, respectively). According to the authors, this result may be related both to DOTATATEs higher affinity for SSTR2 (which is the type most overexpressed in PPGLs) and to the longer half-life of 177Lu and, consequently, prolonged residence time within the tumor lesions. Moreover, this study reports that sympathetic functioning PPGLs were associated to a shorter median PFS compared to non-functioning PPGLs. Prado-Wohlwend et al. treated nine patients with [177Lu]Lu-DOTATATE and eight with [131I]MIBG, obtaining a PFS of 29 and 18.5 months and a DCR of 88.8% and 62.5%, respectively, for each therapy. Despite the low number of patients involved, a trend for a longer PFS was found for adrenal primary PPGLs treated with [177Lu]Lu-DOTATATE. Nevertheless, the authors suggest performing both [68Ga]Ga-DOTA-SSA PET/CT and [123I]MIBG SPECT/CT in each patient in order to offer a personalized theranostic approach based on the highest uptake intensity in one of the two imaging scans. This is consistent with the results by Jaiswal et al. who found that a baseline SUVmax > 21 at [68Ga]Ga-DOTA-SSA PET/CT is a very strong predictor of response to [177Lu]Lu-DOTATATE (p < 0.0001).
Click to Show/Hide
|
||||
| Experiment 331 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Paraganglioma | ||||
| Efficacy Data | Median overall survival (mOS) |
142.6 months
|
|||
| Patients Enrolled |
46 patients with Pheochromocytomas (PPGL) and paragangliomas (PHEO).
|
||||
| Administration Time | 4-5 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
As a consequence, we saw a rising interest in a potential radiolabeled-SST agonist theranostic approach for PPGLs, with several spontaneous studies reporting of the treatment of metastatic or inoperable tumors with Lutathera and other similar radiocompounds, with promising preliminary results. The first RLT experience in PPGLs was reported by van Essen et al. who treated a heterogeneous cohort of patients, including 12 PGLs. Despite the authors reporting lower response rates than those obtained in GEP-NET patients, ORR and DCR were 18% and 73%, respectively. These results are consistent with other reports in the literature. Kong et al. treated 20 PPGLs (8 PHEOs and 12 PGLs) with [117Lu]Lu-DOTATATE, 14 due to uncontrolled secondary hypertension and 6 to radiological PD. A DCR equal to 86% was reached, including five patients with ORR (36%). Of note, 62% of symptomatic patients required a dose reduction of antihypertensive medications, and 57% reported a subjective therapeutic benefit in terms of tumor-related symptoms. Comprehensive PFS was 39 months, including two patients with early recurrence (2 and 5 months post-RLT, respectively) and one patient with remarkable tumor downsizing that allowed a second-step curative liver surgery. Of note, the patient was still disease-free at the time of the study. The results of the largest cohort of PPGLs treated with RLT were reported by Severi et al., who treated 46 patients reaching a comprehensive DCR of 80%. Interestingly, 34 patients treated with [177Lu]Lu-DOTATATE obtained a longer median OS in comparison to those treated with [90Y]Y-DOTATOC (143 vs. 92 months, respectively). According to the authors, this result may be related both to DOTATATEs higher affinity for SSTR2 (which is the type most overexpressed in PPGLs) and to the longer half-life of 177Lu and, consequently, prolonged residence time within the tumor lesions. Moreover, this study reports that sympathetic functioning PPGLs were associated to a shorter median PFS compared to non-functioning PPGLs. Prado-Wohlwend et al. treated nine patients with [177Lu]Lu-DOTATATE and eight with [131I]MIBG, obtaining a PFS of 29 and 18.5 months and a DCR of 88.8% and 62.5%, respectively, for each therapy. Despite the low number of patients involved, a trend for a longer PFS was found for adrenal primary PPGLs treated with [177Lu]Lu-DOTATATE. Nevertheless, the authors suggest performing both [68Ga]Ga-DOTA-SSA PET/CT and [123I]MIBG SPECT/CT in each patient in order to offer a personalized theranostic approach based on the highest uptake intensity in one of the two imaging scans. This is consistent with the results by Jaiswal et al. who found that a baseline SUVmax > 21 at [68Ga]Ga-DOTA-SSA PET/CT is a very strong predictor of response to [177Lu]Lu-DOTATATE (p < 0.0001).
Click to Show/Hide
|
||||
| Experiment 332 Reporting the Activity Data of This PDC | [31] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumour | ||||
| Efficacy Data | Median latency period |
41 months
|
|||
| Patients Enrolled |
Patients with gastroenteropancreatic neuroendocrine tumors.
|
||||
| Description |
The median latency period at diagnosis (or first suspicion of a PHD) was 41 mo (range, 15-84 mo).
|
||||
| Experiment 333 Reporting the Activity Data of This PDC | [25] | ||||
| Indication | SDHB positive metastatic paraganglioma | ||||
| Efficacy Data | Median follow-up |
38 months
|
|||
| Patients Enrolled |
37 patients affected by neuroendocrine tumors.
|
||||
| Administration Time | Five cycles of 5.5 gbq each every 8 weeks | ||||
| Administration Dosage | Cumulative activity of 27.5 GBq | ||||
| Description |
Thirty-three of 37 patients were evaluable for response. Objective responses included partial response (PR) in 10 patients (30%), stable disease (SD) in 18 patients (55%), with a DCR of 85%. Median follow up was 38 months (range 4.6-51.1 months). Median PFS was 31.4 months (17.6-45.4) and mOS was not reached.
|
||||
| Experiment 334 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | High-grade (WHO G3) neuroendocrine tumour | ||||
| Efficacy Data | Mean progression-free survival (mPFS) |
38 months
|
|||
| Patients Enrolled |
Patients with neuroendocrine tumours including high-grade (WHO G3) neuroendocrine tumours.
|
||||
| Evaluation Method | Kaplan-Meier estimated assay | ||||
| Description |
The Kaplan-Meier estimated median PFS was 36.4 months, mean PFS was 38 months and the mean OS was 55 months.
|
||||
| Experiment 335 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | High-grade (WHO G3) neuroendocrine tumour | ||||
| Efficacy Data | Mean overall survival (mOS) |
55 months
|
|||
| Patients Enrolled |
Patients with neuroendocrine tumours including high-grade (WHO G3) neuroendocrine tumours.
|
||||
| Evaluation Method | Kaplan-Meier estimated assay | ||||
| Description |
The Kaplan-Meier estimated median PFS was 36.4 months, mean PFS was 38 months and the mean OS was 55 months.
|
||||
| Experiment 336 Reporting the Activity Data of This PDC | [62] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Malondialdehyde increase rate |
0.44 µM
|
|||
| Patients Enrolled |
Prostate cancer and neuroendocrine tumor patients referred to therapy with <sup>177</sup>Lu-PSMA and <sup>177</sup>Lu-DOTATATE, respectively.
|
||||
| Administration Time | 48 h | ||||
| Administration Dosage | 7.4 GBq | ||||
| MOA of PDC |
Although RNT with 177Lu-DOTATATE/PSMA is known as a novel and effective therapy option for cancer that significantly improves the quality of life and survival of patients, it may have acute or chronic side effects. Therefore, any method that can ameliorate these side effects is useful in the RNT process. For this purpose, a few clinical studies have reported that antioxidants as free radical scavengers such as amifostine and vitamins C and E can reduce radioiodine-related side effects, particularly in salivary glands in thyroid cancer patients.
Click to Show/Hide
|
||||
| Description |
In total, 61 RNT cycles were evaluated in 34 patients with age of 65 ± 2.83 (median ± SE) years (range of 27-99); this total included 20 (59%) prostate cancer patients [35 cycles (57.4%)] and 14 patients (41%) with NET [26 cycles (42.6%)]. Of the 61 evaluated cycles, 27 cycles were given in the control group and 34 cycles were given in the intervention group. The serum level of MDA was significantly increased after treatment compared to before treatment (P = 0.02) in the control group, while no significant change in the serum level of MDA was observed in the intervention group (P = 0.52). The serum level of GSH was insignificantly decreased after treatment compared to before treatment in the control group and slightly increased after treatment in the intervention group (P > 0.05). The serum level of glutathione reductase was insignificantly increased in all groups of patients after treatment (P > 0.05).
Click to Show/Hide
|
||||
| Experiment 337 Reporting the Activity Data of This PDC | [30] | ||||
| Indication | Sinonasal neuroendocrine carcinomas | ||||
| Efficacy Data | Lymph node decrease rate |
42.30%
|
|||
| Administration Dosage | ˜7.4 GBq [200 mCi] | ||||
| Description |
On follow-up for a second PRRT cycle, there was a complete symptomatic response. Follow-up scans showed a significant decrease in the size of the sinonasal mass (˜1.9 0.8 cm vs. 7.0 4.6 5.0 cm at baseline), with a significant decrease in the size of the left cervical level II lymph node (1.5 1.1 cm vs. 2.2 1.3 cm at baseline) and complete resolution of the right hilar lymph node.
Click to Show/Hide
|
||||
| In Vivo Model | A 52-year-old man with SNC. | ||||
| Experiment 338 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Leukopenia |
10%
|
|||
| Patients Enrolled |
30 patients with pheocromocytomas (PCCs) and paragangliomas (PGLs) (3 PCC, 27PGL).
|
||||
| Administration Time | 73% of patients received 4 cycles | ||||
| Administration Dosage | 7.4 GBq/cycle | ||||
| MOA of PDC |
We showed that PRRT based on 177Lu-DOTATATE and 90-YDOTATOC without radiosensitizers are efficacious therapeutic options (DCR of 83% and 76%, respectively) and can be considered in the multidisciplinary treatment of PCCs and PGLs as alternatives to I-131 MIBG and chemotherapy.
|
||||
| Description |
In detail, we report pooled data on 149 177Lu- and 64 90Y-treated patients (total: 213 patients). Most studies were retrospective. The largest one included 46 patients (34 treated with 177Lu-, 12 with 90Y- PRRTs). Median ages ranged from 32.5 to 60.4 years. The female gender was slightly predominant (83 women, 74 men, gender not specified in 3 studies). When reported, mutations of SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B) were the most frequent genetic alterations. According to the predominant retrospective and real practice nature of the selected studies, sites of metastases and previous treatments were heterogeneous, with bone the metastatic site most often involved, and surgery the most frequent previous therapy. Paramount descriptive information on PRRT doses per cycle, number of cycles, response criteria, best responses to treatments, and frequency of G3/G4 hematologic toxicities is reported in Table 2. The lowest applied dose was 3.4 GBq per cycle; the number of administered cycles was heterogeneous (at least 3 cycles in 7 studies). The most used response criteria were the RECIST 1.1. Only one study evaluated the response with SWOG criteria, which are more conservative with regard to the definition of PR (50% or greater reduction in the sum of the product of the perpendicular diameters of target lesions). The majority of the patients experienced PR or SD. Hematologic toxicity was manageable. Funnel plot of selected studies did not show publication bias. The MINORS and Newcastle-Ottawa Scale scoring for the selected articles are reported in Table 3.
Click to Show/Hide
|
||||
| Experiment 339 Reporting the Activity Data of This PDC | [39] | ||||
| Indication | SSR positive lung neuroendocrine tumor | ||||
| Efficacy Data | Less progressive disease |
p Values
|
|||
| Patients Enrolled |
Patients with somatostatin-expressing neuroendocrine tumors who underwent Lu-DOTATATE PRRT; with CRHE.
|
||||
| Description |
Following PRRT, 76.5% of patients with CRHE experienced benefit (partial response + stable disease), whereas 23.4% experienced progressive disease. Patients with CRHE showed more stable disease (P = 0.048) and less progressive disease (P = 0.046) following PRRT compared with no CRHE.
|
||||
| Experiment 340 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Kidney toxicity |
5.06%
|
|||
| Patients Enrolled |
534 male patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (25.9-29.6 GBq) (800 mCi [700-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 341 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Kidney toxicity |
7.34%
|
|||
| Patients Enrolled |
439 female patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (22.2-29.6 GBq) (800 mCi [600-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 342 Reporting the Activity Data of This PDC | [48] | ||||
| Indication | Functioning pancreatic neuroendocrine tumour | ||||
| Efficacy Data | Hormonal crisis |
9%
|
|||
| Patients Enrolled |
Patients with functioning pancreatic neuroendocrine tumors.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | 7.4 GBq/cycle | ||||
| Description |
Subacute hematological toxicity, grade 3 or 4 occurred in 4 patients (12%) and a hormonal crisis in 3 patients (9%). PRRT resulted in partial or complete response in 59% of patients and the disease control rate was 78% in patients with baseline progression. 71% of patients with uncontrolled symptoms had a reduction of symptoms and a more than 80% decrease of circulating hormone levels was measured during follow-up. After PRRT, median progression-free survival was 18.1 months (interquartile range: 3.3 to 35.7) with a concurrent increase in QOL.
Click to Show/Hide
|
||||
| Experiment 343 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Hepatic treatment-related serious adverse events |
0.40%
|
|||
| Patients Enrolled |
Patients with neuroendocrine tumours.
|
||||
| Description |
177Lu-DOTATATE was well tolerated with haematological, renal and hepatic treatment-related serious adverse events experienced by 9.7, 0.4 and 0.4% of patients respectively.
|
||||
| Experiment 344 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Metastatic midgut neuroendocrine tumor | ||||
| Efficacy Data | Hemoglobin level |
85 g/dL
|
|||
| Administration Dosage | 7.4 GBq | ||||
| Description |
Initial laboratory tests showed normocytic regenerative anemia (hemoglobin 85g/dL), thrombocytopenia (54 109/L), no signs of hemolysis, no schistocytes, prolonged prothrombin time (25.8seconds, 35%) and prolonged activated partial thromboplastin time (1.63), low fibrinogen (0.3g/L), elevated fibrin monomer (>400ug/mL) and elevated fibrin degradation products (>20ug/mL). Coagulation factors II (49%) and V (44%) were low; factors VII (97%) and X (81%) were normal. Liver function tests were normal with the exception of moderate elevation of gamma Glutamyl Transferase (γGT=100UI/L, N<38UI/L). C-reactive protein was <5mg/L. There was no sign of tumor lysis syndrome. The bone marrow aspirate and medullar karyotype were normal. Computed tomography showed stable metastatic disease, including a large stable necrotic liver metastasis.
Click to Show/Hide
|
||||
| In Vivo Model | 55-year-old woman with metastatic midgut neuroendocrine tumor. | ||||
| Experiment 345 Reporting the Activity Data of This PDC | [45] | ||||
| Indication | Primary cardiac paragangliomas | ||||
| Efficacy Data | Hemoglobin increase rate |
14 g/L
|
|||
| Patients Enrolled |
47 years old primary cardiac paragangliomas patient.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity of 40.7 GBq (1100 mCi) | ||||
| MOA of PDC |
Cardiac PGL is an exceedingly rare tumor and may lead to hemodynamic compromise. Surgical resection, if indicated, is technically challenging and frequently not feasible. This case demonstrates that personalized 177Lu-DOTATATE PRRT can be considered as a safe and effective palliative treatment for unresectable MIBG negative tumor which can substantially improve quality of life.
Click to Show/Hide
|
||||
| Description |
At the end of the 4 cycles, fatigue, chest pain and dyspnea resolved, and episodes of tachyarrhythmia disappeared. The dosage of metoprolol and prazocin was considerably reduced, decreasing respectively from 125 mg to 50 mg and 2 mg to 1 mg TID. The chromogranin A decreased from 585 to 205 ng/mL and the serum norepinephrine decreased from 104 to 37 nmol/L after 4 cycles.
Click to Show/Hide
|
||||
| Experiment 346 Reporting the Activity Data of This PDC | [31] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumour | ||||
| Efficacy Data | Hematopoietic neoplasm |
2.90%
|
|||
| Patients Enrolled |
Patients with gastroenteropancreatic neuroendocrine tumors.
|
||||
| Description |
8 patients (2.9%) developed a hematopoietic neoplasm (4 MDS, 1 AML, 1 MPN, and 2 MDS/MPN) and 3 patients (1.1%) developed BM failure characterized by cytopenia and BM aplasia.
|
||||
| Experiment 347 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Haematological treatment-related serious adverse events |
9.70%
|
|||
| Patients Enrolled |
Patients with neuroendocrine tumours.
|
||||
| Description |
177Lu-DOTATATE was well tolerated with haematological, renal and hepatic treatment-related serious adverse events experienced by 9.7, 0.4 and 0.4% of patients respectively.
|
||||
| Experiment 348 Reporting the Activity Data of This PDC | [19] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumour | ||||
| Efficacy Data | Grade 5 adverse events |
15.40%
|
|||
| Patients Enrolled |
13 GEP-neuroendocrine tumour patients.
|
||||
| Administration Time | 1-4 cycles | ||||
| Administration Dosage | 7.4 GBq (200mCi) | ||||
| MOA of PDC |
From our initial experience, Lutathera has been well tolerated in patients with GEP-NET. Additional studies are needed to examine long-term clinical and patient-reported outcomes associated with treatment of GEP-NETs as well as financial considerations for hospitals embarking on a PRRT program. A multidisciplinary team and complete collaboration between hospital administration and clinical teams are required for successful implementation of a PRRT program.
Click to Show/Hide
|
||||
| Description |
AE was recorded in 13 patients out of the entire 35 patients treated with PRRT. A grade 5 AE was reported in 2 patients (15.4%) and grade 3 AEs were reported in 5 patients (38.5%). Of the 5 grade 3 AE patients, 1 patient developed ascites, pleural effusion, and acute kidney injury; the second patient developed hematoma at the injection site and pain in the lower extremities; the third patient had shortness of breath, cough, and hemoptysis; the fourth patient had nausea, vomiting, and deep vein thrombosis; and the fifth patient developed obstructive jaundice. All 5 grade 3 AE patients were hospitalized and treated. One patient (7.7%) with a grade 2 AE developed an upper respiratory infection and required antibiotics. Five patients (38.5%) with grade 1 AEs had nausea, vomiting, abdominal pain, and/or diarrhea. No patients developed a grade 4 AE.
Click to Show/Hide
|
||||
| Experiment 349 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade 4 thrombocytopenia |
0%
|
|||
| Patients Enrolled |
363 patients with neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Safety data was available in 363 patients. A total of 28 patients developed grade 3 or 4 bone marrow toxicity (8%) (excluding lymphopenia). Of these, 21 patients (6%) had anaemia, 10 patients (3%) had leukopenia and five patients (1%) had thrombocytopenia. In addition, grade 3 or 4 lymphopenia was recorded in 79 patients (22%) (Table 3). One patient (0.3%) developed grade 4 nephrotoxicity. This patient developed gradual decline in kidney function and was commenced on haemodialysis. The decline in kidney function was attributed to persistent vomiting with poor oral intake but a contributory role of PRRT cannot be excluded. This patient had a baseline eGFR of 39 ml/min/1.73 m2. No further grade 3/4 renal toxicities were encountered. Myelodysplastic disease (MDS) occurred in one patient (0.3%). This patient had undergone chemotherapy for diffuse large B cell lymphoma with chemotherapy (rituximab, cyclophosphamide, vincristine, prednisolone), followed by etoposide, methylprednisolone, cytarabine, cisplatin 6 months which resulted in a complete metabolic response. The patient's MDS was diagnosed 48 months after the start of chemotherapy and 17 months after commencing PRRT. No acute leukaemia occurred.
Click to Show/Hide
|
||||
| Experiment 350 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade 4 nephrotoxicity |
0.30%
|
|||
| Patients Enrolled |
363 patients with neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Safety data was available in 363 patients. A total of 28 patients developed grade 3 or 4 bone marrow toxicity (8%) (excluding lymphopenia). Of these, 21 patients (6%) had anaemia, 10 patients (3%) had leukopenia and five patients (1%) had thrombocytopenia. In addition, grade 3 or 4 lymphopenia was recorded in 79 patients (22%) (Table 3). One patient (0.3%) developed grade 4 nephrotoxicity. This patient developed gradual decline in kidney function and was commenced on haemodialysis. The decline in kidney function was attributed to persistent vomiting with poor oral intake but a contributory role of PRRT cannot be excluded. This patient had a baseline eGFR of 39 ml/min/1.73 m2. No further grade 3/4 renal toxicities were encountered. Myelodysplastic disease (MDS) occurred in one patient (0.3%). This patient had undergone chemotherapy for diffuse large B cell lymphoma with chemotherapy (rituximab, cyclophosphamide, vincristine, prednisolone), followed by etoposide, methylprednisolone, cytarabine, cisplatin 6 months which resulted in a complete metabolic response. The patient's MDS was diagnosed 48 months after the start of chemotherapy and 17 months after commencing PRRT. No acute leukaemia occurred.
Click to Show/Hide
|
||||
| Experiment 351 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade 4 lymphopenia |
3%
|
|||
| Patients Enrolled |
363 patients with neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Safety data was available in 363 patients. A total of 28 patients developed grade 3 or 4 bone marrow toxicity (8%) (excluding lymphopenia). Of these, 21 patients (6%) had anaemia, 10 patients (3%) had leukopenia and five patients (1%) had thrombocytopenia. In addition, grade 3 or 4 lymphopenia was recorded in 79 patients (22%) (Table 3). One patient (0.3%) developed grade 4 nephrotoxicity. This patient developed gradual decline in kidney function and was commenced on haemodialysis. The decline in kidney function was attributed to persistent vomiting with poor oral intake but a contributory role of PRRT cannot be excluded. This patient had a baseline eGFR of 39 ml/min/1.73 m2. No further grade 3/4 renal toxicities were encountered. Myelodysplastic disease (MDS) occurred in one patient (0.3%). This patient had undergone chemotherapy for diffuse large B cell lymphoma with chemotherapy (rituximab, cyclophosphamide, vincristine, prednisolone), followed by etoposide, methylprednisolone, cytarabine, cisplatin 6 months which resulted in a complete metabolic response. The patient's MDS was diagnosed 48 months after the start of chemotherapy and 17 months after commencing PRRT. No acute leukaemia occurred.
Click to Show/Hide
|
||||
| Experiment 352 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade 4 leukopenia |
0.30%
|
|||
| Patients Enrolled |
363 patients with neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Safety data was available in 363 patients. A total of 28 patients developed grade 3 or 4 bone marrow toxicity (8%) (excluding lymphopenia). Of these, 21 patients (6%) had anaemia, 10 patients (3%) had leukopenia and five patients (1%) had thrombocytopenia. In addition, grade 3 or 4 lymphopenia was recorded in 79 patients (22%) (Table 3). One patient (0.3%) developed grade 4 nephrotoxicity. This patient developed gradual decline in kidney function and was commenced on haemodialysis. The decline in kidney function was attributed to persistent vomiting with poor oral intake but a contributory role of PRRT cannot be excluded. This patient had a baseline eGFR of 39 ml/min/1.73 m2. No further grade 3/4 renal toxicities were encountered. Myelodysplastic disease (MDS) occurred in one patient (0.3%). This patient had undergone chemotherapy for diffuse large B cell lymphoma with chemotherapy (rituximab, cyclophosphamide, vincristine, prednisolone), followed by etoposide, methylprednisolone, cytarabine, cisplatin 6 months which resulted in a complete metabolic response. The patient's MDS was diagnosed 48 months after the start of chemotherapy and 17 months after commencing PRRT. No acute leukaemia occurred.
Click to Show/Hide
|
||||
| Experiment 353 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade 4 anaemia |
0%
|
|||
| Patients Enrolled |
363 patients with neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Safety data was available in 363 patients. A total of 28 patients developed grade 3 or 4 bone marrow toxicity (8%) (excluding lymphopenia). Of these, 21 patients (6%) had anaemia, 10 patients (3%) had leukopenia and five patients (1%) had thrombocytopenia. In addition, grade 3 or 4 lymphopenia was recorded in 79 patients (22%) (Table 3). One patient (0.3%) developed grade 4 nephrotoxicity. This patient developed gradual decline in kidney function and was commenced on haemodialysis. The decline in kidney function was attributed to persistent vomiting with poor oral intake but a contributory role of PRRT cannot be excluded. This patient had a baseline eGFR of 39 ml/min/1.73 m2. No further grade 3/4 renal toxicities were encountered. Myelodysplastic disease (MDS) occurred in one patient (0.3%). This patient had undergone chemotherapy for diffuse large B cell lymphoma with chemotherapy (rituximab, cyclophosphamide, vincristine, prednisolone), followed by etoposide, methylprednisolone, cytarabine, cisplatin 6 months which resulted in a complete metabolic response. The patient's MDS was diagnosed 48 months after the start of chemotherapy and 17 months after commencing PRRT. No acute leukaemia occurred.
Click to Show/Hide
|
||||
| Experiment 354 Reporting the Activity Data of This PDC | [48] | ||||
| Indication | Functioning pancreatic neuroendocrine tumour | ||||
| Efficacy Data | Grade 3/4 subacute hematological toxicity |
12%
|
|||
| Patients Enrolled |
Patients with functioning pancreatic neuroendocrine tumors.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | 7.4 GBq/cycle | ||||
| Description |
Subacute hematological toxicity, grade 3 or 4 occurred in 4 patients (12%) and a hormonal crisis in 3 patients (9%). PRRT resulted in partial or complete response in 59% of patients and the disease control rate was 78% in patients with baseline progression. 71% of patients with uncontrolled symptoms had a reduction of symptoms and a more than 80% decrease of circulating hormone levels was measured during follow-up. After PRRT, median progression-free survival was 18.1 months (interquartile range: 3.3 to 35.7) with a concurrent increase in QOL.
Click to Show/Hide
|
||||
| Experiment 355 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Inoperable or metastatic paragangliomas | ||||
| Efficacy Data | Grade 3/4 subacute haematotoxicity |
20%
|
|||
| Patients Enrolled |
Patients with inoperable or metastatic paragangliomas and pheochromocytomas.
|
||||
| Administration Time | Four cycles | ||||
| Administration Dosage | 7.4 Gb per cycle | ||||
| Description |
Grade 3/4 subacute haematotoxicity occurred in 6 (20%) of patients.
|
||||
| Experiment 356 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Inoperable or metastatic pheochromocytomas | ||||
| Efficacy Data | Grade 3/4 subacute haematotoxicity |
20%
|
|||
| Patients Enrolled |
Patients with inoperable or metastatic paragangliomas and pheochromocytomas.
|
||||
| Administration Time | Four cycles | ||||
| Administration Dosage | 7.4 Gb per cycle | ||||
| Description |
Grade 3/4 subacute haematotoxicity occurred in 6 (20%) of patients.
|
||||
| Experiment 357 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Advanced paraganglioma | ||||
| Efficacy Data | Grade 3/4 lymphopenia |
11%
|
|||
| Patients Enrolled |
A total of 201 patients with advanced paragangliomas.
|
||||
| MOA of PDC |
The clinical applications of radiolabeled SSAs and DOTA-SSA compounds in imaging and radionuclide therapy have been rapidly increasing over the past 2 decades and have been integrated into the current management guidelines of NETs. Several studies support the safety and clinical efficacy of FDA-approved radionuclide therapies including 177Lu-DOTATATE in advanced GEP NETs and 131I-MIBG in advanced paragangliomas and pheochromocytomas. The utility of SSTR-based imaging in PRRT response assessment and surveillance of NETs remains to be established.
Click to Show/Hide
|
||||
| Description |
Over the past decade, a small number of studies have evaluated the efficacy and safety of PRRT with 177Lu-DOTA-SSA in patients with metastatic or inoperable paragangliomas [66, 67]. A recent meta-analysis of 12 studies including a total of 201 patients with advanced paragangliomas showed an objective response rate of 25%, biochemical response rate of 64%, and disease control rate of 84% after treatment with PRRT (either 90Y- or 177Lu-based agents) [67]. The following minimal treatment-related adverse effects were observed: grade 3 or 4 lymphopenia, thrombocytopenia, neutropenia, and nephrotoxicity in 11%, 9%, 3%, and 4% of the patients, respectively.
Click to Show/Hide
|
||||
| Experiment 358 Reporting the Activity Data of This PDC | [63] | ||||
| Indication | Inoperable grade I/II neuroendocrine tumor | ||||
| Efficacy Data | Grade 3/4 hematologic toxicity |
3.20%
|
|||
| Patients Enrolled |
31 patients treated using the fast-infusion protocol.
|
||||
| Administration Time | > 5 min | ||||
| Administration Dosage | 7.4 GBq | ||||
| Description |
Thirty-one patients, treated using the fast-infusion protocol, were included. Clinical toxicity mainly consisted of grade 1/2 fatigue (87.1%) and grade 1 nausea or vomiting (67.7%) during follow-up. No acute or long-term clinical toxicity possibly related to the fast-infusion protocol was reported. Grade 3/4 hematologic toxicity occurred after PRRT in 1 patient (3.2%). No grade 3/4 renal toxicity occurred. The laboratory experiment showed that when using the gravity method for infusion, half of the activity is infused after 3.5 min, and 95% is infused within 15 min.
Click to Show/Hide
|
||||
| Experiment 359 Reporting the Activity Data of This PDC | [38] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade 3/4 haematotoxicity |
8%
|
|||
| Patients Enrolled |
100 neuroendocrine tumour patients.
|
||||
| Administration Time | 4 cycles every 10 weeks | ||||
| Administration Dosage | 7.4 GBq | ||||
| MOA of PDC |
The incidence of severe haematotoxicity was low with extensive screening and monitoring. The vast majority of patients (96/100) was not restricted in treatment continuation by haematotoxicity; therefore, our selection criteria appeared appropriate for safe PRRT treatment. Baseline parameters showed limited correlation with the degree of decline in haematological values.
Click to Show/Hide
|
||||
| Description |
Four cycles were completed by 86/100 of patients, 4/100 patients discontinued due to haematotoxicity, and 10/100 patients due to progressive disease. The treatment course was adjusted due to haematotoxicity in 24/100 patients, including postponed next cycle (n = 17), reduced administered activity (n = 13), and both adjustments (n = 10). The most observed haematotoxicity grade was grade 0-1 in 54/100 patients, grade 2 in 38/100 and grade 3-4 in 8/100. Significant differences in baseline leucocyte, neutrophil and platelet counts were observed between grade 0-1 and grade 2. However, the correlation between baseline and lowest observed values was poor to moderate. No differences between haematotoxicity grades and baseline parameters or somatostatin receptor positive tumour volume was observed.
Click to Show/Hide
|
||||
| Experiment 360 Reporting the Activity Data of This PDC | [49] | ||||
| Indication | Bronchial and gastroenteropancreatic neuroendocrine tumour | ||||
| Efficacy Data | Grade 3/4 bone marrow toxicity |
6.60%
|
|||
| Patients Enrolled |
Patients with bronchial and gastroenteropancreatic neuroendocrine tumours.
|
||||
| Administration Time | Over two cycles | ||||
| Administration Dosage | 14.8 GBq | ||||
| Description |
Grade III/IV bone marrow toxicity occurred in 6.6% and 7.7% of patients after R-PRRT and RR-PRRT, respectively.
|
||||
| Experiment 361 Reporting the Activity Data of This PDC | [49] | ||||
| Indication | Bronchial and gastroenteropancreatic neuroendocrine tumour | ||||
| Efficacy Data | Grade 3/4 bone marrow toxicity |
7.70%
|
|||
| Patients Enrolled |
Patients with bronchial and gastroenteropancreatic neuroendocrine tumours.
|
||||
| Administration Time | Over two cycles | ||||
| Administration Dosage | 14.8 GBq | ||||
| Description |
Grade III/IV bone marrow toxicity occurred in 6.6% and 7.7% of patients after R-PRRT and RR-PRRT, respectively.
|
||||
| Experiment 362 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | High-grade (WHO G3) neuroendocrine tumour | ||||
| Efficacy Data | Grade 3 toxicity |
1.20%
|
|||
| Patients Enrolled |
Patients with neuroendocrine tumours including high-grade (WHO G3) neuroendocrine tumours.
|
||||
| Description |
We observed no major renal toxicity except a minor increase (11.1%) in average serum creatinine levels. In 33.9% (n=56) of the patients, grade I toxicity; in 9.1% (n=15), grade II; and in 1.2% (n=2), grade III toxicity were observed.
|
||||
| Experiment 363 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade 3 thrombocytopenia |
1%
|
|||
| Patients Enrolled |
363 patients with neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Safety data was available in 363 patients. A total of 28 patients developed grade 3 or 4 bone marrow toxicity (8%) (excluding lymphopenia). Of these, 21 patients (6%) had anaemia, 10 patients (3%) had leukopenia and five patients (1%) had thrombocytopenia. In addition, grade 3 or 4 lymphopenia was recorded in 79 patients (22%) (Table 3). One patient (0.3%) developed grade 4 nephrotoxicity. This patient developed gradual decline in kidney function and was commenced on haemodialysis. The decline in kidney function was attributed to persistent vomiting with poor oral intake but a contributory role of PRRT cannot be excluded. This patient had a baseline eGFR of 39 ml/min/1.73 m2. No further grade 3/4 renal toxicities were encountered. Myelodysplastic disease (MDS) occurred in one patient (0.3%). This patient had undergone chemotherapy for diffuse large B cell lymphoma with chemotherapy (rituximab, cyclophosphamide, vincristine, prednisolone), followed by etoposide, methylprednisolone, cytarabine, cisplatin 6 months which resulted in a complete metabolic response. The patient's MDS was diagnosed 48 months after the start of chemotherapy and 17 months after commencing PRRT. No acute leukaemia occurred.
Click to Show/Hide
|
||||
| Experiment 364 Reporting the Activity Data of This PDC | [17] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade 3 thrombocytopenia |
14.30%
|
|||
| Patients Enrolled |
7 patients with inoperable, progressive, metastatic, or locally advanced, somatostatin receptor-positive neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | 21.3-30.1 GBq total dose | ||||
| MOA of PDC |
In conclusion, for the first time in Korea, we have conducted a clinical trial of PRRT with SNU-KB-01, a NCA 177Lu-DOTATATE, in patients with SSTR-positive NET. Treatment with SNU-KB-01 was safe and resulted in control of disease in most of the patients with high SSTR expression. The recommended dose per cycle of SNU-KB-01 was determined to be 7.40 GBq for the phase 2 trial. Our results indicate SNU-KB-01 as a potentially safe and efficacious treatment option for NET patients and are expected to provide a wider choice of treatment opportunities for patients with various types of SSTR-positive tumors in Korea.
Click to Show/Hide
|
||||
| Description |
There was grade 3 thrombocytopenia in one patient (14.3%), but no other grade 3 or 4 major hematologic, renal, hepatotoxicity was observed. Grade 1 or 2 hematologic toxicities were anemia (28.6%), thrombocytopenia (57.1%), leukopenia (71.4%), and neutropenia (42.9%). Grade 1 renal toxicity occurred in one patient (14.3%). Grade 1 or 2 hepatotoxicities were observed in two patients (28.6%). The trend of the progressive decline of hematological parameters was observed during the treatment cycles.
Click to Show/Hide
|
||||
| Experiment 365 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Grade 3 thrombocytopenia |
16%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Treatment-related toxicities are listed in Table 3. Five patients (26%) experienced dose modifying toxicity during treatment with 177Lu-DOTATATE. The most common treatment-related toxicities were thrombocytopenia (9 patients, 47%; grade 3/4 in 1 patient, 5%), anemia (7 patients, 37%; grade 3/4 in 2 patients, 11%), leukopenia (6 patients, 32%; grade 3/4 in 0 patients), and elevation of the liver function tests (4 patients, 21%; grade 3/4 in 0 patients).
Click to Show/Hide
|
||||
| Experiment 366 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade 3 nephrotoxicity |
0.60%
|
|||
| Patients Enrolled |
363 patients with neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Safety data was available in 363 patients. A total of 28 patients developed grade 3 or 4 bone marrow toxicity (8%) (excluding lymphopenia). Of these, 21 patients (6%) had anaemia, 10 patients (3%) had leukopenia and five patients (1%) had thrombocytopenia. In addition, grade 3 or 4 lymphopenia was recorded in 79 patients (22%) (Table 3). One patient (0.3%) developed grade 4 nephrotoxicity. This patient developed gradual decline in kidney function and was commenced on haemodialysis. The decline in kidney function was attributed to persistent vomiting with poor oral intake but a contributory role of PRRT cannot be excluded. This patient had a baseline eGFR of 39 ml/min/1.73 m2. No further grade 3/4 renal toxicities were encountered. Myelodysplastic disease (MDS) occurred in one patient (0.3%). This patient had undergone chemotherapy for diffuse large B cell lymphoma with chemotherapy (rituximab, cyclophosphamide, vincristine, prednisolone), followed by etoposide, methylprednisolone, cytarabine, cisplatin 6 months which resulted in a complete metabolic response. The patient's MDS was diagnosed 48 months after the start of chemotherapy and 17 months after commencing PRRT. No acute leukaemia occurred.
Click to Show/Hide
|
||||
| Experiment 367 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Grade 3 nausea |
5%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Treatment-related toxicities are listed in Table 3. Five patients (26%) experienced dose modifying toxicity during treatment with 177Lu-DOTATATE. The most common treatment-related toxicities were thrombocytopenia (9 patients, 47%; grade 3/4 in 1 patient, 5%), anemia (7 patients, 37%; grade 3/4 in 2 patients, 11%), leukopenia (6 patients, 32%; grade 3/4 in 0 patients), and elevation of the liver function tests (4 patients, 21%; grade 3/4 in 0 patients).
Click to Show/Hide
|
||||
| Experiment 368 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade 3 lymphopenia |
22%
|
|||
| Patients Enrolled |
363 patients with neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Safety data was available in 363 patients. A total of 28 patients developed grade 3 or 4 bone marrow toxicity (8%) (excluding lymphopenia). Of these, 21 patients (6%) had anaemia, 10 patients (3%) had leukopenia and five patients (1%) had thrombocytopenia. In addition, grade 3 or 4 lymphopenia was recorded in 79 patients (22%) (Table 3). One patient (0.3%) developed grade 4 nephrotoxicity. This patient developed gradual decline in kidney function and was commenced on haemodialysis. The decline in kidney function was attributed to persistent vomiting with poor oral intake but a contributory role of PRRT cannot be excluded. This patient had a baseline eGFR of 39 ml/min/1.73 m2. No further grade 3/4 renal toxicities were encountered. Myelodysplastic disease (MDS) occurred in one patient (0.3%). This patient had undergone chemotherapy for diffuse large B cell lymphoma with chemotherapy (rituximab, cyclophosphamide, vincristine, prednisolone), followed by etoposide, methylprednisolone, cytarabine, cisplatin 6 months which resulted in a complete metabolic response. The patient's MDS was diagnosed 48 months after the start of chemotherapy and 17 months after commencing PRRT. No acute leukaemia occurred.
Click to Show/Hide
|
||||
| Experiment 369 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade 3 leukopenia |
2%
|
|||
| Patients Enrolled |
363 patients with neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Safety data was available in 363 patients. A total of 28 patients developed grade 3 or 4 bone marrow toxicity (8%) (excluding lymphopenia). Of these, 21 patients (6%) had anaemia, 10 patients (3%) had leukopenia and five patients (1%) had thrombocytopenia. In addition, grade 3 or 4 lymphopenia was recorded in 79 patients (22%) (Table 3). One patient (0.3%) developed grade 4 nephrotoxicity. This patient developed gradual decline in kidney function and was commenced on haemodialysis. The decline in kidney function was attributed to persistent vomiting with poor oral intake but a contributory role of PRRT cannot be excluded. This patient had a baseline eGFR of 39 ml/min/1.73 m2. No further grade 3/4 renal toxicities were encountered. Myelodysplastic disease (MDS) occurred in one patient (0.3%). This patient had undergone chemotherapy for diffuse large B cell lymphoma with chemotherapy (rituximab, cyclophosphamide, vincristine, prednisolone), followed by etoposide, methylprednisolone, cytarabine, cisplatin 6 months which resulted in a complete metabolic response. The patient's MDS was diagnosed 48 months after the start of chemotherapy and 17 months after commencing PRRT. No acute leukaemia occurred.
Click to Show/Hide
|
||||
| Experiment 370 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Grade 3 hypertension |
5%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Treatment-related toxicities are listed in Table 3. Five patients (26%) experienced dose modifying toxicity during treatment with 177Lu-DOTATATE. The most common treatment-related toxicities were thrombocytopenia (9 patients, 47%; grade 3/4 in 1 patient, 5%), anemia (7 patients, 37%; grade 3/4 in 2 patients, 11%), leukopenia (6 patients, 32%; grade 3/4 in 0 patients), and elevation of the liver function tests (4 patients, 21%; grade 3/4 in 0 patients).
Click to Show/Hide
|
||||
| Experiment 371 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Grade 3 diarrhea |
5%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Treatment-related toxicities are listed in Table 3. Five patients (26%) experienced dose modifying toxicity during treatment with 177Lu-DOTATATE. The most common treatment-related toxicities were thrombocytopenia (9 patients, 47%; grade 3/4 in 1 patient, 5%), anemia (7 patients, 37%; grade 3/4 in 2 patients, 11%), leukopenia (6 patients, 32%; grade 3/4 in 0 patients), and elevation of the liver function tests (4 patients, 21%; grade 3/4 in 0 patients).
Click to Show/Hide
|
||||
| Experiment 372 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade 3 anemia |
6%
|
|||
| Patients Enrolled |
363 patients with neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Safety data was available in 363 patients. A total of 28 patients developed grade 3 or 4 bone marrow toxicity (8%) (excluding lymphopenia). Of these, 21 patients (6%) had anaemia, 10 patients (3%) had leukopenia and five patients (1%) had thrombocytopenia. In addition, grade 3 or 4 lymphopenia was recorded in 79 patients (22%) (Table 3). One patient (0.3%) developed grade 4 nephrotoxicity. This patient developed gradual decline in kidney function and was commenced on haemodialysis. The decline in kidney function was attributed to persistent vomiting with poor oral intake but a contributory role of PRRT cannot be excluded. This patient had a baseline eGFR of 39 ml/min/1.73 m2. No further grade 3/4 renal toxicities were encountered. Myelodysplastic disease (MDS) occurred in one patient (0.3%). This patient had undergone chemotherapy for diffuse large B cell lymphoma with chemotherapy (rituximab, cyclophosphamide, vincristine, prednisolone), followed by etoposide, methylprednisolone, cytarabine, cisplatin 6 months which resulted in a complete metabolic response. The patient's MDS was diagnosed 48 months after the start of chemotherapy and 17 months after commencing PRRT. No acute leukaemia occurred.
Click to Show/Hide
|
||||
| Experiment 373 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Grade 3 anemia |
11%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Treatment-related toxicities are listed in Table 3. Five patients (26%) experienced dose modifying toxicity during treatment with 177Lu-DOTATATE. The most common treatment-related toxicities were thrombocytopenia (9 patients, 47%; grade 3/4 in 1 patient, 5%), anemia (7 patients, 37%; grade 3/4 in 2 patients, 11%), leukopenia (6 patients, 32%; grade 3/4 in 0 patients), and elevation of the liver function tests (4 patients, 21%; grade 3/4 in 0 patients).
Click to Show/Hide
|
||||
| Experiment 374 Reporting the Activity Data of This PDC | [19] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumour | ||||
| Efficacy Data | Grade 3 adverse events |
38.50%
|
|||
| Patients Enrolled |
13 GEP-neuroendocrine tumour patients.
|
||||
| Administration Time | 1-4 cycles | ||||
| Administration Dosage | 7.4 GBq (200mCi) | ||||
| MOA of PDC |
From our initial experience, Lutathera has been well tolerated in patients with GEP-NET. Additional studies are needed to examine long-term clinical and patient-reported outcomes associated with treatment of GEP-NETs as well as financial considerations for hospitals embarking on a PRRT program. A multidisciplinary team and complete collaboration between hospital administration and clinical teams are required for successful implementation of a PRRT program.
Click to Show/Hide
|
||||
| Description |
AE was recorded in 13 patients out of the entire 35 patients treated with PRRT. A grade 5 AE was reported in 2 patients (15.4%) and grade 3 AEs were reported in 5 patients (38.5%). Of the 5 grade 3 AE patients, 1 patient developed ascites, pleural effusion, and acute kidney injury; the second patient developed hematoma at the injection site and pain in the lower extremities; the third patient had shortness of breath, cough, and hemoptysis; the fourth patient had nausea, vomiting, and deep vein thrombosis; and the fifth patient developed obstructive jaundice. All 5 grade 3 AE patients were hospitalized and treated. One patient (7.7%) with a grade 2 AE developed an upper respiratory infection and required antibiotics. Five patients (38.5%) with grade 1 AEs had nausea, vomiting, abdominal pain, and/or diarrhea. No patients developed a grade 4 AE.
Click to Show/Hide
|
||||
| Experiment 375 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Grade 3 abdominal distension |
5%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Treatment-related toxicities are listed in Table 3. Five patients (26%) experienced dose modifying toxicity during treatment with 177Lu-DOTATATE. The most common treatment-related toxicities were thrombocytopenia (9 patients, 47%; grade 3/4 in 1 patient, 5%), anemia (7 patients, 37%; grade 3/4 in 2 patients, 11%), leukopenia (6 patients, 32%; grade 3/4 in 0 patients), and elevation of the liver function tests (4 patients, 21%; grade 3/4 in 0 patients).
Click to Show/Hide
|
||||
| Experiment 376 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | High-grade (WHO G3) neuroendocrine tumour | ||||
| Efficacy Data | Grade 2 toxicity |
9.10%
|
|||
| Patients Enrolled |
Patients with neuroendocrine tumours including high-grade (WHO G3) neuroendocrine tumours.
|
||||
| Description |
We observed no major renal toxicity except a minor increase (11.1%) in average serum creatinine levels. In 33.9% (n=56) of the patients, grade I toxicity; in 9.1% (n=15), grade II; and in 1.2% (n=2), grade III toxicity were observed.
|
||||
| Experiment 377 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade 2 thrombocytopenia |
3%
|
|||
| Patients Enrolled |
363 patients with neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Safety data was available in 363 patients. A total of 28 patients developed grade 3 or 4 bone marrow toxicity (8%) (excluding lymphopenia). Of these, 21 patients (6%) had anaemia, 10 patients (3%) had leukopenia and five patients (1%) had thrombocytopenia. In addition, grade 3 or 4 lymphopenia was recorded in 79 patients (22%) (Table 3). One patient (0.3%) developed grade 4 nephrotoxicity. This patient developed gradual decline in kidney function and was commenced on haemodialysis. The decline in kidney function was attributed to persistent vomiting with poor oral intake but a contributory role of PRRT cannot be excluded. This patient had a baseline eGFR of 39 ml/min/1.73 m2. No further grade 3/4 renal toxicities were encountered. Myelodysplastic disease (MDS) occurred in one patient (0.3%). This patient had undergone chemotherapy for diffuse large B cell lymphoma with chemotherapy (rituximab, cyclophosphamide, vincristine, prednisolone), followed by etoposide, methylprednisolone, cytarabine, cisplatin 6 months which resulted in a complete metabolic response. The patient's MDS was diagnosed 48 months after the start of chemotherapy and 17 months after commencing PRRT. No acute leukaemia occurred.
Click to Show/Hide
|
||||
| Experiment 378 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Grade 2 thrombocytopenia |
11%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Treatment-related toxicities are listed in Table 3. Five patients (26%) experienced dose modifying toxicity during treatment with 177Lu-DOTATATE. The most common treatment-related toxicities were thrombocytopenia (9 patients, 47%; grade 3/4 in 1 patient, 5%), anemia (7 patients, 37%; grade 3/4 in 2 patients, 11%), leukopenia (6 patients, 32%; grade 3/4 in 0 patients), and elevation of the liver function tests (4 patients, 21%; grade 3/4 in 0 patients).
Click to Show/Hide
|
||||
| Experiment 379 Reporting the Activity Data of This PDC | [28] | ||||
| Indication | Phaeochromocytoma/Paraganglioma | ||||
| Efficacy Data | Grade 2 platelets toxicity |
0%
|
|||
| Patients Enrolled |
34 patients with SSTr2-positive progressive locally advanced or metastatic PPGLs.
|
||||
| Administration Time | 5 cycles | ||||
| Administration Dosage | 18.5/27.5 GBq | ||||
| MOA of PDC |
In a long-term follow-up of patients with mPPGL, PRRT demonstrated a tolerability and efficacy comparable to those obtained in GEP-NETs. Our results highlight the need for an appropriate cumulative administered dosage to achieve a very prolonged result and underline the importance of specific symptoms and presence of bone lesions as predictive factors. Given the substantial number of patients evaluated over time, the superior mOS of 177Lu-DOTATATE with respect to 90Y-DOTATOC indicates the former radiopharmaceutical as the better candidate for new prospective studies of single or associated therapies.
Click to Show/Hide
|
||||
| Description |
No cases of grade (G) 3/4 bone marrow toxicity were observed in any of the treated patients. G1 and G2 bone marrow toxicity was observed in 19 and 4 patients, respectively. All patients recovered during the interval between cycles, without the need for interference in the treatment schedule. Two patients had G1 renal toxicity and one patient G2 toxicity. The latter patient started treatment (2008) with 1.53 creatinine at baseline and registered a 1.88 creatinine level at the third cycle, at which point therapy was interrupted (cumulative dosage 11.1 GBq 177Lu-DOTATATE). She still has stable disease after >10 years, but a worsening of renal function led to the need for weekly hemodialysis from 2019. No significant differences in toxicity were reported in patients treated with 90Y-DOTATOC or 177Lu-DOTATATE.
Click to Show/Hide
|
||||
| Experiment 380 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Grade 2 pancytopenia |
5%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Treatment-related toxicities are listed in Table 3. Five patients (26%) experienced dose modifying toxicity during treatment with 177Lu-DOTATATE. The most common treatment-related toxicities were thrombocytopenia (9 patients, 47%; grade 3/4 in 1 patient, 5%), anemia (7 patients, 37%; grade 3/4 in 2 patients, 11%), leukopenia (6 patients, 32%; grade 3/4 in 0 patients), and elevation of the liver function tests (4 patients, 21%; grade 3/4 in 0 patients).
Click to Show/Hide
|
||||
| Experiment 381 Reporting the Activity Data of This PDC | [28] | ||||
| Indication | Phaeochromocytoma/Paraganglioma | ||||
| Efficacy Data | Grade 2 neutrophils toxicity |
0%
|
|||
| Patients Enrolled |
34 patients with SSTr2-positive progressive locally advanced or metastatic PPGLs.
|
||||
| Administration Time | 5 cycles | ||||
| Administration Dosage | 18.5/27.5 GBq | ||||
| MOA of PDC |
In a long-term follow-up of patients with mPPGL, PRRT demonstrated a tolerability and efficacy comparable to those obtained in GEP-NETs. Our results highlight the need for an appropriate cumulative administered dosage to achieve a very prolonged result and underline the importance of specific symptoms and presence of bone lesions as predictive factors. Given the substantial number of patients evaluated over time, the superior mOS of 177Lu-DOTATATE with respect to 90Y-DOTATOC indicates the former radiopharmaceutical as the better candidate for new prospective studies of single or associated therapies.
Click to Show/Hide
|
||||
| Description |
No cases of grade (G) 3/4 bone marrow toxicity were observed in any of the treated patients. G1 and G2 bone marrow toxicity was observed in 19 and 4 patients, respectively. All patients recovered during the interval between cycles, without the need for interference in the treatment schedule. Two patients had G1 renal toxicity and one patient G2 toxicity. The latter patient started treatment (2008) with 1.53 creatinine at baseline and registered a 1.88 creatinine level at the third cycle, at which point therapy was interrupted (cumulative dosage 11.1 GBq 177Lu-DOTATATE). She still has stable disease after >10 years, but a worsening of renal function led to the need for weekly hemodialysis from 2019. No significant differences in toxicity were reported in patients treated with 90Y-DOTATOC or 177Lu-DOTATATE.
Click to Show/Hide
|
||||
| Experiment 382 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Grade 2 neutropenia |
5%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Treatment-related toxicities are listed in Table 3. Five patients (26%) experienced dose modifying toxicity during treatment with 177Lu-DOTATATE. The most common treatment-related toxicities were thrombocytopenia (9 patients, 47%; grade 3/4 in 1 patient, 5%), anemia (7 patients, 37%; grade 3/4 in 2 patients, 11%), leukopenia (6 patients, 32%; grade 3/4 in 0 patients), and elevation of the liver function tests (4 patients, 21%; grade 3/4 in 0 patients).
Click to Show/Hide
|
||||
| Experiment 383 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade 2 nephrotoxicity |
2%
|
|||
| Patients Enrolled |
363 patients with neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Safety data was available in 363 patients. A total of 28 patients developed grade 3 or 4 bone marrow toxicity (8%) (excluding lymphopenia). Of these, 21 patients (6%) had anaemia, 10 patients (3%) had leukopenia and five patients (1%) had thrombocytopenia. In addition, grade 3 or 4 lymphopenia was recorded in 79 patients (22%) (Table 3). One patient (0.3%) developed grade 4 nephrotoxicity. This patient developed gradual decline in kidney function and was commenced on haemodialysis. The decline in kidney function was attributed to persistent vomiting with poor oral intake but a contributory role of PRRT cannot be excluded. This patient had a baseline eGFR of 39 ml/min/1.73 m2. No further grade 3/4 renal toxicities were encountered. Myelodysplastic disease (MDS) occurred in one patient (0.3%). This patient had undergone chemotherapy for diffuse large B cell lymphoma with chemotherapy (rituximab, cyclophosphamide, vincristine, prednisolone), followed by etoposide, methylprednisolone, cytarabine, cisplatin 6 months which resulted in a complete metabolic response. The patient's MDS was diagnosed 48 months after the start of chemotherapy and 17 months after commencing PRRT. No acute leukaemia occurred.
Click to Show/Hide
|
||||
| Experiment 384 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade 2 lymphopenia |
25%
|
|||
| Patients Enrolled |
363 patients with neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Safety data was available in 363 patients. A total of 28 patients developed grade 3 or 4 bone marrow toxicity (8%) (excluding lymphopenia). Of these, 21 patients (6%) had anaemia, 10 patients (3%) had leukopenia and five patients (1%) had thrombocytopenia. In addition, grade 3 or 4 lymphopenia was recorded in 79 patients (22%) (Table 3). One patient (0.3%) developed grade 4 nephrotoxicity. This patient developed gradual decline in kidney function and was commenced on haemodialysis. The decline in kidney function was attributed to persistent vomiting with poor oral intake but a contributory role of PRRT cannot be excluded. This patient had a baseline eGFR of 39 ml/min/1.73 m2. No further grade 3/4 renal toxicities were encountered. Myelodysplastic disease (MDS) occurred in one patient (0.3%). This patient had undergone chemotherapy for diffuse large B cell lymphoma with chemotherapy (rituximab, cyclophosphamide, vincristine, prednisolone), followed by etoposide, methylprednisolone, cytarabine, cisplatin 6 months which resulted in a complete metabolic response. The patient's MDS was diagnosed 48 months after the start of chemotherapy and 17 months after commencing PRRT. No acute leukaemia occurred.
Click to Show/Hide
|
||||
| Experiment 385 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade 2 leukopenia |
9%
|
|||
| Patients Enrolled |
363 patients with neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Safety data was available in 363 patients. A total of 28 patients developed grade 3 or 4 bone marrow toxicity (8%) (excluding lymphopenia). Of these, 21 patients (6%) had anaemia, 10 patients (3%) had leukopenia and five patients (1%) had thrombocytopenia. In addition, grade 3 or 4 lymphopenia was recorded in 79 patients (22%) (Table 3). One patient (0.3%) developed grade 4 nephrotoxicity. This patient developed gradual decline in kidney function and was commenced on haemodialysis. The decline in kidney function was attributed to persistent vomiting with poor oral intake but a contributory role of PRRT cannot be excluded. This patient had a baseline eGFR of 39 ml/min/1.73 m2. No further grade 3/4 renal toxicities were encountered. Myelodysplastic disease (MDS) occurred in one patient (0.3%). This patient had undergone chemotherapy for diffuse large B cell lymphoma with chemotherapy (rituximab, cyclophosphamide, vincristine, prednisolone), followed by etoposide, methylprednisolone, cytarabine, cisplatin 6 months which resulted in a complete metabolic response. The patient's MDS was diagnosed 48 months after the start of chemotherapy and 17 months after commencing PRRT. No acute leukaemia occurred.
Click to Show/Hide
|
||||
| Experiment 386 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Grade 2 leukopenia |
16%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Treatment-related toxicities are listed in Table 3. Five patients (26%) experienced dose modifying toxicity during treatment with 177Lu-DOTATATE. The most common treatment-related toxicities were thrombocytopenia (9 patients, 47%; grade 3/4 in 1 patient, 5%), anemia (7 patients, 37%; grade 3/4 in 2 patients, 11%), leukopenia (6 patients, 32%; grade 3/4 in 0 patients), and elevation of the liver function tests (4 patients, 21%; grade 3/4 in 0 patients).
Click to Show/Hide
|
||||
| Experiment 387 Reporting the Activity Data of This PDC | [28] | ||||
| Indication | Phaeochromocytoma/Paraganglioma | ||||
| Efficacy Data | Grade 2 hemoglobin toxicity |
0%
|
|||
| Patients Enrolled |
34 patients with SSTr2-positive progressive locally advanced or metastatic PPGLs.
|
||||
| Administration Time | 5 cycles | ||||
| Administration Dosage | 18.5/27.5 GBq | ||||
| MOA of PDC |
In a long-term follow-up of patients with mPPGL, PRRT demonstrated a tolerability and efficacy comparable to those obtained in GEP-NETs. Our results highlight the need for an appropriate cumulative administered dosage to achieve a very prolonged result and underline the importance of specific symptoms and presence of bone lesions as predictive factors. Given the substantial number of patients evaluated over time, the superior mOS of 177Lu-DOTATATE with respect to 90Y-DOTATOC indicates the former radiopharmaceutical as the better candidate for new prospective studies of single or associated therapies.
Click to Show/Hide
|
||||
| Description |
No cases of grade (G) 3/4 bone marrow toxicity were observed in any of the treated patients. G1 and G2 bone marrow toxicity was observed in 19 and 4 patients, respectively. All patients recovered during the interval between cycles, without the need for interference in the treatment schedule. Two patients had G1 renal toxicity and one patient G2 toxicity. The latter patient started treatment (2008) with 1.53 creatinine at baseline and registered a 1.88 creatinine level at the third cycle, at which point therapy was interrupted (cumulative dosage 11.1 GBq 177Lu-DOTATATE). She still has stable disease after >10 years, but a worsening of renal function led to the need for weekly hemodialysis from 2019. No significant differences in toxicity were reported in patients treated with 90Y-DOTATOC or 177Lu-DOTATATE.
Click to Show/Hide
|
||||
| Experiment 388 Reporting the Activity Data of This PDC | [38] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade 2 haematotoxicity |
38%
|
|||
| Patients Enrolled |
100 neuroendocrine tumour patients.
|
||||
| Administration Time | 4 cycles every 10 weeks | ||||
| Administration Dosage | 7.4 GBq | ||||
| MOA of PDC |
The incidence of severe haematotoxicity was low with extensive screening and monitoring. The vast majority of patients (96/100) was not restricted in treatment continuation by haematotoxicity; therefore, our selection criteria appeared appropriate for safe PRRT treatment. Baseline parameters showed limited correlation with the degree of decline in haematological values.
Click to Show/Hide
|
||||
| Description |
Four cycles were completed by 86/100 of patients, 4/100 patients discontinued due to haematotoxicity, and 10/100 patients due to progressive disease. The treatment course was adjusted due to haematotoxicity in 24/100 patients, including postponed next cycle (n = 17), reduced administered activity (n = 13), and both adjustments (n = 10). The most observed haematotoxicity grade was grade 0-1 in 54/100 patients, grade 2 in 38/100 and grade 3-4 in 8/100. Significant differences in baseline leucocyte, neutrophil and platelet counts were observed between grade 0-1 and grade 2. However, the correlation between baseline and lowest observed values was poor to moderate. No differences between haematotoxicity grades and baseline parameters or somatostatin receptor positive tumour volume was observed.
Click to Show/Hide
|
||||
| Experiment 389 Reporting the Activity Data of This PDC | [28] | ||||
| Indication | Phaeochromocytoma/Paraganglioma | ||||
| Efficacy Data | Grade 2 creatinine toxicity |
0%
|
|||
| Patients Enrolled |
34 patients with SSTr2-positive progressive locally advanced or metastatic PPGLs.
|
||||
| Administration Time | 5 cycles | ||||
| Administration Dosage | 18.5/27.5 GBq | ||||
| MOA of PDC |
In a long-term follow-up of patients with mPPGL, PRRT demonstrated a tolerability and efficacy comparable to those obtained in GEP-NETs. Our results highlight the need for an appropriate cumulative administered dosage to achieve a very prolonged result and underline the importance of specific symptoms and presence of bone lesions as predictive factors. Given the substantial number of patients evaluated over time, the superior mOS of 177Lu-DOTATATE with respect to 90Y-DOTATOC indicates the former radiopharmaceutical as the better candidate for new prospective studies of single or associated therapies.
Click to Show/Hide
|
||||
| Description |
No cases of grade (G) 3/4 bone marrow toxicity were observed in any of the treated patients. G1 and G2 bone marrow toxicity was observed in 19 and 4 patients, respectively. All patients recovered during the interval between cycles, without the need for interference in the treatment schedule. Two patients had G1 renal toxicity and one patient G2 toxicity. The latter patient started treatment (2008) with 1.53 creatinine at baseline and registered a 1.88 creatinine level at the third cycle, at which point therapy was interrupted (cumulative dosage 11.1 GBq 177Lu-DOTATATE). She still has stable disease after >10 years, but a worsening of renal function led to the need for weekly hemodialysis from 2019. No significant differences in toxicity were reported in patients treated with 90Y-DOTATOC or 177Lu-DOTATATE.
Click to Show/Hide
|
||||
| Experiment 390 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade 2 anemia |
9%
|
|||
| Patients Enrolled |
363 patients with neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Safety data was available in 363 patients. A total of 28 patients developed grade 3 or 4 bone marrow toxicity (8%) (excluding lymphopenia). Of these, 21 patients (6%) had anaemia, 10 patients (3%) had leukopenia and five patients (1%) had thrombocytopenia. In addition, grade 3 or 4 lymphopenia was recorded in 79 patients (22%) (Table 3). One patient (0.3%) developed grade 4 nephrotoxicity. This patient developed gradual decline in kidney function and was commenced on haemodialysis. The decline in kidney function was attributed to persistent vomiting with poor oral intake but a contributory role of PRRT cannot be excluded. This patient had a baseline eGFR of 39 ml/min/1.73 m2. No further grade 3/4 renal toxicities were encountered. Myelodysplastic disease (MDS) occurred in one patient (0.3%). This patient had undergone chemotherapy for diffuse large B cell lymphoma with chemotherapy (rituximab, cyclophosphamide, vincristine, prednisolone), followed by etoposide, methylprednisolone, cytarabine, cisplatin 6 months which resulted in a complete metabolic response. The patient's MDS was diagnosed 48 months after the start of chemotherapy and 17 months after commencing PRRT. No acute leukaemia occurred.
Click to Show/Hide
|
||||
| Experiment 391 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Grade 2 anemia |
21%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Treatment-related toxicities are listed in Table 3. Five patients (26%) experienced dose modifying toxicity during treatment with 177Lu-DOTATATE. The most common treatment-related toxicities were thrombocytopenia (9 patients, 47%; grade 3/4 in 1 patient, 5%), anemia (7 patients, 37%; grade 3/4 in 2 patients, 11%), leukopenia (6 patients, 32%; grade 3/4 in 0 patients), and elevation of the liver function tests (4 patients, 21%; grade 3/4 in 0 patients).
Click to Show/Hide
|
||||
| Experiment 392 Reporting the Activity Data of This PDC | [19] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumour | ||||
| Efficacy Data | Grade 2 adverse events |
7.70%
|
|||
| Patients Enrolled |
13 GEP-neuroendocrine tumour patients.
|
||||
| Administration Time | 1-4 cycles | ||||
| Administration Dosage | 7.4 GBq (200mCi) | ||||
| MOA of PDC |
From our initial experience, Lutathera has been well tolerated in patients with GEP-NET. Additional studies are needed to examine long-term clinical and patient-reported outcomes associated with treatment of GEP-NETs as well as financial considerations for hospitals embarking on a PRRT program. A multidisciplinary team and complete collaboration between hospital administration and clinical teams are required for successful implementation of a PRRT program.
Click to Show/Hide
|
||||
| Description |
AE was recorded in 13 patients out of the entire 35 patients treated with PRRT. A grade 5 AE was reported in 2 patients (15.4%) and grade 3 AEs were reported in 5 patients (38.5%). Of the 5 grade 3 AE patients, 1 patient developed ascites, pleural effusion, and acute kidney injury; the second patient developed hematoma at the injection site and pain in the lower extremities; the third patient had shortness of breath, cough, and hemoptysis; the fourth patient had nausea, vomiting, and deep vein thrombosis; and the fifth patient developed obstructive jaundice. All 5 grade 3 AE patients were hospitalized and treated. One patient (7.7%) with a grade 2 AE developed an upper respiratory infection and required antibiotics. Five patients (38.5%) with grade 1 AEs had nausea, vomiting, abdominal pain, and/or diarrhea. No patients developed a grade 4 AE.
Click to Show/Hide
|
||||
| Experiment 393 Reporting the Activity Data of This PDC | [58] | ||||
| Indication | Small bowel neuroendocrine tumor | ||||
| Efficacy Data | Grade 1/2 toxicity |
41%
|
|||
| Patients Enrolled |
Patients with consecutive patients with small bowel neuroendocrine tumour.
|
||||
| Administration Time | Every eight to 12 weeks | ||||
| Administration Dosage | 7.4 GBq (200 mCi) | ||||
| Evaluation Method | 68Ga-DOTATATE PET/CT assay | ||||
| MOA of PDC |
Surgery combined with 177Lu PRRT is safe and provides favourable PFS and OS in selected patients with advanced SBNEN. Liquid biopsy (NETest) has the potential to accurately delineate disease status.
|
||||
| Description |
A surgical cohort of 39 SBNEN patients met eligibility criteria. Thirty-two patients underwent ileal resection and 7 right hemicolectomy. The mean number of 177Lu PRRT cycles was 4. Mortality was nil. Surgical morbidity was 10.3%. Transient grade 1/2 toxicity occurred in 41% (PRRT). NETest scores (n=9 patients) decreased in 100% following treatment and correlated with diminished tumour volume and disease stabilization following surgery and PRRT. Median follow-up: 78 months. Median PFS and OS: 42.7 and 110 months, respectively. Progression-free survival at 1-, 3-, and 5-years was 79.4%, 57.1% and 40.5%, respectively. Overall survival at 1-, 3-, and 5-years was 97.4%, 97.4%, and 94.1%, respectively
Click to Show/Hide
|
||||
| Experiment 394 Reporting the Activity Data of This PDC | [17] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade 1/2 thrombocytopenia |
57.10%
|
|||
| Patients Enrolled |
7 patients with inoperable, progressive, metastatic, or locally advanced, somatostatin receptor-positive neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | 21.3-30.1 GBq total dose | ||||
| MOA of PDC |
In conclusion, for the first time in Korea, we have conducted a clinical trial of PRRT with SNU-KB-01, a NCA 177Lu-DOTATATE, in patients with SSTR-positive NET. Treatment with SNU-KB-01 was safe and resulted in control of disease in most of the patients with high SSTR expression. The recommended dose per cycle of SNU-KB-01 was determined to be 7.40 GBq for the phase 2 trial. Our results indicate SNU-KB-01 as a potentially safe and efficacious treatment option for NET patients and are expected to provide a wider choice of treatment opportunities for patients with various types of SSTR-positive tumors in Korea.
Click to Show/Hide
|
||||
| Description |
There was grade 3 thrombocytopenia in one patient (14.3%), but no other grade 3 or 4 major hematologic, renal, hepatotoxicity was observed. Grade 1 or 2 hematologic toxicities were anemia (28.6%), thrombocytopenia (57.1%), leukopenia (71.4%), and neutropenia (42.9%). Grade 1 renal toxicity occurred in one patient (14.3%). Grade 1 or 2 hepatotoxicities were observed in two patients (28.6%). The trend of the progressive decline of hematological parameters was observed during the treatment cycles.
Click to Show/Hide
|
||||
| Experiment 395 Reporting the Activity Data of This PDC | [17] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade 1/2 neutropenia |
42.90%
|
|||
| Patients Enrolled |
7 patients with inoperable, progressive, metastatic, or locally advanced, somatostatin receptor-positive neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | 21.3-30.1 GBq total dose | ||||
| MOA of PDC |
In conclusion, for the first time in Korea, we have conducted a clinical trial of PRRT with SNU-KB-01, a NCA 177Lu-DOTATATE, in patients with SSTR-positive NET. Treatment with SNU-KB-01 was safe and resulted in control of disease in most of the patients with high SSTR expression. The recommended dose per cycle of SNU-KB-01 was determined to be 7.40 GBq for the phase 2 trial. Our results indicate SNU-KB-01 as a potentially safe and efficacious treatment option for NET patients and are expected to provide a wider choice of treatment opportunities for patients with various types of SSTR-positive tumors in Korea.
Click to Show/Hide
|
||||
| Description |
There was grade 3 thrombocytopenia in one patient (14.3%), but no other grade 3 or 4 major hematologic, renal, hepatotoxicity was observed. Grade 1 or 2 hematologic toxicities were anemia (28.6%), thrombocytopenia (57.1%), leukopenia (71.4%), and neutropenia (42.9%). Grade 1 renal toxicity occurred in one patient (14.3%). Grade 1 or 2 hepatotoxicities were observed in two patients (28.6%). The trend of the progressive decline of hematological parameters was observed during the treatment cycles.
Click to Show/Hide
|
||||
| Experiment 396 Reporting the Activity Data of This PDC | [17] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade 1/2 leukopenia |
71.40%
|
|||
| Patients Enrolled |
7 patients with inoperable, progressive, metastatic, or locally advanced, somatostatin receptor-positive neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | 21.3-30.1 GBq total dose | ||||
| MOA of PDC |
In conclusion, for the first time in Korea, we have conducted a clinical trial of PRRT with SNU-KB-01, a NCA 177Lu-DOTATATE, in patients with SSTR-positive NET. Treatment with SNU-KB-01 was safe and resulted in control of disease in most of the patients with high SSTR expression. The recommended dose per cycle of SNU-KB-01 was determined to be 7.40 GBq for the phase 2 trial. Our results indicate SNU-KB-01 as a potentially safe and efficacious treatment option for NET patients and are expected to provide a wider choice of treatment opportunities for patients with various types of SSTR-positive tumors in Korea.
Click to Show/Hide
|
||||
| Description |
There was grade 3 thrombocytopenia in one patient (14.3%), but no other grade 3 or 4 major hematologic, renal, hepatotoxicity was observed. Grade 1 or 2 hematologic toxicities were anemia (28.6%), thrombocytopenia (57.1%), leukopenia (71.4%), and neutropenia (42.9%). Grade 1 renal toxicity occurred in one patient (14.3%). Grade 1 or 2 hepatotoxicities were observed in two patients (28.6%). The trend of the progressive decline of hematological parameters was observed during the treatment cycles.
Click to Show/Hide
|
||||
| Experiment 397 Reporting the Activity Data of This PDC | [17] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade 1/2 hepatotoxicities |
28.60%
|
|||
| Patients Enrolled |
7 patients with inoperable, progressive, metastatic, or locally advanced, somatostatin receptor-positive neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | 21.3-30.1 GBq total dose | ||||
| MOA of PDC |
In conclusion, for the first time in Korea, we have conducted a clinical trial of PRRT with SNU-KB-01, a NCA 177Lu-DOTATATE, in patients with SSTR-positive NET. Treatment with SNU-KB-01 was safe and resulted in control of disease in most of the patients with high SSTR expression. The recommended dose per cycle of SNU-KB-01 was determined to be 7.40 GBq for the phase 2 trial. Our results indicate SNU-KB-01 as a potentially safe and efficacious treatment option for NET patients and are expected to provide a wider choice of treatment opportunities for patients with various types of SSTR-positive tumors in Korea.
Click to Show/Hide
|
||||
| Description |
There was grade 3 thrombocytopenia in one patient (14.3%), but no other grade 3 or 4 major hematologic, renal, hepatotoxicity was observed. Grade 1 or 2 hematologic toxicities were anemia (28.6%), thrombocytopenia (57.1%), leukopenia (71.4%), and neutropenia (42.9%). Grade 1 renal toxicity occurred in one patient (14.3%). Grade 1 or 2 hepatotoxicities were observed in two patients (28.6%). The trend of the progressive decline of hematological parameters was observed during the treatment cycles.
Click to Show/Hide
|
||||
| Experiment 398 Reporting the Activity Data of This PDC | [63] | ||||
| Indication | Inoperable grade I/II neuroendocrine tumor | ||||
| Efficacy Data | Grade 1/2 fatigue |
87.10%
|
|||
| Patients Enrolled |
31 patients treated using the fast-infusion protocol.
|
||||
| Administration Time | > 5 min | ||||
| Administration Dosage | 7.4 GBq | ||||
| Description |
Thirty-one patients, treated using the fast-infusion protocol, were included. Clinical toxicity mainly consisted of grade 1/2 fatigue (87.1%) and grade 1 nausea or vomiting (67.7%) during follow-up. No acute or long-term clinical toxicity possibly related to the fast-infusion protocol was reported. Grade 3/4 hematologic toxicity occurred after PRRT in 1 patient (3.2%). No grade 3/4 renal toxicity occurred. The laboratory experiment showed that when using the gravity method for infusion, half of the activity is infused after 3.5 min, and 95% is infused within 15 min.
Click to Show/Hide
|
||||
| Experiment 399 Reporting the Activity Data of This PDC | [17] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade 1/2 anemia |
28.60%
|
|||
| Patients Enrolled |
7 patients with inoperable, progressive, metastatic, or locally advanced, somatostatin receptor-positive neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | 21.3-30.1 GBq total dose | ||||
| MOA of PDC |
In conclusion, for the first time in Korea, we have conducted a clinical trial of PRRT with SNU-KB-01, a NCA 177Lu-DOTATATE, in patients with SSTR-positive NET. Treatment with SNU-KB-01 was safe and resulted in control of disease in most of the patients with high SSTR expression. The recommended dose per cycle of SNU-KB-01 was determined to be 7.40 GBq for the phase 2 trial. Our results indicate SNU-KB-01 as a potentially safe and efficacious treatment option for NET patients and are expected to provide a wider choice of treatment opportunities for patients with various types of SSTR-positive tumors in Korea.
Click to Show/Hide
|
||||
| Description |
There was grade 3 thrombocytopenia in one patient (14.3%), but no other grade 3 or 4 major hematologic, renal, hepatotoxicity was observed. Grade 1 or 2 hematologic toxicities were anemia (28.6%), thrombocytopenia (57.1%), leukopenia (71.4%), and neutropenia (42.9%). Grade 1 renal toxicity occurred in one patient (14.3%). Grade 1 or 2 hepatotoxicities were observed in two patients (28.6%). The trend of the progressive decline of hematological parameters was observed during the treatment cycles.
Click to Show/Hide
|
||||
| Experiment 400 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Grade 1 weight loss |
5%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Treatment-related toxicities are listed in Table 3. Five patients (26%) experienced dose modifying toxicity during treatment with 177Lu-DOTATATE. The most common treatment-related toxicities were thrombocytopenia (9 patients, 47%; grade 3/4 in 1 patient, 5%), anemia (7 patients, 37%; grade 3/4 in 2 patients, 11%), leukopenia (6 patients, 32%; grade 3/4 in 0 patients), and elevation of the liver function tests (4 patients, 21%; grade 3/4 in 0 patients).
Click to Show/Hide
|
||||
| Experiment 401 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | High-grade (WHO G3) neuroendocrine tumour | ||||
| Efficacy Data | Grade 1 toxicity |
33.90%
|
|||
| Patients Enrolled |
Patients with neuroendocrine tumours including high-grade (WHO G3) neuroendocrine tumours.
|
||||
| Description |
We observed no major renal toxicity except a minor increase (11.1%) in average serum creatinine levels. In 33.9% (n=56) of the patients, grade I toxicity; in 9.1% (n=15), grade II; and in 1.2% (n=2), grade III toxicity were observed.
|
||||
| Experiment 402 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Grade 1 thrombocytosis |
11%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Treatment-related toxicities are listed in Table 3. Five patients (26%) experienced dose modifying toxicity during treatment with 177Lu-DOTATATE. The most common treatment-related toxicities were thrombocytopenia (9 patients, 47%; grade 3/4 in 1 patient, 5%), anemia (7 patients, 37%; grade 3/4 in 2 patients, 11%), leukopenia (6 patients, 32%; grade 3/4 in 0 patients), and elevation of the liver function tests (4 patients, 21%; grade 3/4 in 0 patients).
Click to Show/Hide
|
||||
| Experiment 403 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade 1 thrombocytosis |
18%
|
|||
| Patients Enrolled |
363 patients with neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Safety data was available in 363 patients. A total of 28 patients developed grade 3 or 4 bone marrow toxicity (8%) (excluding lymphopenia). Of these, 21 patients (6%) had anaemia, 10 patients (3%) had leukopenia and five patients (1%) had thrombocytopenia. In addition, grade 3 or 4 lymphopenia was recorded in 79 patients (22%) (Table 3). One patient (0.3%) developed grade 4 nephrotoxicity. This patient developed gradual decline in kidney function and was commenced on haemodialysis. The decline in kidney function was attributed to persistent vomiting with poor oral intake but a contributory role of PRRT cannot be excluded. This patient had a baseline eGFR of 39 ml/min/1.73 m2. No further grade 3/4 renal toxicities were encountered. Myelodysplastic disease (MDS) occurred in one patient (0.3%). This patient had undergone chemotherapy for diffuse large B cell lymphoma with chemotherapy (rituximab, cyclophosphamide, vincristine, prednisolone), followed by etoposide, methylprednisolone, cytarabine, cisplatin 6 months which resulted in a complete metabolic response. The patient's MDS was diagnosed 48 months after the start of chemotherapy and 17 months after commencing PRRT. No acute leukaemia occurred.
Click to Show/Hide
|
||||
| Experiment 404 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Grade 1 thrombocytosis |
21%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Treatment-related toxicities are listed in Table 3. Five patients (26%) experienced dose modifying toxicity during treatment with 177Lu-DOTATATE. The most common treatment-related toxicities were thrombocytopenia (9 patients, 47%; grade 3/4 in 1 patient, 5%), anemia (7 patients, 37%; grade 3/4 in 2 patients, 11%), leukopenia (6 patients, 32%; grade 3/4 in 0 patients), and elevation of the liver function tests (4 patients, 21%; grade 3/4 in 0 patients).
Click to Show/Hide
|
||||
| Experiment 405 Reporting the Activity Data of This PDC | [17] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade 1 renal toxicity |
14.30%
|
|||
| Patients Enrolled |
7 patients with inoperable, progressive, metastatic, or locally advanced, somatostatin receptor-positive neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | 21.3-30.1 GBq total dose | ||||
| MOA of PDC |
In conclusion, for the first time in Korea, we have conducted a clinical trial of PRRT with SNU-KB-01, a NCA 177Lu-DOTATATE, in patients with SSTR-positive NET. Treatment with SNU-KB-01 was safe and resulted in control of disease in most of the patients with high SSTR expression. The recommended dose per cycle of SNU-KB-01 was determined to be 7.40 GBq for the phase 2 trial. Our results indicate SNU-KB-01 as a potentially safe and efficacious treatment option for NET patients and are expected to provide a wider choice of treatment opportunities for patients with various types of SSTR-positive tumors in Korea.
Click to Show/Hide
|
||||
| Description |
There was grade 3 thrombocytopenia in one patient (14.3%), but no other grade 3 or 4 major hematologic, renal, hepatotoxicity was observed. Grade 1 or 2 hematologic toxicities were anemia (28.6%), thrombocytopenia (57.1%), leukopenia (71.4%), and neutropenia (42.9%). Grade 1 renal toxicity occurred in one patient (14.3%). Grade 1 or 2 hepatotoxicities were observed in two patients (28.6%). The trend of the progressive decline of hematological parameters was observed during the treatment cycles.
Click to Show/Hide
|
||||
| Experiment 406 Reporting the Activity Data of This PDC | [28] | ||||
| Indication | Phaeochromocytoma/Paraganglioma | ||||
| Efficacy Data | Grade 1 platelets toxicity |
0%
|
|||
| Patients Enrolled |
34 patients with SSTr2-positive progressive locally advanced or metastatic PPGLs.
|
||||
| Administration Time | 5 cycles | ||||
| Administration Dosage | 18.5/27.5 GBq | ||||
| MOA of PDC |
In a long-term follow-up of patients with mPPGL, PRRT demonstrated a tolerability and efficacy comparable to those obtained in GEP-NETs. Our results highlight the need for an appropriate cumulative administered dosage to achieve a very prolonged result and underline the importance of specific symptoms and presence of bone lesions as predictive factors. Given the substantial number of patients evaluated over time, the superior mOS of 177Lu-DOTATATE with respect to 90Y-DOTATOC indicates the former radiopharmaceutical as the better candidate for new prospective studies of single or associated therapies.
Click to Show/Hide
|
||||
| Description |
No cases of grade (G) 3/4 bone marrow toxicity were observed in any of the treated patients. G1 and G2 bone marrow toxicity was observed in 19 and 4 patients, respectively. All patients recovered during the interval between cycles, without the need for interference in the treatment schedule. Two patients had G1 renal toxicity and one patient G2 toxicity. The latter patient started treatment (2008) with 1.53 creatinine at baseline and registered a 1.88 creatinine level at the third cycle, at which point therapy was interrupted (cumulative dosage 11.1 GBq 177Lu-DOTATATE). She still has stable disease after >10 years, but a worsening of renal function led to the need for weekly hemodialysis from 2019. No significant differences in toxicity were reported in patients treated with 90Y-DOTATOC or 177Lu-DOTATATE.
Click to Show/Hide
|
||||
| Experiment 407 Reporting the Activity Data of This PDC | [28] | ||||
| Indication | Phaeochromocytoma/Paraganglioma | ||||
| Efficacy Data | Grade 1 neutrophils toxicity |
17%
|
|||
| Patients Enrolled |
34 patients with SSTr2-positive progressive locally advanced or metastatic PPGLs.
|
||||
| Administration Time | 5 cycles | ||||
| Administration Dosage | 18.5/27.5 GBq | ||||
| MOA of PDC |
In a long-term follow-up of patients with mPPGL, PRRT demonstrated a tolerability and efficacy comparable to those obtained in GEP-NETs. Our results highlight the need for an appropriate cumulative administered dosage to achieve a very prolonged result and underline the importance of specific symptoms and presence of bone lesions as predictive factors. Given the substantial number of patients evaluated over time, the superior mOS of 177Lu-DOTATATE with respect to 90Y-DOTATOC indicates the former radiopharmaceutical as the better candidate for new prospective studies of single or associated therapies.
Click to Show/Hide
|
||||
| Description |
No cases of grade (G) 3/4 bone marrow toxicity were observed in any of the treated patients. G1 and G2 bone marrow toxicity was observed in 19 and 4 patients, respectively. All patients recovered during the interval between cycles, without the need for interference in the treatment schedule. Two patients had G1 renal toxicity and one patient G2 toxicity. The latter patient started treatment (2008) with 1.53 creatinine at baseline and registered a 1.88 creatinine level at the third cycle, at which point therapy was interrupted (cumulative dosage 11.1 GBq 177Lu-DOTATATE). She still has stable disease after >10 years, but a worsening of renal function led to the need for weekly hemodialysis from 2019. No significant differences in toxicity were reported in patients treated with 90Y-DOTATOC or 177Lu-DOTATATE.
Click to Show/Hide
|
||||
| Experiment 408 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade 1 nephrotoxicity |
9%
|
|||
| Patients Enrolled |
363 patients with neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Safety data was available in 363 patients. A total of 28 patients developed grade 3 or 4 bone marrow toxicity (8%) (excluding lymphopenia). Of these, 21 patients (6%) had anaemia, 10 patients (3%) had leukopenia and five patients (1%) had thrombocytopenia. In addition, grade 3 or 4 lymphopenia was recorded in 79 patients (22%) (Table 3). One patient (0.3%) developed grade 4 nephrotoxicity. This patient developed gradual decline in kidney function and was commenced on haemodialysis. The decline in kidney function was attributed to persistent vomiting with poor oral intake but a contributory role of PRRT cannot be excluded. This patient had a baseline eGFR of 39 ml/min/1.73 m2. No further grade 3/4 renal toxicities were encountered. Myelodysplastic disease (MDS) occurred in one patient (0.3%). This patient had undergone chemotherapy for diffuse large B cell lymphoma with chemotherapy (rituximab, cyclophosphamide, vincristine, prednisolone), followed by etoposide, methylprednisolone, cytarabine, cisplatin 6 months which resulted in a complete metabolic response. The patient's MDS was diagnosed 48 months after the start of chemotherapy and 17 months after commencing PRRT. No acute leukaemia occurred.
Click to Show/Hide
|
||||
| Experiment 409 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Grade 1 nausea |
5%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Treatment-related toxicities are listed in Table 3. Five patients (26%) experienced dose modifying toxicity during treatment with 177Lu-DOTATATE. The most common treatment-related toxicities were thrombocytopenia (9 patients, 47%; grade 3/4 in 1 patient, 5%), anemia (7 patients, 37%; grade 3/4 in 2 patients, 11%), leukopenia (6 patients, 32%; grade 3/4 in 0 patients), and elevation of the liver function tests (4 patients, 21%; grade 3/4 in 0 patients).
Click to Show/Hide
|
||||
| Experiment 410 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Grade 1 nausea |
11%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Treatment-related toxicities are listed in Table 3. Five patients (26%) experienced dose modifying toxicity during treatment with 177Lu-DOTATATE. The most common treatment-related toxicities were thrombocytopenia (9 patients, 47%; grade 3/4 in 1 patient, 5%), anemia (7 patients, 37%; grade 3/4 in 2 patients, 11%), leukopenia (6 patients, 32%; grade 3/4 in 0 patients), and elevation of the liver function tests (4 patients, 21%; grade 3/4 in 0 patients).
Click to Show/Hide
|
||||
| Experiment 411 Reporting the Activity Data of This PDC | [63] | ||||
| Indication | Inoperable grade I/II neuroendocrine tumor | ||||
| Efficacy Data | Grade 1 nausea |
67.70%
|
|||
| Patients Enrolled |
31 patients treated using the fast-infusion protocol.
|
||||
| Administration Time | > 5 min | ||||
| Administration Dosage | 7.4 GBq | ||||
| Description |
Thirty-one patients, treated using the fast-infusion protocol, were included. Clinical toxicity mainly consisted of grade 1/2 fatigue (87.1%) and grade 1 nausea or vomiting (67.7%) during follow-up. No acute or long-term clinical toxicity possibly related to the fast-infusion protocol was reported. Grade 3/4 hematologic toxicity occurred after PRRT in 1 patient (3.2%). No grade 3/4 renal toxicity occurred. The laboratory experiment showed that when using the gravity method for infusion, half of the activity is infused after 3.5 min, and 95% is infused within 15 min.
Click to Show/Hide
|
||||
| Experiment 412 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Grade 1 myopathy |
5%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Treatment-related toxicities are listed in Table 3. Five patients (26%) experienced dose modifying toxicity during treatment with 177Lu-DOTATATE. The most common treatment-related toxicities were thrombocytopenia (9 patients, 47%; grade 3/4 in 1 patient, 5%), anemia (7 patients, 37%; grade 3/4 in 2 patients, 11%), leukopenia (6 patients, 32%; grade 3/4 in 0 patients), and elevation of the liver function tests (4 patients, 21%; grade 3/4 in 0 patients).
Click to Show/Hide
|
||||
| Experiment 413 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade 1 lymphopenia |
12%
|
|||
| Patients Enrolled |
363 patients with neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Safety data was available in 363 patients. A total of 28 patients developed grade 3 or 4 bone marrow toxicity (8%) (excluding lymphopenia). Of these, 21 patients (6%) had anaemia, 10 patients (3%) had leukopenia and five patients (1%) had thrombocytopenia. In addition, grade 3 or 4 lymphopenia was recorded in 79 patients (22%) (Table 3). One patient (0.3%) developed grade 4 nephrotoxicity. This patient developed gradual decline in kidney function and was commenced on haemodialysis. The decline in kidney function was attributed to persistent vomiting with poor oral intake but a contributory role of PRRT cannot be excluded. This patient had a baseline eGFR of 39 ml/min/1.73 m2. No further grade 3/4 renal toxicities were encountered. Myelodysplastic disease (MDS) occurred in one patient (0.3%). This patient had undergone chemotherapy for diffuse large B cell lymphoma with chemotherapy (rituximab, cyclophosphamide, vincristine, prednisolone), followed by etoposide, methylprednisolone, cytarabine, cisplatin 6 months which resulted in a complete metabolic response. The patient's MDS was diagnosed 48 months after the start of chemotherapy and 17 months after commencing PRRT. No acute leukaemia occurred.
Click to Show/Hide
|
||||
| Experiment 414 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Grade 1 liver injury |
21%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Treatment-related toxicities are listed in Table 3. Five patients (26%) experienced dose modifying toxicity during treatment with 177Lu-DOTATATE. The most common treatment-related toxicities were thrombocytopenia (9 patients, 47%; grade 3/4 in 1 patient, 5%), anemia (7 patients, 37%; grade 3/4 in 2 patients, 11%), leukopenia (6 patients, 32%; grade 3/4 in 0 patients), and elevation of the liver function tests (4 patients, 21%; grade 3/4 in 0 patients).
Click to Show/Hide
|
||||
| Experiment 415 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade 1 leukopenia |
8%
|
|||
| Patients Enrolled |
363 patients with neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Safety data was available in 363 patients. A total of 28 patients developed grade 3 or 4 bone marrow toxicity (8%) (excluding lymphopenia). Of these, 21 patients (6%) had anaemia, 10 patients (3%) had leukopenia and five patients (1%) had thrombocytopenia. In addition, grade 3 or 4 lymphopenia was recorded in 79 patients (22%) (Table 3). One patient (0.3%) developed grade 4 nephrotoxicity. This patient developed gradual decline in kidney function and was commenced on haemodialysis. The decline in kidney function was attributed to persistent vomiting with poor oral intake but a contributory role of PRRT cannot be excluded. This patient had a baseline eGFR of 39 ml/min/1.73 m2. No further grade 3/4 renal toxicities were encountered. Myelodysplastic disease (MDS) occurred in one patient (0.3%). This patient had undergone chemotherapy for diffuse large B cell lymphoma with chemotherapy (rituximab, cyclophosphamide, vincristine, prednisolone), followed by etoposide, methylprednisolone, cytarabine, cisplatin 6 months which resulted in a complete metabolic response. The patient's MDS was diagnosed 48 months after the start of chemotherapy and 17 months after commencing PRRT. No acute leukaemia occurred.
Click to Show/Hide
|
||||
| Experiment 416 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Grade 1 leukopenia |
11%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Treatment-related toxicities are listed in Table 3. Five patients (26%) experienced dose modifying toxicity during treatment with 177Lu-DOTATATE. The most common treatment-related toxicities were thrombocytopenia (9 patients, 47%; grade 3/4 in 1 patient, 5%), anemia (7 patients, 37%; grade 3/4 in 2 patients, 11%), leukopenia (6 patients, 32%; grade 3/4 in 0 patients), and elevation of the liver function tests (4 patients, 21%; grade 3/4 in 0 patients).
Click to Show/Hide
|
||||
| Experiment 417 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Grade 1 leukopenia |
16%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Treatment-related toxicities are listed in Table 3. Five patients (26%) experienced dose modifying toxicity during treatment with 177Lu-DOTATATE. The most common treatment-related toxicities were thrombocytopenia (9 patients, 47%; grade 3/4 in 1 patient, 5%), anemia (7 patients, 37%; grade 3/4 in 2 patients, 11%), leukopenia (6 patients, 32%; grade 3/4 in 0 patients), and elevation of the liver function tests (4 patients, 21%; grade 3/4 in 0 patients).
Click to Show/Hide
|
||||
| Experiment 418 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Grade 1 irregular menstruation |
5%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Treatment-related toxicities are listed in Table 3. Five patients (26%) experienced dose modifying toxicity during treatment with 177Lu-DOTATATE. The most common treatment-related toxicities were thrombocytopenia (9 patients, 47%; grade 3/4 in 1 patient, 5%), anemia (7 patients, 37%; grade 3/4 in 2 patients, 11%), leukopenia (6 patients, 32%; grade 3/4 in 0 patients), and elevation of the liver function tests (4 patients, 21%; grade 3/4 in 0 patients).
Click to Show/Hide
|
||||
| Experiment 419 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Grade 1 hypertension |
5%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Treatment-related toxicities are listed in Table 3. Five patients (26%) experienced dose modifying toxicity during treatment with 177Lu-DOTATATE. The most common treatment-related toxicities were thrombocytopenia (9 patients, 47%; grade 3/4 in 1 patient, 5%), anemia (7 patients, 37%; grade 3/4 in 2 patients, 11%), leukopenia (6 patients, 32%; grade 3/4 in 0 patients), and elevation of the liver function tests (4 patients, 21%; grade 3/4 in 0 patients).
Click to Show/Hide
|
||||
| Experiment 420 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Grade 1 hyperglycemia |
16%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Treatment-related toxicities are listed in Table 3. Five patients (26%) experienced dose modifying toxicity during treatment with 177Lu-DOTATATE. The most common treatment-related toxicities were thrombocytopenia (9 patients, 47%; grade 3/4 in 1 patient, 5%), anemia (7 patients, 37%; grade 3/4 in 2 patients, 11%), leukopenia (6 patients, 32%; grade 3/4 in 0 patients), and elevation of the liver function tests (4 patients, 21%; grade 3/4 in 0 patients).
Click to Show/Hide
|
||||
| Experiment 421 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Grade 1 hemorrhage |
5%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Treatment-related toxicities are listed in Table 3. Five patients (26%) experienced dose modifying toxicity during treatment with 177Lu-DOTATATE. The most common treatment-related toxicities were thrombocytopenia (9 patients, 47%; grade 3/4 in 1 patient, 5%), anemia (7 patients, 37%; grade 3/4 in 2 patients, 11%), leukopenia (6 patients, 32%; grade 3/4 in 0 patients), and elevation of the liver function tests (4 patients, 21%; grade 3/4 in 0 patients).
Click to Show/Hide
|
||||
| Experiment 422 Reporting the Activity Data of This PDC | [28] | ||||
| Indication | Phaeochromocytoma/Paraganglioma | ||||
| Efficacy Data | Grade 1 hemoglobin toxicity |
17%
|
|||
| Patients Enrolled |
34 patients with SSTr2-positive progressive locally advanced or metastatic PPGLs.
|
||||
| Administration Time | 5 cycles | ||||
| Administration Dosage | 18.5/27.5 GBq | ||||
| MOA of PDC |
In a long-term follow-up of patients with mPPGL, PRRT demonstrated a tolerability and efficacy comparable to those obtained in GEP-NETs. Our results highlight the need for an appropriate cumulative administered dosage to achieve a very prolonged result and underline the importance of specific symptoms and presence of bone lesions as predictive factors. Given the substantial number of patients evaluated over time, the superior mOS of 177Lu-DOTATATE with respect to 90Y-DOTATOC indicates the former radiopharmaceutical as the better candidate for new prospective studies of single or associated therapies.
Click to Show/Hide
|
||||
| Description |
No cases of grade (G) 3/4 bone marrow toxicity were observed in any of the treated patients. G1 and G2 bone marrow toxicity was observed in 19 and 4 patients, respectively. All patients recovered during the interval between cycles, without the need for interference in the treatment schedule. Two patients had G1 renal toxicity and one patient G2 toxicity. The latter patient started treatment (2008) with 1.53 creatinine at baseline and registered a 1.88 creatinine level at the third cycle, at which point therapy was interrupted (cumulative dosage 11.1 GBq 177Lu-DOTATATE). She still has stable disease after >10 years, but a worsening of renal function led to the need for weekly hemodialysis from 2019. No significant differences in toxicity were reported in patients treated with 90Y-DOTATOC or 177Lu-DOTATATE.
Click to Show/Hide
|
||||
| Experiment 423 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Grade 1 edema |
5%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Treatment-related toxicities are listed in Table 3. Five patients (26%) experienced dose modifying toxicity during treatment with 177Lu-DOTATATE. The most common treatment-related toxicities were thrombocytopenia (9 patients, 47%; grade 3/4 in 1 patient, 5%), anemia (7 patients, 37%; grade 3/4 in 2 patients, 11%), leukopenia (6 patients, 32%; grade 3/4 in 0 patients), and elevation of the liver function tests (4 patients, 21%; grade 3/4 in 0 patients).
Click to Show/Hide
|
||||
| Experiment 424 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Grade 1 dizziness |
5%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Treatment-related toxicities are listed in Table 3. Five patients (26%) experienced dose modifying toxicity during treatment with 177Lu-DOTATATE. The most common treatment-related toxicities were thrombocytopenia (9 patients, 47%; grade 3/4 in 1 patient, 5%), anemia (7 patients, 37%; grade 3/4 in 2 patients, 11%), leukopenia (6 patients, 32%; grade 3/4 in 0 patients), and elevation of the liver function tests (4 patients, 21%; grade 3/4 in 0 patients).
Click to Show/Hide
|
||||
| Experiment 425 Reporting the Activity Data of This PDC | [28] | ||||
| Indication | Phaeochromocytoma/Paraganglioma | ||||
| Efficacy Data | Grade 1 creatinine toxicity |
0%
|
|||
| Patients Enrolled |
34 patients with SSTr2-positive progressive locally advanced or metastatic PPGLs.
|
||||
| Administration Time | 5 cycles | ||||
| Administration Dosage | 18.5/27.5 GBq | ||||
| MOA of PDC |
In a long-term follow-up of patients with mPPGL, PRRT demonstrated a tolerability and efficacy comparable to those obtained in GEP-NETs. Our results highlight the need for an appropriate cumulative administered dosage to achieve a very prolonged result and underline the importance of specific symptoms and presence of bone lesions as predictive factors. Given the substantial number of patients evaluated over time, the superior mOS of 177Lu-DOTATATE with respect to 90Y-DOTATOC indicates the former radiopharmaceutical as the better candidate for new prospective studies of single or associated therapies.
Click to Show/Hide
|
||||
| Description |
No cases of grade (G) 3/4 bone marrow toxicity were observed in any of the treated patients. G1 and G2 bone marrow toxicity was observed in 19 and 4 patients, respectively. All patients recovered during the interval between cycles, without the need for interference in the treatment schedule. Two patients had G1 renal toxicity and one patient G2 toxicity. The latter patient started treatment (2008) with 1.53 creatinine at baseline and registered a 1.88 creatinine level at the third cycle, at which point therapy was interrupted (cumulative dosage 11.1 GBq 177Lu-DOTATATE). She still has stable disease after >10 years, but a worsening of renal function led to the need for weekly hemodialysis from 2019. No significant differences in toxicity were reported in patients treated with 90Y-DOTATOC or 177Lu-DOTATATE.
Click to Show/Hide
|
||||
| Experiment 426 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Grade 1 ascites |
5%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Treatment-related toxicities are listed in Table 3. Five patients (26%) experienced dose modifying toxicity during treatment with 177Lu-DOTATATE. The most common treatment-related toxicities were thrombocytopenia (9 patients, 47%; grade 3/4 in 1 patient, 5%), anemia (7 patients, 37%; grade 3/4 in 2 patients, 11%), leukopenia (6 patients, 32%; grade 3/4 in 0 patients), and elevation of the liver function tests (4 patients, 21%; grade 3/4 in 0 patients).
Click to Show/Hide
|
||||
| Experiment 427 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Grade 1 anemia |
5%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Treatment-related toxicities are listed in Table 3. Five patients (26%) experienced dose modifying toxicity during treatment with 177Lu-DOTATATE. The most common treatment-related toxicities were thrombocytopenia (9 patients, 47%; grade 3/4 in 1 patient, 5%), anemia (7 patients, 37%; grade 3/4 in 2 patients, 11%), leukopenia (6 patients, 32%; grade 3/4 in 0 patients), and elevation of the liver function tests (4 patients, 21%; grade 3/4 in 0 patients).
Click to Show/Hide
|
||||
| Experiment 428 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade 1 anemia |
38%
|
|||
| Patients Enrolled |
363 patients with neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Average activity of 7.477 GBq (SD ±0.02) per cycle | ||||
| MOA of PDC |
In this single centre study of NET patients with generally moderate or large tumour burden, we found that 177Lu-DOTATATE is an effective therapeutic modality for SSR expressing NETs. 177Lu-DOTATATE resulted in DCR of 86% and median PFS and OS at 33 and 46 months, respectively. We found Ki-67, CgA and BMI to be independent predictive factors of PFS on multivariate analysis. 177Lu-DOTATATE was generally well-tolerated, with grade 3 or 4 toxicity renal toxicity <1%, grade 3 or 4 bone marrow toxicity at 8% and MDS recorded in <1%.
Click to Show/Hide
|
||||
| Description |
Safety data was available in 363 patients. A total of 28 patients developed grade 3 or 4 bone marrow toxicity (8%) (excluding lymphopenia). Of these, 21 patients (6%) had anaemia, 10 patients (3%) had leukopenia and five patients (1%) had thrombocytopenia. In addition, grade 3 or 4 lymphopenia was recorded in 79 patients (22%) (Table 3). One patient (0.3%) developed grade 4 nephrotoxicity. This patient developed gradual decline in kidney function and was commenced on haemodialysis. The decline in kidney function was attributed to persistent vomiting with poor oral intake but a contributory role of PRRT cannot be excluded. This patient had a baseline eGFR of 39 ml/min/1.73 m2. No further grade 3/4 renal toxicities were encountered. Myelodysplastic disease (MDS) occurred in one patient (0.3%). This patient had undergone chemotherapy for diffuse large B cell lymphoma with chemotherapy (rituximab, cyclophosphamide, vincristine, prednisolone), followed by etoposide, methylprednisolone, cytarabine, cisplatin 6 months which resulted in a complete metabolic response. The patient's MDS was diagnosed 48 months after the start of chemotherapy and 17 months after commencing PRRT. No acute leukaemia occurred.
Click to Show/Hide
|
||||
| Experiment 429 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Grade 1 alopecia |
26%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Treatment-related toxicities are listed in Table 3. Five patients (26%) experienced dose modifying toxicity during treatment with 177Lu-DOTATATE. The most common treatment-related toxicities were thrombocytopenia (9 patients, 47%; grade 3/4 in 1 patient, 5%), anemia (7 patients, 37%; grade 3/4 in 2 patients, 11%), leukopenia (6 patients, 32%; grade 3/4 in 0 patients), and elevation of the liver function tests (4 patients, 21%; grade 3/4 in 0 patients).
Click to Show/Hide
|
||||
| Experiment 430 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Grade ≥3 toxicity |
Hematologic: n=5
|
|||
| Patients Enrolled |
33 patients with neuroendocrine tumour.
|
||||
| Administration Time | 2 cycles | ||||
| Administration Dosage | 7.4 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 431 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Grade ≥3 toxicity |
Hematologic: n=7
|
|||
| Patients Enrolled |
33 patients with neuroendocrine tumour.
|
||||
| Administration Time | 2-4 cycles | ||||
| Administration Dosage | Mean administered activity during re-treatment: 17.7 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 432 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Grade ≥3 toxicity |
Renal: n=1; Hematologic: n=1
|
|||
| Patients Enrolled |
26 patients with neuroendocrine tumour.
|
||||
| Administration Time | 2-5 cycles | ||||
| Administration Dosage | Median activity for re-treatment: 16.5 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 433 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Grade ≥3 toxicity |
Hematologic: n=2
|
|||
| Patients Enrolled |
15 patients with neuroendocrine tumour.
|
||||
| Administration Time | 3-6 cycles | ||||
| Administration Dosage | Median cumulative activity: 63.9 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 434 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Grade ≥3 toxicity |
Renal: n=1; Hematologic: n=2; Myelodysplastic syndrome: n=1
|
|||
| Patients Enrolled |
18 patients with neuroendocrine tumour.
|
||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 435 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Grade ≥3 toxicity |
Renal: n=1; Hematologic: n=2; Myelodysplastic syndrome: n=1
|
|||
| Patients Enrolled |
Patients with neuroendocrine tumour (Retreatment).
|
||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 436 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Grade ≥3 toxicity |
Hematologic: n=14; Myelodysplastic syndrome: n=2; Acute myeloid leukemia: n=2
|
|||
| Patients Enrolled |
168 patients with neuroendocrine tumour.
|
||||
| Administration Time | 2 cycles | ||||
| Administration Dosage | Median cumulative dose: 44.7 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 437 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Grade ≥3 toxicity |
Hematologic: n=14; Myelodysplastic syndrome: n=2; Acute myeloid leukemia: n=2
|
|||
| Patients Enrolled |
13 re-retreatment neuroendocrine tumour patients.
|
||||
| Administration Time | 2 cycles | ||||
| Administration Dosage | Median cumulative dose: 59.7 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 438 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Grade ≥3 toxicity |
Hematologic: n=1
|
|||
| Patients Enrolled |
35 patients with neuroendocrine tumour.
|
||||
| Administration Time | 1-4 cycles | ||||
| Administration Dosage | Median cumulative activity 44 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 439 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade ≥3 neutropenia |
2.84%
|
|||
| Patients Enrolled |
534 male patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (25.9-29.6 GBq) (800 mCi [700-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 440 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade ≥3 neutropenia |
3.74%
|
|||
| Patients Enrolled |
439 female patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (22.2-29.6 GBq) (800 mCi [600-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 441 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade ≥3 lymphocytopenia |
48.01%
|
|||
| Patients Enrolled |
439 female patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (22.2-29.6 GBq) (800 mCi [600-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 442 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade ≥3 lymphocytopenia |
48.85%
|
|||
| Patients Enrolled |
534 male patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (25.9-29.6 GBq) (800 mCi [700-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 443 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade ≥3 leucopenia |
5.63%
|
|||
| Patients Enrolled |
534 male patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (25.9-29.6 GBq) (800 mCi [700-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 444 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade ≥3 leucopenia |
7.53%
|
|||
| Patients Enrolled |
439 female patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (22.2-29.6 GBq) (800 mCi [600-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 445 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade ≥3 Creatinine |
0%
|
|||
| Patients Enrolled |
439 female patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (22.2-29.6 GBq) (800 mCi [600-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 446 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade ≥3 Creatinine |
0.37%
|
|||
| Patients Enrolled |
534 male patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (25.9-29.6 GBq) (800 mCi [700-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 447 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade ≥3 bilirubin |
0.76%
|
|||
| Patients Enrolled |
534 male patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (25.9-29.6 GBq) (800 mCi [700-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 448 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade ≥3 bilirubin |
1.39%
|
|||
| Patients Enrolled |
439 female patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (22.2-29.6 GBq) (800 mCi [600-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 449 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade ≥3 aspartate aminotransferase |
1.51%
|
|||
| Patients Enrolled |
534 male patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (25.9-29.6 GBq) (800 mCi [700-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 450 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade ≥3 aspartate aminotransferase |
2.53%
|
|||
| Patients Enrolled |
439 female patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (22.2-29.6 GBq) (800 mCi [600-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 451 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade ≥3 anemia |
3.00%
|
|||
| Patients Enrolled |
534 male patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (25.9-29.6 GBq) (800 mCi [700-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 452 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade ≥3 anemia |
7.29%
|
|||
| Patients Enrolled |
439 female patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (22.2-29.6 GBq) (800 mCi [600-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 453 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade ≥ 3 thrombocytopenia |
6.19%
|
|||
| Patients Enrolled |
534 male patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (25.9-29.6 GBq) (800 mCi [700-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 454 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade ≥ 3 thrombocytopenia |
8.91%
|
|||
| Patients Enrolled |
439 female patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (22.2-29.6 GBq) (800 mCi [600-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 455 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade ≥ 2 thrombocytopenia |
17.67%
|
|||
| Patients Enrolled |
534 male patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (25.9-29.6 GBq) (800 mCi [700-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 456 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Grade ≥ 2 thrombocytopenia |
25.28%
|
|||
| Patients Enrolled |
439 female patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (22.2-29.6 GBq) (800 mCi [600-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 457 Reporting the Activity Data of This PDC | [62] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Glutathione reductase increase rate |
4.04 nmol/min/mg
|
|||
| Patients Enrolled |
Prostate cancer and neuroendocrine tumor patients referred to therapy with <sup>177</sup>Lu-PSMA and <sup>177</sup>Lu-DOTATATE, respectively.
|
||||
| Administration Time | 48 h | ||||
| Administration Dosage | 7.4 GBq | ||||
| Description |
In total, 61 RNT cycles were evaluated in 34 patients with age of 65 ± 2.83 (median ± SE) years (range of 27-99); this total included 20 (59%) prostate cancer patients [35 cycles (57.4%)] and 14 patients (41%) with NET [26 cycles (42.6%)]. Of the 61 evaluated cycles, 27 cycles were given in the control group and 34 cycles were given in the intervention group. The serum level of MDA was significantly increased after treatment compared to before treatment (P = 0.02) in the control group, while no significant change in the serum level of MDA was observed in the intervention group (P = 0.52). The serum level of GSH was insignificantly decreased after treatment compared to before treatment in the control group and slightly increased after treatment in the intervention group (P > 0.05). The serum level of glutathione reductase was insignificantly increased in all groups of patients after treatment (P > 0.05).
Click to Show/Hide
|
||||
| Experiment 458 Reporting the Activity Data of This PDC | [62] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Glutathione decrease rate |
0.001 mg/mL
|
|||
| Patients Enrolled |
Prostate cancer and neuroendocrine tumor patients referred to therapy with <sup>177</sup>Lu-PSMA and <sup>177</sup>Lu-DOTATATE, respectively.
|
||||
| Administration Time | 48 h | ||||
| Administration Dosage | 7.4 GBq | ||||
| Description |
In total, 61 RNT cycles were evaluated in 34 patients with age of 65 ± 2.83 (median ± SE) years (range of 27-99); this total included 20 (59%) prostate cancer patients [35 cycles (57.4%)] and 14 patients (41%) with NET [26 cycles (42.6%)]. Of the 61 evaluated cycles, 27 cycles were given in the control group and 34 cycles were given in the intervention group. The serum level of MDA was significantly increased after treatment compared to before treatment (P = 0.02) in the control group, while no significant change in the serum level of MDA was observed in the intervention group (P = 0.52). The serum level of GSH was insignificantly decreased after treatment compared to before treatment in the control group and slightly increased after treatment in the intervention group (P > 0.05). The serum level of glutathione reductase was insignificantly increased in all groups of patients after treatment (P > 0.05).
Click to Show/Hide
|
||||
| Experiment 459 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Metastatic midgut neuroendocrine tumor | ||||
| Efficacy Data | Glutamyl Transferase N level | < 38 UI/L | |||
| Administration Dosage | 7.4 GBq | ||||
| Description |
Initial laboratory tests showed normocytic regenerative anemia (hemoglobin 85g/dL), thrombocytopenia (54 109/L), no signs of hemolysis, no schistocytes, prolonged prothrombin time (25.8seconds, 35%) and prolonged activated partial thromboplastin time (1.63), low fibrinogen (0.3g/L), elevated fibrin monomer (>400ug/mL) and elevated fibrin degradation products (>20ug/mL). Coagulation factors II (49%) and V (44%) were low; factors VII (97%) and X (81%) were normal. Liver function tests were normal with the exception of moderate elevation of gamma Glutamyl Transferase (γGT=100UI/L, N<38UI/L). C-reactive protein was <5mg/L. There was no sign of tumor lysis syndrome. The bone marrow aspirate and medullar karyotype were normal. Computed tomography showed stable metastatic disease, including a large stable necrotic liver metastasis.
Click to Show/Hide
|
||||
| In Vivo Model | 55-year-old woman with metastatic midgut neuroendocrine tumor. | ||||
| Experiment 460 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Metastatic midgut neuroendocrine tumor | ||||
| Efficacy Data | Glutamyl Transferase γGT |
100 UI/L
|
|||
| Administration Dosage | 7.4 GBq | ||||
| Description |
Initial laboratory tests showed normocytic regenerative anemia (hemoglobin 85g/dL), thrombocytopenia (54 109/L), no signs of hemolysis, no schistocytes, prolonged prothrombin time (25.8seconds, 35%) and prolonged activated partial thromboplastin time (1.63), low fibrinogen (0.3g/L), elevated fibrin monomer (>400ug/mL) and elevated fibrin degradation products (>20ug/mL). Coagulation factors II (49%) and V (44%) were low; factors VII (97%) and X (81%) were normal. Liver function tests were normal with the exception of moderate elevation of gamma Glutamyl Transferase (γGT=100UI/L, N<38UI/L). C-reactive protein was <5mg/L. There was no sign of tumor lysis syndrome. The bone marrow aspirate and medullar karyotype were normal. Computed tomography showed stable metastatic disease, including a large stable necrotic liver metastasis.
Click to Show/Hide
|
||||
| In Vivo Model | 55-year-old woman with metastatic midgut neuroendocrine tumor. | ||||
| Experiment 461 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Well-differentiated pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Frequency of N0 |
61%
|
|||
| Patients Enrolled |
24 patients with morphologically WD-PanNETs who underwent PRRT preoperatively.
|
||||
| Administration Time | A median of 6 months (interquartile range [iqr] 6; 8 months) from the last cycle of prrt | ||||
| Evaluation Method | Stained with hematoxylin and eosin (H&E) & Formalin-fixed and paraffin-embedded (FFPE) assay | ||||
| Description |
Patients who underwent PRRT had a trend towards a higher frequency of N0 as compared to control group (61% vs 33%, p = 0.059).
|
||||
| Experiment 462 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | SDHB positive metastatic paraganglioma | ||||
| Efficacy Data | Free thyroxine level |
0.3 ng/dl
|
|||
| Administration Time | 1 month | ||||
| Administration Dosage | 200mCi | ||||
| Evaluation Method | Immunoassay | ||||
| Description |
The patient was diagnosed with Hashimoto's thyrotoxicosis. However, the patient was asymptomatic. One month after the first dose of 200mCi 177Lu-DOTATATE, the patient noted fatigue and a 2.6 Kg weight gain. The TSH (73.04 μIU/ml), anti-TPO antibodies (>1,000 IU/ml), and anti-Tg antibodies (668 IU/ml) had substantially increased, with reductions in FT4 (0.3 ng/dl) and TT3 [54 ng/dl (87-169 ng/dl)]. Diagnostic gallium 68 - DOTATATE positron emission tomography-computed tomography performed prior to 177Lu-DOTATATE treatment revealed diffuse thyroid uptake. Post-therapy single-photon emission computed tomography also revealed diffuse uptake of 177Lu-DOTATATE in the thyroid gland. Levothyroxine therapy was initiated, and the patient's symptoms resolved.
Click to Show/Hide
|
||||
| In Vivo Model | A 29-year-old male with SDHB positive metastatic paraganglioma. | ||||
| Related Clinical Trial | |||||
| NCT Number | NCT03206060 | Clinical Status | Phase 2 | ||
| Clinical Description | Background:Pheochromocytoma and paraganglioma are rare tumors. They usually form inside and near the adrenal gland or in the neck region. Not all these tumors can be removed with surgery, and there are no good treatments if the disease has spread. Researchers think a new drug may be able to help.Objective:To learn the safety and tolerability of Lu-177-DOTATATE. Also, to see if it improves the length of time it takes for the cancer to return.Eligibility:Adults who have an inoperable tumor of the study cancer that can be detected with Ga-68-DOTATATE PET/CT imagingDesign:Participants will be screened with a medical history, physical exam, and blood tests.Eligible participants will be admitted to the NIH Clinical Center.Participants will get the study drug in an intravenous infusion. They will get 4 doses, given about 8 weeks apart.Between 4 and 24 hours after each study drug dose, participants will have scans taken. They will lie on their back on a scanner table.Participants will have vital signs taken. They will give blood and urine samples.During the study, participants will have other scans taken. Some scans will use a radioactive tracer.Participants will complete quality of life questionnaires.Participants will be contacted by phone 1-3 days after they leave the Clinical Center. They will then be followed every 3 to 6 months for 3 years or until their disease gets worse. | ||||
| Experiment 463 Reporting the Activity Data of This PDC | [64] | ||||
| Indication | Progressive, well-differentiated neuroendocrine tumor | ||||
| Efficacy Data | Flushes decrease rate |
1.9
|
|||
| Patients Enrolled |
22 patients with a metastatic midgut neuroendocrine tumour.
|
||||
| Administration Dosage | Intended cumulative dose: 29.6 GBq | ||||
| MOA of PDC |
PRRT with 177Lu-DOTATATE effectively reduced diarrhea and flushing in patients with carcinoid syndrome and can be considered for symptomatic treatment of carcinoid syndrome insufficiently controlled with somatostatin analogs.
|
||||
| Description |
After PRRT, mean bowel movement frequency (BMF) decreased from 6.1 ± 3.4 to 4.6 ± 3.6 per day (P = 0.009). Flushes decreased from 4.3 ± 2.9 to 2.4 ± 2.7 flushes per day (P = 0.002). A decrease of BMF of more than 30% occurred in 47% of patients with baseline BMF of 4 or more (n = 17). In patients with ≥2 episodes of flushing a day (n = 15), 67% of patients had more than 50% decrease of daily flushing. A decrease in urinary 5-hydroxyindolacetic acid excretion of more than 30% was seen in 56% of patients. The European Organization for Research and Treatment of Cancer-Core Module diarrhea subscale score showed a trend toward improvement by an average of 16.7 ± 33.3 points (P = 0.11).
Click to Show/Hide
|
||||
| Experiment 464 Reporting the Activity Data of This PDC | [65] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumour | ||||
| Efficacy Data | Flare reaction rate |
42%
|
|||
| Patients Enrolled |
12 GEP neuroendocrine tumours patients.
|
||||
| Description |
In our first 12 patients who received Lutetium Lu-177 dotatate, tumor flare reactions occurred in 5 patients due to worsening symptoms of bone or soft tissue metastasis. This flare reaction can be mitigated with short course of corticosteroid therapy or other strategies.
|
||||
| Experiment 465 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Metastatic midgut neuroendocrine tumor | ||||
| Efficacy Data | Fibrin degradation increase rate | > 20 µg/mL | |||
| Administration Dosage | 7.4 GBq | ||||
| Description |
Initial laboratory tests showed normocytic regenerative anemia (hemoglobin 85g/dL), thrombocytopenia (54 109/L), no signs of hemolysis, no schistocytes, prolonged prothrombin time (25.8seconds, 35%) and prolonged activated partial thromboplastin time (1.63), low fibrinogen (0.3g/L), elevated fibrin monomer (>400ug/mL) and elevated fibrin degradation products (>20ug/mL). Coagulation factors II (49%) and V (44%) were low; factors VII (97%) and X (81%) were normal. Liver function tests were normal with the exception of moderate elevation of gamma Glutamyl Transferase (γGT=100UI/L, N<38UI/L). C-reactive protein was <5mg/L. There was no sign of tumor lysis syndrome. The bone marrow aspirate and medullar karyotype were normal. Computed tomography showed stable metastatic disease, including a large stable necrotic liver metastasis.
Click to Show/Hide
|
||||
| In Vivo Model | 55-year-old woman with metastatic midgut neuroendocrine tumor. | ||||
| Experiment 466 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Metastatic midgut neuroendocrine tumor | ||||
| Efficacy Data | Fibrin degradation increase rate | > 400 µg/mL | |||
| Administration Dosage | 7.4 GBq | ||||
| Description |
Initial laboratory tests showed normocytic regenerative anemia (hemoglobin 85g/dL), thrombocytopenia (54 109/L), no signs of hemolysis, no schistocytes, prolonged prothrombin time (25.8seconds, 35%) and prolonged activated partial thromboplastin time (1.63), low fibrinogen (0.3g/L), elevated fibrin monomer (>400ug/mL) and elevated fibrin degradation products (>20ug/mL). Coagulation factors II (49%) and V (44%) were low; factors VII (97%) and X (81%) were normal. Liver function tests were normal with the exception of moderate elevation of gamma Glutamyl Transferase (γGT=100UI/L, N<38UI/L). C-reactive protein was <5mg/L. There was no sign of tumor lysis syndrome. The bone marrow aspirate and medullar karyotype were normal. Computed tomography showed stable metastatic disease, including a large stable necrotic liver metastasis.
Click to Show/Hide
|
||||
| In Vivo Model | 55-year-old woman with metastatic midgut neuroendocrine tumor. | ||||
| Experiment 467 Reporting the Activity Data of This PDC | [40] | ||||
| Indication | High-grade gliomas | ||||
| Efficacy Data | Disease stable rate |
18.70%
|
|||
| Patients Enrolled |
16 patients with high-grade gliomas (10 males and 6 females).
|
||||
| Administration Time | 1 to 4 cycles | ||||
| Administration Dosage | 3.7 to 29.6 GBq (median, 10.45 GBq) | ||||
| Evaluation Method | MRI assay | ||||
| Description |
In total, 16 subjects (10 males and 6 females) with a mean age of 55.68 ± 13.17 years (26-73 years) participated in the study. Of them, 8 patients were new HGG cases, and 8 patients had recurrent tumors. The participants' responses to treatments were complete remission in 12.5% of (n = 2), partial remission in 31.25% (n = 5), disease stability in 18.7% (n = 3), and disease progression in 37.5% (n = 6). In total, pretreatment and posttreatment Karnofsky Performance Score and Eastern Cooperative Oncology Group scores did not improved (P > 0.05). The patients were followed up from 1 month to 26 months (median, 3 months).
Click to Show/Hide
|
||||
| Experiment 468 Reporting the Activity Data of This PDC | [66] | ||||
| Indication | Metastatic neuroendocrine tumor | ||||
| Efficacy Data | Disease response rate (DRR) |
20.59%
|
|||
| Patients Enrolled |
Patients with metastatic neuroendocrine tumours.
|
||||
| Evaluation Method | Southwest Oncology Group criteria | ||||
| Description |
The pooled effect in the RECIST group (13 studies) was 27.58% (95% confidence interval (CI) 21.03-35.27%) for the DRR and 79.14% (95% CI 75.83-82.1%) for the DCR. In the SWOG criteria group (7 studies), the pooled effect was 20.59% (95% CI 10.89-35.51%) for the DRR and 78.28% (95% CI 74.39-81.72%) for the DCR.
|
||||
| Experiment 469 Reporting the Activity Data of This PDC | [66] | ||||
| Indication | Metastatic neuroendocrine tumor | ||||
| Efficacy Data | Disease response rate (DRR) |
27.58%
|
|||
| Patients Enrolled |
Patients with metastatic neuroendocrine tumours.
|
||||
| Evaluation Method | Response Evaluation Criteria in Solid Tumours | ||||
| Description |
The pooled effect in the RECIST group (13 studies) was 27.58% (95% confidence interval (CI) 21.03-35.27%) for the DRR and 79.14% (95% CI 75.83-82.1%) for the DCR. In the SWOG criteria group (7 studies), the pooled effect was 20.59% (95% CI 10.89-35.51%) for the DRR and 78.28% (95% CI 74.39-81.72%) for the DCR.
|
||||
| Experiment 470 Reporting the Activity Data of This PDC | [49] | ||||
| Indication | Bronchial and gastroenteropancreatic neuroendocrine tumour | ||||
| Efficacy Data | Disease occurrence rate |
2.20%
|
|||
| Patients Enrolled |
Patients with bronchial and gastroenteropancreatic neuroendocrine tumours.
|
||||
| Administration Time | Over two cycles | ||||
| Administration Dosage | 14.8 GBq | ||||
| Description |
The total incidence of acute myeloid leukaemia (AML) and myelodysplastic syndrome (MDS) was 2.2%.
|
||||
| Experiment 471 Reporting the Activity Data of This PDC | [28] | ||||
| Indication | Phaeochromocytoma/Paraganglioma | ||||
| Efficacy Data | Disease control rate (DCR) |
18%
|
|||
| Patients Enrolled |
34 patients with SSTr2-positive progressive locally advanced or metastatic PPGLs.
|
||||
| Administration Time | 5 cycles | ||||
| Administration Dosage | 18.5/27.5 GBq | ||||
| MOA of PDC |
In a long-term follow-up of patients with mPPGL, PRRT demonstrated a tolerability and efficacy comparable to those obtained in GEP-NETs. Our results highlight the need for an appropriate cumulative administered dosage to achieve a very prolonged result and underline the importance of specific symptoms and presence of bone lesions as predictive factors. Given the substantial number of patients evaluated over time, the superior mOS of 177Lu-DOTATATE with respect to 90Y-DOTATOC indicates the former radiopharmaceutical as the better candidate for new prospective studies of single or associated therapies.
Click to Show/Hide
|
||||
| Description |
Both 90Y-DOTATOC and 177Lu-DOTATATE PRRT were well tolerated by patients without significant renal or bone marrow toxicity. The median follow-up was 73 months (range 5-146 months). The overall disease control rate (DCR) was 80% [95% confidence interval (CI) 68.9% to 91.9%] with a mean five cycles of therapy. However, 177Lu-DOTATATE patients showed a longer median overall survival (mOS) than those receiving 90Y-Dotatoc and a better DCR when higher dosages were administered, even if a direct comparison was not carried out. Syndromic patients had a poorer mOS. SDHx mutations did not interfere with treatment efficacy.
Click to Show/Hide
|
||||
| Experiment 472 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Meningiomas | ||||
| Efficacy Data | Disease control rate (DCR) |
40%
|
|||
| Patients Enrolled |
15 patients with meningiomas.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
A few studies evaluated the efficacy of RLT in meningiomas, administrating 2-5 cycles of [90Y]Y-DOTATATE/-DOTATOC or [177Lu]Lu-DOTATATE/-DOTATOC. In a meta-analysis performed by Mirian et al., RLToffered as mono-therapy or in combination with other oncological treatmentsallowed a comprehensive DCR of 63% in refractory meningiomas. The 6-month PFS rates were 94%, 48%, and 0% for patients with WHO grade I, II, and III meningiomas, respectively, whereas the corresponding 1-year OS rates were 88%, 71%, and 52%, respectively. In a study by Seystahl et al. RLT, offered mainly with [177Lu]Lu-DOTATATE (85% of patients), obtained 6-month PFS rates of 100%, 57%, and 0% for grade I, II, and III refractory meningiomas, respectively. In a recent study on a small group of selected patients affected by WHO grade II refractory meningiomas, 6-month PFS was 85.7% and 1-year PFS was 66.7%. The treatment was safe and well tolerated.
Click to Show/Hide
|
||||
| Experiment 473 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Disease control rate (DCR) |
48.50%
|
|||
| Patients Enrolled |
33 patients with neuroendocrine tumour.
|
||||
| Administration Time | 2 cycles | ||||
| Administration Dosage | 7.4 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 474 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Meningiomas | ||||
| Efficacy Data | Disease control rate (DCR) |
50%
|
|||
| Patients Enrolled |
20 patients with meningiomas.
|
||||
| Administration Time | Median 3 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
A few studies evaluated the efficacy of RLT in meningiomas, administrating 2-5 cycles of [90Y]Y-DOTATATE/-DOTATOC or [177Lu]Lu-DOTATATE/-DOTATOC. In a meta-analysis performed by Mirian et al., RLToffered as mono-therapy or in combination with other oncological treatmentsallowed a comprehensive DCR of 63% in refractory meningiomas. The 6-month PFS rates were 94%, 48%, and 0% for patients with WHO grade I, II, and III meningiomas, respectively, whereas the corresponding 1-year OS rates were 88%, 71%, and 52%, respectively. In a study by Seystahl et al. RLT, offered mainly with [177Lu]Lu-DOTATATE (85% of patients), obtained 6-month PFS rates of 100%, 57%, and 0% for grade I, II, and III refractory meningiomas, respectively. In a recent study on a small group of selected patients affected by WHO grade II refractory meningiomas, 6-month PFS was 85.7% and 1-year PFS was 66.7%. The treatment was safe and well tolerated.
Click to Show/Hide
|
||||
| Experiment 475 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | High-grade (WHO G3) neuroendocrine tumour | ||||
| Efficacy Data | Disease control rate (DCR) |
60%
|
|||
| Patients Enrolled |
Patients with WHO grades III.
|
||||
| Description |
The disease control rates in patients with WHO grades I, II and III were 74, 73 and 60%, respectively, and the OS rates were 61.9, 52.2 and 38.4 months, respectively.
|
||||
| Experiment 476 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Bronchial carcinoids | ||||
| Efficacy Data | Disease control rate (DCR) |
60%
|
|||
| Patients Enrolled |
443 patients with bronchial carcinoids.
|
||||
| Administration Dosage | Cumulative dose: 27.8-29.6 GBq | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
Treatment of bronchial carcinoids is not simple and requires a multidisciplinary approach. Surgery remains the mainstay of treatment for local disease, while in advanced disease, management includes chemotherapy, cold somatostatin analogues, immunotherapy, everolimus, and others target therapies. RLT is an option for selected patients with advanced or metastatic BCs overexpressing SSTR in progression to cold SSAs therapy. Although the experience of [177Lu]Lu-DOTATATE is more limited in BCs than GEP-NET, off-label use can be considered with promising results. A prospective phase II trial in 34 patients with stage IV BCs treated with cumulative activity of 18.5-27.8 GBq in four or five cycles of [177Lu]Lu-DOTATATE documented a DCR of 80% (6% complete response, 27% partial response, and 47% stable disease) and a median PFS of 20 months. In this study, negative prognostic factors were AC histology, tumor thyroid transcription factor-1 (TTF-1) expression, and a positive [18F]FDG PET imaging. Comparable results (median PFS of 17 months) were reported in a group of patients with diffuse extrahepatic metastases treated with RLT after several lines of therapies. Higher activities were administered in a study by van Essen et al., who treated nine patients with metastatic BCs, delivering a cumulative dose of 22.2-29.6 GBq of [177Lu]Lu-DOTATATE, demonstrating an overall response rate comparable to that of other GEP-NET (50% vs. 47%, respectively for BCs and GEP-NET). Moreover, tumor regression was reported in 66.6% of patients, without any outcome discrepancies between TCs and ACs. Comparable median OS and response rate between BCs and others GEP-NETs were reported also in another recent retrospective study. Likewise, the efficacy of RLT in BCs was demonstrated by Brabander et al. in a study involving 443 patients affected by midgut, bronchial, and CUP-NETs treated with 7.4 GBq of [177Lu]Lu-DOTATATE every 8 weeks. The authors found that objective response rate (ORR) for BCs was 30%, and an additional 30% of patients had a stable disease. Nevertheless, median OS for BCs was 52 months versus 71 months for pancreatic-NET. Long term results were analyzed by Mariniello et al. in 114 patients with advanced BCs treated with different RLT protocols. They documented a median PFS and OS of 28.0 and 58.8 months, respectively, while morphological responses were observed in 26.5% of patients. [177Lu]Lu-DOTATATE monotherapy protocol resulted in the highest 5-year OS (61.4%), despite tandem protocol ([90Y]Y-DOTATOC + [177Lu]Lu-DOTATATE) provided the highest response rates (38.1%). Best outcome was reached in patients who underwent RLT in early stage of disease, suggesting that this treatment should also be considered for newly diagnosed unresectable BCs. Moreover, despite most patients having only mild and transient adverse events, patients with hematologic toxicity showed worse survival outcomes.
Click to Show/Hide
|
||||
| Experiment 477 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Disease control rate (DCR) |
66.60%
|
|||
| Patients Enrolled |
33 patients with neuroendocrine tumour.
|
||||
| Administration Time | 2-4 cycles | ||||
| Administration Dosage | Mean administered activity during re-treatment: 17.7 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 478 Reporting the Activity Data of This PDC | [51] | ||||
| Indication | Negative progressive/Symptomatic locally advanced/Metastatic paragangliomas | ||||
| Efficacy Data | Disease control rate (DCR) |
67%
|
|||
| Patients Enrolled |
9 patients with paragangliomas.
|
||||
| Administration Time | ≥ 4cycles | ||||
| Administration Dosage | > 22.2 GBq) | ||||
| Experiment 479 Reporting the Activity Data of This PDC | [26] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumor | ||||
| Efficacy Data | Disease control rate (DCR) |
70%
|
|||
| Patients Enrolled |
Patients with advanced GEP-neuroendocrine tumour.
|
||||
| Administration Time | Four or five cycles | ||||
| Administration Dosage | 18.5 GBq/27.8 GBq | ||||
| Description |
Two patients (6%) achieved a partial response and 21 (64%) showed stable disease, giving a disease control rate (DCR) of 70%.
|
||||
| Experiment 480 Reporting the Activity Data of This PDC | [21] | ||||
| Indication | Well-differentiated high-grade neuroendocrine tumor | ||||
| Efficacy Data | Disease control rate (DCR) |
72%
|
|||
| Patients Enrolled |
18 patients with well-differentiated high-grade neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | Cumulative activity: 25-28 GBq | ||||
| MOA of PDC |
We observed a meaningful disease control rate of 72% during treatment of WD HG NETs with 177Lu-DOTATATE. In this heavily pre-treated population, more than half of patients received all four treatment cycles with toxicities largely bone marrow-related. As would be expected in WD NETs, the vast majority had alterations in chromatin remodeling genes and no RB1 alterations.
Click to Show/Hide
|
||||
| Description |
Nineteen patients, all with progressive, heavily treated disease, were identified. Sites of tumor origin were: pancreas (74%), small bowel (11%), rectum (11%), and lung (5%); median Ki-67 was 32% (range 22-56). Thirteen patients (68%) completed all four 177Lu-DOTATATE cycles. Best response (N = 18 evaluable) was: 5/18 (28%) partial response, 8/18 (44%) stable disease, and 5/18 (28%) disease progression. Median PFS was 13.1 months (95% CI: 8.7-20.9).
Click to Show/Hide
|
||||
| Experiment 481 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Disease control rate (DCR) |
72.20%
|
|||
| Patients Enrolled |
18 patients with paraganglioma.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The efficacy and safety data of the interim analysis of an open-label, single-arm phase 2 study evaluating 177Lu-DOTATATE in paraganglioma patients conducted at the National Institutes of Health (NCT03206060) in 36 patients [(n=18, apparently sporadic cohort and n=18 in patients having germline variant in one of the genes encoding subunits of succinate dehydrogenase complex (SDHA=2, SDHB=15, SDHD=1)), SDHx cohort] after 177LuDOTATATE therapy with 7.4 GBq every 8 weeks for 4 cycles. The progression-free survival at 6 months since initiation of 177Lu-DOTATATE was 19.1 months (22.7 months in apparently sporadic cohort and 15.4 months in SDHx cohort) and overall survival was not reached in both cohorts (80). Per RECIST 1.1, overall, 86.1% patients showed partial response (13.9%) or stable disease (72.2%). In sporadic cohort; partial response was observed in 11.1% and stable disease in 88.9% patients whereas those in SDHx cohort were, 16.7% and 55.5%, respectively at the end of 6 months.
Click to Show/Hide
|
||||
| Experiment 482 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | High-grade (WHO G3) neuroendocrine tumour | ||||
| Efficacy Data | Disease control rate (DCR) |
73%
|
|||
| Patients Enrolled |
Patients with WHO grades II.
|
||||
| Description |
The disease control rates in patients with WHO grades I, II and III were 74, 73 and 60%, respectively, and the OS rates were 61.9, 52.2 and 38.4 months, respectively.
|
||||
| Experiment 483 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Phaeochromocytoma | ||||
| Efficacy Data | Disease control rate (DCR) |
73%
|
|||
| Patients Enrolled |
12 patients with pheocromocytomas and paragangliomas.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
As a consequence, we saw a rising interest in a potential radiolabeled-SST agonist theranostic approach for PPGLs, with several spontaneous studies reporting of the treatment of metastatic or inoperable tumors with Lutathera and other similar radiocompounds, with promising preliminary results. The first RLT experience in PPGLs was reported by van Essen et al. who treated a heterogeneous cohort of patients, including 12 PGLs. Despite the authors reporting lower response rates than those obtained in GEP-NET patients, ORR and DCR were 18% and 73%, respectively. These results are consistent with other reports in the literature. Kong et al. treated 20 PPGLs (8 PHEOs and 12 PGLs) with [117Lu]Lu-DOTATATE, 14 due to uncontrolled secondary hypertension and 6 to radiological PD. A DCR equal to 86% was reached, including five patients with ORR (36%). Of note, 62% of symptomatic patients required a dose reduction of antihypertensive medications, and 57% reported a subjective therapeutic benefit in terms of tumor-related symptoms. Comprehensive PFS was 39 months, including two patients with early recurrence (2 and 5 months post-RLT, respectively) and one patient with remarkable tumor downsizing that allowed a second-step curative liver surgery. Of note, the patient was still disease-free at the time of the study. The results of the largest cohort of PPGLs treated with RLT were reported by Severi et al., who treated 46 patients reaching a comprehensive DCR of 80%. Interestingly, 34 patients treated with [177Lu]Lu-DOTATATE obtained a longer median OS in comparison to those treated with [90Y]Y-DOTATOC (143 vs. 92 months, respectively). According to the authors, this result may be related both to DOTATATEs higher affinity for SSTR2 (which is the type most overexpressed in PPGLs) and to the longer half-life of 177Lu and, consequently, prolonged residence time within the tumor lesions. Moreover, this study reports that sympathetic functioning PPGLs were associated to a shorter median PFS compared to non-functioning PPGLs. Prado-Wohlwend et al. treated nine patients with [177Lu]Lu-DOTATATE and eight with [131I]MIBG, obtaining a PFS of 29 and 18.5 months and a DCR of 88.8% and 62.5%, respectively, for each therapy. Despite the low number of patients involved, a trend for a longer PFS was found for adrenal primary PPGLs treated with [177Lu]Lu-DOTATATE. Nevertheless, the authors suggest performing both [68Ga]Ga-DOTA-SSA PET/CT and [123I]MIBG SPECT/CT in each patient in order to offer a personalized theranostic approach based on the highest uptake intensity in one of the two imaging scans. This is consistent with the results by Jaiswal et al. who found that a baseline SUVmax > 21 at [68Ga]Ga-DOTA-SSA PET/CT is a very strong predictor of response to [177Lu]Lu-DOTATATE (p < 0.0001).
Click to Show/Hide
|
||||
| Experiment 484 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Paraganglioma | ||||
| Efficacy Data | Disease control rate (DCR) |
73%
|
|||
| Patients Enrolled |
12 patients with pheocromocytomas and paragangliomas.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
As a consequence, we saw a rising interest in a potential radiolabeled-SST agonist theranostic approach for PPGLs, with several spontaneous studies reporting of the treatment of metastatic or inoperable tumors with Lutathera and other similar radiocompounds, with promising preliminary results. The first RLT experience in PPGLs was reported by van Essen et al. who treated a heterogeneous cohort of patients, including 12 PGLs. Despite the authors reporting lower response rates than those obtained in GEP-NET patients, ORR and DCR were 18% and 73%, respectively. These results are consistent with other reports in the literature. Kong et al. treated 20 PPGLs (8 PHEOs and 12 PGLs) with [117Lu]Lu-DOTATATE, 14 due to uncontrolled secondary hypertension and 6 to radiological PD. A DCR equal to 86% was reached, including five patients with ORR (36%). Of note, 62% of symptomatic patients required a dose reduction of antihypertensive medications, and 57% reported a subjective therapeutic benefit in terms of tumor-related symptoms. Comprehensive PFS was 39 months, including two patients with early recurrence (2 and 5 months post-RLT, respectively) and one patient with remarkable tumor downsizing that allowed a second-step curative liver surgery. Of note, the patient was still disease-free at the time of the study. The results of the largest cohort of PPGLs treated with RLT were reported by Severi et al., who treated 46 patients reaching a comprehensive DCR of 80%. Interestingly, 34 patients treated with [177Lu]Lu-DOTATATE obtained a longer median OS in comparison to those treated with [90Y]Y-DOTATOC (143 vs. 92 months, respectively). According to the authors, this result may be related both to DOTATATEs higher affinity for SSTR2 (which is the type most overexpressed in PPGLs) and to the longer half-life of 177Lu and, consequently, prolonged residence time within the tumor lesions. Moreover, this study reports that sympathetic functioning PPGLs were associated to a shorter median PFS compared to non-functioning PPGLs. Prado-Wohlwend et al. treated nine patients with [177Lu]Lu-DOTATATE and eight with [131I]MIBG, obtaining a PFS of 29 and 18.5 months and a DCR of 88.8% and 62.5%, respectively, for each therapy. Despite the low number of patients involved, a trend for a longer PFS was found for adrenal primary PPGLs treated with [177Lu]Lu-DOTATATE. Nevertheless, the authors suggest performing both [68Ga]Ga-DOTA-SSA PET/CT and [123I]MIBG SPECT/CT in each patient in order to offer a personalized theranostic approach based on the highest uptake intensity in one of the two imaging scans. This is consistent with the results by Jaiswal et al. who found that a baseline SUVmax > 21 at [68Ga]Ga-DOTA-SSA PET/CT is a very strong predictor of response to [177Lu]Lu-DOTATATE (p < 0.0001).
Click to Show/Hide
|
||||
| Experiment 485 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | High-grade (WHO G3) neuroendocrine tumour | ||||
| Efficacy Data | Disease control rate (DCR) |
74%
|
|||
| Patients Enrolled |
Patients with WHO grades I.
|
||||
| Description |
The disease control rates in patients with WHO grades I, II and III were 74, 73 and 60%, respectively, and the OS rates were 61.9, 52.2 and 38.4 months, respectively.
|
||||
| Experiment 486 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Disease control rate (DCR) |
75%
|
|||
| Patients Enrolled |
Patients with neuroendocrine tumour (Retreatment).
|
||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 487 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Disease control rate (DCR) |
75%
|
|||
| Patients Enrolled |
168 patients with neuroendocrine tumour.
|
||||
| Administration Time | 2 cycles | ||||
| Administration Dosage | Median cumulative dose: 44.7 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 488 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Bronchial carcinoids | ||||
| Efficacy Data | Disease control rate (DCR) |
75%
|
|||
| Patients Enrolled |
114 patients with bronchial carcinoids.
|
||||
| Administration Time | 4-6 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
Treatment of bronchial carcinoids is not simple and requires a multidisciplinary approach. Surgery remains the mainstay of treatment for local disease, while in advanced disease, management includes chemotherapy, cold somatostatin analogues, immunotherapy, everolimus, and others target therapies. RLT is an option for selected patients with advanced or metastatic BCs overexpressing SSTR in progression to cold SSAs therapy. Although the experience of [177Lu]Lu-DOTATATE is more limited in BCs than GEP-NET, off-label use can be considered with promising results. A prospective phase II trial in 34 patients with stage IV BCs treated with cumulative activity of 18.5-27.8 GBq in four or five cycles of [177Lu]Lu-DOTATATE documented a DCR of 80% (6% complete response, 27% partial response, and 47% stable disease) and a median PFS of 20 months. In this study, negative prognostic factors were AC histology, tumor thyroid transcription factor-1 (TTF-1) expression, and a positive [18F]FDG PET imaging. Comparable results (median PFS of 17 months) were reported in a group of patients with diffuse extrahepatic metastases treated with RLT after several lines of therapies. Higher activities were administered in a study by van Essen et al., who treated nine patients with metastatic BCs, delivering a cumulative dose of 22.2-29.6 GBq of [177Lu]Lu-DOTATATE, demonstrating an overall response rate comparable to that of other GEP-NET (50% vs. 47%, respectively for BCs and GEP-NET). Moreover, tumor regression was reported in 66.6% of patients, without any outcome discrepancies between TCs and ACs. Comparable median OS and response rate between BCs and others GEP-NETs were reported also in another recent retrospective study. Likewise, the efficacy of RLT in BCs was demonstrated by Brabander et al. in a study involving 443 patients affected by midgut, bronchial, and CUP-NETs treated with 7.4 GBq of [177Lu]Lu-DOTATATE every 8 weeks. The authors found that objective response rate (ORR) for BCs was 30%, and an additional 30% of patients had a stable disease. Nevertheless, median OS for BCs was 52 months versus 71 months for pancreatic-NET. Long term results were analyzed by Mariniello et al. in 114 patients with advanced BCs treated with different RLT protocols. They documented a median PFS and OS of 28.0 and 58.8 months, respectively, while morphological responses were observed in 26.5% of patients. [177Lu]Lu-DOTATATE monotherapy protocol resulted in the highest 5-year OS (61.4%), despite tandem protocol ([90Y]Y-DOTATOC + [177Lu]Lu-DOTATATE) provided the highest response rates (38.1%). Best outcome was reached in patients who underwent RLT in early stage of disease, suggesting that this treatment should also be considered for newly diagnosed unresectable BCs. Moreover, despite most patients having only mild and transient adverse events, patients with hematologic toxicity showed worse survival outcomes.
Click to Show/Hide
|
||||
| Experiment 489 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Disease control rate (DCR) |
77.10%
|
|||
| Patients Enrolled |
35 patients with neuroendocrine tumour.
|
||||
| Administration Time | 1-4 cycles | ||||
| Administration Dosage | Median cumulative activity 44 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 490 Reporting the Activity Data of This PDC | [48] | ||||
| Indication | Functioning pancreatic neuroendocrine tumour | ||||
| Efficacy Data | Disease control rate (DCR) |
78%
|
|||
| Patients Enrolled |
Patients with functioning pancreatic neuroendocrine tumors.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | 7.4 GBq/cycle | ||||
| Description |
Subacute hematological toxicity, grade 3 or 4 occurred in 4 patients (12%) and a hormonal crisis in 3 patients (9%). PRRT resulted in partial or complete response in 59% of patients and the disease control rate was 78% in patients with baseline progression. 71% of patients with uncontrolled symptoms had a reduction of symptoms and a more than 80% decrease of circulating hormone levels was measured during follow-up. After PRRT, median progression-free survival was 18.1 months (interquartile range: 3.3 to 35.7) with a concurrent increase in QOL.
Click to Show/Hide
|
||||
| Experiment 491 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Disease control rate (DCR) |
78%
|
|||
| Patients Enrolled |
82 patients with CUP-neuroendocrine tumour.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
Unfortunately, the literature lacks specific trials assessing RLT efficacy and safety on CUP-NETs, and the only available literature evidence is provided by mixed trials also including a few patients with SSTR-positive CUP-NETs. In a large cohort of patients treated with [177Lu]Lu-DOTATATE, Brabander et al.reported of 82 CUP-NET patients. Overall, DCR was reached in 78% of patients and median PFS and OS were 29 and 53 months, respectively. These results show that RLT may be effective in CUP-NETs patients, with a response rate intermediate between that obtained in GEP-NETswho showed longer median OS (60 vs. 53 months, respectively)and BCswho showed shorter median PFS (20 vs. 29 months, respectively). These results are consistent with those reported by Demirci et al. who treated 19 CUP-NETs, with an overall DCR of 84.2% and mean PFS and OS of 40 and 48 months, respectively. Once again, RLT outcomes in CUP-NETs were intermediate between those in GEP-NETs and BCs. In a large mixed cohort, Baum et al. treated 151 CUP-NETs (mostly with [177Lu]Lu-DOTATATE, although a small percentage of patients was treated with [90Y]Y-DOTATATE/DOTATOC in the study), obtaining median PFS and OS of 13 and 46 months, respectively. CUP-NETStogether with BCswere associated with a significantly shorter PFS at multivariate analysis if compared to pancreatic NETs despite showing a longer OS (50 vs. 43 months, respectively). Future prospective trials assessing efficacy and safety of RLT in CUP-NETs are desirable.
Click to Show/Hide
|
||||
| Experiment 492 Reporting the Activity Data of This PDC | [66] | ||||
| Indication | Metastatic neuroendocrine tumor | ||||
| Efficacy Data | Disease control rate (DCR) |
78.28%
|
|||
| Patients Enrolled |
Patients with metastatic neuroendocrine tumours.
|
||||
| Evaluation Method | Southwest Oncology Group criteria | ||||
| Description |
The pooled effect in the RECIST group (13 studies) was 27.58% (95% confidence interval (CI) 21.03-35.27%) for the DRR and 79.14% (95% CI 75.83-82.1%) for the DCR. In the SWOG criteria group (7 studies), the pooled effect was 20.59% (95% CI 10.89-35.51%) for the DRR and 78.28% (95% CI 74.39-81.72%) for the DCR.
|
||||
| Experiment 493 Reporting the Activity Data of This PDC | [66] | ||||
| Indication | Metastatic neuroendocrine tumor | ||||
| Efficacy Data | Disease control rate (DCR) |
79.14%
|
|||
| Patients Enrolled |
Patients with metastatic neuroendocrine tumours.
|
||||
| Evaluation Method | Response Evaluation Criteria in Solid Tumours | ||||
| Description |
The pooled effect in the RECIST group (13 studies) was 27.58% (95% confidence interval (CI) 21.03-35.27%) for the DRR and 79.14% (95% CI 75.83-82.1%) for the DCR. In the SWOG criteria group (7 studies), the pooled effect was 20.59% (95% CI 10.89-35.51%) for the DRR and 78.28% (95% CI 74.39-81.72%) for the DCR.
|
||||
| Experiment 494 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Bronchial carcinoids | ||||
| Efficacy Data | Disease control rate (DCR) |
80%
|
|||
| Patients Enrolled |
34 patients with bronchial carcinoids.
|
||||
| Administration Time | 4-5 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
Treatment of bronchial carcinoids is not simple and requires a multidisciplinary approach. Surgery remains the mainstay of treatment for local disease, while in advanced disease, management includes chemotherapy, cold somatostatin analogues, immunotherapy, everolimus, and others target therapies. RLT is an option for selected patients with advanced or metastatic BCs overexpressing SSTR in progression to cold SSAs therapy. Although the experience of [177Lu]Lu-DOTATATE is more limited in BCs than GEP-NET, off-label use can be considered with promising results. A prospective phase II trial in 34 patients with stage IV BCs treated with cumulative activity of 18.5-27.8 GBq in four or five cycles of [177Lu]Lu-DOTATATE documented a DCR of 80% (6% complete response, 27% partial response, and 47% stable disease) and a median PFS of 20 months. In this study, negative prognostic factors were AC histology, tumor thyroid transcription factor-1 (TTF-1) expression, and a positive [18F]FDG PET imaging. Comparable results (median PFS of 17 months) were reported in a group of patients with diffuse extrahepatic metastases treated with RLT after several lines of therapies. Higher activities were administered in a study by van Essen et al., who treated nine patients with metastatic BCs, delivering a cumulative dose of 22.2-29.6 GBq of [177Lu]Lu-DOTATATE, demonstrating an overall response rate comparable to that of other GEP-NET (50% vs. 47%, respectively for BCs and GEP-NET). Moreover, tumor regression was reported in 66.6% of patients, without any outcome discrepancies between TCs and ACs. Comparable median OS and response rate between BCs and others GEP-NETs were reported also in another recent retrospective study. Likewise, the efficacy of RLT in BCs was demonstrated by Brabander et al. in a study involving 443 patients affected by midgut, bronchial, and CUP-NETs treated with 7.4 GBq of [177Lu]Lu-DOTATATE every 8 weeks. The authors found that objective response rate (ORR) for BCs was 30%, and an additional 30% of patients had a stable disease. Nevertheless, median OS for BCs was 52 months versus 71 months for pancreatic-NET. Long term results were analyzed by Mariniello et al. in 114 patients with advanced BCs treated with different RLT protocols. They documented a median PFS and OS of 28.0 and 58.8 months, respectively, while morphological responses were observed in 26.5% of patients. [177Lu]Lu-DOTATATE monotherapy protocol resulted in the highest 5-year OS (61.4%), despite tandem protocol ([90Y]Y-DOTATOC + [177Lu]Lu-DOTATATE) provided the highest response rates (38.1%). Best outcome was reached in patients who underwent RLT in early stage of disease, suggesting that this treatment should also be considered for newly diagnosed unresectable BCs. Moreover, despite most patients having only mild and transient adverse events, patients with hematologic toxicity showed worse survival outcomes.
Click to Show/Hide
|
||||
| Experiment 495 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Phaeochromocytoma | ||||
| Efficacy Data | Disease control rate (DCR) |
80.40%
|
|||
| Patients Enrolled |
46 patients with Pheochromocytomas (PPGL) and paragangliomas (PHEO).
|
||||
| Administration Time | 4-5 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
As a consequence, we saw a rising interest in a potential radiolabeled-SST agonist theranostic approach for PPGLs, with several spontaneous studies reporting of the treatment of metastatic or inoperable tumors with Lutathera and other similar radiocompounds, with promising preliminary results. The first RLT experience in PPGLs was reported by van Essen et al. who treated a heterogeneous cohort of patients, including 12 PGLs. Despite the authors reporting lower response rates than those obtained in GEP-NET patients, ORR and DCR were 18% and 73%, respectively. These results are consistent with other reports in the literature. Kong et al. treated 20 PPGLs (8 PHEOs and 12 PGLs) with [117Lu]Lu-DOTATATE, 14 due to uncontrolled secondary hypertension and 6 to radiological PD. A DCR equal to 86% was reached, including five patients with ORR (36%). Of note, 62% of symptomatic patients required a dose reduction of antihypertensive medications, and 57% reported a subjective therapeutic benefit in terms of tumor-related symptoms. Comprehensive PFS was 39 months, including two patients with early recurrence (2 and 5 months post-RLT, respectively) and one patient with remarkable tumor downsizing that allowed a second-step curative liver surgery. Of note, the patient was still disease-free at the time of the study. The results of the largest cohort of PPGLs treated with RLT were reported by Severi et al., who treated 46 patients reaching a comprehensive DCR of 80%. Interestingly, 34 patients treated with [177Lu]Lu-DOTATATE obtained a longer median OS in comparison to those treated with [90Y]Y-DOTATOC (143 vs. 92 months, respectively). According to the authors, this result may be related both to DOTATATEs higher affinity for SSTR2 (which is the type most overexpressed in PPGLs) and to the longer half-life of 177Lu and, consequently, prolonged residence time within the tumor lesions. Moreover, this study reports that sympathetic functioning PPGLs were associated to a shorter median PFS compared to non-functioning PPGLs. Prado-Wohlwend et al. treated nine patients with [177Lu]Lu-DOTATATE and eight with [131I]MIBG, obtaining a PFS of 29 and 18.5 months and a DCR of 88.8% and 62.5%, respectively, for each therapy. Despite the low number of patients involved, a trend for a longer PFS was found for adrenal primary PPGLs treated with [177Lu]Lu-DOTATATE. Nevertheless, the authors suggest performing both [68Ga]Ga-DOTA-SSA PET/CT and [123I]MIBG SPECT/CT in each patient in order to offer a personalized theranostic approach based on the highest uptake intensity in one of the two imaging scans. This is consistent with the results by Jaiswal et al. who found that a baseline SUVmax > 21 at [68Ga]Ga-DOTA-SSA PET/CT is a very strong predictor of response to [177Lu]Lu-DOTATATE (p < 0.0001).
Click to Show/Hide
|
||||
| Experiment 496 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Paraganglioma | ||||
| Efficacy Data | Disease control rate (DCR) |
80.40%
|
|||
| Patients Enrolled |
46 patients with Pheochromocytomas (PPGL) and paragangliomas (PHEO).
|
||||
| Administration Time | 4-5 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
As a consequence, we saw a rising interest in a potential radiolabeled-SST agonist theranostic approach for PPGLs, with several spontaneous studies reporting of the treatment of metastatic or inoperable tumors with Lutathera and other similar radiocompounds, with promising preliminary results. The first RLT experience in PPGLs was reported by van Essen et al. who treated a heterogeneous cohort of patients, including 12 PGLs. Despite the authors reporting lower response rates than those obtained in GEP-NET patients, ORR and DCR were 18% and 73%, respectively. These results are consistent with other reports in the literature. Kong et al. treated 20 PPGLs (8 PHEOs and 12 PGLs) with [117Lu]Lu-DOTATATE, 14 due to uncontrolled secondary hypertension and 6 to radiological PD. A DCR equal to 86% was reached, including five patients with ORR (36%). Of note, 62% of symptomatic patients required a dose reduction of antihypertensive medications, and 57% reported a subjective therapeutic benefit in terms of tumor-related symptoms. Comprehensive PFS was 39 months, including two patients with early recurrence (2 and 5 months post-RLT, respectively) and one patient with remarkable tumor downsizing that allowed a second-step curative liver surgery. Of note, the patient was still disease-free at the time of the study. The results of the largest cohort of PPGLs treated with RLT were reported by Severi et al., who treated 46 patients reaching a comprehensive DCR of 80%. Interestingly, 34 patients treated with [177Lu]Lu-DOTATATE obtained a longer median OS in comparison to those treated with [90Y]Y-DOTATOC (143 vs. 92 months, respectively). According to the authors, this result may be related both to DOTATATEs higher affinity for SSTR2 (which is the type most overexpressed in PPGLs) and to the longer half-life of 177Lu and, consequently, prolonged residence time within the tumor lesions. Moreover, this study reports that sympathetic functioning PPGLs were associated to a shorter median PFS compared to non-functioning PPGLs. Prado-Wohlwend et al. treated nine patients with [177Lu]Lu-DOTATATE and eight with [131I]MIBG, obtaining a PFS of 29 and 18.5 months and a DCR of 88.8% and 62.5%, respectively, for each therapy. Despite the low number of patients involved, a trend for a longer PFS was found for adrenal primary PPGLs treated with [177Lu]Lu-DOTATATE. Nevertheless, the authors suggest performing both [68Ga]Ga-DOTA-SSA PET/CT and [123I]MIBG SPECT/CT in each patient in order to offer a personalized theranostic approach based on the highest uptake intensity in one of the two imaging scans. This is consistent with the results by Jaiswal et al. who found that a baseline SUVmax > 21 at [68Ga]Ga-DOTA-SSA PET/CT is a very strong predictor of response to [177Lu]Lu-DOTATATE (p < 0.0001).
Click to Show/Hide
|
||||
| Experiment 497 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Disease control rate (DCR) |
83%
|
|||
| Patients Enrolled |
Patients with pheocromocytomas (PCCs) and paragangliomas (PGLs) (1 PCC, 11PGL).
|
||||
| MOA of PDC |
We showed that PRRT based on 177Lu-DOTATATE and 90-YDOTATOC without radiosensitizers are efficacious therapeutic options (DCR of 83% and 76%, respectively) and can be considered in the multidisciplinary treatment of PCCs and PGLs as alternatives to I-131 MIBG and chemotherapy.
|
||||
| Description |
There was no statistically significant heterogeneity among the 177Lu- and 90Y- PRRTs studies (I2 value = 0%, p = 0.68 and I2 value = 0%, p = 0.91, respectively). The pooled DCRs were 0.83 (95% CI: 0.75-0.88) and 0.76 (95% CI: 0.56-0.89) for 177Lu- and 90Y-PRRT treatments, respectively. The pooled DCR for PRRT agents was 0.81 (95% CI: 0.74-0.87). Although the present study is the largest description of DCR in PRRT therapy of PCCs and PGLs, given the small sample size, no formal attempts were made to compare efficacy between 177Lu- and 90Y-PRRT treatments. In detail, we report pooled data on 149 177Lu- and 64 90Y-treated patients (total: 213 patients). Most studies were retrospective. The largest one included 46 patients (34 treated with 177Lu-, 12 with 90Y- PRRTs). Median ages ranged from 32.5 to 60.4 years. The female gender was slightly predominant (83 women, 74 men, gender not specified in 3 studies). When reported, mutations of SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B) were the most frequent genetic alterations. According to the predominant retrospective and real practice nature of the selected studies, sites of metastases and previous treatments were heterogeneous, with bone the metastatic site most often involved, and surgery the most frequent previous therapy. Paramount descriptive information on PRRT doses per cycle, number of cycles, response criteria, best responses to treatments, and frequency of G3/G4 hematologic toxicities is reported in Table 2. The lowest applied dose was 3.4 GBq per cycle; the number of administered cycles was heterogeneous (at least 3 cycles in 7 studies). The most used response criteria were the RECIST 1.1. Only one study evaluated the response with SWOG criteria, which are more conservative with regard to the definition of PR (50% or greater reduction in the sum of the product of the perpendicular diameters of target lesions). The majority of the patients experienced PR or SD. Hematologic toxicity was manageable. Funnel plot of selected studies did not show publication bias. The MINORS and Newcastle-Ottawa Scale scoring for the selected articles are reported in Table 3.
Click to Show/Hide
|
||||
| Experiment 498 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Bronchial carcinoids | ||||
| Efficacy Data | Disease control rate (DCR) |
83%
|
|||
| Patients Enrolled |
48 patients with bronchial carcinoids.
|
||||
| Administration Time | Median 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
Treatment of bronchial carcinoids is not simple and requires a multidisciplinary approach. Surgery remains the mainstay of treatment for local disease, while in advanced disease, management includes chemotherapy, cold somatostatin analogues, immunotherapy, everolimus, and others target therapies. RLT is an option for selected patients with advanced or metastatic BCs overexpressing SSTR in progression to cold SSAs therapy. Although the experience of [177Lu]Lu-DOTATATE is more limited in BCs than GEP-NET, off-label use can be considered with promising results. A prospective phase II trial in 34 patients with stage IV BCs treated with cumulative activity of 18.5-27.8 GBq in four or five cycles of [177Lu]Lu-DOTATATE documented a DCR of 80% (6% complete response, 27% partial response, and 47% stable disease) and a median PFS of 20 months. In this study, negative prognostic factors were AC histology, tumor thyroid transcription factor-1 (TTF-1) expression, and a positive [18F]FDG PET imaging. Comparable results (median PFS of 17 months) were reported in a group of patients with diffuse extrahepatic metastases treated with RLT after several lines of therapies. Higher activities were administered in a study by van Essen et al., who treated nine patients with metastatic BCs, delivering a cumulative dose of 22.2-29.6 GBq of [177Lu]Lu-DOTATATE, demonstrating an overall response rate comparable to that of other GEP-NET (50% vs. 47%, respectively for BCs and GEP-NET). Moreover, tumor regression was reported in 66.6% of patients, without any outcome discrepancies between TCs and ACs. Comparable median OS and response rate between BCs and others GEP-NETs were reported also in another recent retrospective study. Likewise, the efficacy of RLT in BCs was demonstrated by Brabander et al. in a study involving 443 patients affected by midgut, bronchial, and CUP-NETs treated with 7.4 GBq of [177Lu]Lu-DOTATATE every 8 weeks. The authors found that objective response rate (ORR) for BCs was 30%, and an additional 30% of patients had a stable disease. Nevertheless, median OS for BCs was 52 months versus 71 months for pancreatic-NET. Long term results were analyzed by Mariniello et al. in 114 patients with advanced BCs treated with different RLT protocols. They documented a median PFS and OS of 28.0 and 58.8 months, respectively, while morphological responses were observed in 26.5% of patients. [177Lu]Lu-DOTATATE monotherapy protocol resulted in the highest 5-year OS (61.4%), despite tandem protocol ([90Y]Y-DOTATOC + [177Lu]Lu-DOTATATE) provided the highest response rates (38.1%). Best outcome was reached in patients who underwent RLT in early stage of disease, suggesting that this treatment should also be considered for newly diagnosed unresectable BCs. Moreover, despite most patients having only mild and transient adverse events, patients with hematologic toxicity showed worse survival outcomes.
Click to Show/Hide
|
||||
| Experiment 499 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Disease control rate (DCR) |
83%
|
|||
| Patients Enrolled |
149 patients with paraganglioma.
|
||||
| Administration Time | 1-5 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The use of PRRT for patients with PPGLs who had high tumor SSTR expression has mainly been reported in retrospective small case series and the protocols varied in all these studies. So far, four meta-analyses (Taieb et al. and Satapathy et al. in 2019 as well as Su et al. and Marretta et al. in 2023) based on PRRT (177Lu/90Y-DOTAT-TATE/TOC) therapy have been reported. The reported number of pooled patients ranged from 179-330 without any complete response. The reported partial response ranged from 20-25% and stable disease from 59-70% giving an objective response rate range of 20-25% and disease control rate range of 8190%. The survival rates were reported by only 3 meta-analyses. The reported progression-free survival were 17-39 months in seven studies by Taieb et al., 10-91 months in 4 studies and a mean of 37.1 months in 2 studies by Satapathy et al., and a median of 31.8 months in 13 studies by Su et al. whereas overall survival were 49.6-68.0 months in 3 studies by Taieb et al., a mean of 54.5 months in 4 studies by Satapathy et al. and a median of 74.3 months in 11 studies by Su et al. Su et al. reports a pooled adverse events data in 274/330 patients which mentions a grade 3/4 hematotoxicity in 4.3% (grade 1-4, 22.3%) and grade 3/4 nephrotoxicity in 1.9% (grade 1-4, 1.9%), and myelodysplastic syndrome in 1 patient. Treatment discontinuation occurred in 4 patients due to thrombocytopenia (n=3) or nephrotoxicity (n=1).
Click to Show/Hide
|
||||
| Experiment 500 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Advanced paraganglioma | ||||
| Efficacy Data | Disease control rate (DCR) |
84%
|
|||
| Patients Enrolled |
A total of 201 patients with advanced paragangliomas.
|
||||
| MOA of PDC |
The clinical applications of radiolabeled SSAs and DOTA-SSA compounds in imaging and radionuclide therapy have been rapidly increasing over the past 2 decades and have been integrated into the current management guidelines of NETs. Several studies support the safety and clinical efficacy of FDA-approved radionuclide therapies including 177Lu-DOTATATE in advanced GEP NETs and 131I-MIBG in advanced paragangliomas and pheochromocytomas. The utility of SSTR-based imaging in PRRT response assessment and surveillance of NETs remains to be established.
Click to Show/Hide
|
||||
| Description |
Over the past decade, a small number of studies have evaluated the efficacy and safety of PRRT with 177Lu-DOTA-SSA in patients with metastatic or inoperable paragangliomas [66, 67]. A recent meta-analysis of 12 studies including a total of 201 patients with advanced paragangliomas showed an objective response rate of 25%, biochemical response rate of 64%, and disease control rate of 84% after treatment with PRRT (either 90Y- or 177Lu-based agents) [67]. The following minimal treatment-related adverse effects were observed: grade 3 or 4 lymphopenia, thrombocytopenia, neutropenia, and nephrotoxicity in 11%, 9%, 3%, and 4% of the patients, respectively.
Click to Show/Hide
|
||||
| Experiment 501 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Disease control rate (DCR) |
84%
|
|||
| Patients Enrolled |
201 patients with paraganglioma.
|
||||
| Administration Time | 1-10 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The use of PRRT for patients with PPGLs who had high tumor SSTR expression has mainly been reported in retrospective small case series and the protocols varied in all these studies. So far, four meta-analyses (Taieb et al. and Satapathy et al. in 2019 as well as Su et al. and Marretta et al. in 2023) based on PRRT (177Lu/90Y-DOTAT-TATE/TOC) therapy have been reported. The reported number of pooled patients ranged from 179-330 without any complete response. The reported partial response ranged from 20-25% and stable disease from 59-70% giving an objective response rate range of 20-25% and disease control rate range of 8190%. The survival rates were reported by only 3 meta-analyses. The reported progression-free survival were 17-39 months in seven studies by Taieb et al., 10-91 months in 4 studies and a mean of 37.1 months in 2 studies by Satapathy et al., and a median of 31.8 months in 13 studies by Su et al. whereas overall survival were 49.6-68.0 months in 3 studies by Taieb et al., a mean of 54.5 months in 4 studies by Satapathy et al. and a median of 74.3 months in 11 studies by Su et al. Su et al. reports a pooled adverse events data in 274/330 patients which mentions a grade 3/4 hematotoxicity in 4.3% (grade 1-4, 22.3%) and grade 3/4 nephrotoxicity in 1.9% (grade 1-4, 1.9%), and myelodysplastic syndrome in 1 patient. Treatment discontinuation occurred in 4 patients due to thrombocytopenia (n=3) or nephrotoxicity (n=1).
Click to Show/Hide
|
||||
| Experiment 502 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Disease control rate (DCR) |
84.20%
|
|||
| Patients Enrolled |
19 patients with CUP-neuroendocrine tumor.
|
||||
| Administration Time | Median 6 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
Unfortunately, the literature lacks specific trials assessing RLT efficacy and safety on CUP-NETs, and the only available literature evidence is provided by mixed trials also including a few patients with SSTR-positive CUP-NETs. In a large cohort of patients treated with [177Lu]Lu-DOTATATE, Brabander et al.reported of 82 CUP-NET patients. Overall, DCR was reached in 78% of patients and median PFS and OS were 29 and 53 months, respectively. These results show that RLT may be effective in CUP-NETs patients, with a response rate intermediate between that obtained in GEP-NETswho showed longer median OS (60 vs. 53 months, respectively)and BCswho showed shorter median PFS (20 vs. 29 months, respectively). These results are consistent with those reported by Demirci et al. who treated 19 CUP-NETs, with an overall DCR of 84.2% and mean PFS and OS of 40 and 48 months, respectively. Once again, RLT outcomes in CUP-NETs were intermediate between those in GEP-NETs and BCs. In a large mixed cohort, Baum et al. treated 151 CUP-NETs (mostly with [177Lu]Lu-DOTATATE, although a small percentage of patients was treated with [90Y]Y-DOTATATE/DOTATOC in the study), obtaining median PFS and OS of 13 and 46 months, respectively. CUP-NETStogether with BCswere associated with a significantly shorter PFS at multivariate analysis if compared to pancreatic NETs despite showing a longer OS (50 vs. 43 months, respectively). Future prospective trials assessing efficacy and safety of RLT in CUP-NETs are desirable.
Click to Show/Hide
|
||||
| Experiment 503 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Disease control rate (DCR) |
84.60%
|
|||
| Patients Enrolled |
26 patients with neuroendocrine tumour.
|
||||
| Administration Time | 2-5 cycles | ||||
| Administration Dosage | Median activity for re-treatment: 16.5 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 504 Reporting the Activity Data of This PDC | [25] | ||||
| Indication | SDHB positive metastatic paraganglioma | ||||
| Efficacy Data | Disease control rate (DCR) |
85%
|
|||
| Patients Enrolled |
37 patients affected by neuroendocrine tumors.
|
||||
| Administration Time | Five cycles of 5.5 gbq each every 8 weeks | ||||
| Administration Dosage | Cumulative activity of 27.5 GBq | ||||
| Description |
Thirty-three of 37 patients were evaluable for response. Objective responses included partial response (PR) in 10 patients (30%), stable disease (SD) in 18 patients (55%), with a DCR of 85%. Median follow up was 38 months (range 4.6-51.1 months). Median PFS was 31.4 months (17.6-45.4) and mOS was not reached.
|
||||
| Experiment 505 Reporting the Activity Data of This PDC | [17] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Disease control rate (DCR) |
85.70%
|
|||
| Patients Enrolled |
7 patients with inoperable, progressive, metastatic, or locally advanced, somatostatin receptor-positive neuroendocrine tumours.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | 21.3-30.1 GBq total dose | ||||
| MOA of PDC |
In conclusion, for the first time in Korea, we have conducted a clinical trial of PRRT with SNU-KB-01, a NCA 177Lu-DOTATATE, in patients with SSTR-positive NET. Treatment with SNU-KB-01 was safe and resulted in control of disease in most of the patients with high SSTR expression. The recommended dose per cycle of SNU-KB-01 was determined to be 7.40 GBq for the phase 2 trial. Our results indicate SNU-KB-01 as a potentially safe and efficacious treatment option for NET patients and are expected to provide a wider choice of treatment opportunities for patients with various types of SSTR-positive tumors in Korea.
Click to Show/Hide
|
||||
| Description |
Grade 3 thrombocytopenia occurred in one patient, but no other grade 3 or 4 major hematologic or renal toxicity was observed. The best objective response to SNU-KB-01 was partial response. Overall response rate was 42.9% and disease control rate was 85.7%.
|
||||
| Experiment 506 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Phaeochromocytoma | ||||
| Efficacy Data | Disease control rate (DCR) |
86%
|
|||
| Patients Enrolled |
20 patients with phaeochromocytoma and paraganglioma.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
As a consequence, we saw a rising interest in a potential radiolabeled-SST agonist theranostic approach for PPGLs, with several spontaneous studies reporting of the treatment of metastatic or inoperable tumors with Lutathera and other similar radiocompounds, with promising preliminary results. The first RLT experience in PPGLs was reported by van Essen et al. who treated a heterogeneous cohort of patients, including 12 PGLs. Despite the authors reporting lower response rates than those obtained in GEP-NET patients, ORR and DCR were 18% and 73%, respectively. These results are consistent with other reports in the literature. Kong et al. treated 20 PPGLs (8 PHEOs and 12 PGLs) with [117Lu]Lu-DOTATATE, 14 due to uncontrolled secondary hypertension and 6 to radiological PD. A DCR equal to 86% was reached, including five patients with ORR (36%). Of note, 62% of symptomatic patients required a dose reduction of antihypertensive medications, and 57% reported a subjective therapeutic benefit in terms of tumor-related symptoms. Comprehensive PFS was 39 months, including two patients with early recurrence (2 and 5 months post-RLT, respectively) and one patient with remarkable tumor downsizing that allowed a second-step curative liver surgery. Of note, the patient was still disease-free at the time of the study. The results of the largest cohort of PPGLs treated with RLT were reported by Severi et al., who treated 46 patients reaching a comprehensive DCR of 80%. Interestingly, 34 patients treated with [177Lu]Lu-DOTATATE obtained a longer median OS in comparison to those treated with [90Y]Y-DOTATOC (143 vs. 92 months, respectively). According to the authors, this result may be related both to DOTATATEs higher affinity for SSTR2 (which is the type most overexpressed in PPGLs) and to the longer half-life of 177Lu and, consequently, prolonged residence time within the tumor lesions. Moreover, this study reports that sympathetic functioning PPGLs were associated to a shorter median PFS compared to non-functioning PPGLs. Prado-Wohlwend et al. treated nine patients with [177Lu]Lu-DOTATATE and eight with [131I]MIBG, obtaining a PFS of 29 and 18.5 months and a DCR of 88.8% and 62.5%, respectively, for each therapy. Despite the low number of patients involved, a trend for a longer PFS was found for adrenal primary PPGLs treated with [177Lu]Lu-DOTATATE. Nevertheless, the authors suggest performing both [68Ga]Ga-DOTA-SSA PET/CT and [123I]MIBG SPECT/CT in each patient in order to offer a personalized theranostic approach based on the highest uptake intensity in one of the two imaging scans. This is consistent with the results by Jaiswal et al. who found that a baseline SUVmax > 21 at [68Ga]Ga-DOTA-SSA PET/CT is a very strong predictor of response to [177Lu]Lu-DOTATATE (p < 0.0001).
Click to Show/Hide
|
||||
| Experiment 507 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Paraganglioma | ||||
| Efficacy Data | Disease control rate (DCR) |
86%
|
|||
| Patients Enrolled |
20 patients with phaeochromocytoma and paraganglioma.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
As a consequence, we saw a rising interest in a potential radiolabeled-SST agonist theranostic approach for PPGLs, with several spontaneous studies reporting of the treatment of metastatic or inoperable tumors with Lutathera and other similar radiocompounds, with promising preliminary results. The first RLT experience in PPGLs was reported by van Essen et al. who treated a heterogeneous cohort of patients, including 12 PGLs. Despite the authors reporting lower response rates than those obtained in GEP-NET patients, ORR and DCR were 18% and 73%, respectively. These results are consistent with other reports in the literature. Kong et al. treated 20 PPGLs (8 PHEOs and 12 PGLs) with [117Lu]Lu-DOTATATE, 14 due to uncontrolled secondary hypertension and 6 to radiological PD. A DCR equal to 86% was reached, including five patients with ORR (36%). Of note, 62% of symptomatic patients required a dose reduction of antihypertensive medications, and 57% reported a subjective therapeutic benefit in terms of tumor-related symptoms. Comprehensive PFS was 39 months, including two patients with early recurrence (2 and 5 months post-RLT, respectively) and one patient with remarkable tumor downsizing that allowed a second-step curative liver surgery. Of note, the patient was still disease-free at the time of the study. The results of the largest cohort of PPGLs treated with RLT were reported by Severi et al., who treated 46 patients reaching a comprehensive DCR of 80%. Interestingly, 34 patients treated with [177Lu]Lu-DOTATATE obtained a longer median OS in comparison to those treated with [90Y]Y-DOTATOC (143 vs. 92 months, respectively). According to the authors, this result may be related both to DOTATATEs higher affinity for SSTR2 (which is the type most overexpressed in PPGLs) and to the longer half-life of 177Lu and, consequently, prolonged residence time within the tumor lesions. Moreover, this study reports that sympathetic functioning PPGLs were associated to a shorter median PFS compared to non-functioning PPGLs. Prado-Wohlwend et al. treated nine patients with [177Lu]Lu-DOTATATE and eight with [131I]MIBG, obtaining a PFS of 29 and 18.5 months and a DCR of 88.8% and 62.5%, respectively, for each therapy. Despite the low number of patients involved, a trend for a longer PFS was found for adrenal primary PPGLs treated with [177Lu]Lu-DOTATATE. Nevertheless, the authors suggest performing both [68Ga]Ga-DOTA-SSA PET/CT and [123I]MIBG SPECT/CT in each patient in order to offer a personalized theranostic approach based on the highest uptake intensity in one of the two imaging scans. This is consistent with the results by Jaiswal et al. who found that a baseline SUVmax > 21 at [68Ga]Ga-DOTA-SSA PET/CT is a very strong predictor of response to [177Lu]Lu-DOTATATE (p < 0.0001).
Click to Show/Hide
|
||||
| Experiment 508 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Disease control rate (DCR) |
86.10%
|
|||
| Patients Enrolled |
36 patients with paraganglioma.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The efficacy and safety data of the interim analysis of an open-label, single-arm phase 2 study evaluating 177Lu-DOTATATE in paraganglioma patients conducted at the National Institutes of Health (NCT03206060) in 36 patients [(n=18, apparently sporadic cohort and n=18 in patients having germline variant in one of the genes encoding subunits of succinate dehydrogenase complex (SDHA=2, SDHB=15, SDHD=1)), SDHx cohort] after 177LuDOTATATE therapy with 7.4 GBq every 8 weeks for 4 cycles. The progression-free survival at 6 months since initiation of 177Lu-DOTATATE was 19.1 months (22.7 months in apparently sporadic cohort and 15.4 months in SDHx cohort) and overall survival was not reached in both cohorts (80). Per RECIST 1.1, overall, 86.1% patients showed partial response (13.9%) or stable disease (72.2%). In sporadic cohort; partial response was observed in 11.1% and stable disease in 88.9% patients whereas those in SDHx cohort were, 16.7% and 55.5%, respectively at the end of 6 months.
Click to Show/Hide
|
||||
| Experiment 509 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Phaeochromocytoma | ||||
| Efficacy Data | Disease control rate (DCR) |
88.80%
|
|||
| Patients Enrolled |
9 patients with Pheochromocytomas (PPGL) and paragangliomas (PHEO).
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
As a consequence, we saw a rising interest in a potential radiolabeled-SST agonist theranostic approach for PPGLs, with several spontaneous studies reporting of the treatment of metastatic or inoperable tumors with Lutathera and other similar radiocompounds, with promising preliminary results. The first RLT experience in PPGLs was reported by van Essen et al. who treated a heterogeneous cohort of patients, including 12 PGLs. Despite the authors reporting lower response rates than those obtained in GEP-NET patients, ORR and DCR were 18% and 73%, respectively. These results are consistent with other reports in the literature. Kong et al. treated 20 PPGLs (8 PHEOs and 12 PGLs) with [117Lu]Lu-DOTATATE, 14 due to uncontrolled secondary hypertension and 6 to radiological PD. A DCR equal to 86% was reached, including five patients with ORR (36%). Of note, 62% of symptomatic patients required a dose reduction of antihypertensive medications, and 57% reported a subjective therapeutic benefit in terms of tumor-related symptoms. Comprehensive PFS was 39 months, including two patients with early recurrence (2 and 5 months post-RLT, respectively) and one patient with remarkable tumor downsizing that allowed a second-step curative liver surgery. Of note, the patient was still disease-free at the time of the study. The results of the largest cohort of PPGLs treated with RLT were reported by Severi et al., who treated 46 patients reaching a comprehensive DCR of 80%. Interestingly, 34 patients treated with [177Lu]Lu-DOTATATE obtained a longer median OS in comparison to those treated with [90Y]Y-DOTATOC (143 vs. 92 months, respectively). According to the authors, this result may be related both to DOTATATEs higher affinity for SSTR2 (which is the type most overexpressed in PPGLs) and to the longer half-life of 177Lu and, consequently, prolonged residence time within the tumor lesions. Moreover, this study reports that sympathetic functioning PPGLs were associated to a shorter median PFS compared to non-functioning PPGLs. Prado-Wohlwend et al. treated nine patients with [177Lu]Lu-DOTATATE and eight with [131I]MIBG, obtaining a PFS of 29 and 18.5 months and a DCR of 88.8% and 62.5%, respectively, for each therapy. Despite the low number of patients involved, a trend for a longer PFS was found for adrenal primary PPGLs treated with [177Lu]Lu-DOTATATE. Nevertheless, the authors suggest performing both [68Ga]Ga-DOTA-SSA PET/CT and [123I]MIBG SPECT/CT in each patient in order to offer a personalized theranostic approach based on the highest uptake intensity in one of the two imaging scans. This is consistent with the results by Jaiswal et al. who found that a baseline SUVmax > 21 at [68Ga]Ga-DOTA-SSA PET/CT is a very strong predictor of response to [177Lu]Lu-DOTATATE (p < 0.0001).
Click to Show/Hide
|
||||
| Experiment 510 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Paraganglioma | ||||
| Efficacy Data | Disease control rate (DCR) |
88.80%
|
|||
| Patients Enrolled |
9 patients with Pheochromocytomas (PPGL) and paragangliomas (PHEO).
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
As a consequence, we saw a rising interest in a potential radiolabeled-SST agonist theranostic approach for PPGLs, with several spontaneous studies reporting of the treatment of metastatic or inoperable tumors with Lutathera and other similar radiocompounds, with promising preliminary results. The first RLT experience in PPGLs was reported by van Essen et al. who treated a heterogeneous cohort of patients, including 12 PGLs. Despite the authors reporting lower response rates than those obtained in GEP-NET patients, ORR and DCR were 18% and 73%, respectively. These results are consistent with other reports in the literature. Kong et al. treated 20 PPGLs (8 PHEOs and 12 PGLs) with [117Lu]Lu-DOTATATE, 14 due to uncontrolled secondary hypertension and 6 to radiological PD. A DCR equal to 86% was reached, including five patients with ORR (36%). Of note, 62% of symptomatic patients required a dose reduction of antihypertensive medications, and 57% reported a subjective therapeutic benefit in terms of tumor-related symptoms. Comprehensive PFS was 39 months, including two patients with early recurrence (2 and 5 months post-RLT, respectively) and one patient with remarkable tumor downsizing that allowed a second-step curative liver surgery. Of note, the patient was still disease-free at the time of the study. The results of the largest cohort of PPGLs treated with RLT were reported by Severi et al., who treated 46 patients reaching a comprehensive DCR of 80%. Interestingly, 34 patients treated with [177Lu]Lu-DOTATATE obtained a longer median OS in comparison to those treated with [90Y]Y-DOTATOC (143 vs. 92 months, respectively). According to the authors, this result may be related both to DOTATATEs higher affinity for SSTR2 (which is the type most overexpressed in PPGLs) and to the longer half-life of 177Lu and, consequently, prolonged residence time within the tumor lesions. Moreover, this study reports that sympathetic functioning PPGLs were associated to a shorter median PFS compared to non-functioning PPGLs. Prado-Wohlwend et al. treated nine patients with [177Lu]Lu-DOTATATE and eight with [131I]MIBG, obtaining a PFS of 29 and 18.5 months and a DCR of 88.8% and 62.5%, respectively, for each therapy. Despite the low number of patients involved, a trend for a longer PFS was found for adrenal primary PPGLs treated with [177Lu]Lu-DOTATATE. Nevertheless, the authors suggest performing both [68Ga]Ga-DOTA-SSA PET/CT and [123I]MIBG SPECT/CT in each patient in order to offer a personalized theranostic approach based on the highest uptake intensity in one of the two imaging scans. This is consistent with the results by Jaiswal et al. who found that a baseline SUVmax > 21 at [68Ga]Ga-DOTA-SSA PET/CT is a very strong predictor of response to [177Lu]Lu-DOTATATE (p < 0.0001).
Click to Show/Hide
|
||||
| Experiment 511 Reporting the Activity Data of This PDC | [22] | ||||
| Indication | Bronchial carcinoids | ||||
| Efficacy Data | Disease control rate (DCR) |
89%
|
|||
| Patients Enrolled |
9 patients with bronchial carcinoids.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Radioligand therapy (RLT) with radiolabeled somatostatin analogues (SSA) is currently a mainstay in advanced gastroenteropancreatic (GEP) neuroendocrine tumor (NET) treatment, as it represents an ideal model of a modern system of personalized medicine. However, to reach this achievement required quite a long and challenging scientific journey. The first experiences with radiolabeled SSA, dating back to the late 1990s, employed yttrium-90 labelled SSA as radiopharmaceutical for RLT. However, renal toxicities were not negligible, hindering the widespread of this therapeutic option. Afterwards, the attention shifted to lutetium-177 labelled SSA, due to their more favorable toxicity profile and to the first positive experiences obtained with [177Lu][Lu-DOTA,Tyr3]octreotate ([177Lu]Lu-DOTATATE). The growing interest in this radiopharmaceutical led to the NETTER-1 study, a phase III multicenter trial whose results started a new era, demonstrating the most favorable outcomes of [177Lu]Lu-DOTATATE RLT plus octreotide LAR 30 mg versus octreotide LAR 60 mg alone. As a consequence of NETTER-1 results, [177Lu]Lu-DOTATATE was finally approved by the European Medicines Agency (EMA) in September 2017, the Food and Drug Administration (FDA) in January 2018, the Canada Health in January 2019, and the State of Israel Ministry of Health in July 2019 (Lutathera). Currently, Lutathera is approved in 23 countries worldwide. However, this should be considered only a partial achievement as a large portion of tumors overexpressing somatostatin receptors (SSTR) still cannot be treated with Lutathera, giving rise to the so-called Lutathera Orphans. Indeed, Lutathera is currently administered in a protected hospitalization regime and is indicated in adult patients diagnosed with well-differentiated (G1 and G2) gastroenteropancreatic neuroendocrine tumors (GEP-NET) that are progressive, non-removable or metastatic, and positive to the receptors for somatostatin. Therefore, paediatric patients cannot be treated with Lutathera.
Click to Show/Hide
|
||||
| Description |
Treatment of bronchial carcinoids is not simple and requires a multidisciplinary approach. Surgery remains the mainstay of treatment for local disease, while in advanced disease, management includes chemotherapy, cold somatostatin analogues, immunotherapy, everolimus, and others target therapies. RLT is an option for selected patients with advanced or metastatic BCs overexpressing SSTR in progression to cold SSAs therapy. Although the experience of [177Lu]Lu-DOTATATE is more limited in BCs than GEP-NET, off-label use can be considered with promising results. A prospective phase II trial in 34 patients with stage IV BCs treated with cumulative activity of 18.5-27.8 GBq in four or five cycles of [177Lu]Lu-DOTATATE documented a DCR of 80% (6% complete response, 27% partial response, and 47% stable disease) and a median PFS of 20 months. In this study, negative prognostic factors were AC histology, tumor thyroid transcription factor-1 (TTF-1) expression, and a positive [18F]FDG PET imaging. Comparable results (median PFS of 17 months) were reported in a group of patients with diffuse extrahepatic metastases treated with RLT after several lines of therapies. Higher activities were administered in a study by van Essen et al., who treated nine patients with metastatic BCs, delivering a cumulative dose of 22.2-29.6 GBq of [177Lu]Lu-DOTATATE, demonstrating an overall response rate comparable to that of other GEP-NET (50% vs. 47%, respectively for BCs and GEP-NET). Moreover, tumor regression was reported in 66.6% of patients, without any outcome discrepancies between TCs and ACs. Comparable median OS and response rate between BCs and others GEP-NETs were reported also in another recent retrospective study. Likewise, the efficacy of RLT in BCs was demonstrated by Brabander et al. in a study involving 443 patients affected by midgut, bronchial, and CUP-NETs treated with 7.4 GBq of [177Lu]Lu-DOTATATE every 8 weeks. The authors found that objective response rate (ORR) for BCs was 30%, and an additional 30% of patients had a stable disease. Nevertheless, median OS for BCs was 52 months versus 71 months for pancreatic-NET. Long term results were analyzed by Mariniello et al. in 114 patients with advanced BCs treated with different RLT protocols. They documented a median PFS and OS of 28.0 and 58.8 months, respectively, while morphological responses were observed in 26.5% of patients. [177Lu]Lu-DOTATATE monotherapy protocol resulted in the highest 5-year OS (61.4%), despite tandem protocol ([90Y]Y-DOTATOC + [177Lu]Lu-DOTATATE) provided the highest response rates (38.1%). Best outcome was reached in patients who underwent RLT in early stage of disease, suggesting that this treatment should also be considered for newly diagnosed unresectable BCs. Moreover, despite most patients having only mild and transient adverse events, patients with hematologic toxicity showed worse survival outcomes.
Click to Show/Hide
|
||||
| Experiment 512 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Disease control rate (DCR) |
89.60%
|
|||
| Patients Enrolled |
179 patients with paraganglioma.
|
||||
| Administration Time | 1-11 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The use of PRRT for patients with PPGLs who had high tumor SSTR expression has mainly been reported in retrospective small case series and the protocols varied in all these studies. So far, four meta-analyses (Taieb et al. and Satapathy et al. in 2019 as well as Su et al. and Marretta et al. in 2023) based on PRRT (177Lu/90Y-DOTAT-TATE/TOC) therapy have been reported. The reported number of pooled patients ranged from 179-330 without any complete response. The reported partial response ranged from 20-25% and stable disease from 59-70% giving an objective response rate range of 20-25% and disease control rate range of 8190%. The survival rates were reported by only 3 meta-analyses. The reported progression-free survival were 17-39 months in seven studies by Taieb et al., 10-91 months in 4 studies and a mean of 37.1 months in 2 studies by Satapathy et al., and a median of 31.8 months in 13 studies by Su et al. whereas overall survival were 49.6-68.0 months in 3 studies by Taieb et al., a mean of 54.5 months in 4 studies by Satapathy et al. and a median of 74.3 months in 11 studies by Su et al. Su et al. reports a pooled adverse events data in 274/330 patients which mentions a grade 3/4 hematotoxicity in 4.3% (grade 1-4, 22.3%) and grade 3/4 nephrotoxicity in 1.9% (grade 1-4, 1.9%), and myelodysplastic syndrome in 1 patient. Treatment discontinuation occurred in 4 patients due to thrombocytopenia (n=3) or nephrotoxicity (n=1).
Click to Show/Hide
|
||||
| Experiment 513 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Disease control rate (DCR) |
90%
|
|||
| Patients Enrolled |
330 patients with paraganglioma.
|
||||
| Administration Time | 1-10 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The use of PRRT for patients with PPGLs who had high tumor SSTR expression has mainly been reported in retrospective small case series and the protocols varied in all these studies. So far, four meta-analyses (Taieb et al. and Satapathy et al. in 2019 as well as Su et al. and Marretta et al. in 2023) based on PRRT (177Lu/90Y-DOTAT-TATE/TOC) therapy have been reported. The reported number of pooled patients ranged from 179-330 without any complete response. The reported partial response ranged from 20-25% and stable disease from 59-70% giving an objective response rate range of 20-25% and disease control rate range of 8190%. The survival rates were reported by only 3 meta-analyses. The reported progression-free survival were 17-39 months in seven studies by Taieb et al., 10-91 months in 4 studies and a mean of 37.1 months in 2 studies by Satapathy et al., and a median of 31.8 months in 13 studies by Su et al. whereas overall survival were 49.6-68.0 months in 3 studies by Taieb et al., a mean of 54.5 months in 4 studies by Satapathy et al. and a median of 74.3 months in 11 studies by Su et al. Su et al. reports a pooled adverse events data in 274/330 patients which mentions a grade 3/4 hematotoxicity in 4.3% (grade 1-4, 22.3%) and grade 3/4 nephrotoxicity in 1.9% (grade 1-4, 1.9%), and myelodysplastic syndrome in 1 patient. Treatment discontinuation occurred in 4 patients due to thrombocytopenia (n=3) or nephrotoxicity (n=1).
Click to Show/Hide
|
||||
| Experiment 514 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Disease control rate (DCR) |
92.30%
|
|||
| Patients Enrolled |
13 re-retreatment neuroendocrine tumour patients.
|
||||
| Administration Time | 2 cycles | ||||
| Administration Dosage | Median cumulative dose: 59.7 GBq | ||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 515 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Advanced neuroendocrine tumour | ||||
| Efficacy Data | Disease control rate (DCR) |
100%
|
|||
| Patients Enrolled |
18 patients with neuroendocrine tumour.
|
||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 516 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Disease control rate (DCR) |
100%
|
|||
| Patients Enrolled |
18 patients with paraganglioma.
|
||||
| Administration Time | 4 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The efficacy and safety data of the interim analysis of an open-label, single-arm phase 2 study evaluating 177Lu-DOTATATE in paraganglioma patients conducted at the National Institutes of Health (NCT03206060) in 36 patients [(n=18, apparently sporadic cohort and n=18 in patients having germline variant in one of the genes encoding subunits of succinate dehydrogenase complex (SDHA=2, SDHB=15, SDHD=1)), SDHx cohort] after 177LuDOTATATE therapy with 7.4 GBq every 8 weeks for 4 cycles. The progression-free survival at 6 months since initiation of 177Lu-DOTATATE was 19.1 months (22.7 months in apparently sporadic cohort and 15.4 months in SDHx cohort) and overall survival was not reached in both cohorts (80). Per RECIST 1.1, overall, 86.1% patients showed partial response (13.9%) or stable disease (72.2%). In sporadic cohort; partial response was observed in 11.1% and stable disease in 88.9% patients whereas those in SDHx cohort were, 16.7% and 55.5%, respectively at the end of 6 months.
Click to Show/Hide
|
||||
| Experiment 517 Reporting the Activity Data of This PDC | [50] | ||||
| Indication | Medullary thyroid carcinoma | ||||
| Efficacy Data | Death rate |
43.75%
|
|||
| Patients Enrolled |
144 patients with metastatic medullary thyroid carcinoma.
|
||||
| MOA of PDC |
Findings of the study revealed that in the absence of the established treatment for the patients with RR-DTC and metastatic MTC, PRRT could be effective with few adverse events.
|
||||
| Description |
Among 2284 related papers, 41 papers met the inclusion criteria. A total of 157 patients with RR-DTC were treated with PPRT. Biochemical and objective responses (partial and complete) were observed in 25.3 and 10.5% of patients, respectively. Among 220 patients with metastatic MTC, biochemical and objective responses were observed in 37.2 and 10.6% of the patients, respectively. Forty-six deaths were reported in 95 patients with advanced RR-DTC. In addition, 63 deaths were observed in 144 patients with metastatic MTC. Major side effects were reported in 124 patients treated with 90Y -based agent. In the patients treated with 177Lu-DOTA-TATE and 111In-Octreotide, mild and transient hematologic or renal complications were reported.
Click to Show/Hide
|
||||
| Experiment 518 Reporting the Activity Data of This PDC | [50] | ||||
| Indication | Differentiated thyroid cancer | ||||
| Efficacy Data | Death rate |
48.42%
|
|||
| Patients Enrolled |
95 patients with advanced radioiodine-refractory differentiated thyroid cancer.
|
||||
| MOA of PDC |
Findings of the study revealed that in the absence of the established treatment for the patients with RR-DTC and metastatic MTC, PRRT could be effective with few adverse events.
|
||||
| Description |
Among 2284 related papers, 41 papers met the inclusion criteria. A total of 157 patients with RR-DTC were treated with PPRT. Biochemical and objective responses (partial and complete) were observed in 25.3 and 10.5% of patients, respectively. Among 220 patients with metastatic MTC, biochemical and objective responses were observed in 37.2 and 10.6% of the patients, respectively. Forty-six deaths were reported in 95 patients with advanced RR-DTC. In addition, 63 deaths were observed in 144 patients with metastatic MTC. Major side effects were reported in 124 patients treated with 90Y -based agent. In the patients treated with 177Lu-DOTA-TATE and 111In-Octreotide, mild and transient hematologic or renal complications were reported.
Click to Show/Hide
|
||||
| Experiment 519 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Creatinine increase ≥40% |
3.75%
|
|||
| Patients Enrolled |
534 male patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (25.9-29.6 GBq) (800 mCi [700-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 520 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Creatinine increase ≥40% |
5.26%
|
|||
| Patients Enrolled |
439 female patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (22.2-29.6 GBq) (800 mCi [600-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 521 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Creatinine |
1.13%
|
|||
| Patients Enrolled |
534 male patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (25.9-29.6 GBq) (800 mCi [700-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 522 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Creatinine |
2.29%
|
|||
| Patients Enrolled |
439 female patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (22.2-29.6 GBq) (800 mCi [600-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 523 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Creatinine |
2.46%
|
|||
| Patients Enrolled |
534 male patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (25.9-29.6 GBq) (800 mCi [700-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 524 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Creatinine |
3.93%
|
|||
| Patients Enrolled |
439 female patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (22.2-29.6 GBq) (800 mCi [600-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 525 Reporting the Activity Data of This PDC | [14] | ||||
| Indication | Metastatic/Advanced neuroendocrine tumour | ||||
| Efficacy Data | Complete response (CR) |
3%
|
|||
| Patients Enrolled |
468 patients with metastatic/advanced neuroendocrine tumor.
|
||||
| Administration Time | At least two cycles at 10-12 weeks interval | ||||
| Administration Dosage | 5.55 to 7.4 GBq per patient | ||||
| Description |
On RECIST 1.1 criteria, CR was observed in 14 patients (3%), PR in 126 patients (27%), SD in 282 patients (60%) and PD in 46 patients (10%).
|
||||
| Experiment 526 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | High-grade (WHO G3) neuroendocrine tumour | ||||
| Efficacy Data | Complete response (CR) |
3.10%
|
|||
| Patients Enrolled |
Patients with neuroendocrine tumours including high-grade (WHO G3) neuroendocrine tumours.
|
||||
| Evaluation Method | RECIST criteria assay | ||||
| Description |
Among 160 patients whose responses to treatment could be evaluated according to the RECIST criteria, 28.1% (n=45) had a progressive disease, 21.9% (n=35) had a stable disease, 46.9% (n=75) had a partial response and 3.1% (n=5) had a complete response.
|
||||
| Experiment 527 Reporting the Activity Data of This PDC | [14] | ||||
| Indication | Metastatic/Advanced neuroendocrine tumour | ||||
| Efficacy Data | Complete response (CR) |
6%
|
|||
| Patients Enrolled |
468 patients with metastatic/advanced neuroendocrine tumor.
|
||||
| Administration Time | At least two cycles at 10-12 weeks interval | ||||
| Administration Dosage | 5.55 to 7.4 GBq per patient | ||||
| Evaluation Method | PERCIST criteria assay | ||||
| Description |
Molecular imaging response (by PERCIST criteria) showed CR in 29 (6%), PR in 116 (25%), SD in 267 (57%) and PD in 56 (12%) patients.
|
||||
| Experiment 528 Reporting the Activity Data of This PDC | [14] | ||||
| Indication | Metastatic/Advanced neuroendocrine tumour | ||||
| Efficacy Data | Complete response (CR) |
12%
|
|||
| Patients Enrolled |
468 patients with metastatic/advanced neuroendocrine tumor.
|
||||
| Administration Time | At least two cycles at 10-12 weeks interval | ||||
| Administration Dosage | 5.55 to 7.4 GBq per patient | ||||
| Description |
Biochemical response evaluation showed CR in 52 (12%), PR in 172 (40%), SD in 161 (38%), and PD in 42 patients (10%).
|
||||
| Experiment 529 Reporting the Activity Data of This PDC | [40] | ||||
| Indication | High-grade gliomas | ||||
| Efficacy Data | Complete response (CR) |
12.50%
|
|||
| Patients Enrolled |
16 patients with high-grade gliomas (10 males and 6 females).
|
||||
| Administration Time | 1 to 4 cycles | ||||
| Administration Dosage | 3.7 to 29.6 GBq (median, 10.45 GBq) | ||||
| Evaluation Method | MRI assay | ||||
| Description |
In total, 16 subjects (10 males and 6 females) with a mean age of 55.68 ± 13.17 years (26-73 years) participated in the study. Of them, 8 patients were new HGG cases, and 8 patients had recurrent tumors. The participants' responses to treatments were complete remission in 12.5% of (n = 2), partial remission in 31.25% (n = 5), disease stability in 18.7% (n = 3), and disease progression in 37.5% (n = 6). In total, pretreatment and posttreatment Karnofsky Performance Score and Eastern Cooperative Oncology Group scores did not improved (P > 0.05). The patients were followed up from 1 month to 26 months (median, 3 months).
Click to Show/Hide
|
||||
| Experiment 530 Reporting the Activity Data of This PDC | [14] | ||||
| Indication | Metastatic/Advanced neuroendocrine tumour | ||||
| Efficacy Data | Complete response (CR) |
45.70%
|
|||
| Patients Enrolled |
468 patients with metastatic/advanced neuroendocrine tumor.
|
||||
| Administration Time | At least two cycles at 10-12 weeks interval | ||||
| Administration Dosage | 5.55 to 7.4 GBq per patient | ||||
| Description |
Long-term outcome (follow-up ranging from 4 to 97.6 months; median period:46 months following first 177Lu-DOTATATE PRRT) results showed, (i) on symptomatic response evaluation scale, complete response (CR) in 214 patients (45.7%), partial response (PR) in 108 (23.1%), stable disease (SD) in 118 (25.2%), progressive disease (PD) in 28 (6%).
Click to Show/Hide
|
||||
| Experiment 531 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Metastatic midgut neuroendocrine tumor | ||||
| Efficacy Data | Coagulation factors X decrease rate |
81%
|
|||
| Administration Dosage | 7.4 GBq | ||||
| Description |
Initial laboratory tests showed normocytic regenerative anemia (hemoglobin 85g/dL), thrombocytopenia (54 109/L), no signs of hemolysis, no schistocytes, prolonged prothrombin time (25.8seconds, 35%) and prolonged activated partial thromboplastin time (1.63), low fibrinogen (0.3g/L), elevated fibrin monomer (>400ug/mL) and elevated fibrin degradation products (>20ug/mL). Coagulation factors II (49%) and V (44%) were low; factors VII (97%) and X (81%) were normal. Liver function tests were normal with the exception of moderate elevation of gamma Glutamyl Transferase (γGT=100UI/L, N<38UI/L). C-reactive protein was <5mg/L. There was no sign of tumor lysis syndrome. The bone marrow aspirate and medullar karyotype were normal. Computed tomography showed stable metastatic disease, including a large stable necrotic liver metastasis.
Click to Show/Hide
|
||||
| In Vivo Model | 55-year-old woman with metastatic midgut neuroendocrine tumor. | ||||
| Experiment 532 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Metastatic midgut neuroendocrine tumor | ||||
| Efficacy Data | Coagulation factors VII decrease rate |
97%
|
|||
| Administration Dosage | 7.4 GBq | ||||
| Description |
Initial laboratory tests showed normocytic regenerative anemia (hemoglobin 85g/dL), thrombocytopenia (54 109/L), no signs of hemolysis, no schistocytes, prolonged prothrombin time (25.8seconds, 35%) and prolonged activated partial thromboplastin time (1.63), low fibrinogen (0.3g/L), elevated fibrin monomer (>400ug/mL) and elevated fibrin degradation products (>20ug/mL). Coagulation factors II (49%) and V (44%) were low; factors VII (97%) and X (81%) were normal. Liver function tests were normal with the exception of moderate elevation of gamma Glutamyl Transferase (γGT=100UI/L, N<38UI/L). C-reactive protein was <5mg/L. There was no sign of tumor lysis syndrome. The bone marrow aspirate and medullar karyotype were normal. Computed tomography showed stable metastatic disease, including a large stable necrotic liver metastasis.
Click to Show/Hide
|
||||
| In Vivo Model | 55-year-old woman with metastatic midgut neuroendocrine tumor. | ||||
| Experiment 533 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Metastatic midgut neuroendocrine tumor | ||||
| Efficacy Data | Coagulation factors V decrease rate |
44%
|
|||
| Administration Dosage | 7.4 GBq | ||||
| Description |
Initial laboratory tests showed normocytic regenerative anemia (hemoglobin 85g/dL), thrombocytopenia (54 109/L), no signs of hemolysis, no schistocytes, prolonged prothrombin time (25.8seconds, 35%) and prolonged activated partial thromboplastin time (1.63), low fibrinogen (0.3g/L), elevated fibrin monomer (>400ug/mL) and elevated fibrin degradation products (>20ug/mL). Coagulation factors II (49%) and V (44%) were low; factors VII (97%) and X (81%) were normal. Liver function tests were normal with the exception of moderate elevation of gamma Glutamyl Transferase (γGT=100UI/L, N<38UI/L). C-reactive protein was <5mg/L. There was no sign of tumor lysis syndrome. The bone marrow aspirate and medullar karyotype were normal. Computed tomography showed stable metastatic disease, including a large stable necrotic liver metastasis.
Click to Show/Hide
|
||||
| In Vivo Model | 55-year-old woman with metastatic midgut neuroendocrine tumor. | ||||
| Experiment 534 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Metastatic midgut neuroendocrine tumor | ||||
| Efficacy Data | Coagulation factors II decrease rate |
49%
|
|||
| Administration Dosage | 7.4 GBq | ||||
| Description |
Initial laboratory tests showed normocytic regenerative anemia (hemoglobin 85g/dL), thrombocytopenia (54 109/L), no signs of hemolysis, no schistocytes, prolonged prothrombin time (25.8seconds, 35%) and prolonged activated partial thromboplastin time (1.63), low fibrinogen (0.3g/L), elevated fibrin monomer (>400ug/mL) and elevated fibrin degradation products (>20ug/mL). Coagulation factors II (49%) and V (44%) were low; factors VII (97%) and X (81%) were normal. Liver function tests were normal with the exception of moderate elevation of gamma Glutamyl Transferase (γGT=100UI/L, N<38UI/L). C-reactive protein was <5mg/L. There was no sign of tumor lysis syndrome. The bone marrow aspirate and medullar karyotype were normal. Computed tomography showed stable metastatic disease, including a large stable necrotic liver metastasis.
Click to Show/Hide
|
||||
| In Vivo Model | 55-year-old woman with metastatic midgut neuroendocrine tumor. | ||||
| Experiment 535 Reporting the Activity Data of This PDC | [19] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumour | ||||
| Efficacy Data | CgA levels (95-1000 ng/mL) |
66.70%
|
|||
| Patients Enrolled |
3 patients with GEP neuroendocrine tumour.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | 7.4 GBq (200mCi) | ||||
| MOA of PDC |
From our initial experience, Lutathera has been well tolerated in patients with GEP-NET. Additional studies are needed to examine long-term clinical and patient-reported outcomes associated with treatment of GEP-NETs as well as financial considerations for hospitals embarking on a PRRT program. A multidisciplinary team and complete collaboration between hospital administration and clinical teams are required for successful implementation of a PRRT program.
Click to Show/Hide
|
||||
| Description |
CgA is a marker used in the diagnosis of NETs. Pre-treatment CgA levels were measured in 25 patients. Of the 25 patients, 6 (24.0%), 13 (52.0%), and 6 (24.0%) patients had pretreatment CgA blood level less than 95ng/mL, between 95 and 1000ng/mL, and greater than 1000ng/mL, respectively. After all 4 PRRT cycles were completed; CgA levels were measured in 3 patients. Two (66.7%) patients had post-treatment CgA between 95 and 1000 ng/mL while 1 (33.3%) patient had levels above 1000 ng/mL.
Click to Show/Hide
|
||||
| Experiment 536 Reporting the Activity Data of This PDC | [19] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumour | ||||
| Efficacy Data | CgA levels (≥1000 ng/mL) |
33.30%
|
|||
| Patients Enrolled |
3 patients with GEP neuroendocrine tumour.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | 7.4 GBq (200mCi) | ||||
| MOA of PDC |
From our initial experience, Lutathera has been well tolerated in patients with GEP-NET. Additional studies are needed to examine long-term clinical and patient-reported outcomes associated with treatment of GEP-NETs as well as financial considerations for hospitals embarking on a PRRT program. A multidisciplinary team and complete collaboration between hospital administration and clinical teams are required for successful implementation of a PRRT program.
Click to Show/Hide
|
||||
| Description |
CgA is a marker used in the diagnosis of NETs. Pre-treatment CgA levels were measured in 25 patients. Of the 25 patients, 6 (24.0%), 13 (52.0%), and 6 (24.0%) patients had pretreatment CgA blood level less than 95ng/mL, between 95 and 1000ng/mL, and greater than 1000ng/mL, respectively. After all 4 PRRT cycles were completed; CgA levels were measured in 3 patients. Two (66.7%) patients had post-treatment CgA between 95 and 1000 ng/mL while 1 (33.3%) patient had levels above 1000 ng/mL.
Click to Show/Hide
|
||||
| Experiment 537 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Metastatic midgut neuroendocrine tumor | ||||
| Efficacy Data | C-reactive protein level | < 5 mg/L | |||
| Administration Dosage | 7.4 GBq | ||||
| Description |
Initial laboratory tests showed normocytic regenerative anemia (hemoglobin 85g/dL), thrombocytopenia (54 109/L), no signs of hemolysis, no schistocytes, prolonged prothrombin time (25.8seconds, 35%) and prolonged activated partial thromboplastin time (1.63), low fibrinogen (0.3g/L), elevated fibrin monomer (>400ug/mL) and elevated fibrin degradation products (>20ug/mL). Coagulation factors II (49%) and V (44%) were low; factors VII (97%) and X (81%) were normal. Liver function tests were normal with the exception of moderate elevation of gamma Glutamyl Transferase (γGT=100UI/L, N<38UI/L). C-reactive protein was <5mg/L. There was no sign of tumor lysis syndrome. The bone marrow aspirate and medullar karyotype were normal. Computed tomography showed stable metastatic disease, including a large stable necrotic liver metastasis.
Click to Show/Hide
|
||||
| In Vivo Model | 55-year-old woman with metastatic midgut neuroendocrine tumor. | ||||
| Experiment 538 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Bone marrow, liver or renal toxicity rate |
60.86%
|
|||
| Patients Enrolled |
534 male patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (25.9-29.6 GBq) (800 mCi [700-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 539 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Bone marrow, liver or renal toxicity rate |
65.38%
|
|||
| Patients Enrolled |
439 female patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (22.2-29.6 GBq) (800 mCi [600-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 540 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Bone marrow toxicity |
56.01%
|
|||
| Patients Enrolled |
534 male patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (25.9-29.6 GBq) (800 mCi [700-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 541 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Bone marrow toxicity |
58.77%
|
|||
| Patients Enrolled |
439 female patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (22.2-29.6 GBq) (800 mCi [600-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 542 Reporting the Activity Data of This PDC | [31] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumour | ||||
| Efficacy Data | Bone marrow failure rate |
1.10%
|
|||
| Patients Enrolled |
Patients with gastroenteropancreatic neuroendocrine tumors.
|
||||
| Description |
8 patients (2.9%) developed a hematopoietic neoplasm (4 MDS, 1 AML, 1 MPN, and 2 MDS/MPN) and 3 patients (1.1%) developed BM failure characterized by cytopenia and BM aplasia.
|
||||
| Experiment 543 Reporting the Activity Data of This PDC | [50] | ||||
| Indication | Differentiated thyroid cancer | ||||
| Efficacy Data | Biochemical response rate |
25.30%
|
|||
| Patients Enrolled |
157 patients with RR-DTC treated with PPRT.
|
||||
| MOA of PDC |
Findings of the study revealed that in the absence of the established treatment for the patients with RR-DTC and metastatic MTC, PRRT could be effective with few adverse events.
|
||||
| Description |
Among 2284 related papers, 41 papers met the inclusion criteria. A total of 157 patients with RR-DTC were treated with PPRT. Biochemical and objective responses (partial and complete) were observed in 25.3 and 10.5% of patients, respectively. Among 220 patients with metastatic MTC, biochemical and objective responses were observed in 37.2 and 10.6% of the patients, respectively. Forty-six deaths were reported in 95 patients with advanced RR-DTC. In addition, 63 deaths were observed in 144 patients with metastatic MTC. Major side effects were reported in 124 patients treated with 90Y -based agent. In the patients treated with 177Lu-DOTA-TATE and 111In-Octreotide, mild and transient hematologic or renal complications were reported.
Click to Show/Hide
|
||||
| Experiment 544 Reporting the Activity Data of This PDC | [50] | ||||
| Indication | Medullary thyroid carcinoma | ||||
| Efficacy Data | Biochemical response rate |
37.20%
|
|||
| Patients Enrolled |
220 patients with metastatic medullary thyroid cancer.
|
||||
| MOA of PDC |
Findings of the study revealed that in the absence of the established treatment for the patients with RR-DTC and metastatic MTC, PRRT could be effective with few adverse events.
|
||||
| Description |
Among 2284 related papers, 41 papers met the inclusion criteria. A total of 157 patients with RR-DTC were treated with PPRT. Biochemical and objective responses (partial and complete) were observed in 25.3 and 10.5% of patients, respectively. Among 220 patients with metastatic MTC, biochemical and objective responses were observed in 37.2 and 10.6% of the patients, respectively. Forty-six deaths were reported in 95 patients with advanced RR-DTC. In addition, 63 deaths were observed in 144 patients with metastatic MTC. Major side effects were reported in 124 patients treated with 90Y -based agent. In the patients treated with 177Lu-DOTA-TATE and 111In-Octreotide, mild and transient hematologic or renal complications were reported.
Click to Show/Hide
|
||||
| Experiment 545 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Advanced paraganglioma | ||||
| Efficacy Data | Biochemical response rate |
64%
|
|||
| Patients Enrolled |
A total of 201 patients with advanced paragangliomas.
|
||||
| MOA of PDC |
The clinical applications of radiolabeled SSAs and DOTA-SSA compounds in imaging and radionuclide therapy have been rapidly increasing over the past 2 decades and have been integrated into the current management guidelines of NETs. Several studies support the safety and clinical efficacy of FDA-approved radionuclide therapies including 177Lu-DOTATATE in advanced GEP NETs and 131I-MIBG in advanced paragangliomas and pheochromocytomas. The utility of SSTR-based imaging in PRRT response assessment and surveillance of NETs remains to be established.
Click to Show/Hide
|
||||
| Description |
Over the past decade, a small number of studies have evaluated the efficacy and safety of PRRT with 177Lu-DOTA-SSA in patients with metastatic or inoperable paragangliomas [66, 67]. A recent meta-analysis of 12 studies including a total of 201 patients with advanced paragangliomas showed an objective response rate of 25%, biochemical response rate of 64%, and disease control rate of 84% after treatment with PRRT (either 90Y- or 177Lu-based agents) [67]. The following minimal treatment-related adverse effects were observed: grade 3 or 4 lymphopenia, thrombocytopenia, neutropenia, and nephrotoxicity in 11%, 9%, 3%, and 4% of the patients, respectively.
Click to Show/Hide
|
||||
| Experiment 546 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Inoperable or metastatic paragangliomas | ||||
| Efficacy Data | Best tumour response |
23%
|
|||
| Patients Enrolled |
Patients with inoperable or metastatic paragangliomas and pheochromocytomas.
|
||||
| Administration Time | Four cycles | ||||
| Administration Dosage | 7.4 Gb per cycle | ||||
| Description |
Best tumour response was partial response in 7 (23%) and stable disease in 20 (67%)
|
||||
| Experiment 547 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Inoperable or metastatic pheochromocytomas | ||||
| Efficacy Data | Best tumour response |
23%
|
|||
| Patients Enrolled |
Patients with inoperable or metastatic paragangliomas and pheochromocytomas.
|
||||
| Administration Time | Four cycles | ||||
| Administration Dosage | 7.4 Gb per cycle | ||||
| Description |
Best tumour response was partial response in 7 (23%) and stable disease in 20 (67%)
|
||||
| Experiment 548 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | SDHB positive metastatic paraganglioma | ||||
| Efficacy Data | Anti-thyroid peroxidase antibody level | > 1,000 IU/ml | |||
| Administration Time | 1 month | ||||
| Administration Dosage | 200mCi | ||||
| Description |
The patient was diagnosed with Hashimoto's thyrotoxicosis. However, the patient was asymptomatic. One month after the first dose of 200mCi 177Lu-DOTATATE, the patient noted fatigue and a 2.6 Kg weight gain. The TSH (73.04 μIU/ml), anti-TPO antibodies (>1,000 IU/ml), and anti-Tg antibodies (668 IU/ml) had substantially increased, with reductions in FT4 (0.3 ng/dl) and TT3 [54 ng/dl (87-169 ng/dl)]. Diagnostic gallium 68 - DOTATATE positron emission tomography-computed tomography performed prior to 177Lu-DOTATATE treatment revealed diffuse thyroid uptake. Post-therapy single-photon emission computed tomography also revealed diffuse uptake of 177Lu-DOTATATE in the thyroid gland. Levothyroxine therapy was initiated, and the patient's symptoms resolved.
Click to Show/Hide
|
||||
| In Vivo Model | A 29-year-old male with SDHB positive metastatic paraganglioma. | ||||
| Related Clinical Trial | |||||
| NCT Number | NCT03206060 | Clinical Status | Phase 2 | ||
| Clinical Description | Background:Pheochromocytoma and paraganglioma are rare tumors. They usually form inside and near the adrenal gland or in the neck region. Not all these tumors can be removed with surgery, and there are no good treatments if the disease has spread. Researchers think a new drug may be able to help.Objective:To learn the safety and tolerability of Lu-177-DOTATATE. Also, to see if it improves the length of time it takes for the cancer to return.Eligibility:Adults who have an inoperable tumor of the study cancer that can be detected with Ga-68-DOTATATE PET/CT imagingDesign:Participants will be screened with a medical history, physical exam, and blood tests.Eligible participants will be admitted to the NIH Clinical Center.Participants will get the study drug in an intravenous infusion. They will get 4 doses, given about 8 weeks apart.Between 4 and 24 hours after each study drug dose, participants will have scans taken. They will lie on their back on a scanner table.Participants will have vital signs taken. They will give blood and urine samples.During the study, participants will have other scans taken. Some scans will use a radioactive tracer.Participants will complete quality of life questionnaires.Participants will be contacted by phone 1-3 days after they leave the Clinical Center. They will then be followed every 3 to 6 months for 3 years or until their disease gets worse. | ||||
| Experiment 549 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | SDHB positive metastatic paraganglioma | ||||
| Efficacy Data | Anti-thyroglobulin antibody level |
668 IU/ml
|
|||
| Administration Time | 1 month | ||||
| Administration Dosage | 200mCi | ||||
| Description |
The patient was diagnosed with Hashimoto's thyrotoxicosis. However, the patient was asymptomatic. One month after the first dose of 200mCi 177Lu-DOTATATE, the patient noted fatigue and a 2.6 Kg weight gain. The TSH (73.04 μIU/ml), anti-TPO antibodies (>1,000 IU/ml), and anti-Tg antibodies (668 IU/ml) had substantially increased, with reductions in FT4 (0.3 ng/dl) and TT3 [54 ng/dl (87-169 ng/dl)]. Diagnostic gallium 68 - DOTATATE positron emission tomography-computed tomography performed prior to 177Lu-DOTATATE treatment revealed diffuse thyroid uptake. Post-therapy single-photon emission computed tomography also revealed diffuse uptake of 177Lu-DOTATATE in the thyroid gland. Levothyroxine therapy was initiated, and the patient's symptoms resolved.
Click to Show/Hide
|
||||
| In Vivo Model | A 29-year-old male with SDHB positive metastatic paraganglioma. | ||||
| Related Clinical Trial | |||||
| NCT Number | NCT03206060 | Clinical Status | Phase 2 | ||
| Clinical Description | Background:Pheochromocytoma and paraganglioma are rare tumors. They usually form inside and near the adrenal gland or in the neck region. Not all these tumors can be removed with surgery, and there are no good treatments if the disease has spread. Researchers think a new drug may be able to help.Objective:To learn the safety and tolerability of Lu-177-DOTATATE. Also, to see if it improves the length of time it takes for the cancer to return.Eligibility:Adults who have an inoperable tumor of the study cancer that can be detected with Ga-68-DOTATATE PET/CT imagingDesign:Participants will be screened with a medical history, physical exam, and blood tests.Eligible participants will be admitted to the NIH Clinical Center.Participants will get the study drug in an intravenous infusion. They will get 4 doses, given about 8 weeks apart.Between 4 and 24 hours after each study drug dose, participants will have scans taken. They will lie on their back on a scanner table.Participants will have vital signs taken. They will give blood and urine samples.During the study, participants will have other scans taken. Some scans will use a radioactive tracer.Participants will complete quality of life questionnaires.Participants will be contacted by phone 1-3 days after they leave the Clinical Center. They will then be followed every 3 to 6 months for 3 years or until their disease gets worse. | ||||
| Experiment 550 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Anemia |
6.67%
|
|||
| Patients Enrolled |
30 patients with pheocromocytomas (PCCs) and paragangliomas (PGLs) (3 PCC, 27PGL).
|
||||
| Administration Time | 73% of patients received 4 cycles | ||||
| Administration Dosage | 7.4 GBq/cycle | ||||
| MOA of PDC |
We showed that PRRT based on 177Lu-DOTATATE and 90-YDOTATOC without radiosensitizers are efficacious therapeutic options (DCR of 83% and 76%, respectively) and can be considered in the multidisciplinary treatment of PCCs and PGLs as alternatives to I-131 MIBG and chemotherapy.
|
||||
| Description |
In detail, we report pooled data on 149 177Lu- and 64 90Y-treated patients (total: 213 patients). Most studies were retrospective. The largest one included 46 patients (34 treated with 177Lu-, 12 with 90Y- PRRTs). Median ages ranged from 32.5 to 60.4 years. The female gender was slightly predominant (83 women, 74 men, gender not specified in 3 studies). When reported, mutations of SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B) were the most frequent genetic alterations. According to the predominant retrospective and real practice nature of the selected studies, sites of metastases and previous treatments were heterogeneous, with bone the metastatic site most often involved, and surgery the most frequent previous therapy. Paramount descriptive information on PRRT doses per cycle, number of cycles, response criteria, best responses to treatments, and frequency of G3/G4 hematologic toxicities is reported in Table 2. The lowest applied dose was 3.4 GBq per cycle; the number of administered cycles was heterogeneous (at least 3 cycles in 7 studies). The most used response criteria were the RECIST 1.1. Only one study evaluated the response with SWOG criteria, which are more conservative with regard to the definition of PR (50% or greater reduction in the sum of the product of the perpendicular diameters of target lesions). The majority of the patients experienced PR or SD. Hematologic toxicity was manageable. Funnel plot of selected studies did not show publication bias. The MINORS and Newcastle-Ottawa Scale scoring for the selected articles are reported in Table 3.
Click to Show/Hide
|
||||
| Experiment 551 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Alkaline phosphatase grade ≥3 |
2.31%
|
|||
| Patients Enrolled |
534 male patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (25.9-29.6 GBq) (800 mCi [700-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 552 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Alkaline phosphatase grade ≥3 |
3.54%
|
|||
| Patients Enrolled |
439 female patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (22.2-29.6 GBq) (800 mCi [600-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 553 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Albumin grade ≥3 |
0.19%
|
|||
| Patients Enrolled |
534 male patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (25.9-29.6 GBq) (800 mCi [700-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 554 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Albumin grade ≥3 |
0.23%
|
|||
| Patients Enrolled |
439 female patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (22.2-29.6 GBq) (800 mCi [600-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 555 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Alanine aminotransferase grade ≥3 |
1.50%
|
|||
| Patients Enrolled |
534 male patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (25.9-29.6 GBq) (800 mCi [700-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 556 Reporting the Activity Data of This PDC | [33] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Alanine aminotransferase grade ≥3 |
3.02%
|
|||
| Patients Enrolled |
439 female patients with neuroendocrine tumour.
|
||||
| Administration Time | Median of 4 cycles | ||||
| Administration Dosage | Cumulative administered activity 29.6 GBq (22.2-29.6 GBq) (800 mCi [600-800 mCi]) | ||||
| MOA of PDC |
Subacute PRRT-related toxicities are commonly detected and can lead to dose adjustments, treatment delay, and treatment discontinuation in a limited number of patients. Severe platelet and hemoglobin toxicities were more frequently observed in female than male NET patients, without evidently leading to a lower cumulative 177Lu-DOTATATE activity. Future research should focus on the etiology and on the impact on the treatment efficacy and long-term safety of PRRT between sexes.
Click to Show/Hide
|
||||
| Description |
Grade 2, 3, or 4 thrombocytopenia occurred more often in women (25%) than in men (18%, P = 0.004), which was driven by a significant increase in grade ≥3 thrombocytopenia in women (11% vs 6%, P = 0.008). Grade ≥3 anemia was likewise more prevalent in women (7%) than in men (3%, P = 0.002). A ≥ 25% decrease from the baseline hemoglobin level occurred in 55 women (13%) and 38 men (7%) (P = 0.004). Of the patients with grade ≥3 anemia, 21 (66%) female and 10 (63%) male patients also developed grade ≥2 thrombocytopenia (P = 0.831). Grade ≥3 pancytopenia (thrombocytopenia, leukopenia, and anemia) occurred in 1% of both sexes. Of all study patients, 52 female patients (12%) and 47 male patients (9%) received a cumulative activity of <27.8 GBq (750 mCi) as a result of a bone marrow DLT (P = 0.122).
Click to Show/Hide
|
||||
| Experiment 557 Reporting the Activity Data of This PDC | [58] | ||||
| Indication | Small bowel neuroendocrine tumor | ||||
| Efficacy Data | 5-years overall survival overall survival (OS) |
94.10%
|
|||
| Patients Enrolled |
Patients with consecutive patients with small bowel neuroendocrine tumour.
|
||||
| Administration Time | Every eight to 12 weeks | ||||
| Administration Dosage | 7.4 GBq (200 mCi) | ||||
| Evaluation Method | 68Ga-DOTATATE PET/CT assay | ||||
| MOA of PDC |
Surgery combined with 177Lu PRRT is safe and provides favourable PFS and OS in selected patients with advanced SBNEN. Liquid biopsy (NETest) has the potential to accurately delineate disease status.
|
||||
| Description |
A surgical cohort of 39 SBNEN patients met eligibility criteria. Thirty-two patients underwent ileal resection and 7 right hemicolectomy. The mean number of 177Lu PRRT cycles was 4. Mortality was nil. Surgical morbidity was 10.3%. Transient grade 1/2 toxicity occurred in 41% (PRRT). NETest scores (n=9 patients) decreased in 100% following treatment and correlated with diminished tumour volume and disease stabilization following surgery and PRRT. Median follow-up: 78 months. Median PFS and OS: 42.7 and 110 months, respectively. Progression-free survival at 1-, 3-, and 5-years was 79.4%, 57.1% and 40.5%, respectively. Overall survival at 1-, 3-, and 5-years was 97.4%, 97.4%, and 94.1%, respectively
Click to Show/Hide
|
||||
| Experiment 558 Reporting the Activity Data of This PDC | [64] | ||||
| Indication | Progressive, well-differentiated neuroendocrine tumor | ||||
| Efficacy Data | 5-hydroxyindolacetic acid decrease rate (≥30%) |
56.00%
|
|||
| Patients Enrolled |
22 patients with a metastatic midgut neuroendocrine tumour.
|
||||
| Administration Dosage | Intended cumulative dose: 29.6 GBq | ||||
| MOA of PDC |
PRRT with 177Lu-DOTATATE effectively reduced diarrhea and flushing in patients with carcinoid syndrome and can be considered for symptomatic treatment of carcinoid syndrome insufficiently controlled with somatostatin analogs.
|
||||
| Description |
After PRRT, mean bowel movement frequency (BMF) decreased from 6.1 ± 3.4 to 4.6 ± 3.6 per day (P = 0.009). Flushes decreased from 4.3 ± 2.9 to 2.4 ± 2.7 flushes per day (P = 0.002). A decrease of BMF of more than 30% occurred in 47% of patients with baseline BMF of 4 or more (n = 17). In patients with ≥2 episodes of flushing a day (n = 15), 67% of patients had more than 50% decrease of daily flushing. A decrease in urinary 5-hydroxyindolacetic acid excretion of more than 30% was seen in 56% of patients. The European Organization for Research and Treatment of Cancer-Core Module diarrhea subscale score showed a trend toward improvement by an average of 16.7 ± 33.3 points (P = 0.11).
Click to Show/Hide
|
||||
| Experiment 559 Reporting the Activity Data of This PDC | [60] | ||||
| Indication | Metastatic/Advanced pulmonary neuroendocrine tumor | ||||
| Efficacy Data | 4-year objective response rate (ORR) |
38.50%
|
|||
| Patients Enrolled |
Patients treated with Lu-DOTATATE.
|
||||
| Administration Time | 12-16 weeks' intervals [range, 1-5 cycles; average, 4 cycles] | ||||
| Administration Dosage | 150 mCi [5.55 GBq]/cycle | ||||
| Description |
The observed annual OS rates were as follows: 1 year: 94.7% (95% CI, 68.1%-99.2%), 2 years: 66% (95% CI, 35.5%-84.5%), 3 years: 57.7% (95% CI, 28-78.9%), and 4 years: 38.5% (95% CI, 8.1%-69.5%); median OS was 40 months with 39% (95% CI, 13.1%-64.8%).
|
||||
| Experiment 560 Reporting the Activity Data of This PDC | [58] | ||||
| Indication | Small bowel neuroendocrine tumor | ||||
| Efficacy Data | 3-years overall survival (OS) |
97.40%
|
|||
| Patients Enrolled |
Patients with consecutive patients with small bowel neuroendocrine tumour.
|
||||
| Administration Time | Every eight to 12 weeks | ||||
| Administration Dosage | 7.4 GBq (200 mCi) | ||||
| Evaluation Method | 68Ga-DOTATATE PET/CT assay | ||||
| MOA of PDC |
Surgery combined with 177Lu PRRT is safe and provides favourable PFS and OS in selected patients with advanced SBNEN. Liquid biopsy (NETest) has the potential to accurately delineate disease status.
|
||||
| Description |
A surgical cohort of 39 SBNEN patients met eligibility criteria. Thirty-two patients underwent ileal resection and 7 right hemicolectomy. The mean number of 177Lu PRRT cycles was 4. Mortality was nil. Surgical morbidity was 10.3%. Transient grade 1/2 toxicity occurred in 41% (PRRT). NETest scores (n=9 patients) decreased in 100% following treatment and correlated with diminished tumour volume and disease stabilization following surgery and PRRT. Median follow-up: 78 months. Median PFS and OS: 42.7 and 110 months, respectively. Progression-free survival at 1-, 3-, and 5-years was 79.4%, 57.1% and 40.5%, respectively. Overall survival at 1-, 3-, and 5-years was 97.4%, 97.4%, and 94.1%, respectively
Click to Show/Hide
|
||||
| Experiment 561 Reporting the Activity Data of This PDC | [60] | ||||
| Indication | Metastatic/Advanced pulmonary neuroendocrine tumor | ||||
| Efficacy Data | 3-year objective response rate (ORR) |
57.70%
|
|||
| Patients Enrolled |
Patients treated with Lu-DOTATATE.
|
||||
| Administration Time | 12-16 weeks' intervals [range, 1-5 cycles; average, 4 cycles] | ||||
| Administration Dosage | 150 mCi [5.55 GBq]/cycle | ||||
| Description |
The observed annual OS rates were as follows: 1 year: 94.7% (95% CI, 68.1%-99.2%), 2 years: 66% (95% CI, 35.5%-84.5%), 3 years: 57.7% (95% CI, 28-78.9%), and 4 years: 38.5% (95% CI, 8.1%-69.5%); median OS was 40 months with 39% (95% CI, 13.1%-64.8%).
|
||||
| Experiment 562 Reporting the Activity Data of This PDC | [60] | ||||
| Indication | Metastatic/Advanced pulmonary neuroendocrine tumor | ||||
| Efficacy Data | 2-year objective response rate (ORR) |
66.00%
|
|||
| Patients Enrolled |
Patients treated with Lu-DOTATATE.
|
||||
| Administration Time | 12-16 weeks' intervals [range, 1-5 cycles; average, 4 cycles] | ||||
| Administration Dosage | 150 mCi [5.55 GBq]/cycle | ||||
| Description |
The observed annual OS rates were as follows: 1 year: 94.7% (95% CI, 68.1%-99.2%), 2 years: 66% (95% CI, 35.5%-84.5%), 3 years: 57.7% (95% CI, 28-78.9%), and 4 years: 38.5% (95% CI, 8.1%-69.5%); median OS was 40 months with 39% (95% CI, 13.1%-64.8%).
|
||||
| Experiment 563 Reporting the Activity Data of This PDC | [58] | ||||
| Indication | Small bowel neuroendocrine tumor | ||||
| Efficacy Data | 1-year overall survival (OS) |
97.40%
|
|||
| Patients Enrolled |
Patients with consecutive patients with small bowel neuroendocrine tumour.
|
||||
| Administration Time | Every eight to 12 weeks | ||||
| Administration Dosage | 7.4 GBq (200 mCi) | ||||
| Evaluation Method | 68Ga-DOTATATE PET/CT assay | ||||
| MOA of PDC |
Surgery combined with 177Lu PRRT is safe and provides favourable PFS and OS in selected patients with advanced SBNEN. Liquid biopsy (NETest) has the potential to accurately delineate disease status.
|
||||
| Description |
A surgical cohort of 39 SBNEN patients met eligibility criteria. Thirty-two patients underwent ileal resection and 7 right hemicolectomy. The mean number of 177Lu PRRT cycles was 4. Mortality was nil. Surgical morbidity was 10.3%. Transient grade 1/2 toxicity occurred in 41% (PRRT). NETest scores (n=9 patients) decreased in 100% following treatment and correlated with diminished tumour volume and disease stabilization following surgery and PRRT. Median follow-up: 78 months. Median PFS and OS: 42.7 and 110 months, respectively. Progression-free survival at 1-, 3-, and 5-years was 79.4%, 57.1% and 40.5%, respectively. Overall survival at 1-, 3-, and 5-years was 97.4%, 97.4%, and 94.1%, respectively
Click to Show/Hide
|
||||
| Experiment 564 Reporting the Activity Data of This PDC | [60] | ||||
| Indication | Metastatic/Advanced pulmonary neuroendocrine tumor | ||||
| Efficacy Data | 1-year objective response rate (ORR) |
94.70%
|
|||
| Patients Enrolled |
Patients treated with Lu-DOTATATE.
|
||||
| Administration Time | 12-16 weeks' intervals [range, 1-5 cycles; average, 4 cycles] | ||||
| Administration Dosage | 150 mCi [5.55 GBq]/cycle | ||||
| Description |
The observed annual OS rates were as follows: 1 year: 94.7% (95% CI, 68.1%-99.2%), 2 years: 66% (95% CI, 35.5%-84.5%), 3 years: 57.7% (95% CI, 28-78.9%), and 4 years: 38.5% (95% CI, 8.1%-69.5%); median OS was 40 months with 39% (95% CI, 13.1%-64.8%).
|
||||
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [67] | ||||
| Indication | Pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Tumor volume |
800 mm3
|
|||
| Administration Time | 15 days | ||||
| Administration Dosage | 30 MBq/mouse | ||||
| MOA of PDC |
In conclusion, our preclinical data demonstrate that pretreatment with the HDACi CI-994 improves 177Lu-DOTATATE therapy, compared with PRRT alone in models of SSTR2-deficient NETs. Our study forms the basis for a clinical trial testing the therapeutic efficacy of HDACi CI-994 pretreatment in combination with 177Lu-DOTATATE therapy in patients with high-grade, SSTR2-negative metastatic PNETs.
Click to Show/Hide
|
||||
| Description |
Using an identical 10-day CI-994 pretreatment model, the mice that received a single intravenous administration of 30 MBq of 177Lu-DOTATATE after CI-994 pretreatment demonstrated a significant reduction in tumor growth compared with the control group (P < 0.0001) and to the group receiving standard therapy, that is, 30 MBq 177Lu-DOTATATE alone (P = 0.0028). This was confirmed 11 and 15 days after 177Lu-DOTATATE injections. And, the effects of combination therapy were additive, that is, there was no interaction effect between CI-994 and 177Lu-DOTATATE. The clear difference in tumor growth after 5 days of 177Lu-DOTATATE therapy between the two CI-994-pretreated groups signaled a strong response to 177Lu-DOTATATE. Tumor growth was not slowed in the 177Lu-DOTATATE-only treatment group, potentially due to SSTR2 deficiency. Notably, the combined CI-994 and 177Lu-DOTATATE treatment appeared well-tolerated, with limited toxicity as evidenced by some changes in mouse body weight as observed in other studies, with no visible toxicity signs at the time of euthanasia. Tumors of mice treated with combined CI-994 and 177Lu-DOTATATE were significantly smaller compared with tumors of mice treated 177Lu-DOTATATE alone. Furthermore, Pan H3 staining revealed increased open chromatin (red foci, Pan H3) in tumors treated with CI-994 in comparison to control tumors. And increased DNA damage (green foci, γH2AX) was observed in these CI-994-pretreated tumors compared with control after 177Lu-DOTATATE therapy. This increase in DNA damage was found to be significant (P < 0.001).
Click to Show/Hide
|
||||
| In Vivo Model | QGP-1 cells athymic nude male mice xenograft model. | ||||
| In Vitro Model | Pancreatic somatostatinoma | QGP-1 cell | CVCL_3143 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [68] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Time of tumour stasis |
12 Day
|
|||
| Administration Dosage | 20 MBq | ||||
| Evaluation Method | PET imaging studies | ||||
| Description |
Cell lines that expressed SSTR2 in vivo were implanted into nude mice and once the tumours became well-established the animals were injected intravenously with 20 MBq LuTate and the tumour response evaluated. Most tumour models showed similar robust growth kinetics but their response to LuTate varied widely. LuTate treatment of the H1299-7 and AR42J models induced tumour stasis for sixteen and twelve days post dosing, respectively, after which tumour growth rapidly resumed. In contrast, the D341 model, which showed similar SSTR2 expression and GaTate uptake to that of the AR42J model, was highly sensitive to LuTate with complete tumour regression observed for 65 days. The SK-N-BE(2) and BON tumour models which demonstrated low SSTR2 expression and GaTate binding were highly resistant to LuTate treatment.
Click to Show/Hide
|
||||
| In Vivo Model | Balb/c nude mice H1299-7 cells xenograft model. | ||||
| In Vitro Model | Digestive system neoplasms | AR42J cell | CVCL_0143 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [68] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Time of tumour stasis |
16 Day
|
|||
| Administration Dosage | 20 MBq | ||||
| Evaluation Method | PET imaging studies | ||||
| Description |
Cell lines that expressed SSTR2 in vivo were implanted into nude mice and once the tumours became well-established the animals were injected intravenously with 20 MBq LuTate and the tumour response evaluated. Most tumour models showed similar robust growth kinetics but their response to LuTate varied widely. LuTate treatment of the H1299-7 and AR42J models induced tumour stasis for sixteen and twelve days post dosing, respectively, after which tumour growth rapidly resumed. In contrast, the D341 model, which showed similar SSTR2 expression and GaTate uptake to that of the AR42J model, was highly sensitive to LuTate with complete tumour regression observed for 65 days. The SK-N-BE(2) and BON tumour models which demonstrated low SSTR2 expression and GaTate binding were highly resistant to LuTate treatment.
Click to Show/Hide
|
||||
| In Vivo Model | Balb/c nude mice H1299-7 cells xenograft model. | ||||
| In Vitro Model | Lung large cell carcinoma | H1299-7 cell | CVCL_0060 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [68] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Time of tumour stasis |
65 Day
|
|||
| Administration Dosage | 20 MBq | ||||
| Evaluation Method | PET imaging studies | ||||
| Description |
Cell lines that expressed SSTR2 in vivo were implanted into nude mice and once the tumours became well-established the animals were injected intravenously with 20 MBq LuTate and the tumour response evaluated. Most tumour models showed similar robust growth kinetics but their response to LuTate varied widely. LuTate treatment of the H1299-7 and AR42J models induced tumour stasis for sixteen and twelve days post dosing, respectively, after which tumour growth rapidly resumed. In contrast, the D341 model, which showed similar SSTR2 expression and GaTate uptake to that of the AR42J model, was highly sensitive to LuTate with complete tumour regression observed for 65 days. The SK-N-BE(2) and BON tumour models which demonstrated low SSTR2 expression and GaTate binding were highly resistant to LuTate treatment.
Click to Show/Hide
|
||||
| In Vivo Model | Balb/c nude mice H1299-7 cells xenograft model. | ||||
| In Vitro Model | Medulloblastoma | D341 cell | CVCL_0018 | ||
Copper dotatate Cu-64 [Approved]
Identified from the Human Clinical Data
| Experiment 1 Reporting the Activity Data of This PDC | [47] | ||||
| Indication | Neuroendocrine neoplasms | ||||
| Efficacy Data | PET75% overall specificities |
92%
|
|||
| Patients Enrolled |
38 patients with NEN.
|
||||
| Administration Dosage | 142 MBq | ||||
| Evaluation Method | PET/CT | ||||
| MOA of PDC |
Somatostatin analogues radiolabeled with the radioisotope 68Ga, e.g. [68Ga]Ga-DOTATATE and [68Ga]Ga-DOTATOC, are currently the predominately used tracers for PET imaging of patients with NEN. However, the 64Cu-labeled SSR targeting somatostatin analogue [64Cu]Cu-DOTATATE is emerging as an alternative to 68Ga-labeled radiotracers. Although 64Cu has a lower branching ratio for + decay than 68Ga (18% vs 89%), 64Cu has the advantage of a longer half-life (12.7 h vs 68 min) and shorter positron range (0.7 mm vs 3.5 mm) where the latter at least in theory should lead to better spatial resolution. The effective whole-body dose is 6.3 mSv for a 200 MBq injection of [64Cu]Cu-DOTATATE or 4.7 mSv for the United States Food and Drug Administration (FDA) recommended dose of 148 MBq.
Click to Show/Hide
|
||||
| Description |
The median [64Cu]Cu-DOTATATE activity dose could be reduced from 191 to 142 MBq without decline in diagnostic image quality (P = 0.62), diagnostic lesion confidence (P = 1.0), or number of lesions detected in major organs or regions (P = 0.19-0.71). Sensitivity and specificity for detection of liver disease were 100% (26/26 patients) and 100% (12/12), respectively, for both PET75% and PET50%. Overall sensitivity for detection of NEN was 100% (26/26) for both PET75% and PET50%, and overall specificities were 92% (11/12) and 100% (12/12) for PET75 and PET50, respectively. Following dose-blinded post hoc review, the PET75% specificity was adjusted to 100% (12/12).
Click to Show/Hide
|
||||
| Experiment 2 Reporting the Activity Data of This PDC | [47] | ||||
| Indication | Neuroendocrine neoplasms | ||||
| Efficacy Data | PET75% overall sensitivity |
100%
|
|||
| Patients Enrolled |
38 patients with NEN.
|
||||
| Administration Dosage | 142 MBq | ||||
| Evaluation Method | PET/CT | ||||
| MOA of PDC |
Somatostatin analogues radiolabeled with the radioisotope 68Ga, e.g. [68Ga]Ga-DOTATATE and [68Ga]Ga-DOTATOC, are currently the predominately used tracers for PET imaging of patients with NEN. However, the 64Cu-labeled SSR targeting somatostatin analogue [64Cu]Cu-DOTATATE is emerging as an alternative to 68Ga-labeled radiotracers. Although 64Cu has a lower branching ratio for + decay than 68Ga (18% vs 89%), 64Cu has the advantage of a longer half-life (12.7 h vs 68 min) and shorter positron range (0.7 mm vs 3.5 mm) where the latter at least in theory should lead to better spatial resolution. The effective whole-body dose is 6.3 mSv for a 200 MBq injection of [64Cu]Cu-DOTATATE or 4.7 mSv for the United States Food and Drug Administration (FDA) recommended dose of 148 MBq.
Click to Show/Hide
|
||||
| Description |
The median [64Cu]Cu-DOTATATE activity dose could be reduced from 191 to 142 MBq without decline in diagnostic image quality (P = 0.62), diagnostic lesion confidence (P = 1.0), or number of lesions detected in major organs or regions (P = 0.19-0.71). Sensitivity and specificity for detection of liver disease were 100% (26/26 patients) and 100% (12/12), respectively, for both PET75% and PET50%. Overall sensitivity for detection of NEN was 100% (26/26) for both PET75% and PET50%, and overall specificities were 92% (11/12) and 100% (12/12) for PET75 and PET50, respectively. Following dose-blinded post hoc review, the PET75% specificity was adjusted to 100% (12/12).
Click to Show/Hide
|
||||
| Experiment 3 Reporting the Activity Data of This PDC | [47] | ||||
| Indication | Neuroendocrine neoplasms | ||||
| Efficacy Data | PET50% overall specificities |
100%
|
|||
| Patients Enrolled |
38 patients with NEN.
|
||||
| Administration Dosage | 142 MBq | ||||
| Evaluation Method | PET/CT | ||||
| MOA of PDC |
Somatostatin analogues radiolabeled with the radioisotope 68Ga, e.g. [68Ga]Ga-DOTATATE and [68Ga]Ga-DOTATOC, are currently the predominately used tracers for PET imaging of patients with NEN. However, the 64Cu-labeled SSR targeting somatostatin analogue [64Cu]Cu-DOTATATE is emerging as an alternative to 68Ga-labeled radiotracers. Although 64Cu has a lower branching ratio for + decay than 68Ga (18% vs 89%), 64Cu has the advantage of a longer half-life (12.7 h vs 68 min) and shorter positron range (0.7 mm vs 3.5 mm) where the latter at least in theory should lead to better spatial resolution. The effective whole-body dose is 6.3 mSv for a 200 MBq injection of [64Cu]Cu-DOTATATE or 4.7 mSv for the United States Food and Drug Administration (FDA) recommended dose of 148 MBq.
Click to Show/Hide
|
||||
| Description |
The median [64Cu]Cu-DOTATATE activity dose could be reduced from 191 to 142 MBq without decline in diagnostic image quality (P = 0.62), diagnostic lesion confidence (P = 1.0), or number of lesions detected in major organs or regions (P = 0.19-0.71). Sensitivity and specificity for detection of liver disease were 100% (26/26 patients) and 100% (12/12), respectively, for both PET75% and PET50%. Overall sensitivity for detection of NEN was 100% (26/26) for both PET75% and PET50%, and overall specificities were 92% (11/12) and 100% (12/12) for PET75 and PET50, respectively. Following dose-blinded post hoc review, the PET75% specificity was adjusted to 100% (12/12).
Click to Show/Hide
|
||||
| Experiment 4 Reporting the Activity Data of This PDC | [47] | ||||
| Indication | Neuroendocrine neoplasms | ||||
| Efficacy Data | PET50% overall sensitivity |
100%
|
|||
| Patients Enrolled |
38 patients with NEN.
|
||||
| Administration Dosage | 142 MBq | ||||
| Evaluation Method | PET/CT | ||||
| MOA of PDC |
Somatostatin analogues radiolabeled with the radioisotope 68Ga, e.g. [68Ga]Ga-DOTATATE and [68Ga]Ga-DOTATOC, are currently the predominately used tracers for PET imaging of patients with NEN. However, the 64Cu-labeled SSR targeting somatostatin analogue [64Cu]Cu-DOTATATE is emerging as an alternative to 68Ga-labeled radiotracers. Although 64Cu has a lower branching ratio for + decay than 68Ga (18% vs 89%), 64Cu has the advantage of a longer half-life (12.7 h vs 68 min) and shorter positron range (0.7 mm vs 3.5 mm) where the latter at least in theory should lead to better spatial resolution. The effective whole-body dose is 6.3 mSv for a 200 MBq injection of [64Cu]Cu-DOTATATE or 4.7 mSv for the United States Food and Drug Administration (FDA) recommended dose of 148 MBq.
Click to Show/Hide
|
||||
| Description |
The median [64Cu]Cu-DOTATATE activity dose could be reduced from 191 to 142 MBq without decline in diagnostic image quality (P = 0.62), diagnostic lesion confidence (P = 1.0), or number of lesions detected in major organs or regions (P = 0.19-0.71). Sensitivity and specificity for detection of liver disease were 100% (26/26 patients) and 100% (12/12), respectively, for both PET75% and PET50%. Overall sensitivity for detection of NEN was 100% (26/26) for both PET75% and PET50%, and overall specificities were 92% (11/12) and 100% (12/12) for PET75 and PET50, respectively. Following dose-blinded post hoc review, the PET75% specificity was adjusted to 100% (12/12).
Click to Show/Hide
|
||||
68Ga-DOTATATE [Approved]
Identified from the Human Clinical Data
| Experiment 1 Reporting the Activity Data of This PDC | [61] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Median detection ratio |
75.30%
|
|||
| Patients Enrolled |
75 patients with histologically confirmed neuroendocrine tumours and routine clinical.
|
||||
| MOA of PDC |
18F-AlF-OC is noninferior and even superior to 68Ga-DOTATATE/NOC PET in NET patients. This validates 18F-AlF-OC as an option for clinical practice somatostatin receptor PET.
|
||||
| Description |
In total, 4,709 different tumor lesions were detected: 3,454 with 68Ga-DOTATATE/NOC and 4,278 with 18F-AlF-OC. The mean DR with 18F-AlF-OC was significantly higher than with 68Ga-DOTATATE/NOC (91.1% vs. 75.3%; P < 10-5).
|
||||
| Experiment 2 Reporting the Activity Data of This PDC | [61] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Difference in detection ratio |
11.80%
|
|||
| Patients Enrolled |
75 patients with histologically confirmed neuroendocrine tumours and routine clinical.
|
||||
| MOA of PDC |
18F-AlF-OC is noninferior and even superior to 68Ga-DOTATATE/NOC PET in NET patients. This validates 18F-AlF-OC as an option for clinical practice somatostatin receptor PET.
|
||||
| Description |
The resulting mean DDR was 15.8%, with a lower margin of the 95% CI (95% CI, 9.6%-22.0%) higher than -15%, which is the prespecified boundary for noninferiority. The mean DDRs for the 68Ga-DOTATATE and 68Ga-DOTANOC subgroups were 11.8% (95% CI, 4.3-19.3) and 27.5% (95% CI, 17.8-37.1), respectively.
|
||||
111In-DTPA-octreotide [Approved]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [69] | ||||
| Indication | Digestive system neoplasms | ||||
| Efficacy Data | Tumor cell uptake rate |
47%
|
|||
| Administration Time | 4 h | ||||
| Administration Dosage | 10 nM | ||||
| MOA of PDC |
In our previous study, d-Phe at position 1 in 111In-DTPA-d-Phe1-octreotide was replaced with Asp to minimize the change in molecular size. While the Phe3D-Trp4Lys5Thr6 sequence in octreotide is important for binding to the somatostatin receptor, the majority of octreotide derivatives with high somatostatin receptor affinity possess an N-terminal amino acid with an aromatic side chain, such as d-Phe or naphthylalanine. With these findings in mind, we designed, synthesized and evaluated not only the previously reported 111In-DTPA-Asp1-octreotide but also 111In-DTPA-Asp0-d-Phe1-octreotide, in which an Asp was incorporated between DTPA and N-terminal d-Phe in 111In-DTPA-d-Phe1-octreotide.
Click to Show/Hide
|
||||
| Description |
Figure 3 shows the results of cellular uptake experiments. When incubated with111In-DTPA-Asp1-octreotide, the uptake levels of radioactivity by AR42J cells were extremely low compared with those for111In-DTPA-d-Phe1-octreotide. In contrast, the uptake levels of111In-DTPA-Asp0-d-Phe1-octreotide increased in a time-dependent manner. The uptake levels of111In-DTPA-Asp0-d-Phe1-octreotide were not significantly different from those for111In-DTPA-d-Phe1-octreotide at 4h. For 111In-DTPA-Asp0-d-Phe1-octreotide, cell uptake experiments were conducted in the presence of varying concentrations of an unmodified octreotide. In Figure 4, uptake levels are shown as a percentage of the radioactivity level in the absence of the unmodified octreotide. The uptake levels of 111In-DTPA-Asp0-d-Phe1-octreotide were reduced in association with the concentration of the unmodified octreotide. The radioactivity uptake level was reduced by approximately 90% at 1 M concentration of the octreotide.
Click to Show/Hide
|
||||
| In Vivo Model | AR42J tumor-bearing mice model. | ||||
90Y-DOTATOC [Phase 2]
Identified from the Human Clinical Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Well-differentiated pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
7 mm reduction in tumor diameter
|
|||
| Patients Enrolled |
24 patients with morphologically WD-PanNETs who underwent PRRT preoperatively.
|
||||
| Administration Time | A median of 6 months (interquartile range [iqr] 6; 8 months) from the last cycle of prrt | ||||
| Evaluation Method | Stained with hematoxylin and eosin (H&E) & Formalin-fixed and paraffin-embedded (FFPE) assay | ||||
| Description |
In the neoadjuvant group, the average reduction in tumor diameter, before and after PRRT, assessed by computerized tomography (CT), was 7 mm. The average volume reduction showed a statistically significant correlation (p < 0.022) with the percentage of stroma after PRRT.
|
||||
| Experiment 2 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Medullary thyroid carcinoma | ||||
| Efficacy Data | Thrombocytopenia |
9%
|
|||
| Patients Enrolled |
A total of 201 patients with advanced paragangliomas.
|
||||
| MOA of PDC |
The clinical applications of radiolabeled SSAs and DOTA-SSA compounds in imaging and radionuclide therapy have been rapidly increasing over the past 2 decades and have been integrated into the current management guidelines of NETs. Several studies support the safety and clinical efficacy of FDA-approved radionuclide therapies including 177Lu-DOTATATE in advanced GEP NETs and 131I-MIBG in advanced paragangliomas and pheochromocytomas. The utility of SSTR-based imaging in PRRT response assessment and surveillance of NETs remains to be established.
Click to Show/Hide
|
||||
| Description |
Over the past decade, a small number of studies have evaluated the efficacy and safety of PRRT with 177Lu-DOTA-SSA in patients with metastatic or inoperable paragangliomas [66, 67]. A recent meta-analysis of 12 studies including a total of 201 patients with advanced paragangliomas showed an objective response rate of 25%, biochemical response rate of 64%, and disease control rate of 84% after treatment with PRRT (either 90Y- or 177Lu-based agents) [67]. The following minimal treatment-related adverse effects were observed: grade 3 or 4 lymphopenia, thrombocytopenia, neutropenia, and nephrotoxicity in 11%, 9%, 3%, and 4% of the patients, respectively.
Click to Show/Hide
|
||||
| Experiment 3 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Small bowel neuroendocrine tumor | ||||
| Efficacy Data | Stable disease (SD) |
59.10%
|
|||
| Patients Enrolled |
Patients with neuroendocrine tumour (Retreatment).
|
||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 4 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Well-differentiated pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Stable disease (SD) |
66.67%
|
|||
| Patients Enrolled |
12 patients with pheocromocytomas and paragangliomas.
|
||||
| Administration Time | 5 cycles | ||||
| Administration Dosage | 1.1-1.85 GBq/cycle | ||||
| MOA of PDC |
We showed that PRRT based on 177Lu-DOTATATE and 90-YDOTATOC without radiosensitizers are efficacious therapeutic options (DCR of 83% and 76%, respectively) and can be considered in the multidisciplinary treatment of PCCs and PGLs as alternatives to I-131 MIBG and chemotherapy.
|
||||
| Description |
There was no statistically significant heterogeneity among the 177Lu- and 90Y- PRRTs studies (I2 value = 0%, p = 0.68 and I2 value = 0%, p = 0.91, respectively). The pooled DCRs were 0.83 (95% CI: 0.75-0.88) and 0.76 (95% CI: 0.56-0.89) for 177Lu- and 90Y-PRRT treatments, respectively. The pooled DCR for PRRT agents was 0.81 (95% CI: 0.74-0.87). Although the present study is the largest description of DCR in PRRT therapy of PCCs and PGLs, given the small sample size, no formal attempts were made to compare efficacy between 177Lu- and 90Y-PRRT treatments. In detail, we report pooled data on 149 177Lu- and 64 90Y-treated patients (total: 213 patients). Most studies were retrospective. The largest one included 46 patients (34 treated with 177Lu-, 12 with 90Y- PRRTs). Median ages ranged from 32.5 to 60.4 years. The female gender was slightly predominant (83 women, 74 men, gender not specified in 3 studies). When reported, mutations of SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B) were the most frequent genetic alterations. According to the predominant retrospective and real practice nature of the selected studies, sites of metastases and previous treatments were heterogeneous, with bone the metastatic site most often involved, and surgery the most frequent previous therapy. Paramount descriptive information on PRRT doses per cycle, number of cycles, response criteria, best responses to treatments, and frequency of G3/G4 hematologic toxicities is reported in Table 2. The lowest applied dose was 3.4 GBq per cycle; the number of administered cycles was heterogeneous (at least 3 cycles in 7 studies). The most used response criteria were the RECIST 1.1. Only one study evaluated the response with SWOG criteria, which are more conservative with regard to the definition of PR (50% or greater reduction in the sum of the product of the perpendicular diameters of target lesions). The majority of the patients experienced PR or SD. Hematologic toxicity was manageable. Funnel plot of selected studies did not show publication bias. The MINORS and Newcastle-Ottawa Scale scoring for the selected articles are reported in Table 3.
Click to Show/Hide
|
||||
| Experiment 5 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Well-differentiated pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Stable disease (SD) |
69.23%
|
|||
| Patients Enrolled |
13 patients with pheocromocytomas (PCCs) and paragangliomas (PGLs) (4 PCC , 9PGL).
|
||||
| Administration Time | 61% of patients received 2 cycles | ||||
| Administration Dosage | 3.4 GBq/cycle | ||||
| MOA of PDC |
We showed that PRRT based on 177Lu-DOTATATE and 90-YDOTATOC without radiosensitizers are efficacious therapeutic options (DCR of 83% and 76%, respectively) and can be considered in the multidisciplinary treatment of PCCs and PGLs as alternatives to I-131 MIBG and chemotherapy.
|
||||
| Description |
There was no statistically significant heterogeneity among the 177Lu- and 90Y- PRRTs studies (I2 value = 0%, p = 0.68 and I2 value = 0%, p = 0.91, respectively). The pooled DCRs were 0.83 (95% CI: 0.75-0.88) and 0.76 (95% CI: 0.56-0.89) for 177Lu- and 90Y-PRRT treatments, respectively. The pooled DCR for PRRT agents was 0.81 (95% CI: 0.74-0.87). Although the present study is the largest description of DCR in PRRT therapy of PCCs and PGLs, given the small sample size, no formal attempts were made to compare efficacy between 177Lu- and 90Y-PRRT treatments. In detail, we report pooled data on 149 177Lu- and 64 90Y-treated patients (total: 213 patients). Most studies were retrospective. The largest one included 46 patients (34 treated with 177Lu-, 12 with 90Y- PRRTs). Median ages ranged from 32.5 to 60.4 years. The female gender was slightly predominant (83 women, 74 men, gender not specified in 3 studies). When reported, mutations of SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B) were the most frequent genetic alterations. According to the predominant retrospective and real practice nature of the selected studies, sites of metastases and previous treatments were heterogeneous, with bone the metastatic site most often involved, and surgery the most frequent previous therapy. Paramount descriptive information on PRRT doses per cycle, number of cycles, response criteria, best responses to treatments, and frequency of G3/G4 hematologic toxicities is reported in Table 2. The lowest applied dose was 3.4 GBq per cycle; the number of administered cycles was heterogeneous (at least 3 cycles in 7 studies). The most used response criteria were the RECIST 1.1. Only one study evaluated the response with SWOG criteria, which are more conservative with regard to the definition of PR (50% or greater reduction in the sum of the product of the perpendicular diameters of target lesions). The majority of the patients experienced PR or SD. Hematologic toxicity was manageable. Funnel plot of selected studies did not show publication bias. The MINORS and Newcastle-Ottawa Scale scoring for the selected articles are reported in Table 3.
Click to Show/Hide
|
||||
| Experiment 6 Reporting the Activity Data of This PDC | [28] | ||||
| Indication | Phaeochromocytoma/Paraganglioma | ||||
| Efficacy Data | Stable disease (SD) |
72%
|
|||
| Patients Enrolled |
12 patients with SSTr2-positive progressive locally advanced or metastatic paragangliomas.
|
||||
| Administration Time | 5 cycles | ||||
| Administration Dosage | 7.4 to 11 GBq | ||||
| MOA of PDC |
In a long-term follow-up of patients with mPPGL, PRRT demonstrated a tolerability and efficacy comparable to those obtained in GEP-NETs. Our results highlight the need for an appropriate cumulative administered dosage to achieve a very prolonged result and underline the importance of specific symptoms and presence of bone lesions as predictive factors. Given the substantial number of patients evaluated over time, the superior mOS of 177Lu-DOTATATE with respect to 90Y-DOTATOC indicates the former radiopharmaceutical as the better candidate for new prospective studies of single or associated therapies.
Click to Show/Hide
|
||||
| Description |
Both 90Y-DOTATOC and 177Lu-DOTATATE PRRT were well tolerated by patients without significant renal or bone marrow toxicity. The median follow-up was 73 months (range 5-146 months). The overall disease control rate (DCR) was 80% [95% confidence interval (CI) 68.9% to 91.9%] with a mean five cycles of therapy. However, 177Lu-DOTATATE patients showed a longer median overall survival (mOS) than those receiving 90Y-Dotatoc and a better DCR when higher dosages were administered, even if a direct comparison was not carried out. Syndromic patients had a poorer mOS. SDHx mutations did not interfere with treatment efficacy.
Click to Show/Hide
|
||||
| Experiment 7 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Small bowel neuroendocrine tumor | ||||
| Efficacy Data | Stable disease (SD) |
78.72%
|
|||
| Patients Enrolled |
29 patients with neuroendocrine tumour.
|
||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 8 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Small bowel neuroendocrine tumor | ||||
| Efficacy Data | Progressive Disease (PD) |
25%
|
|||
| Patients Enrolled |
Patients with neuroendocrine tumour (Retreatment).
|
||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 9 Reporting the Activity Data of This PDC | [28] | ||||
| Indication | Phaeochromocytoma/Paraganglioma | ||||
| Efficacy Data | Progressive Disease (PD) |
80%
|
|||
| Patients Enrolled |
12 patients with SSTr2-positive progressive locally advanced or metastatic paragangliomas.
|
||||
| Administration Time | 5 cycles | ||||
| Administration Dosage | 7.4 to 11 GBq | ||||
| MOA of PDC |
In a long-term follow-up of patients with mPPGL, PRRT demonstrated a tolerability and efficacy comparable to those obtained in GEP-NETs. Our results highlight the need for an appropriate cumulative administered dosage to achieve a very prolonged result and underline the importance of specific symptoms and presence of bone lesions as predictive factors. Given the substantial number of patients evaluated over time, the superior mOS of 177Lu-DOTATATE with respect to 90Y-DOTATOC indicates the former radiopharmaceutical as the better candidate for new prospective studies of single or associated therapies.
Click to Show/Hide
|
||||
| Description |
Both 90Y-DOTATOC and 177Lu-DOTATATE PRRT were well tolerated by patients without significant renal or bone marrow toxicity. The median follow-up was 73 months (range 5-146 months). The overall disease control rate (DCR) was 80% [95% confidence interval (CI) 68.9% to 91.9%] with a mean five cycles of therapy. However, 177Lu-DOTATATE patients showed a longer median overall survival (mOS) than those receiving 90Y-Dotatoc and a better DCR when higher dosages were administered, even if a direct comparison was not carried out. Syndromic patients had a poorer mOS. SDHx mutations did not interfere with treatment efficacy.
Click to Show/Hide
|
||||
| Experiment 10 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Well-differentiated pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Progression-free survival (PFS) |
10-91 months
|
|||
| Patients Enrolled |
4 patients with paraganglioma.
|
||||
| Administration Time | 1-10 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The use of PRRT for patients with PPGLs who had high tumor SSTR expression has mainly been reported in retrospective small case series and the protocols varied in all these studies. So far, four meta-analyses (Taieb et al. and Satapathy et al. in 2019 as well as Su et al. and Marretta et al. in 2023) based on PRRT (177Lu/90Y-DOTAT-TATE/TOC) therapy have been reported. The reported number of pooled patients ranged from 179-330 without any complete response. The reported partial response ranged from 20-25% and stable disease from 59-70% giving an objective response rate range of 20-25% and disease control rate range of 8190%. The survival rates were reported by only 3 meta-analyses. The reported progression-free survival were 17-39 months in seven studies by Taieb et al., 10-91 months in 4 studies and a mean of 37.1 months in 2 studies by Satapathy et al., and a median of 31.8 months in 13 studies by Su et al. whereas overall survival were 49.6-68.0 months in 3 studies by Taieb et al., a mean of 54.5 months in 4 studies by Satapathy et al. and a median of 74.3 months in 11 studies by Su et al. Su et al. reports a pooled adverse events data in 274/330 patients which mentions a grade 3/4 hematotoxicity in 4.3% (grade 1-4, 22.3%) and grade 3/4 nephrotoxicity in 1.9% (grade 1-4, 1.9%), and myelodysplastic syndrome in 1 patient. Treatment discontinuation occurred in 4 patients due to thrombocytopenia (n=3) or nephrotoxicity (n=1).
Click to Show/Hide
|
||||
| Experiment 11 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Well-differentiated pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Progression-free survival (PFS) |
17-39 months
|
|||
| Patients Enrolled |
179 patients with paraganglioma.
|
||||
| Administration Time | 1-11 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The use of PRRT for patients with PPGLs who had high tumor SSTR expression has mainly been reported in retrospective small case series and the protocols varied in all these studies. So far, four meta-analyses (Taieb et al. and Satapathy et al. in 2019 as well as Su et al. and Marretta et al. in 2023) based on PRRT (177Lu/90Y-DOTAT-TATE/TOC) therapy have been reported. The reported number of pooled patients ranged from 179-330 without any complete response. The reported partial response ranged from 20-25% and stable disease from 59-70% giving an objective response rate range of 20-25% and disease control rate range of 8190%. The survival rates were reported by only 3 meta-analyses. The reported progression-free survival were 17-39 months in seven studies by Taieb et al., 10-91 months in 4 studies and a mean of 37.1 months in 2 studies by Satapathy et al., and a median of 31.8 months in 13 studies by Su et al. whereas overall survival were 49.6-68.0 months in 3 studies by Taieb et al., a mean of 54.5 months in 4 studies by Satapathy et al. and a median of 74.3 months in 11 studies by Su et al. Su et al. reports a pooled adverse events data in 274/330 patients which mentions a grade 3/4 hematotoxicity in 4.3% (grade 1-4, 22.3%) and grade 3/4 nephrotoxicity in 1.9% (grade 1-4, 1.9%), and myelodysplastic syndrome in 1 patient. Treatment discontinuation occurred in 4 patients due to thrombocytopenia (n=3) or nephrotoxicity (n=1).
Click to Show/Hide
|
||||
| Experiment 12 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Well-differentiated pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Progression-free survival (PFS) |
31.8 months
|
|||
| Patients Enrolled |
330 patients with paraganglioma.
|
||||
| Administration Time | 1-10 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The use of PRRT for patients with PPGLs who had high tumor SSTR expression has mainly been reported in retrospective small case series and the protocols varied in all these studies. So far, four meta-analyses (Taieb et al. and Satapathy et al. in 2019 as well as Su et al. and Marretta et al. in 2023) based on PRRT (177Lu/90Y-DOTAT-TATE/TOC) therapy have been reported. The reported number of pooled patients ranged from 179-330 without any complete response. The reported partial response ranged from 20-25% and stable disease from 59-70% giving an objective response rate range of 20-25% and disease control rate range of 8190%. The survival rates were reported by only 3 meta-analyses. The reported progression-free survival were 17-39 months in seven studies by Taieb et al., 10-91 months in 4 studies and a mean of 37.1 months in 2 studies by Satapathy et al., and a median of 31.8 months in 13 studies by Su et al. whereas overall survival were 49.6-68.0 months in 3 studies by Taieb et al., a mean of 54.5 months in 4 studies by Satapathy et al. and a median of 74.3 months in 11 studies by Su et al. Su et al. reports a pooled adverse events data in 274/330 patients which mentions a grade 3/4 hematotoxicity in 4.3% (grade 1-4, 22.3%) and grade 3/4 nephrotoxicity in 1.9% (grade 1-4, 1.9%), and myelodysplastic syndrome in 1 patient. Treatment discontinuation occurred in 4 patients due to thrombocytopenia (n=3) or nephrotoxicity (n=1).
Click to Show/Hide
|
||||
| Experiment 13 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Well-differentiated pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Progression-free survival (PFS) |
37.1 months
|
|||
| Patients Enrolled |
2 patients with paraganglioma.
|
||||
| Administration Time | 1-10 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The use of PRRT for patients with PPGLs who had high tumor SSTR expression has mainly been reported in retrospective small case series and the protocols varied in all these studies. So far, four meta-analyses (Taieb et al. and Satapathy et al. in 2019 as well as Su et al. and Marretta et al. in 2023) based on PRRT (177Lu/90Y-DOTAT-TATE/TOC) therapy have been reported. The reported number of pooled patients ranged from 179-330 without any complete response. The reported partial response ranged from 20-25% and stable disease from 59-70% giving an objective response rate range of 20-25% and disease control rate range of 8190%. The survival rates were reported by only 3 meta-analyses. The reported progression-free survival were 17-39 months in seven studies by Taieb et al., 10-91 months in 4 studies and a mean of 37.1 months in 2 studies by Satapathy et al., and a median of 31.8 months in 13 studies by Su et al. whereas overall survival were 49.6-68.0 months in 3 studies by Taieb et al., a mean of 54.5 months in 4 studies by Satapathy et al. and a median of 74.3 months in 11 studies by Su et al. Su et al. reports a pooled adverse events data in 274/330 patients which mentions a grade 3/4 hematotoxicity in 4.3% (grade 1-4, 22.3%) and grade 3/4 nephrotoxicity in 1.9% (grade 1-4, 1.9%), and myelodysplastic syndrome in 1 patient. Treatment discontinuation occurred in 4 patients due to thrombocytopenia (n=3) or nephrotoxicity (n=1).
Click to Show/Hide
|
||||
| Experiment 14 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Well-differentiated pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Partial response (PR) |
7.69%
|
|||
| Patients Enrolled |
13 patients with pheocromocytomas (PCCs) and paragangliomas (PGLs) (4 PCC , 9PGL).
|
||||
| Administration Time | 61% of patients received 2 cycles | ||||
| Administration Dosage | 3.4 GBq/cycle | ||||
| MOA of PDC |
We showed that PRRT based on 177Lu-DOTATATE and 90-YDOTATOC without radiosensitizers are efficacious therapeutic options (DCR of 83% and 76%, respectively) and can be considered in the multidisciplinary treatment of PCCs and PGLs as alternatives to I-131 MIBG and chemotherapy.
|
||||
| Description |
There was no statistically significant heterogeneity among the 177Lu- and 90Y- PRRTs studies (I2 value = 0%, p = 0.68 and I2 value = 0%, p = 0.91, respectively). The pooled DCRs were 0.83 (95% CI: 0.75-0.88) and 0.76 (95% CI: 0.56-0.89) for 177Lu- and 90Y-PRRT treatments, respectively. The pooled DCR for PRRT agents was 0.81 (95% CI: 0.74-0.87). Although the present study is the largest description of DCR in PRRT therapy of PCCs and PGLs, given the small sample size, no formal attempts were made to compare efficacy between 177Lu- and 90Y-PRRT treatments. In detail, we report pooled data on 149 177Lu- and 64 90Y-treated patients (total: 213 patients). Most studies were retrospective. The largest one included 46 patients (34 treated with 177Lu-, 12 with 90Y- PRRTs). Median ages ranged from 32.5 to 60.4 years. The female gender was slightly predominant (83 women, 74 men, gender not specified in 3 studies). When reported, mutations of SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B) were the most frequent genetic alterations. According to the predominant retrospective and real practice nature of the selected studies, sites of metastases and previous treatments were heterogeneous, with bone the metastatic site most often involved, and surgery the most frequent previous therapy. Paramount descriptive information on PRRT doses per cycle, number of cycles, response criteria, best responses to treatments, and frequency of G3/G4 hematologic toxicities is reported in Table 2. The lowest applied dose was 3.4 GBq per cycle; the number of administered cycles was heterogeneous (at least 3 cycles in 7 studies). The most used response criteria were the RECIST 1.1. Only one study evaluated the response with SWOG criteria, which are more conservative with regard to the definition of PR (50% or greater reduction in the sum of the product of the perpendicular diameters of target lesions). The majority of the patients experienced PR or SD. Hematologic toxicity was manageable. Funnel plot of selected studies did not show publication bias. The MINORS and Newcastle-Ottawa Scale scoring for the selected articles are reported in Table 3.
Click to Show/Hide
|
||||
| Experiment 15 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Well-differentiated pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Partial response (PR) |
8.33%
|
|||
| Patients Enrolled |
12 patients with pheocromocytomas and paragangliomas.
|
||||
| Administration Time | 5 cycles | ||||
| Administration Dosage | 1.1-1.85 GBq/cycle | ||||
| MOA of PDC |
We showed that PRRT based on 177Lu-DOTATATE and 90-YDOTATOC without radiosensitizers are efficacious therapeutic options (DCR of 83% and 76%, respectively) and can be considered in the multidisciplinary treatment of PCCs and PGLs as alternatives to I-131 MIBG and chemotherapy.
|
||||
| Description |
There was no statistically significant heterogeneity among the 177Lu- and 90Y- PRRTs studies (I2 value = 0%, p = 0.68 and I2 value = 0%, p = 0.91, respectively). The pooled DCRs were 0.83 (95% CI: 0.75-0.88) and 0.76 (95% CI: 0.56-0.89) for 177Lu- and 90Y-PRRT treatments, respectively. The pooled DCR for PRRT agents was 0.81 (95% CI: 0.74-0.87). Although the present study is the largest description of DCR in PRRT therapy of PCCs and PGLs, given the small sample size, no formal attempts were made to compare efficacy between 177Lu- and 90Y-PRRT treatments. In detail, we report pooled data on 149 177Lu- and 64 90Y-treated patients (total: 213 patients). Most studies were retrospective. The largest one included 46 patients (34 treated with 177Lu-, 12 with 90Y- PRRTs). Median ages ranged from 32.5 to 60.4 years. The female gender was slightly predominant (83 women, 74 men, gender not specified in 3 studies). When reported, mutations of SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B) were the most frequent genetic alterations. According to the predominant retrospective and real practice nature of the selected studies, sites of metastases and previous treatments were heterogeneous, with bone the metastatic site most often involved, and surgery the most frequent previous therapy. Paramount descriptive information on PRRT doses per cycle, number of cycles, response criteria, best responses to treatments, and frequency of G3/G4 hematologic toxicities is reported in Table 2. The lowest applied dose was 3.4 GBq per cycle; the number of administered cycles was heterogeneous (at least 3 cycles in 7 studies). The most used response criteria were the RECIST 1.1. Only one study evaluated the response with SWOG criteria, which are more conservative with regard to the definition of PR (50% or greater reduction in the sum of the product of the perpendicular diameters of target lesions). The majority of the patients experienced PR or SD. Hematologic toxicity was manageable. Funnel plot of selected studies did not show publication bias. The MINORS and Newcastle-Ottawa Scale scoring for the selected articles are reported in Table 3.
Click to Show/Hide
|
||||
| Experiment 16 Reporting the Activity Data of This PDC | [28] | ||||
| Indication | Phaeochromocytoma/Paraganglioma | ||||
| Efficacy Data | Partial response (PR) |
9%
|
|||
| Patients Enrolled |
12 patients with SSTr2-positive progressive locally advanced or metastatic paragangliomas.
|
||||
| Administration Time | 5 cycles | ||||
| Administration Dosage | 7.4 to 11 GBq | ||||
| MOA of PDC |
In a long-term follow-up of patients with mPPGL, PRRT demonstrated a tolerability and efficacy comparable to those obtained in GEP-NETs. Our results highlight the need for an appropriate cumulative administered dosage to achieve a very prolonged result and underline the importance of specific symptoms and presence of bone lesions as predictive factors. Given the substantial number of patients evaluated over time, the superior mOS of 177Lu-DOTATATE with respect to 90Y-DOTATOC indicates the former radiopharmaceutical as the better candidate for new prospective studies of single or associated therapies.
Click to Show/Hide
|
||||
| Description |
Both 90Y-DOTATOC and 177Lu-DOTATATE PRRT were well tolerated by patients without significant renal or bone marrow toxicity. The median follow-up was 73 months (range 5-146 months). The overall disease control rate (DCR) was 80% [95% confidence interval (CI) 68.9% to 91.9%] with a mean five cycles of therapy. However, 177Lu-DOTATATE patients showed a longer median overall survival (mOS) than those receiving 90Y-Dotatoc and a better DCR when higher dosages were administered, even if a direct comparison was not carried out. Syndromic patients had a poorer mOS. SDHx mutations did not interfere with treatment efficacy.
Click to Show/Hide
|
||||
| Experiment 17 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Small bowel neuroendocrine tumor | ||||
| Efficacy Data | Partial response (PR) |
15.90%
|
|||
| Patients Enrolled |
Patients with neuroendocrine tumour (Retreatment).
|
||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 18 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Well-differentiated pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Partial response (PR) |
17.95%
|
|||
| Patients Enrolled |
39 patients with pheocromocytomas (PCCs) and paragangliomas (PGLs) (11 PCC, 28PGL).
|
||||
| Administration Time | 2 cycles (range: 1-10) | ||||
| Administration Dosage | 3.7 GBq/mq per cycle | ||||
| MOA of PDC |
We showed that PRRT based on 177Lu-DOTATATE and 90-YDOTATOC without radiosensitizers are efficacious therapeutic options (DCR of 83% and 76%, respectively) and can be considered in the multidisciplinary treatment of PCCs and PGLs as alternatives to I-131 MIBG and chemotherapy.
|
||||
| Description |
There was no statistically significant heterogeneity among the 177Lu- and 90Y- PRRTs studies (I2 value = 0%, p = 0.68 and I2 value = 0%, p = 0.91, respectively). The pooled DCRs were 0.83 (95% CI: 0.75-0.88) and 0.76 (95% CI: 0.56-0.89) for 177Lu- and 90Y-PRRT treatments, respectively. The pooled DCR for PRRT agents was 0.81 (95% CI: 0.74-0.87). Although the present study is the largest description of DCR in PRRT therapy of PCCs and PGLs, given the small sample size, no formal attempts were made to compare efficacy between 177Lu- and 90Y-PRRT treatments. In detail, we report pooled data on 149 177Lu- and 64 90Y-treated patients (total: 213 patients). Most studies were retrospective. The largest one included 46 patients (34 treated with 177Lu-, 12 with 90Y- PRRTs). Median ages ranged from 32.5 to 60.4 years. The female gender was slightly predominant (83 women, 74 men, gender not specified in 3 studies). When reported, mutations of SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B) were the most frequent genetic alterations. According to the predominant retrospective and real practice nature of the selected studies, sites of metastases and previous treatments were heterogeneous, with bone the metastatic site most often involved, and surgery the most frequent previous therapy. Paramount descriptive information on PRRT doses per cycle, number of cycles, response criteria, best responses to treatments, and frequency of G3/G4 hematologic toxicities is reported in Table 2. The lowest applied dose was 3.4 GBq per cycle; the number of administered cycles was heterogeneous (at least 3 cycles in 7 studies). The most used response criteria were the RECIST 1.1. Only one study evaluated the response with SWOG criteria, which are more conservative with regard to the definition of PR (50% or greater reduction in the sum of the product of the perpendicular diameters of target lesions). The majority of the patients experienced PR or SD. Hematologic toxicity was manageable. Funnel plot of selected studies did not show publication bias. The MINORS and Newcastle-Ottawa Scale scoring for the selected articles are reported in Table 3.
Click to Show/Hide
|
||||
| Experiment 19 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Small bowel neuroendocrine tumor | ||||
| Efficacy Data | Partial response (PR) |
21.27%
|
|||
| Patients Enrolled |
29 patients with neuroendocrine tumour.
|
||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 20 Reporting the Activity Data of This PDC | [70] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Overall survival (OS) |
22.3 months
|
|||
| Patients Enrolled |
Patients with neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | 1.85 GBq/m2/cycle | ||||
| Description |
Median progression-free survival and overall survival (OS) were 13.9 and 22.3 mo, respectively.
|
||||
| Experiment 21 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Well-differentiated pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Overall survival (OS) |
49.6-68.0 months
|
|||
| Patients Enrolled |
179 patients with paraganglioma.
|
||||
| Administration Time | 1-11 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The use of PRRT for patients with PPGLs who had high tumor SSTR expression has mainly been reported in retrospective small case series and the protocols varied in all these studies. So far, four meta-analyses (Taieb et al. and Satapathy et al. in 2019 as well as Su et al. and Marretta et al. in 2023) based on PRRT (177Lu/90Y-DOTAT-TATE/TOC) therapy have been reported. The reported number of pooled patients ranged from 179-330 without any complete response. The reported partial response ranged from 20-25% and stable disease from 59-70% giving an objective response rate range of 20-25% and disease control rate range of 8190%. The survival rates were reported by only 3 meta-analyses. The reported progression-free survival were 17-39 months in seven studies by Taieb et al., 10-91 months in 4 studies and a mean of 37.1 months in 2 studies by Satapathy et al., and a median of 31.8 months in 13 studies by Su et al. whereas overall survival were 49.6-68.0 months in 3 studies by Taieb et al., a mean of 54.5 months in 4 studies by Satapathy et al. and a median of 74.3 months in 11 studies by Su et al. Su et al. reports a pooled adverse events data in 274/330 patients which mentions a grade 3/4 hematotoxicity in 4.3% (grade 1-4, 22.3%) and grade 3/4 nephrotoxicity in 1.9% (grade 1-4, 1.9%), and myelodysplastic syndrome in 1 patient. Treatment discontinuation occurred in 4 patients due to thrombocytopenia (n=3) or nephrotoxicity (n=1).
Click to Show/Hide
|
||||
| Experiment 22 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Well-differentiated pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Overall survival (OS) |
54.5 months
|
|||
| Patients Enrolled |
201 patients with paraganglioma.
|
||||
| Administration Time | 1-10 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The use of PRRT for patients with PPGLs who had high tumor SSTR expression has mainly been reported in retrospective small case series and the protocols varied in all these studies. So far, four meta-analyses (Taieb et al. and Satapathy et al. in 2019 as well as Su et al. and Marretta et al. in 2023) based on PRRT (177Lu/90Y-DOTAT-TATE/TOC) therapy have been reported. The reported number of pooled patients ranged from 179-330 without any complete response. The reported partial response ranged from 20-25% and stable disease from 59-70% giving an objective response rate range of 20-25% and disease control rate range of 8190%. The survival rates were reported by only 3 meta-analyses. The reported progression-free survival were 17-39 months in seven studies by Taieb et al., 10-91 months in 4 studies and a mean of 37.1 months in 2 studies by Satapathy et al., and a median of 31.8 months in 13 studies by Su et al. whereas overall survival were 49.6-68.0 months in 3 studies by Taieb et al., a mean of 54.5 months in 4 studies by Satapathy et al. and a median of 74.3 months in 11 studies by Su et al. Su et al. reports a pooled adverse events data in 274/330 patients which mentions a grade 3/4 hematotoxicity in 4.3% (grade 1-4, 22.3%) and grade 3/4 nephrotoxicity in 1.9% (grade 1-4, 1.9%), and myelodysplastic syndrome in 1 patient. Treatment discontinuation occurred in 4 patients due to thrombocytopenia (n=3) or nephrotoxicity (n=1).
Click to Show/Hide
|
||||
| Experiment 23 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Well-differentiated pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Overall survival (OS) |
74.3 months
|
|||
| Patients Enrolled |
330 patients with paraganglioma.
|
||||
| Administration Time | 1-10 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The use of PRRT for patients with PPGLs who had high tumor SSTR expression has mainly been reported in retrospective small case series and the protocols varied in all these studies. So far, four meta-analyses (Taieb et al. and Satapathy et al. in 2019 as well as Su et al. and Marretta et al. in 2023) based on PRRT (177Lu/90Y-DOTAT-TATE/TOC) therapy have been reported. The reported number of pooled patients ranged from 179-330 without any complete response. The reported partial response ranged from 20-25% and stable disease from 59-70% giving an objective response rate range of 20-25% and disease control rate range of 8190%. The survival rates were reported by only 3 meta-analyses. The reported progression-free survival were 17-39 months in seven studies by Taieb et al., 10-91 months in 4 studies and a mean of 37.1 months in 2 studies by Satapathy et al., and a median of 31.8 months in 13 studies by Su et al. whereas overall survival were 49.6-68.0 months in 3 studies by Taieb et al., a mean of 54.5 months in 4 studies by Satapathy et al. and a median of 74.3 months in 11 studies by Su et al. Su et al. reports a pooled adverse events data in 274/330 patients which mentions a grade 3/4 hematotoxicity in 4.3% (grade 1-4, 22.3%) and grade 3/4 nephrotoxicity in 1.9% (grade 1-4, 1.9%), and myelodysplastic syndrome in 1 patient. Treatment discontinuation occurred in 4 patients due to thrombocytopenia (n=3) or nephrotoxicity (n=1).
Click to Show/Hide
|
||||
| Experiment 24 Reporting the Activity Data of This PDC | [50] | ||||
| Indication | Differentiated thyroid cancer | ||||
| Efficacy Data | Objective response rate (ORR) |
10.50%
|
|||
| Patients Enrolled |
157 patients with RR-DTC treated with PPRT.
|
||||
| MOA of PDC |
Findings of the study revealed that in the absence of the established treatment for the patients with RR-DTC and metastatic MTC, PRRT could be effective with few adverse events.
|
||||
| Description |
Among 2284 related papers, 41 papers met the inclusion criteria. A total of 157 patients with RR-DTC were treated with PPRT. Biochemical and objective responses (partial and complete) were observed in 25.3 and 10.5% of patients, respectively. Among 220 patients with metastatic MTC, biochemical and objective responses were observed in 37.2 and 10.6% of the patients, respectively. Forty-six deaths were reported in 95 patients with advanced RR-DTC. In addition, 63 deaths were observed in 144 patients with metastatic MTC. Major side effects were reported in 124 patients treated with 90Y -based agent. In the patients treated with 177Lu-DOTA-TATE and 111In-Octreotide, mild and transient hematologic or renal complications were reported.
Click to Show/Hide
|
||||
| Experiment 25 Reporting the Activity Data of This PDC | [50] | ||||
| Indication | Medullary thyroid carcinoma | ||||
| Efficacy Data | Objective response rate (ORR) |
10.60%
|
|||
| Patients Enrolled |
220 patients with metastatic medullary thyroid cancer.
|
||||
| MOA of PDC |
Findings of the study revealed that in the absence of the established treatment for the patients with RR-DTC and metastatic MTC, PRRT could be effective with few adverse events.
|
||||
| Description |
Among 2284 related papers, 41 papers met the inclusion criteria. A total of 157 patients with RR-DTC were treated with PPRT. Biochemical and objective responses (partial and complete) were observed in 25.3 and 10.5% of patients, respectively. Among 220 patients with metastatic MTC, biochemical and objective responses were observed in 37.2 and 10.6% of the patients, respectively. Forty-six deaths were reported in 95 patients with advanced RR-DTC. In addition, 63 deaths were observed in 144 patients with metastatic MTC. Major side effects were reported in 124 patients treated with 90Y -based agent. In the patients treated with 177Lu-DOTA-TATE and 111In-Octreotide, mild and transient hematologic or renal complications were reported.
Click to Show/Hide
|
||||
| Experiment 26 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Well-differentiated pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Objective response rate (ORR) |
20%
|
|||
| Patients Enrolled |
330 patients with paraganglioma.
|
||||
| Administration Time | 1-10 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The use of PRRT for patients with PPGLs who had high tumor SSTR expression has mainly been reported in retrospective small case series and the protocols varied in all these studies. So far, four meta-analyses (Taieb et al. and Satapathy et al. in 2019 as well as Su et al. and Marretta et al. in 2023) based on PRRT (177Lu/90Y-DOTAT-TATE/TOC) therapy have been reported. The reported number of pooled patients ranged from 179-330 without any complete response. The reported partial response ranged from 20-25% and stable disease from 59-70% giving an objective response rate range of 20-25% and disease control rate range of 8190%. The survival rates were reported by only 3 meta-analyses. The reported progression-free survival were 17-39 months in seven studies by Taieb et al., 10-91 months in 4 studies and a mean of 37.1 months in 2 studies by Satapathy et al., and a median of 31.8 months in 13 studies by Su et al. whereas overall survival were 49.6-68.0 months in 3 studies by Taieb et al., a mean of 54.5 months in 4 studies by Satapathy et al. and a median of 74.3 months in 11 studies by Su et al. Su et al. reports a pooled adverse events data in 274/330 patients which mentions a grade 3/4 hematotoxicity in 4.3% (grade 1-4, 22.3%) and grade 3/4 nephrotoxicity in 1.9% (grade 1-4, 1.9%), and myelodysplastic syndrome in 1 patient. Treatment discontinuation occurred in 4 patients due to thrombocytopenia (n=3) or nephrotoxicity (n=1).
Click to Show/Hide
|
||||
| Experiment 27 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Medullary thyroid carcinoma | ||||
| Efficacy Data | Objective response rate (ORR) |
25%
|
|||
| Patients Enrolled |
A total of 201 patients with advanced paragangliomas.
|
||||
| MOA of PDC |
The clinical applications of radiolabeled SSAs and DOTA-SSA compounds in imaging and radionuclide therapy have been rapidly increasing over the past 2 decades and have been integrated into the current management guidelines of NETs. Several studies support the safety and clinical efficacy of FDA-approved radionuclide therapies including 177Lu-DOTATATE in advanced GEP NETs and 131I-MIBG in advanced paragangliomas and pheochromocytomas. The utility of SSTR-based imaging in PRRT response assessment and surveillance of NETs remains to be established.
Click to Show/Hide
|
||||
| Description |
Over the past decade, a small number of studies have evaluated the efficacy and safety of PRRT with 177Lu-DOTA-SSA in patients with metastatic or inoperable paragangliomas [66, 67]. A recent meta-analysis of 12 studies including a total of 201 patients with advanced paragangliomas showed an objective response rate of 25%, biochemical response rate of 64%, and disease control rate of 84% after treatment with PRRT (either 90Y- or 177Lu-based agents) [67]. The following minimal treatment-related adverse effects were observed: grade 3 or 4 lymphopenia, thrombocytopenia, neutropenia, and nephrotoxicity in 11%, 9%, 3%, and 4% of the patients, respectively.
Click to Show/Hide
|
||||
| Experiment 28 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Well-differentiated pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Objective response rate (ORR) |
25%
|
|||
| Patients Enrolled |
201 patients with paraganglioma.
|
||||
| Administration Time | 1-10 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The use of PRRT for patients with PPGLs who had high tumor SSTR expression has mainly been reported in retrospective small case series and the protocols varied in all these studies. So far, four meta-analyses (Taieb et al. and Satapathy et al. in 2019 as well as Su et al. and Marretta et al. in 2023) based on PRRT (177Lu/90Y-DOTAT-TATE/TOC) therapy have been reported. The reported number of pooled patients ranged from 179-330 without any complete response. The reported partial response ranged from 20-25% and stable disease from 59-70% giving an objective response rate range of 20-25% and disease control rate range of 8190%. The survival rates were reported by only 3 meta-analyses. The reported progression-free survival were 17-39 months in seven studies by Taieb et al., 10-91 months in 4 studies and a mean of 37.1 months in 2 studies by Satapathy et al., and a median of 31.8 months in 13 studies by Su et al. whereas overall survival were 49.6-68.0 months in 3 studies by Taieb et al., a mean of 54.5 months in 4 studies by Satapathy et al. and a median of 74.3 months in 11 studies by Su et al. Su et al. reports a pooled adverse events data in 274/330 patients which mentions a grade 3/4 hematotoxicity in 4.3% (grade 1-4, 22.3%) and grade 3/4 nephrotoxicity in 1.9% (grade 1-4, 1.9%), and myelodysplastic syndrome in 1 patient. Treatment discontinuation occurred in 4 patients due to thrombocytopenia (n=3) or nephrotoxicity (n=1).
Click to Show/Hide
|
||||
| Experiment 29 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Medullary thyroid carcinoma | ||||
| Efficacy Data | Neutropenia |
3%
|
|||
| Patients Enrolled |
A total of 201 patients with advanced paragangliomas.
|
||||
| MOA of PDC |
The clinical applications of radiolabeled SSAs and DOTA-SSA compounds in imaging and radionuclide therapy have been rapidly increasing over the past 2 decades and have been integrated into the current management guidelines of NETs. Several studies support the safety and clinical efficacy of FDA-approved radionuclide therapies including 177Lu-DOTATATE in advanced GEP NETs and 131I-MIBG in advanced paragangliomas and pheochromocytomas. The utility of SSTR-based imaging in PRRT response assessment and surveillance of NETs remains to be established.
Click to Show/Hide
|
||||
| Description |
Over the past decade, a small number of studies have evaluated the efficacy and safety of PRRT with 177Lu-DOTA-SSA in patients with metastatic or inoperable paragangliomas [66, 67]. A recent meta-analysis of 12 studies including a total of 201 patients with advanced paragangliomas showed an objective response rate of 25%, biochemical response rate of 64%, and disease control rate of 84% after treatment with PRRT (either 90Y- or 177Lu-based agents) [67]. The following minimal treatment-related adverse effects were observed: grade 3 or 4 lymphopenia, thrombocytopenia, neutropenia, and nephrotoxicity in 11%, 9%, 3%, and 4% of the patients, respectively.
Click to Show/Hide
|
||||
| Experiment 30 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Medullary thyroid carcinoma | ||||
| Efficacy Data | Nephrotoxicity |
4%
|
|||
| Patients Enrolled |
A total of 201 patients with advanced paragangliomas.
|
||||
| MOA of PDC |
The clinical applications of radiolabeled SSAs and DOTA-SSA compounds in imaging and radionuclide therapy have been rapidly increasing over the past 2 decades and have been integrated into the current management guidelines of NETs. Several studies support the safety and clinical efficacy of FDA-approved radionuclide therapies including 177Lu-DOTATATE in advanced GEP NETs and 131I-MIBG in advanced paragangliomas and pheochromocytomas. The utility of SSTR-based imaging in PRRT response assessment and surveillance of NETs remains to be established.
Click to Show/Hide
|
||||
| Description |
Over the past decade, a small number of studies have evaluated the efficacy and safety of PRRT with 177Lu-DOTA-SSA in patients with metastatic or inoperable paragangliomas [66, 67]. A recent meta-analysis of 12 studies including a total of 201 patients with advanced paragangliomas showed an objective response rate of 25%, biochemical response rate of 64%, and disease control rate of 84% after treatment with PRRT (either 90Y- or 177Lu-based agents) [67]. The following minimal treatment-related adverse effects were observed: grade 3 or 4 lymphopenia, thrombocytopenia, neutropenia, and nephrotoxicity in 11%, 9%, 3%, and 4% of the patients, respectively.
Click to Show/Hide
|
||||
| Experiment 31 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Well-differentiated pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Median stroma percentage |
40%
|
|||
| Patients Enrolled |
24 patients with morphologically WD-PanNETs who underwent PRRT preoperatively.
|
||||
| Administration Time | A median of 6 months (interquartile range [iqr] 6; 8 months) from the last cycle of prrt | ||||
| Evaluation Method | Stained with hematoxylin and eosin (H&E) & Formalin-fixed and paraffin-embedded (FFPE) assay | ||||
| Description |
In the PRRT group, the median stroma percentage was 40% compared to 20% in the control group (p < 0.0001).
|
||||
| Experiment 32 Reporting the Activity Data of This PDC | [70] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
13.9 months
|
|||
| Patients Enrolled |
Patients with neuroendocrine tumor.
|
||||
| Administration Time | 4 cycles | ||||
| Administration Dosage | 1.85 GBq/m2/cycle | ||||
| Description |
Median progression-free survival and overall survival (OS) were 13.9 and 22.3 mo, respectively.
|
||||
| Experiment 33 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Small bowel neuroendocrine tumor | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
17.5 months
|
|||
| Patients Enrolled |
29 patients with neuroendocrine tumour.
|
||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 34 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Small bowel neuroendocrine tumor | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
17.5 months
|
|||
| Patients Enrolled |
Patients with neuroendocrine tumour (Retreatment).
|
||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 35 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Medullary thyroid carcinoma | ||||
| Efficacy Data | Grade 3/4 lymphopenia |
11%
|
|||
| Patients Enrolled |
A total of 201 patients with advanced paragangliomas.
|
||||
| MOA of PDC |
The clinical applications of radiolabeled SSAs and DOTA-SSA compounds in imaging and radionuclide therapy have been rapidly increasing over the past 2 decades and have been integrated into the current management guidelines of NETs. Several studies support the safety and clinical efficacy of FDA-approved radionuclide therapies including 177Lu-DOTATATE in advanced GEP NETs and 131I-MIBG in advanced paragangliomas and pheochromocytomas. The utility of SSTR-based imaging in PRRT response assessment and surveillance of NETs remains to be established.
Click to Show/Hide
|
||||
| Description |
Over the past decade, a small number of studies have evaluated the efficacy and safety of PRRT with 177Lu-DOTA-SSA in patients with metastatic or inoperable paragangliomas [66, 67]. A recent meta-analysis of 12 studies including a total of 201 patients with advanced paragangliomas showed an objective response rate of 25%, biochemical response rate of 64%, and disease control rate of 84% after treatment with PRRT (either 90Y- or 177Lu-based agents) [67]. The following minimal treatment-related adverse effects were observed: grade 3 or 4 lymphopenia, thrombocytopenia, neutropenia, and nephrotoxicity in 11%, 9%, 3%, and 4% of the patients, respectively.
Click to Show/Hide
|
||||
| Experiment 36 Reporting the Activity Data of This PDC | [28] | ||||
| Indication | Phaeochromocytoma/Paraganglioma | ||||
| Efficacy Data | Grade 2 platelets toxicity |
3%
|
|||
| Patients Enrolled |
12 patients with SSTr2-positive progressive locally advanced or metastatic paragangliomas.
|
||||
| Administration Time | 5 cycles | ||||
| Administration Dosage | 7.4 to 11 GBq | ||||
| MOA of PDC |
In a long-term follow-up of patients with mPPGL, PRRT demonstrated a tolerability and efficacy comparable to those obtained in GEP-NETs. Our results highlight the need for an appropriate cumulative administered dosage to achieve a very prolonged result and underline the importance of specific symptoms and presence of bone lesions as predictive factors. Given the substantial number of patients evaluated over time, the superior mOS of 177Lu-DOTATATE with respect to 90Y-DOTATOC indicates the former radiopharmaceutical as the better candidate for new prospective studies of single or associated therapies.
Click to Show/Hide
|
||||
| Description |
No cases of grade (G) 3/4 bone marrow toxicity were observed in any of the treated patients. G1 and G2 bone marrow toxicity was observed in 19 and 4 patients, respectively. All patients recovered during the interval between cycles, without the need for interference in the treatment schedule. Two patients had G1 renal toxicity and one patient G2 toxicity. The latter patient started treatment (2008) with 1.53 creatinine at baseline and registered a 1.88 creatinine level at the third cycle, at which point therapy was interrupted (cumulative dosage 11.1 GBq 177Lu-DOTATATE). She still has stable disease after >10 years, but a worsening of renal function led to the need for weekly hemodialysis from 2019. No significant differences in toxicity were reported in patients treated with 90Y-DOTATOC or 177Lu-DOTATATE.
Click to Show/Hide
|
||||
| Experiment 37 Reporting the Activity Data of This PDC | [28] | ||||
| Indication | Phaeochromocytoma/Paraganglioma | ||||
| Efficacy Data | Grade 2 neutrophils toxicity |
6%
|
|||
| Patients Enrolled |
12 patients with SSTr2-positive progressive locally advanced or metastatic paragangliomas.
|
||||
| Administration Time | 5 cycles | ||||
| Administration Dosage | 7.4 to 11 GBq | ||||
| MOA of PDC |
In a long-term follow-up of patients with mPPGL, PRRT demonstrated a tolerability and efficacy comparable to those obtained in GEP-NETs. Our results highlight the need for an appropriate cumulative administered dosage to achieve a very prolonged result and underline the importance of specific symptoms and presence of bone lesions as predictive factors. Given the substantial number of patients evaluated over time, the superior mOS of 177Lu-DOTATATE with respect to 90Y-DOTATOC indicates the former radiopharmaceutical as the better candidate for new prospective studies of single or associated therapies.
Click to Show/Hide
|
||||
| Description |
No cases of grade (G) 3/4 bone marrow toxicity were observed in any of the treated patients. G1 and G2 bone marrow toxicity was observed in 19 and 4 patients, respectively. All patients recovered during the interval between cycles, without the need for interference in the treatment schedule. Two patients had G1 renal toxicity and one patient G2 toxicity. The latter patient started treatment (2008) with 1.53 creatinine at baseline and registered a 1.88 creatinine level at the third cycle, at which point therapy was interrupted (cumulative dosage 11.1 GBq 177Lu-DOTATATE). She still has stable disease after >10 years, but a worsening of renal function led to the need for weekly hemodialysis from 2019. No significant differences in toxicity were reported in patients treated with 90Y-DOTATOC or 177Lu-DOTATATE.
Click to Show/Hide
|
||||
| Experiment 38 Reporting the Activity Data of This PDC | [28] | ||||
| Indication | Phaeochromocytoma/Paraganglioma | ||||
| Efficacy Data | Grade 2 hemoglobin toxicity |
3%
|
|||
| Patients Enrolled |
12 patients with SSTr2-positive progressive locally advanced or metastatic paragangliomas.
|
||||
| Administration Time | 5 cycles | ||||
| Administration Dosage | 7.4 to 11 GBq | ||||
| MOA of PDC |
In a long-term follow-up of patients with mPPGL, PRRT demonstrated a tolerability and efficacy comparable to those obtained in GEP-NETs. Our results highlight the need for an appropriate cumulative administered dosage to achieve a very prolonged result and underline the importance of specific symptoms and presence of bone lesions as predictive factors. Given the substantial number of patients evaluated over time, the superior mOS of 177Lu-DOTATATE with respect to 90Y-DOTATOC indicates the former radiopharmaceutical as the better candidate for new prospective studies of single or associated therapies.
Click to Show/Hide
|
||||
| Description |
No cases of grade (G) 3/4 bone marrow toxicity were observed in any of the treated patients. G1 and G2 bone marrow toxicity was observed in 19 and 4 patients, respectively. All patients recovered during the interval between cycles, without the need for interference in the treatment schedule. Two patients had G1 renal toxicity and one patient G2 toxicity. The latter patient started treatment (2008) with 1.53 creatinine at baseline and registered a 1.88 creatinine level at the third cycle, at which point therapy was interrupted (cumulative dosage 11.1 GBq 177Lu-DOTATATE). She still has stable disease after >10 years, but a worsening of renal function led to the need for weekly hemodialysis from 2019. No significant differences in toxicity were reported in patients treated with 90Y-DOTATOC or 177Lu-DOTATATE.
Click to Show/Hide
|
||||
| Experiment 39 Reporting the Activity Data of This PDC | [28] | ||||
| Indication | Phaeochromocytoma/Paraganglioma | ||||
| Efficacy Data | Grade 2 creatinine toxicity |
3%
|
|||
| Patients Enrolled |
12 patients with SSTr2-positive progressive locally advanced or metastatic paragangliomas.
|
||||
| Administration Time | 5 cycles | ||||
| Administration Dosage | 7.4 to 11 GBq | ||||
| MOA of PDC |
In a long-term follow-up of patients with mPPGL, PRRT demonstrated a tolerability and efficacy comparable to those obtained in GEP-NETs. Our results highlight the need for an appropriate cumulative administered dosage to achieve a very prolonged result and underline the importance of specific symptoms and presence of bone lesions as predictive factors. Given the substantial number of patients evaluated over time, the superior mOS of 177Lu-DOTATATE with respect to 90Y-DOTATOC indicates the former radiopharmaceutical as the better candidate for new prospective studies of single or associated therapies.
Click to Show/Hide
|
||||
| Description |
No cases of grade (G) 3/4 bone marrow toxicity were observed in any of the treated patients. G1 and G2 bone marrow toxicity was observed in 19 and 4 patients, respectively. All patients recovered during the interval between cycles, without the need for interference in the treatment schedule. Two patients had G1 renal toxicity and one patient G2 toxicity. The latter patient started treatment (2008) with 1.53 creatinine at baseline and registered a 1.88 creatinine level at the third cycle, at which point therapy was interrupted (cumulative dosage 11.1 GBq 177Lu-DOTATATE). She still has stable disease after >10 years, but a worsening of renal function led to the need for weekly hemodialysis from 2019. No significant differences in toxicity were reported in patients treated with 90Y-DOTATOC or 177Lu-DOTATATE.
Click to Show/Hide
|
||||
| Experiment 40 Reporting the Activity Data of This PDC | [28] | ||||
| Indication | Phaeochromocytoma/Paraganglioma | ||||
| Efficacy Data | Grade 1 platelets toxicity |
15%
|
|||
| Patients Enrolled |
12 patients with SSTr2-positive progressive locally advanced or metastatic paragangliomas.
|
||||
| Administration Time | 5 cycles | ||||
| Administration Dosage | 7.4 to 11 GBq | ||||
| MOA of PDC |
In a long-term follow-up of patients with mPPGL, PRRT demonstrated a tolerability and efficacy comparable to those obtained in GEP-NETs. Our results highlight the need for an appropriate cumulative administered dosage to achieve a very prolonged result and underline the importance of specific symptoms and presence of bone lesions as predictive factors. Given the substantial number of patients evaluated over time, the superior mOS of 177Lu-DOTATATE with respect to 90Y-DOTATOC indicates the former radiopharmaceutical as the better candidate for new prospective studies of single or associated therapies.
Click to Show/Hide
|
||||
| Description |
No cases of grade (G) 3/4 bone marrow toxicity were observed in any of the treated patients. G1 and G2 bone marrow toxicity was observed in 19 and 4 patients, respectively. All patients recovered during the interval between cycles, without the need for interference in the treatment schedule. Two patients had G1 renal toxicity and one patient G2 toxicity. The latter patient started treatment (2008) with 1.53 creatinine at baseline and registered a 1.88 creatinine level at the third cycle, at which point therapy was interrupted (cumulative dosage 11.1 GBq 177Lu-DOTATATE). She still has stable disease after >10 years, but a worsening of renal function led to the need for weekly hemodialysis from 2019. No significant differences in toxicity were reported in patients treated with 90Y-DOTATOC or 177Lu-DOTATATE.
Click to Show/Hide
|
||||
| Experiment 41 Reporting the Activity Data of This PDC | [28] | ||||
| Indication | Phaeochromocytoma/Paraganglioma | ||||
| Efficacy Data | Grade 1 neutrophils toxicity |
6%
|
|||
| Patients Enrolled |
12 patients with SSTr2-positive progressive locally advanced or metastatic paragangliomas.
|
||||
| Administration Time | 5 cycles | ||||
| Administration Dosage | 7.4 to 11 GBq | ||||
| MOA of PDC |
In a long-term follow-up of patients with mPPGL, PRRT demonstrated a tolerability and efficacy comparable to those obtained in GEP-NETs. Our results highlight the need for an appropriate cumulative administered dosage to achieve a very prolonged result and underline the importance of specific symptoms and presence of bone lesions as predictive factors. Given the substantial number of patients evaluated over time, the superior mOS of 177Lu-DOTATATE with respect to 90Y-DOTATOC indicates the former radiopharmaceutical as the better candidate for new prospective studies of single or associated therapies.
Click to Show/Hide
|
||||
| Description |
No cases of grade (G) 3/4 bone marrow toxicity were observed in any of the treated patients. G1 and G2 bone marrow toxicity was observed in 19 and 4 patients, respectively. All patients recovered during the interval between cycles, without the need for interference in the treatment schedule. Two patients had G1 renal toxicity and one patient G2 toxicity. The latter patient started treatment (2008) with 1.53 creatinine at baseline and registered a 1.88 creatinine level at the third cycle, at which point therapy was interrupted (cumulative dosage 11.1 GBq 177Lu-DOTATATE). She still has stable disease after >10 years, but a worsening of renal function led to the need for weekly hemodialysis from 2019. No significant differences in toxicity were reported in patients treated with 90Y-DOTATOC or 177Lu-DOTATATE.
Click to Show/Hide
|
||||
| Experiment 42 Reporting the Activity Data of This PDC | [28] | ||||
| Indication | Phaeochromocytoma/Paraganglioma | ||||
| Efficacy Data | Grade 1 hemoglobin toxicity |
24%
|
|||
| Patients Enrolled |
12 patients with SSTr2-positive progressive locally advanced or metastatic paragangliomas.
|
||||
| Administration Time | 5 cycles | ||||
| Administration Dosage | 7.4 to 11 GBq | ||||
| MOA of PDC |
In a long-term follow-up of patients with mPPGL, PRRT demonstrated a tolerability and efficacy comparable to those obtained in GEP-NETs. Our results highlight the need for an appropriate cumulative administered dosage to achieve a very prolonged result and underline the importance of specific symptoms and presence of bone lesions as predictive factors. Given the substantial number of patients evaluated over time, the superior mOS of 177Lu-DOTATATE with respect to 90Y-DOTATOC indicates the former radiopharmaceutical as the better candidate for new prospective studies of single or associated therapies.
Click to Show/Hide
|
||||
| Description |
No cases of grade (G) 3/4 bone marrow toxicity were observed in any of the treated patients. G1 and G2 bone marrow toxicity was observed in 19 and 4 patients, respectively. All patients recovered during the interval between cycles, without the need for interference in the treatment schedule. Two patients had G1 renal toxicity and one patient G2 toxicity. The latter patient started treatment (2008) with 1.53 creatinine at baseline and registered a 1.88 creatinine level at the third cycle, at which point therapy was interrupted (cumulative dosage 11.1 GBq 177Lu-DOTATATE). She still has stable disease after >10 years, but a worsening of renal function led to the need for weekly hemodialysis from 2019. No significant differences in toxicity were reported in patients treated with 90Y-DOTATOC or 177Lu-DOTATATE.
Click to Show/Hide
|
||||
| Experiment 43 Reporting the Activity Data of This PDC | [28] | ||||
| Indication | Phaeochromocytoma/Paraganglioma | ||||
| Efficacy Data | Grade 1 creatinine toxicity |
6%
|
|||
| Patients Enrolled |
12 patients with SSTr2-positive progressive locally advanced or metastatic paragangliomas.
|
||||
| Administration Time | 5 cycles | ||||
| Administration Dosage | 7.4 to 11 GBq | ||||
| MOA of PDC |
In a long-term follow-up of patients with mPPGL, PRRT demonstrated a tolerability and efficacy comparable to those obtained in GEP-NETs. Our results highlight the need for an appropriate cumulative administered dosage to achieve a very prolonged result and underline the importance of specific symptoms and presence of bone lesions as predictive factors. Given the substantial number of patients evaluated over time, the superior mOS of 177Lu-DOTATATE with respect to 90Y-DOTATOC indicates the former radiopharmaceutical as the better candidate for new prospective studies of single or associated therapies.
Click to Show/Hide
|
||||
| Description |
No cases of grade (G) 3/4 bone marrow toxicity were observed in any of the treated patients. G1 and G2 bone marrow toxicity was observed in 19 and 4 patients, respectively. All patients recovered during the interval between cycles, without the need for interference in the treatment schedule. Two patients had G1 renal toxicity and one patient G2 toxicity. The latter patient started treatment (2008) with 1.53 creatinine at baseline and registered a 1.88 creatinine level at the third cycle, at which point therapy was interrupted (cumulative dosage 11.1 GBq 177Lu-DOTATATE). She still has stable disease after >10 years, but a worsening of renal function led to the need for weekly hemodialysis from 2019. No significant differences in toxicity were reported in patients treated with 90Y-DOTATOC or 177Lu-DOTATATE.
Click to Show/Hide
|
||||
| Experiment 44 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Small bowel neuroendocrine tumor | ||||
| Efficacy Data | Grade ≥3 toxicity |
Renal: n=1; Hematologic: n=2; Myelodysplastic syndrome: n=1
|
|||
| Patients Enrolled |
29 patients with neuroendocrine tumour.
|
||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 45 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Small bowel neuroendocrine tumor | ||||
| Efficacy Data | Grade ≥3 toxicity |
Renal: n=1; Hematologic: n=2; Myelodysplastic syndrome: n=1
|
|||
| Patients Enrolled |
Patients with neuroendocrine tumour (Retreatment).
|
||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 46 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Well-differentiated pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Frequency of N0 |
61%
|
|||
| Patients Enrolled |
24 patients with morphologically WD-PanNETs who underwent PRRT preoperatively.
|
||||
| Administration Time | A median of 6 months (interquartile range [iqr] 6; 8 months) from the last cycle of prrt | ||||
| Evaluation Method | Stained with hematoxylin and eosin (H&E) & Formalin-fixed and paraffin-embedded (FFPE) assay | ||||
| Description |
Patients who underwent PRRT had a trend towards a higher frequency of N0 as compared to control group (61% vs 33%, p = 0.059).
|
||||
| Experiment 47 Reporting the Activity Data of This PDC | [71] | ||||
| Indication | Gastroenteropancreatic neuroendocrine tumour | ||||
| Efficacy Data | Disease stable rate |
70%
|
|||
| Patients Enrolled |
10 patients with neuroendocrine tumour.
|
||||
| Administration Dosage | 3.5±0.3 GBq (94.7±7.5 mCi) | ||||
| Evaluation Method | 68Ga-DOTATATE PET/CT assay | ||||
| Description |
During the follow-up period (24 wk), the best response was stable disease in 70% of subjects (7/10) and progressive disease in 20% (2/10)
|
||||
| Experiment 48 Reporting the Activity Data of This PDC | [28] | ||||
| Indication | Phaeochromocytoma/Paraganglioma | ||||
| Efficacy Data | Disease control rate (DCR) |
20%
|
|||
| Patients Enrolled |
12 patients with SSTr2-positive progressive locally advanced or metastatic paragangliomas.
|
||||
| Administration Time | 5 cycles | ||||
| Administration Dosage | 7.4 to 11 GBq | ||||
| MOA of PDC |
In a long-term follow-up of patients with mPPGL, PRRT demonstrated a tolerability and efficacy comparable to those obtained in GEP-NETs. Our results highlight the need for an appropriate cumulative administered dosage to achieve a very prolonged result and underline the importance of specific symptoms and presence of bone lesions as predictive factors. Given the substantial number of patients evaluated over time, the superior mOS of 177Lu-DOTATATE with respect to 90Y-DOTATOC indicates the former radiopharmaceutical as the better candidate for new prospective studies of single or associated therapies.
Click to Show/Hide
|
||||
| Description |
Both 90Y-DOTATOC and 177Lu-DOTATATE PRRT were well tolerated by patients without significant renal or bone marrow toxicity. The median follow-up was 73 months (range 5-146 months). The overall disease control rate (DCR) was 80% [95% confidence interval (CI) 68.9% to 91.9%] with a mean five cycles of therapy. However, 177Lu-DOTATATE patients showed a longer median overall survival (mOS) than those receiving 90Y-Dotatoc and a better DCR when higher dosages were administered, even if a direct comparison was not carried out. Syndromic patients had a poorer mOS. SDHx mutations did not interfere with treatment efficacy.
Click to Show/Hide
|
||||
| Experiment 49 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Small bowel neuroendocrine tumor | ||||
| Efficacy Data | Disease control rate (DCR) |
75%
|
|||
| Patients Enrolled |
Patients with neuroendocrine tumour (Retreatment).
|
||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 50 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Well-differentiated pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Disease control rate (DCR) |
76%
|
|||
| Patients Enrolled |
Patients with pheocromocytomas (PCCs) and paragangliomas (PGLs) (1 PCC, 11PGL).
|
||||
| MOA of PDC |
We showed that PRRT based on 177Lu-DOTATATE and 90-YDOTATOC without radiosensitizers are efficacious therapeutic options (DCR of 83% and 76%, respectively) and can be considered in the multidisciplinary treatment of PCCs and PGLs as alternatives to I-131 MIBG and chemotherapy.
|
||||
| Description |
There was no statistically significant heterogeneity among the 177Lu- and 90Y- PRRTs studies (I2 value = 0%, p = 0.68 and I2 value = 0%, p = 0.91, respectively). The pooled DCRs were 0.83 (95% CI: 0.75-0.88) and 0.76 (95% CI: 0.56-0.89) for 177Lu- and 90Y-PRRT treatments, respectively. The pooled DCR for PRRT agents was 0.81 (95% CI: 0.74-0.87). Although the present study is the largest description of DCR in PRRT therapy of PCCs and PGLs, given the small sample size, no formal attempts were made to compare efficacy between 177Lu- and 90Y-PRRT treatments. In detail, we report pooled data on 149 177Lu- and 64 90Y-treated patients (total: 213 patients). Most studies were retrospective. The largest one included 46 patients (34 treated with 177Lu-, 12 with 90Y- PRRTs). Median ages ranged from 32.5 to 60.4 years. The female gender was slightly predominant (83 women, 74 men, gender not specified in 3 studies). When reported, mutations of SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B) were the most frequent genetic alterations. According to the predominant retrospective and real practice nature of the selected studies, sites of metastases and previous treatments were heterogeneous, with bone the metastatic site most often involved, and surgery the most frequent previous therapy. Paramount descriptive information on PRRT doses per cycle, number of cycles, response criteria, best responses to treatments, and frequency of G3/G4 hematologic toxicities is reported in Table 2. The lowest applied dose was 3.4 GBq per cycle; the number of administered cycles was heterogeneous (at least 3 cycles in 7 studies). The most used response criteria were the RECIST 1.1. Only one study evaluated the response with SWOG criteria, which are more conservative with regard to the definition of PR (50% or greater reduction in the sum of the product of the perpendicular diameters of target lesions). The majority of the patients experienced PR or SD. Hematologic toxicity was manageable. Funnel plot of selected studies did not show publication bias. The MINORS and Newcastle-Ottawa Scale scoring for the selected articles are reported in Table 3.
Click to Show/Hide
|
||||
| Experiment 51 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Well-differentiated pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Disease control rate (DCR) |
76%
|
|||
| Patients Enrolled |
64 patients with paraganglioma.
|
||||
| Administration Time | 1-10 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The use of PRRT for patients with PPGLs who had high tumor SSTR expression has mainly been reported in retrospective small case series and the protocols varied in all these studies. So far, four meta-analyses (Taieb et al. and Satapathy et al. in 2019 as well as Su et al. and Marretta et al. in 2023) based on PRRT (177Lu/90Y-DOTAT-TATE/TOC) therapy have been reported. The reported number of pooled patients ranged from 179-330 without any complete response. The reported partial response ranged from 20-25% and stable disease from 59-70% giving an objective response rate range of 20-25% and disease control rate range of 8190%. The survival rates were reported by only 3 meta-analyses. The reported progression-free survival were 17-39 months in seven studies by Taieb et al., 10-91 months in 4 studies and a mean of 37.1 months in 2 studies by Satapathy et al., and a median of 31.8 months in 13 studies by Su et al. whereas overall survival were 49.6-68.0 months in 3 studies by Taieb et al., a mean of 54.5 months in 4 studies by Satapathy et al. and a median of 74.3 months in 11 studies by Su et al. Su et al. reports a pooled adverse events data in 274/330 patients which mentions a grade 3/4 hematotoxicity in 4.3% (grade 1-4, 22.3%) and grade 3/4 nephrotoxicity in 1.9% (grade 1-4, 1.9%), and myelodysplastic syndrome in 1 patient. Treatment discontinuation occurred in 4 patients due to thrombocytopenia (n=3) or nephrotoxicity (n=1).
Click to Show/Hide
|
||||
| Experiment 52 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Medullary thyroid carcinoma | ||||
| Efficacy Data | Disease control rate (DCR) |
84%
|
|||
| Patients Enrolled |
A total of 201 patients with advanced paragangliomas.
|
||||
| MOA of PDC |
The clinical applications of radiolabeled SSAs and DOTA-SSA compounds in imaging and radionuclide therapy have been rapidly increasing over the past 2 decades and have been integrated into the current management guidelines of NETs. Several studies support the safety and clinical efficacy of FDA-approved radionuclide therapies including 177Lu-DOTATATE in advanced GEP NETs and 131I-MIBG in advanced paragangliomas and pheochromocytomas. The utility of SSTR-based imaging in PRRT response assessment and surveillance of NETs remains to be established.
Click to Show/Hide
|
||||
| Description |
Over the past decade, a small number of studies have evaluated the efficacy and safety of PRRT with 177Lu-DOTA-SSA in patients with metastatic or inoperable paragangliomas [66, 67]. A recent meta-analysis of 12 studies including a total of 201 patients with advanced paragangliomas showed an objective response rate of 25%, biochemical response rate of 64%, and disease control rate of 84% after treatment with PRRT (either 90Y- or 177Lu-based agents) [67]. The following minimal treatment-related adverse effects were observed: grade 3 or 4 lymphopenia, thrombocytopenia, neutropenia, and nephrotoxicity in 11%, 9%, 3%, and 4% of the patients, respectively.
Click to Show/Hide
|
||||
| Experiment 53 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Well-differentiated pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Disease control rate (DCR) |
84%
|
|||
| Patients Enrolled |
201 patients with paraganglioma.
|
||||
| Administration Time | 1-10 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The use of PRRT for patients with PPGLs who had high tumor SSTR expression has mainly been reported in retrospective small case series and the protocols varied in all these studies. So far, four meta-analyses (Taieb et al. and Satapathy et al. in 2019 as well as Su et al. and Marretta et al. in 2023) based on PRRT (177Lu/90Y-DOTAT-TATE/TOC) therapy have been reported. The reported number of pooled patients ranged from 179-330 without any complete response. The reported partial response ranged from 20-25% and stable disease from 59-70% giving an objective response rate range of 20-25% and disease control rate range of 8190%. The survival rates were reported by only 3 meta-analyses. The reported progression-free survival were 17-39 months in seven studies by Taieb et al., 10-91 months in 4 studies and a mean of 37.1 months in 2 studies by Satapathy et al., and a median of 31.8 months in 13 studies by Su et al. whereas overall survival were 49.6-68.0 months in 3 studies by Taieb et al., a mean of 54.5 months in 4 studies by Satapathy et al. and a median of 74.3 months in 11 studies by Su et al. Su et al. reports a pooled adverse events data in 274/330 patients which mentions a grade 3/4 hematotoxicity in 4.3% (grade 1-4, 22.3%) and grade 3/4 nephrotoxicity in 1.9% (grade 1-4, 1.9%), and myelodysplastic syndrome in 1 patient. Treatment discontinuation occurred in 4 patients due to thrombocytopenia (n=3) or nephrotoxicity (n=1).
Click to Show/Hide
|
||||
| Experiment 54 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Well-differentiated pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Disease control rate (DCR) |
89.60%
|
|||
| Patients Enrolled |
179 patients with paraganglioma.
|
||||
| Administration Time | 1-11 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The use of PRRT for patients with PPGLs who had high tumor SSTR expression has mainly been reported in retrospective small case series and the protocols varied in all these studies. So far, four meta-analyses (Taieb et al. and Satapathy et al. in 2019 as well as Su et al. and Marretta et al. in 2023) based on PRRT (177Lu/90Y-DOTAT-TATE/TOC) therapy have been reported. The reported number of pooled patients ranged from 179-330 without any complete response. The reported partial response ranged from 20-25% and stable disease from 59-70% giving an objective response rate range of 20-25% and disease control rate range of 8190%. The survival rates were reported by only 3 meta-analyses. The reported progression-free survival were 17-39 months in seven studies by Taieb et al., 10-91 months in 4 studies and a mean of 37.1 months in 2 studies by Satapathy et al., and a median of 31.8 months in 13 studies by Su et al. whereas overall survival were 49.6-68.0 months in 3 studies by Taieb et al., a mean of 54.5 months in 4 studies by Satapathy et al. and a median of 74.3 months in 11 studies by Su et al. Su et al. reports a pooled adverse events data in 274/330 patients which mentions a grade 3/4 hematotoxicity in 4.3% (grade 1-4, 22.3%) and grade 3/4 nephrotoxicity in 1.9% (grade 1-4, 1.9%), and myelodysplastic syndrome in 1 patient. Treatment discontinuation occurred in 4 patients due to thrombocytopenia (n=3) or nephrotoxicity (n=1).
Click to Show/Hide
|
||||
| Experiment 55 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Well-differentiated pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Disease control rate (DCR) |
90%
|
|||
| Patients Enrolled |
330 patients with paraganglioma.
|
||||
| Administration Time | 1-10 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
The use of PRRT for patients with PPGLs who had high tumor SSTR expression has mainly been reported in retrospective small case series and the protocols varied in all these studies. So far, four meta-analyses (Taieb et al. and Satapathy et al. in 2019 as well as Su et al. and Marretta et al. in 2023) based on PRRT (177Lu/90Y-DOTAT-TATE/TOC) therapy have been reported. The reported number of pooled patients ranged from 179-330 without any complete response. The reported partial response ranged from 20-25% and stable disease from 59-70% giving an objective response rate range of 20-25% and disease control rate range of 8190%. The survival rates were reported by only 3 meta-analyses. The reported progression-free survival were 17-39 months in seven studies by Taieb et al., 10-91 months in 4 studies and a mean of 37.1 months in 2 studies by Satapathy et al., and a median of 31.8 months in 13 studies by Su et al. whereas overall survival were 49.6-68.0 months in 3 studies by Taieb et al., a mean of 54.5 months in 4 studies by Satapathy et al. and a median of 74.3 months in 11 studies by Su et al. Su et al. reports a pooled adverse events data in 274/330 patients which mentions a grade 3/4 hematotoxicity in 4.3% (grade 1-4, 22.3%) and grade 3/4 nephrotoxicity in 1.9% (grade 1-4, 1.9%), and myelodysplastic syndrome in 1 patient. Treatment discontinuation occurred in 4 patients due to thrombocytopenia (n=3) or nephrotoxicity (n=1).
Click to Show/Hide
|
||||
| Experiment 56 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Small bowel neuroendocrine tumor | ||||
| Efficacy Data | Disease control rate (DCR) |
100%
|
|||
| Patients Enrolled |
29 patients with neuroendocrine tumour.
|
||||
| MOA of PDC |
In conclusion, our patient is a good example of re-treatment with PRRT, due to its initial response to 177Lu-DOTATATE, which lasted more than 3 years. In that moment, two additional cycles of PRRT were administered, reaching again partial response without significant toxicity. 177Lu-DOTATATE is an effective therapy in NETs with an excellent safety profile. There is evidence that salvage therapy following progression to PRRT after a long response is an option in these patients, with high disease control rates and acceptable safety profile. Nevertheless, large prospective randomized studies are needed to confirm these findings.
Click to Show/Hide
|
||||
| Description |
This case report is a good example of the efficacy and safety of salvage therapy with 177Lu-DOTATATE in heavily pretreated patients after an initial response to PRRT, an especially challenging context with a limited number of alternatives. Currently, 177Lu-DOTATATE is an established therapeutic option for the treatment of metastatic NETs. However, there is a lack of evidence for salvage therapy. Recently, published studies have shown the efficacy and acceptable tolerance of re-treatment with PRRT. Nevertheless, these studies are heterogeneous and mostly retrospective, with significant differences between them regarding the patients included, the cumulative dose of PRRT, or the radiolabeled drug used.
Click to Show/Hide
|
||||
| Experiment 57 Reporting the Activity Data of This PDC | [50] | ||||
| Indication | Medullary thyroid carcinoma | ||||
| Efficacy Data | Death rate |
43.75%
|
|||
| Patients Enrolled |
144 patients with metastatic medullary thyroid carcinoma.
|
||||
| MOA of PDC |
Findings of the study revealed that in the absence of the established treatment for the patients with RR-DTC and metastatic MTC, PRRT could be effective with few adverse events.
|
||||
| Description |
Among 2284 related papers, 41 papers met the inclusion criteria. A total of 157 patients with RR-DTC were treated with PPRT. Biochemical and objective responses (partial and complete) were observed in 25.3 and 10.5% of patients, respectively. Among 220 patients with metastatic MTC, biochemical and objective responses were observed in 37.2 and 10.6% of the patients, respectively. Forty-six deaths were reported in 95 patients with advanced RR-DTC. In addition, 63 deaths were observed in 144 patients with metastatic MTC. Major side effects were reported in 124 patients treated with 90Y -based agent. In the patients treated with 177Lu-DOTA-TATE and 111In-Octreotide, mild and transient hematologic or renal complications were reported.
Click to Show/Hide
|
||||
| Experiment 58 Reporting the Activity Data of This PDC | [50] | ||||
| Indication | Differentiated thyroid cancer | ||||
| Efficacy Data | Death rate |
48.42%
|
|||
| Patients Enrolled |
95 patients with advanced radioiodine-refractory differentiated thyroid cancer.
|
||||
| MOA of PDC |
Findings of the study revealed that in the absence of the established treatment for the patients with RR-DTC and metastatic MTC, PRRT could be effective with few adverse events.
|
||||
| Description |
Among 2284 related papers, 41 papers met the inclusion criteria. A total of 157 patients with RR-DTC were treated with PPRT. Biochemical and objective responses (partial and complete) were observed in 25.3 and 10.5% of patients, respectively. Among 220 patients with metastatic MTC, biochemical and objective responses were observed in 37.2 and 10.6% of the patients, respectively. Forty-six deaths were reported in 95 patients with advanced RR-DTC. In addition, 63 deaths were observed in 144 patients with metastatic MTC. Major side effects were reported in 124 patients treated with 90Y -based agent. In the patients treated with 177Lu-DOTA-TATE and 111In-Octreotide, mild and transient hematologic or renal complications were reported.
Click to Show/Hide
|
||||
| Experiment 59 Reporting the Activity Data of This PDC | [50] | ||||
| Indication | Differentiated thyroid cancer | ||||
| Efficacy Data | Biochemical response rate |
25.30%
|
|||
| Patients Enrolled |
157 patients with RR-DTC treated with PPRT.
|
||||
| MOA of PDC |
Findings of the study revealed that in the absence of the established treatment for the patients with RR-DTC and metastatic MTC, PRRT could be effective with few adverse events.
|
||||
| Description |
Among 2284 related papers, 41 papers met the inclusion criteria. A total of 157 patients with RR-DTC were treated with PPRT. Biochemical and objective responses (partial and complete) were observed in 25.3 and 10.5% of patients, respectively. Among 220 patients with metastatic MTC, biochemical and objective responses were observed in 37.2 and 10.6% of the patients, respectively. Forty-six deaths were reported in 95 patients with advanced RR-DTC. In addition, 63 deaths were observed in 144 patients with metastatic MTC. Major side effects were reported in 124 patients treated with 90Y -based agent. In the patients treated with 177Lu-DOTA-TATE and 111In-Octreotide, mild and transient hematologic or renal complications were reported.
Click to Show/Hide
|
||||
| Experiment 60 Reporting the Activity Data of This PDC | [50] | ||||
| Indication | Medullary thyroid carcinoma | ||||
| Efficacy Data | Biochemical response rate |
37.20%
|
|||
| Patients Enrolled |
220 patients with metastatic medullary thyroid cancer.
|
||||
| MOA of PDC |
Findings of the study revealed that in the absence of the established treatment for the patients with RR-DTC and metastatic MTC, PRRT could be effective with few adverse events.
|
||||
| Description |
Among 2284 related papers, 41 papers met the inclusion criteria. A total of 157 patients with RR-DTC were treated with PPRT. Biochemical and objective responses (partial and complete) were observed in 25.3 and 10.5% of patients, respectively. Among 220 patients with metastatic MTC, biochemical and objective responses were observed in 37.2 and 10.6% of the patients, respectively. Forty-six deaths were reported in 95 patients with advanced RR-DTC. In addition, 63 deaths were observed in 144 patients with metastatic MTC. Major side effects were reported in 124 patients treated with 90Y -based agent. In the patients treated with 177Lu-DOTA-TATE and 111In-Octreotide, mild and transient hematologic or renal complications were reported.
Click to Show/Hide
|
||||
| Experiment 61 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Medullary thyroid carcinoma | ||||
| Efficacy Data | Biochemical response rate |
64%
|
|||
| Patients Enrolled |
A total of 201 patients with advanced paragangliomas.
|
||||
| MOA of PDC |
The clinical applications of radiolabeled SSAs and DOTA-SSA compounds in imaging and radionuclide therapy have been rapidly increasing over the past 2 decades and have been integrated into the current management guidelines of NETs. Several studies support the safety and clinical efficacy of FDA-approved radionuclide therapies including 177Lu-DOTATATE in advanced GEP NETs and 131I-MIBG in advanced paragangliomas and pheochromocytomas. The utility of SSTR-based imaging in PRRT response assessment and surveillance of NETs remains to be established.
Click to Show/Hide
|
||||
| Description |
Over the past decade, a small number of studies have evaluated the efficacy and safety of PRRT with 177Lu-DOTA-SSA in patients with metastatic or inoperable paragangliomas [66, 67]. A recent meta-analysis of 12 studies including a total of 201 patients with advanced paragangliomas showed an objective response rate of 25%, biochemical response rate of 64%, and disease control rate of 84% after treatment with PRRT (either 90Y- or 177Lu-based agents) [67]. The following minimal treatment-related adverse effects were observed: grade 3 or 4 lymphopenia, thrombocytopenia, neutropenia, and nephrotoxicity in 11%, 9%, 3%, and 4% of the patients, respectively.
Click to Show/Hide
|
||||
| Experiment 62 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Well-differentiated pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Anemia |
15.38%
|
|||
| Patients Enrolled |
13 patients with pheocromocytomas (PCCs) and paragangliomas (PGLs) (4 PCC , 9PGL).
|
||||
| Administration Time | 61% of patients received 2 cycles | ||||
| Administration Dosage | 3.4 GBq/cycle | ||||
| MOA of PDC |
We showed that PRRT based on 177Lu-DOTATATE and 90-YDOTATOC without radiosensitizers are efficacious therapeutic options (DCR of 83% and 76%, respectively) and can be considered in the multidisciplinary treatment of PCCs and PGLs as alternatives to I-131 MIBG and chemotherapy.
|
||||
| Description |
In detail, we report pooled data on 149 177Lu- and 64 90Y-treated patients (total: 213 patients). Most studies were retrospective. The largest one included 46 patients (34 treated with 177Lu-, 12 with 90Y- PRRTs). Median ages ranged from 32.5 to 60.4 years. The female gender was slightly predominant (83 women, 74 men, gender not specified in 3 studies). When reported, mutations of SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B) were the most frequent genetic alterations. According to the predominant retrospective and real practice nature of the selected studies, sites of metastases and previous treatments were heterogeneous, with bone the metastatic site most often involved, and surgery the most frequent previous therapy. Paramount descriptive information on PRRT doses per cycle, number of cycles, response criteria, best responses to treatments, and frequency of G3/G4 hematologic toxicities is reported in Table 2. The lowest applied dose was 3.4 GBq per cycle; the number of administered cycles was heterogeneous (at least 3 cycles in 7 studies). The most used response criteria were the RECIST 1.1. Only one study evaluated the response with SWOG criteria, which are more conservative with regard to the definition of PR (50% or greater reduction in the sum of the product of the perpendicular diameters of target lesions). The majority of the patients experienced PR or SD. Hematologic toxicity was manageable. Funnel plot of selected studies did not show publication bias. The MINORS and Newcastle-Ottawa Scale scoring for the selected articles are reported in Table 3.
Click to Show/Hide
|
||||
68Ga-DOTANOC [Phase 1]
Identified from the Human Clinical Data
| Experiment 1 Reporting the Activity Data of This PDC | [61] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Difference in detection ratio |
27.50%
|
|||
| Patients Enrolled |
75 patients with histologically confirmed neuroendocrine tumours and routine clinical.
|
||||
| MOA of PDC |
18F-AlF-OC is noninferior and even superior to 68Ga-DOTATATE/NOC PET in NET patients. This validates 18F-AlF-OC as an option for clinical practice somatostatin receptor PET.
|
||||
| Description |
The resulting mean DDR was 15.8%, with a lower margin of the 95% CI (95% CI, 9.6%-22.0%) higher than -15%, which is the prespecified boundary for noninferiority. The mean DDRs for the 68Ga-DOTATATE and 68Ga-DOTANOC subgroups were 11.8% (95% CI, 4.3-19.3) and 27.5% (95% CI, 17.8-37.1), respectively.
|
||||
Piflufolastat F-18 [Investigative]
Identified from the Human Clinical Data
| Experiment 1 Reporting the Activity Data of This PDC | [72] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Specificity |
98%
|
|||
| Patients Enrolled |
252 patients with prostate cancer.
|
||||
| Administration Dosage | 8-10 mCi | ||||
| Evaluation Method | PET/CT | ||||
| MOA of PDC |
PSMA, also known as glutamate carboxypeptidase 2 or folate hydrolase 1, is highly expressed in nearly all prostate cancers. Structurally, PSMA is a large extracellular carboxypeptidase domain that is tethered to the cellular membrane by a short transmembrane domain. Physiologically, PSMA is also expressed in normal tissue, including non-malignant prostate tissue, the kidney, the small intestine, and the central nervous system. In most tissues, PSMA likely contributes to folate uptake by cleaving the C-terminal glutamate from folic acid (Figure 1). Within the brain, PSMA regulates the level of N-acetylaspartylglutamate (NAAG) by catalyzing its degradation to glutamate. Structure of PSMA substrates and inhibitors. Left: structure of the PSMA substrate folic acid. The peptide bond (dashed box) linking the P1 glutamate residue (blue) to the remainder of the substrate (P1, red) is cleaved by PSMA. Right: Structure of the PSMA-PET agents 18F-DCFPyL and 68Ga-PSMA-11. The scissile peptide bond has been replaced by a non-hydrolyzable urea motif (red) to generate PMSA inhibitors. The P1 glutamate residue is essential for binding to PSMA, but a wide degree of variability is tolerated in the opposing end of the structure.
Click to Show/Hide
|
||||
| Description |
Within cohort A, the primary end point was the sensitivity and specificity for detection of metastases to the pelvic lymph nodes by 18F-DCFPyL PET/CT. These findings were validated by histopathology after radical proctectomy and lymph node dissection (Figure 6). When all confirmed positive lymph node metastases were considered, the sensitivity of 18F-DCFPyL PET/CT was 40%, with a 98% specificity. It is perhaps unsurprising that histopathology is more sensitivity for the detection of early metastases as a sufficient number of malignant cells must accumulate before any imaging modality will be positive. However, a post-hoc analysis considering only lesions >5 mm found the sensitivity of 18F-DCFPyL PET/CT increased to 60%. Despite this, across all lesions, 18F-DCFPyL PET/CT significantly outperformed conventional CT pelvic imaging with markedly improved specificity (97.7% vs. 65.1%) and positive predictive value (85.1% vs. 28.3%) for pelvic lymph node metastases, as well as sensitivity for disease within the prostate (96.8% vs. 35.9%).
Click to Show/Hide
|
||||
| Half life period | 8 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [72] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Sensitivity |
40%
|
|||
| Patients Enrolled |
252 patients with prostate cancer.
|
||||
| Administration Dosage | 8-10 mCi | ||||
| Evaluation Method | PET/CT | ||||
| MOA of PDC |
PSMA, also known as glutamate carboxypeptidase 2 or folate hydrolase 1, is highly expressed in nearly all prostate cancers. Structurally, PSMA is a large extracellular carboxypeptidase domain that is tethered to the cellular membrane by a short transmembrane domain. Physiologically, PSMA is also expressed in normal tissue, including non-malignant prostate tissue, the kidney, the small intestine, and the central nervous system. In most tissues, PSMA likely contributes to folate uptake by cleaving the C-terminal glutamate from folic acid (Figure 1). Within the brain, PSMA regulates the level of N-acetylaspartylglutamate (NAAG) by catalyzing its degradation to glutamate. Structure of PSMA substrates and inhibitors. Left: structure of the PSMA substrate folic acid. The peptide bond (dashed box) linking the P1 glutamate residue (blue) to the remainder of the substrate (P1, red) is cleaved by PSMA. Right: Structure of the PSMA-PET agents 18F-DCFPyL and 68Ga-PSMA-11. The scissile peptide bond has been replaced by a non-hydrolyzable urea motif (red) to generate PMSA inhibitors. The P1 glutamate residue is essential for binding to PSMA, but a wide degree of variability is tolerated in the opposing end of the structure.
Click to Show/Hide
|
||||
| Description |
Within cohort A, the primary end point was the sensitivity and specificity for detection of metastases to the pelvic lymph nodes by 18F-DCFPyL PET/CT. These findings were validated by histopathology after radical proctectomy and lymph node dissection (Figure 6). When all confirmed positive lymph node metastases were considered, the sensitivity of 18F-DCFPyL PET/CT was 40%, with a 98% specificity. It is perhaps unsurprising that histopathology is more sensitivity for the detection of early metastases as a sufficient number of malignant cells must accumulate before any imaging modality will be positive. However, a post-hoc analysis considering only lesions >5 mm found the sensitivity of 18F-DCFPyL PET/CT increased to 60%. Despite this, across all lesions, 18F-DCFPyL PET/CT significantly outperformed conventional CT pelvic imaging with markedly improved specificity (97.7% vs. 65.1%) and positive predictive value (85.1% vs. 28.3%) for pelvic lymph node metastases, as well as sensitivity for disease within the prostate (96.8% vs. 35.9%).
Click to Show/Hide
|
||||
| Half life period | 8 h | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [72] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Sensitivity |
86%
|
|||
| Patients Enrolled |
208 patients with prostate cancer.
|
||||
| Administration Dosage | 8-10 mCi | ||||
| Evaluation Method | PET/CT | ||||
| MOA of PDC |
PSMA, also known as glutamate carboxypeptidase 2 or folate hydrolase 1, is highly expressed in nearly all prostate cancers. Structurally, PSMA is a large extracellular carboxypeptidase domain that is tethered to the cellular membrane by a short transmembrane domain. Physiologically, PSMA is also expressed in normal tissue, including non-malignant prostate tissue, the kidney, the small intestine, and the central nervous system. In most tissues, PSMA likely contributes to folate uptake by cleaving the C-terminal glutamate from folic acid (Figure 1). Within the brain, PSMA regulates the level of N-acetylaspartylglutamate (NAAG) by catalyzing its degradation to glutamate. Structure of PSMA substrates and inhibitors. Left: structure of the PSMA substrate folic acid. The peptide bond (dashed box) linking the P1 glutamate residue (blue) to the remainder of the substrate (P1, red) is cleaved by PSMA. Right: Structure of the PSMA-PET agents 18F-DCFPyL and 68Ga-PSMA-11. The scissile peptide bond has been replaced by a non-hydrolyzable urea motif (red) to generate PMSA inhibitors. The P1 glutamate residue is essential for binding to PSMA, but a wide degree of variability is tolerated in the opposing end of the structure.
Click to Show/Hide
|
||||
| Description |
All patients were dosed with 9 mCi of 18F-DCFPyL and PET/CT images were obtained. 18F-DCFPyL avid lesions were detected in ~60% of the patients. To confirm the metastatic nature of these lesions, biopsies were obtained, if possible. If the lesion was not amendable to biopsy, validation is obtained by focused conventional follow-up imaging or biochemical response to external beam radiotherapy. The differentiation between the number of true metastases validated by other techniques (i.e. true positives, TP) and false positives (FP) allows for the calculation of the correct localization rate (CLR), where the CLR = TP/(TP + FP) * 100 (essentially, PPV with the added requirement of anatomic co-localization). Across all patients, the CLR ranged from 85% to 87% between readers, however the CLR improved as the baseline PSA increased, from 75% to 96% (PSA < 1.0 to PSA ≥ 5.0, respectively). The PPV of the 18F-DCFPyL PET/CT was found to vary by anatomic region and was highest for prostatic lesions (80%) and pelvic lymph nodes (71%), but lower for extrapelvic visceral/soft tissue masses (29%).
Click to Show/Hide
|
||||
| Half life period | 8 h | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [72] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Sensitivity |
96%
|
|||
| Patients Enrolled |
93 patients with prostate cancer.
|
||||
| Administration Dosage | 8-10 mCi | ||||
| Evaluation Method | PET/CT | ||||
| MOA of PDC |
PSMA, also known as glutamate carboxypeptidase 2 or folate hydrolase 1, is highly expressed in nearly all prostate cancers. Structurally, PSMA is a large extracellular carboxypeptidase domain that is tethered to the cellular membrane by a short transmembrane domain. Physiologically, PSMA is also expressed in normal tissue, including non-malignant prostate tissue, the kidney, the small intestine, and the central nervous system. In most tissues, PSMA likely contributes to folate uptake by cleaving the C-terminal glutamate from folic acid (Figure 1). Within the brain, PSMA regulates the level of N-acetylaspartylglutamate (NAAG) by catalyzing its degradation to glutamate. Structure of PSMA substrates and inhibitors. Left: structure of the PSMA substrate folic acid. The peptide bond (dashed box) linking the P1 glutamate residue (blue) to the remainder of the substrate (P1, red) is cleaved by PSMA. Right: Structure of the PSMA-PET agents 18F-DCFPyL and 68Ga-PSMA-11. The scissile peptide bond has been replaced by a non-hydrolyzable urea motif (red) to generate PMSA inhibitors. The P1 glutamate residue is essential for binding to PSMA, but a wide degree of variability is tolerated in the opposing end of the structure.
Click to Show/Hide
|
||||
| Description |
Within cohort A, the primary end point was the sensitivity and specificity for detection of metastases to the pelvic lymph nodes by 18F-DCFPyL PET/CT. These findings were validated by histopathology after radical proctectomy and lymph node dissection (Figure 6). When all confirmed positive lymph node metastases were considered, the sensitivity of 18F-DCFPyL PET/CT was 40%, with a 98% specificity. It is perhaps unsurprising that histopathology is more sensitivity for the detection of early metastases as a sufficient number of malignant cells must accumulate before any imaging modality will be positive. However, a post-hoc analysis considering only lesions >5 mm found the sensitivity of 18F-DCFPyL PET/CT increased to 60%. Despite this, across all lesions, 18F-DCFPyL PET/CT significantly outperformed conventional CT pelvic imaging with markedly improved specificity (97.7% vs. 65.1%) and positive predictive value (85.1% vs. 28.3%) for pelvic lymph node metastases, as well as sensitivity for disease within the prostate (96.8% vs. 35.9%).
Click to Show/Hide
|
||||
| Half life period | 8 h | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [72] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Negative predictive value |
83.00%
|
|||
| Patients Enrolled |
252 patients with prostate cancer.
|
||||
| Administration Dosage | 8-10 mCi | ||||
| Evaluation Method | PET/CT | ||||
| MOA of PDC |
PSMA, also known as glutamate carboxypeptidase 2 or folate hydrolase 1, is highly expressed in nearly all prostate cancers. Structurally, PSMA is a large extracellular carboxypeptidase domain that is tethered to the cellular membrane by a short transmembrane domain. Physiologically, PSMA is also expressed in normal tissue, including non-malignant prostate tissue, the kidney, the small intestine, and the central nervous system. In most tissues, PSMA likely contributes to folate uptake by cleaving the C-terminal glutamate from folic acid (Figure 1). Within the brain, PSMA regulates the level of N-acetylaspartylglutamate (NAAG) by catalyzing its degradation to glutamate. Structure of PSMA substrates and inhibitors. Left: structure of the PSMA substrate folic acid. The peptide bond (dashed box) linking the P1 glutamate residue (blue) to the remainder of the substrate (P1, red) is cleaved by PSMA. Right: Structure of the PSMA-PET agents 18F-DCFPyL and 68Ga-PSMA-11. The scissile peptide bond has been replaced by a non-hydrolyzable urea motif (red) to generate PMSA inhibitors. The P1 glutamate residue is essential for binding to PSMA, but a wide degree of variability is tolerated in the opposing end of the structure.
Click to Show/Hide
|
||||
| Description |
Within cohort A, the primary end point was the sensitivity and specificity for detection of metastases to the pelvic lymph nodes by 18F-DCFPyL PET/CT. These findings were validated by histopathology after radical proctectomy and lymph node dissection (Figure 6). When all confirmed positive lymph node metastases were considered, the sensitivity of 18F-DCFPyL PET/CT was 40%, with a 98% specificity. It is perhaps unsurprising that histopathology is more sensitivity for the detection of early metastases as a sufficient number of malignant cells must accumulate before any imaging modality will be positive. However, a post-hoc analysis considering only lesions >5 mm found the sensitivity of 18F-DCFPyL PET/CT increased to 60%. Despite this, across all lesions, 18F-DCFPyL PET/CT significantly outperformed conventional CT pelvic imaging with markedly improved specificity (97.7% vs. 65.1%) and positive predictive value (85.1% vs. 28.3%) for pelvic lymph node metastases, as well as sensitivity for disease within the prostate (96.8% vs. 35.9%).
Click to Show/Hide
|
||||
| Half life period | 8 h | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [72] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Hypertension |
1%
|
|||
| Patients Enrolled |
208 patients with prostate cancer.
|
||||
| Administration Dosage | 8-10 mCi | ||||
| Evaluation Method | PET/CT | ||||
| MOA of PDC |
PSMA, also known as glutamate carboxypeptidase 2 or folate hydrolase 1, is highly expressed in nearly all prostate cancers. Structurally, PSMA is a large extracellular carboxypeptidase domain that is tethered to the cellular membrane by a short transmembrane domain. Physiologically, PSMA is also expressed in normal tissue, including non-malignant prostate tissue, the kidney, the small intestine, and the central nervous system. In most tissues, PSMA likely contributes to folate uptake by cleaving the C-terminal glutamate from folic acid (Figure 1). Within the brain, PSMA regulates the level of N-acetylaspartylglutamate (NAAG) by catalyzing its degradation to glutamate. Structure of PSMA substrates and inhibitors. Left: structure of the PSMA substrate folic acid. The peptide bond (dashed box) linking the P1 glutamate residue (blue) to the remainder of the substrate (P1, red) is cleaved by PSMA. Right: Structure of the PSMA-PET agents 18F-DCFPyL and 68Ga-PSMA-11. The scissile peptide bond has been replaced by a non-hydrolyzable urea motif (red) to generate PMSA inhibitors. The P1 glutamate residue is essential for binding to PSMA, but a wide degree of variability is tolerated in the opposing end of the structure.
Click to Show/Hide
|
||||
| Description |
All patients were dosed with 9 mCi of 18F-DCFPyL and PET/CT images were obtained. 18F-DCFPyL avid lesions were detected in ~60% of the patients. To confirm the metastatic nature of these lesions, biopsies were obtained, if possible. If the lesion was not amendable to biopsy, validation is obtained by focused conventional follow-up imaging or biochemical response to external beam radiotherapy. The differentiation between the number of true metastases validated by other techniques (i.e. true positives, TP) and false positives (FP) allows for the calculation of the correct localization rate (CLR), where the CLR = TP/(TP + FP) * 100 (essentially, PPV with the added requirement of anatomic co-localization). Across all patients, the CLR ranged from 85% to 87% between readers, however the CLR improved as the baseline PSA increased, from 75% to 96% (PSA < 1.0 to PSA ≥ 5.0, respectively). The PPV of the 18F-DCFPyL PET/CT was found to vary by anatomic region and was highest for prostatic lesions (80%) and pelvic lymph nodes (71%), but lower for extrapelvic visceral/soft tissue masses (29%).
Click to Show/Hide
|
||||
| Half life period | 8 h | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [72] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Headache rate |
2%
|
|||
| Patients Enrolled |
345 patients with prostate cancer.
|
||||
| Administration Dosage | 8-10 mCi | ||||
| Evaluation Method | PET/CT | ||||
| MOA of PDC |
PSMA, also known as glutamate carboxypeptidase 2 or folate hydrolase 1, is highly expressed in nearly all prostate cancers. Structurally, PSMA is a large extracellular carboxypeptidase domain that is tethered to the cellular membrane by a short transmembrane domain. Physiologically, PSMA is also expressed in normal tissue, including non-malignant prostate tissue, the kidney, the small intestine, and the central nervous system. In most tissues, PSMA likely contributes to folate uptake by cleaving the C-terminal glutamate from folic acid (Figure 1). Within the brain, PSMA regulates the level of N-acetylaspartylglutamate (NAAG) by catalyzing its degradation to glutamate. Structure of PSMA substrates and inhibitors. Left: structure of the PSMA substrate folic acid. The peptide bond (dashed box) linking the P1 glutamate residue (blue) to the remainder of the substrate (P1, red) is cleaved by PSMA. Right: Structure of the PSMA-PET agents 18F-DCFPyL and 68Ga-PSMA-11. The scissile peptide bond has been replaced by a non-hydrolyzable urea motif (red) to generate PMSA inhibitors. The P1 glutamate residue is essential for binding to PSMA, but a wide degree of variability is tolerated in the opposing end of the structure.
Click to Show/Hide
|
||||
| Description |
Within cohort A, the primary end point was the sensitivity and specificity for detection of metastases to the pelvic lymph nodes by 18F-DCFPyL PET/CT. These findings were validated by histopathology after radical proctectomy and lymph node dissection (Figure 6). When all confirmed positive lymph node metastases were considered, the sensitivity of 18F-DCFPyL PET/CT was 40%, with a 98% specificity. It is perhaps unsurprising that histopathology is more sensitivity for the detection of early metastases as a sufficient number of malignant cells must accumulate before any imaging modality will be positive. However, a post-hoc analysis considering only lesions >5 mm found the sensitivity of 18F-DCFPyL PET/CT increased to 60%. Despite this, across all lesions, 18F-DCFPyL PET/CT significantly outperformed conventional CT pelvic imaging with markedly improved specificity (97.7% vs. 65.1%) and positive predictive value (85.1% vs. 28.3%) for pelvic lymph node metastases, as well as sensitivity for disease within the prostate (96.8% vs. 35.9%).
Click to Show/Hide
|
||||
| Half life period | 8 h | ||||
| Experiment 8 Reporting the Activity Data of This PDC | [72] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Headache rate |
2%
|
|||
| Patients Enrolled |
208 patients with prostate cancer.
|
||||
| Administration Dosage | 8-10 mCi | ||||
| Evaluation Method | PET/CT | ||||
| MOA of PDC |
PSMA, also known as glutamate carboxypeptidase 2 or folate hydrolase 1, is highly expressed in nearly all prostate cancers. Structurally, PSMA is a large extracellular carboxypeptidase domain that is tethered to the cellular membrane by a short transmembrane domain. Physiologically, PSMA is also expressed in normal tissue, including non-malignant prostate tissue, the kidney, the small intestine, and the central nervous system. In most tissues, PSMA likely contributes to folate uptake by cleaving the C-terminal glutamate from folic acid (Figure 1). Within the brain, PSMA regulates the level of N-acetylaspartylglutamate (NAAG) by catalyzing its degradation to glutamate. Structure of PSMA substrates and inhibitors. Left: structure of the PSMA substrate folic acid. The peptide bond (dashed box) linking the P1 glutamate residue (blue) to the remainder of the substrate (P1, red) is cleaved by PSMA. Right: Structure of the PSMA-PET agents 18F-DCFPyL and 68Ga-PSMA-11. The scissile peptide bond has been replaced by a non-hydrolyzable urea motif (red) to generate PMSA inhibitors. The P1 glutamate residue is essential for binding to PSMA, but a wide degree of variability is tolerated in the opposing end of the structure.
Click to Show/Hide
|
||||
| Description |
All patients were dosed with 9 mCi of 18F-DCFPyL and PET/CT images were obtained. 18F-DCFPyL avid lesions were detected in ~60% of the patients. To confirm the metastatic nature of these lesions, biopsies were obtained, if possible. If the lesion was not amendable to biopsy, validation is obtained by focused conventional follow-up imaging or biochemical response to external beam radiotherapy. The differentiation between the number of true metastases validated by other techniques (i.e. true positives, TP) and false positives (FP) allows for the calculation of the correct localization rate (CLR), where the CLR = TP/(TP + FP) * 100 (essentially, PPV with the added requirement of anatomic co-localization). Across all patients, the CLR ranged from 85% to 87% between readers, however the CLR improved as the baseline PSA increased, from 75% to 96% (PSA < 1.0 to PSA ≥ 5.0, respectively). The PPV of the 18F-DCFPyL PET/CT was found to vary by anatomic region and was highest for prostatic lesions (80%) and pelvic lymph nodes (71%), but lower for extrapelvic visceral/soft tissue masses (29%).
Click to Show/Hide
|
||||
| Half life period | 8 h | ||||
| Experiment 9 Reporting the Activity Data of This PDC | [72] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Fatigue |
1%
|
|||
| Patients Enrolled |
345 patients with prostate cancer.
|
||||
| Administration Dosage | 8-10 mCi | ||||
| Evaluation Method | PET/CT | ||||
| MOA of PDC |
PSMA, also known as glutamate carboxypeptidase 2 or folate hydrolase 1, is highly expressed in nearly all prostate cancers. Structurally, PSMA is a large extracellular carboxypeptidase domain that is tethered to the cellular membrane by a short transmembrane domain. Physiologically, PSMA is also expressed in normal tissue, including non-malignant prostate tissue, the kidney, the small intestine, and the central nervous system. In most tissues, PSMA likely contributes to folate uptake by cleaving the C-terminal glutamate from folic acid (Figure 1). Within the brain, PSMA regulates the level of N-acetylaspartylglutamate (NAAG) by catalyzing its degradation to glutamate. Structure of PSMA substrates and inhibitors. Left: structure of the PSMA substrate folic acid. The peptide bond (dashed box) linking the P1 glutamate residue (blue) to the remainder of the substrate (P1, red) is cleaved by PSMA. Right: Structure of the PSMA-PET agents 18F-DCFPyL and 68Ga-PSMA-11. The scissile peptide bond has been replaced by a non-hydrolyzable urea motif (red) to generate PMSA inhibitors. The P1 glutamate residue is essential for binding to PSMA, but a wide degree of variability is tolerated in the opposing end of the structure.
Click to Show/Hide
|
||||
| Description |
Within cohort A, the primary end point was the sensitivity and specificity for detection of metastases to the pelvic lymph nodes by 18F-DCFPyL PET/CT. These findings were validated by histopathology after radical proctectomy and lymph node dissection (Figure 6). When all confirmed positive lymph node metastases were considered, the sensitivity of 18F-DCFPyL PET/CT was 40%, with a 98% specificity. It is perhaps unsurprising that histopathology is more sensitivity for the detection of early metastases as a sufficient number of malignant cells must accumulate before any imaging modality will be positive. However, a post-hoc analysis considering only lesions >5 mm found the sensitivity of 18F-DCFPyL PET/CT increased to 60%. Despite this, across all lesions, 18F-DCFPyL PET/CT significantly outperformed conventional CT pelvic imaging with markedly improved specificity (97.7% vs. 65.1%) and positive predictive value (85.1% vs. 28.3%) for pelvic lymph node metastases, as well as sensitivity for disease within the prostate (96.8% vs. 35.9%).
Click to Show/Hide
|
||||
| Half life period | 8 h | ||||
| Experiment 10 Reporting the Activity Data of This PDC | [72] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Fatigue |
1%
|
|||
| Patients Enrolled |
208 patients with prostate cancer.
|
||||
| Administration Dosage | 8-10 mCi | ||||
| Evaluation Method | PET/CT | ||||
| MOA of PDC |
PSMA, also known as glutamate carboxypeptidase 2 or folate hydrolase 1, is highly expressed in nearly all prostate cancers. Structurally, PSMA is a large extracellular carboxypeptidase domain that is tethered to the cellular membrane by a short transmembrane domain. Physiologically, PSMA is also expressed in normal tissue, including non-malignant prostate tissue, the kidney, the small intestine, and the central nervous system. In most tissues, PSMA likely contributes to folate uptake by cleaving the C-terminal glutamate from folic acid (Figure 1). Within the brain, PSMA regulates the level of N-acetylaspartylglutamate (NAAG) by catalyzing its degradation to glutamate. Structure of PSMA substrates and inhibitors. Left: structure of the PSMA substrate folic acid. The peptide bond (dashed box) linking the P1 glutamate residue (blue) to the remainder of the substrate (P1, red) is cleaved by PSMA. Right: Structure of the PSMA-PET agents 18F-DCFPyL and 68Ga-PSMA-11. The scissile peptide bond has been replaced by a non-hydrolyzable urea motif (red) to generate PMSA inhibitors. The P1 glutamate residue is essential for binding to PSMA, but a wide degree of variability is tolerated in the opposing end of the structure.
Click to Show/Hide
|
||||
| Description |
All patients were dosed with 9 mCi of 18F-DCFPyL and PET/CT images were obtained. 18F-DCFPyL avid lesions were detected in ~60% of the patients. To confirm the metastatic nature of these lesions, biopsies were obtained, if possible. If the lesion was not amendable to biopsy, validation is obtained by focused conventional follow-up imaging or biochemical response to external beam radiotherapy. The differentiation between the number of true metastases validated by other techniques (i.e. true positives, TP) and false positives (FP) allows for the calculation of the correct localization rate (CLR), where the CLR = TP/(TP + FP) * 100 (essentially, PPV with the added requirement of anatomic co-localization). Across all patients, the CLR ranged from 85% to 87% between readers, however the CLR improved as the baseline PSA increased, from 75% to 96% (PSA < 1.0 to PSA ≥ 5.0, respectively). The PPV of the 18F-DCFPyL PET/CT was found to vary by anatomic region and was highest for prostatic lesions (80%) and pelvic lymph nodes (71%), but lower for extrapelvic visceral/soft tissue masses (29%).
Click to Show/Hide
|
||||
| Half life period | 8 h | ||||
| Experiment 11 Reporting the Activity Data of This PDC | [72] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Dysgeusia |
3%
|
|||
| Patients Enrolled |
345 patients with prostate cancer.
|
||||
| Administration Dosage | 8-10 mCi | ||||
| Evaluation Method | PET/CT | ||||
| MOA of PDC |
PSMA, also known as glutamate carboxypeptidase 2 or folate hydrolase 1, is highly expressed in nearly all prostate cancers. Structurally, PSMA is a large extracellular carboxypeptidase domain that is tethered to the cellular membrane by a short transmembrane domain. Physiologically, PSMA is also expressed in normal tissue, including non-malignant prostate tissue, the kidney, the small intestine, and the central nervous system. In most tissues, PSMA likely contributes to folate uptake by cleaving the C-terminal glutamate from folic acid (Figure 1). Within the brain, PSMA regulates the level of N-acetylaspartylglutamate (NAAG) by catalyzing its degradation to glutamate. Structure of PSMA substrates and inhibitors. Left: structure of the PSMA substrate folic acid. The peptide bond (dashed box) linking the P1 glutamate residue (blue) to the remainder of the substrate (P1, red) is cleaved by PSMA. Right: Structure of the PSMA-PET agents 18F-DCFPyL and 68Ga-PSMA-11. The scissile peptide bond has been replaced by a non-hydrolyzable urea motif (red) to generate PMSA inhibitors. The P1 glutamate residue is essential for binding to PSMA, but a wide degree of variability is tolerated in the opposing end of the structure.
Click to Show/Hide
|
||||
| Description |
Within cohort A, the primary end point was the sensitivity and specificity for detection of metastases to the pelvic lymph nodes by 18F-DCFPyL PET/CT. These findings were validated by histopathology after radical proctectomy and lymph node dissection (Figure 6). When all confirmed positive lymph node metastases were considered, the sensitivity of 18F-DCFPyL PET/CT was 40%, with a 98% specificity. It is perhaps unsurprising that histopathology is more sensitivity for the detection of early metastases as a sufficient number of malignant cells must accumulate before any imaging modality will be positive. However, a post-hoc analysis considering only lesions >5 mm found the sensitivity of 18F-DCFPyL PET/CT increased to 60%. Despite this, across all lesions, 18F-DCFPyL PET/CT significantly outperformed conventional CT pelvic imaging with markedly improved specificity (97.7% vs. 65.1%) and positive predictive value (85.1% vs. 28.3%) for pelvic lymph node metastases, as well as sensitivity for disease within the prostate (96.8% vs. 35.9%).
Click to Show/Hide
|
||||
| Half life period | 8 h | ||||
[18F]AlF-NOTA-octreotide [Investigative]
Identified from the Human Clinical Data
| Experiment 1 Reporting the Activity Data of This PDC | [73] | ||||
| Indication | Tumor-induced osteomalacia | ||||
| Efficacy Data | Specificity |
100%
|
|||
| Patients Enrolled |
17 patients with hypophosphatemic osteomalacia suspected to be TIO.
|
||||
| Administration Dosage | 3.7 MBq (0.10 mCi) per kilogram of body weight | ||||
| Evaluation Method | 18F-OC PET/CT assay | ||||
| MOA of PDC |
18F-OC PET/CT scan is useful in the detection of tumors causing TIO. Further studies with larger patient populations are needed to validate the result.
|
||||
| Description |
The 18F-OC PET/CT scans were positive in 14 patients. Furthermore, 4 of 14 patients were scanned with both 18F-OC and 68Ga-DOTATATE PET/CT. Both studies were able to localize the tumor in all 4 patients. In total, 14 patients had surgery to remove the lesions. Postsurgical pathological examination confirmed causative tumors in these patients, whose symptoms diminished promptly. Serum phosphate levels normalized, confirming the diagnosis of TIO. 18F-OC PET/CT sensitivity, specificity, and accuracy were 87.5%, 100%, and 88.2% respectively. 18F-OC PET/CT findings affected patient management in 88.2% of cases.
Click to Show/Hide
|
||||
| Experiment 2 Reporting the Activity Data of This PDC | [73] | ||||
| Indication | Tumor-induced osteomalacia | ||||
| Efficacy Data | Sensitivity |
87.50%
|
|||
| Patients Enrolled |
17 patients with hypophosphatemic osteomalacia suspected to be TIO.
|
||||
| Administration Dosage | 3.7 MBq (0.10 mCi) per kilogram of body weight | ||||
| Evaluation Method | 18F-OC PET/CT assay | ||||
| MOA of PDC |
18F-OC PET/CT scan is useful in the detection of tumors causing TIO. Further studies with larger patient populations are needed to validate the result.
|
||||
| Description |
The 18F-OC PET/CT scans were positive in 14 patients. Furthermore, 4 of 14 patients were scanned with both 18F-OC and 68Ga-DOTATATE PET/CT. Both studies were able to localize the tumor in all 4 patients. In total, 14 patients had surgery to remove the lesions. Postsurgical pathological examination confirmed causative tumors in these patients, whose symptoms diminished promptly. Serum phosphate levels normalized, confirming the diagnosis of TIO. 18F-OC PET/CT sensitivity, specificity, and accuracy were 87.5%, 100%, and 88.2% respectively. 18F-OC PET/CT findings affected patient management in 88.2% of cases.
Click to Show/Hide
|
||||
| Experiment 3 Reporting the Activity Data of This PDC | [61] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Median detection ratio |
91.10%
|
|||
| Patients Enrolled |
75 patients with histologically confirmed neuroendocrine tumours and routine clinical.
|
||||
| MOA of PDC |
18F-AlF-OC is noninferior and even superior to 68Ga-DOTATATE/NOC PET in NET patients. This validates 18F-AlF-OC as an option for clinical practice somatostatin receptor PET.
|
||||
| Description |
In total, 4,709 different tumor lesions were detected: 3,454 with 68Ga-DOTATATE/NOC and 4,278 with 18F-AlF-OC. The mean DR with 18F-AlF-OC was significantly higher than with 68Ga-DOTATATE/NOC (91.1% vs. 75.3%; P < 10-5).
|
||||
| Experiment 4 Reporting the Activity Data of This PDC | [61] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Difference in detection ratio |
15.80%
|
|||
| Patients Enrolled |
75 patients with histologically confirmed neuroendocrine tumours and routine clinical.
|
||||
| MOA of PDC |
18F-AlF-OC is noninferior and even superior to 68Ga-DOTATATE/NOC PET in NET patients. This validates 18F-AlF-OC as an option for clinical practice somatostatin receptor PET.
|
||||
| Description |
The resulting mean DDR was 15.8%, with a lower margin of the 95% CI (95% CI, 9.6%-22.0%) higher than -15%, which is the prespecified boundary for noninferiority. The mean DDRs for the 68Ga-DOTATATE and 68Ga-DOTANOC subgroups were 11.8% (95% CI, 4.3-19.3) and 27.5% (95% CI, 17.8-37.1), respectively.
|
||||
| Experiment 5 Reporting the Activity Data of This PDC | [73] | ||||
| Indication | Tumor-induced osteomalacia | ||||
| Efficacy Data | Accuracy |
88.20%
|
|||
| Patients Enrolled |
17 patients with hypophosphatemic osteomalacia suspected to be TIO.
|
||||
| Administration Dosage | 3.7 MBq (0.10 mCi) per kilogram of body weight | ||||
| Evaluation Method | 18F-OC PET/CT assay | ||||
| MOA of PDC |
18F-OC PET/CT scan is useful in the detection of tumors causing TIO. Further studies with larger patient populations are needed to validate the result.
|
||||
| Description |
The 18F-OC PET/CT scans were positive in 14 patients. Furthermore, 4 of 14 patients were scanned with both 18F-OC and 68Ga-DOTATATE PET/CT. Both studies were able to localize the tumor in all 4 patients. In total, 14 patients had surgery to remove the lesions. Postsurgical pathological examination confirmed causative tumors in these patients, whose symptoms diminished promptly. Serum phosphate levels normalized, confirming the diagnosis of TIO. 18F-OC PET/CT sensitivity, specificity, and accuracy were 87.5%, 100%, and 88.2% respectively. 18F-OC PET/CT findings affected patient management in 88.2% of cases.
Click to Show/Hide
|
||||
225Ac-DOTATOC [Investigative]
Identified from the Human Clinical Data
| Experiment 1 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Well-differentiated pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Objective response rate (ORR) |
50%
|
|||
| Patients Enrolled |
9 patients with paraganglioma.
|
||||
| Administration Time | 2-9 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
Recently, a study in 9 patients of metastatic paragangliomas (7 patients received prior 177Lu-DOTATATE and 3 out of 7 patients failed prior 177Lu-DOTATATE) treated with 225Ac-DOTATATE ( particle based radiotherapy targeting SSTRs) and concomitant capecitabine showed a high partial response of 50% (compared to < 25% with particle targeted radiotherapies) and stable disease in 37.5% with a disease control rate of 87.5%, without any grade 3/4 hematotoxicity, nephrotoxicity, or hepatotoxicity.
Click to Show/Hide
|
||||
| Experiment 2 Reporting the Activity Data of This PDC | [44] | ||||
| Indication | Well-differentiated pancreatic neuroendocrine tumor | ||||
| Efficacy Data | Disease control rate (DCR) |
87.50%
|
|||
| Patients Enrolled |
9 patients with paraganglioma.
|
||||
| Administration Time | 2-9 cycles | ||||
| MOA of PDC |
Paragangliomas can metastasize, posing potential challenges in both symptomatic management and disease control. Systemic targeted radiotherapies using 131I-MIBG and 177Lu-DOTATATE are a mainstay in the treatment of metastatic paragangliomas. This clinical scenario and discussion aim to enhance physicians' knowledge of the stepwise approach to treat these patients with paraganglioma targeted radiotherapies. It comprehensively discusses current approaches to selecting paraganglioma patients for targeted radiotherapies and how to choose between the two radiotherapies based on specific patient and tumor characteristics, when either therapy is feasible, or one is superior to another one. The safety, efficacy, toxicity profiles, and optimization of these radiotherapies are also discussed, along with other therapeutic options including radiotherapies, available for patients besides these two therapies. As conclusion, perspectives in radiotherapies of paraganglioma patients are outlined since they hold near future promising approaches that can improve patient outcomes.
Click to Show/Hide
|
||||
| Description |
Recently, a study in 9 patients of metastatic paragangliomas (7 patients received prior 177Lu-DOTATATE and 3 out of 7 patients failed prior 177Lu-DOTATATE) treated with 225Ac-DOTATATE ( particle based radiotherapy targeting SSTRs) and concomitant capecitabine showed a high partial response of 50% (compared to < 25% with particle targeted radiotherapies) and stable disease in 37.5% with a disease control rate of 87.5%, without any grade 3/4 hematotoxicity, nephrotoxicity, or hepatotoxicity.
Click to Show/Hide
|
||||
MPD1 [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [74] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
12.50%
|
|||
| Administration Time | 30 days | ||||
| Administration Dosage | 10 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BxPC-3 cells (KRAS wild type) xenografted mice. | ||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | BxPC-3 cell | CVCL_0186 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [74] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
69.10%
|
|||
| Administration Time | 30 days | ||||
| Administration Dosage | 5 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | AsPC-1 cells (G12D KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | AsPC-1 (KRAS G12D) cell | L-929 cell line | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [74] | ||||
| Indication | Lung cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
80.00%
|
|||
| Administration Time | 30 days | ||||
| Administration Dosage | 5 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | A549 cells (G12S KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Lung adenocarcinoma | A-549 (KRAS G12S) cell | CVCL_0023 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [74] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
86.80%
|
|||
| Administration Time | 30 days | ||||
| Administration Dosage | 5 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | HCT116 cells (G13D KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Colon carcinoma | HCT 116 (KRAS G13D) cell | CVCL_0291 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [74] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
95.10%
|
|||
| Administration Time | 30 days | ||||
| Administration Dosage | 10 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | AsPC-1 cells (G12D KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | AsPC-1 (KRAS G12D) cell | L-929 cell line | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [74] | ||||
| Indication | Lung cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
95.10%
|
|||
| Administration Time | 30 days | ||||
| Administration Dosage | 10 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | A549 cells (G12S KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Lung adenocarcinoma | A-549 (KRAS G12S) cell | CVCL_0023 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [74] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
96.50%
|
|||
| Administration Time | 30 days | ||||
| Administration Dosage | 5 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | MDA-MB-231 cells (G13D KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (KRAS G13D) cell | CVCL_0062 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 8 Reporting the Activity Data of This PDC | [74] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
97.80%
|
|||
| Administration Time | 30 days | ||||
| Administration Dosage | 10 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | HCT116 cells (G13D KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Colon carcinoma | HCT 116 (KRAS G13D) cell | CVCL_0291 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 9 Reporting the Activity Data of This PDC | [74] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
99.90%
|
|||
| Administration Time | 30 days | ||||
| Administration Dosage | 5 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | MIA PaCa-2 cells (G12C KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | MIA PaCa-2 (KRAS G12C) cell | CVCL_0428 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 10 Reporting the Activity Data of This PDC | [74] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
100.00%
|
|||
| Administration Time | 30 days | ||||
| Administration Dosage | 10 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | MIA PaCa-2 cells (G12C KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | MIA PaCa-2 (KRAS G12C) cell | CVCL_0428 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 11 Reporting the Activity Data of This PDC | [74] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
100.00%
|
|||
| Administration Time | 30 days | ||||
| Administration Dosage | 10 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | MDA-MB-231 cells (G13D KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (KRAS G13D) cell | CVCL_0062 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 12 Reporting the Activity Data of This PDC | [74] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Albumin uptake rate |
6.67%
|
|||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | AsPC-1 cells (G12D KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | AsPC-1 (KRAS G12D) cell | L-929 cell line | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 13 Reporting the Activity Data of This PDC | [74] | ||||
| Indication | Lung cancer | ||||
| Efficacy Data | Albumin uptake rate |
12.80%
|
|||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | A549 cells (G12S KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Lung adenocarcinoma | A-549 (KRAS G12S) cell | CVCL_0023 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 14 Reporting the Activity Data of This PDC | [74] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Albumin uptake rate |
51.57%
|
|||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | MDA-MB-231 cells (G13D KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (KRAS G13D) cell | CVCL_0062 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 15 Reporting the Activity Data of This PDC | [74] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Albumin uptake rate |
52.30%
|
|||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | HCT116 cells (G13D KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Colon carcinoma | HCT 116 (KRAS G13D) cell | CVCL_0291 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 16 Reporting the Activity Data of This PDC | [74] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Albumin uptake rate |
68.43%
|
|||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | MIA PaCa-2 cells (G12C KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | MIA PaCa-2 (KRAS G12C) cell | CVCL_0428 | ||
| Half life period | 8.51 ± 0.50 h | ||||
SMAC-FRRG-DOX [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [75] | ||||
| Indication | Drug-resistant cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
76.00%
|
|||
| Administration Dosage | 1 mg/kg of DOX | ||||
| MOA of PDC |
These results suggest that the DD-NPs could improve the in vivo bioavailability and cancer targeting efficiency of SMAC via their stable nanoparticle-derived EPR effect, leading to effective IAPs inhibition in targeted tumor tissues.
|
||||
| Description |
Finally, the tumor volumes were successfully suppressed from 76% to 89% when the intravenous dose of DD-NPs increased from 1 mg/kg of DOX to 5 mg/kg of DOX.
|
||||
| In Vivo Model | MCF-7 tumor-bearing mice. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [75] | ||||
| Indication | Drug-resistant cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
89%-99%
|
|||
| Administration Dosage | 5 mg/kg | ||||
| MOA of PDC |
These results suggest that the DD-NPs could improve the in vivo bioavailability and cancer targeting efficiency of SMAC via their stable nanoparticle-derived EPR effect, leading to effective IAPs inhibition in targeted tumor tissues.
|
||||
| Description |
Finally, the tumor volumes were successfully suppressed from 76% to 89% when the intravenous dose of DD-NPs increased from 1 mg/kg of DOX to 5 mg/kg of DOX.
|
||||
| In Vivo Model | MCF-7 tumor-bearing mice. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [75] | ||||
| Indication | Drug-resistant cancer | ||||
| Efficacy Data | Tumer volume |
10 mm3
|
|||
| Administration Time | 21 days | ||||
| MOA of PDC |
These results suggest that the DD-NPs could improve the in vivo bioavailability and cancer targeting efficiency of SMAC via their stable nanoparticle-derived EPR effect, leading to effective IAPs inhibition in targeted tumor tissues.
|
||||
| In Vivo Model | MCF-7 tumor-bearing mice. | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [75] | ||||
| Indication | Drug-resistant cancer | ||||
| Efficacy Data | Body weight |
23.5g
|
|||
| Administration Time | 13 days | ||||
| MOA of PDC |
These results suggest that the DD-NPs could improve the in vivo bioavailability and cancer targeting efficiency of SMAC via their stable nanoparticle-derived EPR effect, leading to effective IAPs inhibition in targeted tumor tissues.
|
||||
| In Vivo Model | MCF-7 tumor-bearing mice. | ||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [75] | ||||
| Indication | Drug-resistant cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
7.53 μM
|
|||
| MOA of PDC |
These results suggest that the DD-NPs could improve the in vivo bioavailability and cancer targeting efficiency of SMAC via their stable nanoparticle-derived EPR effect, leading to effective IAPs inhibition in targeted tumor tissues.
|
||||
| Description |
When both cells were treated with different concentrations of DD-NPs (0-186 μg/ml), the cytotoxicity was induced only in MCF-7, showing similar cytotoxicity with free DOX at a high concentration (Fig. 2h). However, DD-NPs did not exhibit significant cytotoxicity in H9C2 whereas free DOX showed similar cytotoxicity when treated to MCF-7 (Fig. 2i).
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [75] | ||||
| Indication | Drug-resistant cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
9.2 μM
|
|||
| Administration Time | 48 h | ||||
| MOA of PDC |
These results suggest that the DD-NPs could improve the in vivo bioavailability and cancer targeting efficiency of SMAC via their stable nanoparticle-derived EPR effect, leading to effective IAPs inhibition in targeted tumor tissues.
|
||||
| Description |
However, DD-NP-treated group showed similar IC50 in wild-MCF-7 (7.53 μM) and ADR-MCF-7 (9.2 μM), due to the synergetic pro-apoptotic effect of SMAC molecules in DD-NPs (Fig. 3g).
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7/ADR cell | CVCL_0031 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [75] | ||||
| Indication | Drug-resistant cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | >> 200 μM | |||
| MOA of PDC |
These results suggest that the DD-NPs could improve the in vivo bioavailability and cancer targeting efficiency of SMAC via their stable nanoparticle-derived EPR effect, leading to effective IAPs inhibition in targeted tumor tissues.
|
||||
| Description |
When both cells were treated with different concentrations of DD-NPs (0-186 μg/ml), the cytotoxicity was induced only in MCF-7, showing similar cytotoxicity with free DOX at a high concentration (Fig. 2h). However, DD-NPs did not exhibit significant cytotoxicity in H9C2 whereas free DOX showed similar cytotoxicity when treated to MCF-7 (Fig. 2i).
Click to Show/Hide
|
||||
| In Vitro Model | Normal | H9c2 cell | CVCL_0286 | ||
67Ga-MMC(TMZ)-TOC [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [76] | ||||
| Indication | Pancreatic serotonin-producing neuroendocrine tumor | ||||
| Efficacy Data | Surviving fraction |
80%
|
|||
| Administration Time | 5 days | ||||
| Administration Dosage | 2 μmol/L | ||||
| Evaluation Method | Colony-forming assay | ||||
| MOA of PDC |
In this study, we hypothesized that the clinically utilized radiopharmaceutical 68Ga-DOTA-TOC could be converted to a radiolabeled peptide-drug conjugate (PDC) for selective delivery of TMZ to SSTR2-positive tumor cells. We used the intrinsic radiolabeling utility of the PDC to characterize receptor-binding properties, pharmacokinetics (PK), and tissue biodistribution in cells and animal studies. Receptor-targeted DNA-damaging properties and cytotoxic effects of the PDC were examined in different cell lines, and the effects of accumulated doses on MGMT content were evaluated in MGMT-positive cells.
Click to Show/Hide
|
||||
| Description |
TMZ is an alkylating agent that causes cell death through DNA-damaging mechanisms.?To determine whether MMC(TMZ)-TOC induces the same cytotoxic effects through a receptor-dependent process, we first studied its DNA-damaging properties in IMR-32 cells that endogenously express SSTR2. We performed an alkaline comet assay to evaluate the ability of our drug conjugate to produce single- and double-stranded DNA breaks. As shown in?Figure 3A,B, the targeted agent-induced DNA breaks that were similar to free TMZ, resulting in migrating DNA fragments (comet tail) from the nucleoid (comet head). The damaging effects of MMC(TMZ)-TOC were significantly reduced (P?< 0.0001) when cells were preincubated with blocking doses of the high-affinity SSTR2 antagonist JR11, indicating receptor-dependent cytotoxicity. Next, we used a colony formation assay to assess the long-term survival and reproductive capability of the cells after treatment with the PDC. We treated BON1 (receptor-negative) and BON1-SSTR2 (receptor-positive) cells with 2 mu;mol/L MMC(TMZ)-TOC or 10 mu;mol/L TMZ for 5 consecutive days, seeded the cells at a low-density number, and allowed them to grow and form colonies for 14 days. The clonogenic survival data showed that MMC(TMZ)-TOC produced cytotoxic effects in the receptor-positive BON1-SSTR2 cells (P?< 0.01) similar to clinically relevant doses of TMZ. Conversely, no cytotoxicity was observed in receptor-negative BON1 cells following treatment with the drug conjugate, further illustrating the receptor-dependent effects of the PDC.
Click to Show/Hide
|
||||
| In Vivo Model | Dually implanted HCT116-WT/SSTR2 mice xenograft models. | ||||
| In Vitro Model | Pancreatic serotonin-producing neuroendocrine tumor | BON-1 cell | CVCL_3985 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [76] | ||||
| Indication | Pancreatic serotonin-producing neuroendocrine tumor | ||||
| Efficacy Data | Surviving fraction |
100%
|
|||
| Administration Time | 5 days | ||||
| Administration Dosage | 2 μmol/L | ||||
| Evaluation Method | Colony-forming assay | ||||
| MOA of PDC |
In this study, we hypothesized that the clinically utilized radiopharmaceutical 68Ga-DOTA-TOC could be converted to a radiolabeled peptide-drug conjugate (PDC) for selective delivery of TMZ to SSTR2-positive tumor cells. We used the intrinsic radiolabeling utility of the PDC to characterize receptor-binding properties, pharmacokinetics (PK), and tissue biodistribution in cells and animal studies. Receptor-targeted DNA-damaging properties and cytotoxic effects of the PDC were examined in different cell lines, and the effects of accumulated doses on MGMT content were evaluated in MGMT-positive cells.
Click to Show/Hide
|
||||
| Description |
TMZ is an alkylating agent that causes cell death through DNA-damaging mechanisms.?To determine whether MMC(TMZ)-TOC induces the same cytotoxic effects through a receptor-dependent process, we first studied its DNA-damaging properties in IMR-32 cells that endogenously express SSTR2. We performed an alkaline comet assay to evaluate the ability of our drug conjugate to produce single- and double-stranded DNA breaks. As shown in?Figure 3A,B, the targeted agent-induced DNA breaks that were similar to free TMZ, resulting in migrating DNA fragments (comet tail) from the nucleoid (comet head). The damaging effects of MMC(TMZ)-TOC were significantly reduced (P?< 0.0001) when cells were preincubated with blocking doses of the high-affinity SSTR2 antagonist JR11, indicating receptor-dependent cytotoxicity. Next, we used a colony formation assay to assess the long-term survival and reproductive capability of the cells after treatment with the PDC. We treated BON1 (receptor-negative) and BON1-SSTR2 (receptor-positive) cells with 2 mu;mol/L MMC(TMZ)-TOC or 10 mu;mol/L TMZ for 5 consecutive days, seeded the cells at a low-density number, and allowed them to grow and form colonies for 14 days. The clonogenic survival data showed that MMC(TMZ)-TOC produced cytotoxic effects in the receptor-positive BON1-SSTR2 cells (P?< 0.01) similar to clinically relevant doses of TMZ. Conversely, no cytotoxicity was observed in receptor-negative BON1 cells following treatment with the drug conjugate, further illustrating the receptor-dependent effects of the PDC.
Click to Show/Hide
|
||||
| In Vivo Model | Dually implanted HCT116-WT/SSTR2 mice xenograft models. | ||||
| In Vitro Model | Pancreatic serotonin-producing neuroendocrine tumor | BON-1 cell | CVCL_3985 | ||
De Novo Cyclic Peptide (DNCP)-β-naloxamine (NalA) conjugates 1 [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [77] | ||||
| Indication | Pain | ||||
| Efficacy Data | Half Maximal Effect Concentration (EC50) |
0.1 ± 0.0 x 10-9 mol
|
|||
| MOA of PDC |
Here, we exploit the crystal structure of KOR bound to the dual KOR/delta-opioid receptor (DOR) epoxymorphinan opioid agonist MP11048 and use the Rosetta protein and peptide design software to computationally design peptide-small molecule conjugates targeting KOR. Utilizing the high affinity interaction of MP1104 with KOR as an anchor to initiate the design calculations, we seek to computationally design thioether cyclic peptides that interact with ECL2 and ECL3 of the receptor.
Click to Show/Hide
|
||||
| Description |
The TRUPATH screening platform has been developed as an alternative to cAMP second messenger assays to minimize signal overamplification35. Intriguingly, in contrast to U50,488 and MP1104, but similar to the mixed-action KOR agonist pentazocine, DNCP--NalA(1) exhibited KOR partial agonism at Gi2, Gi3, GoA, GoB and Ggastducin subtypes with Emax values ranging from 48% to 77%, whereas it elicited full agonism at Gi1 and Gz with Emax values of 81% and 101%, respectively. The highest potency in the picomolar range was observed at Gz subtype for DNCP--NalA(1), U50,488 and MP1104, while pentazocine revealed the least variation in potency and efficacy across the transducerome.
Click to Show/Hide
|
||||
| In Vivo Model | Experiments were performed in male SWISS mice. Radioligand binding (n=3) and functional cAMP assays (n=3-4) of DNCP-beta-NalA conjugates were performed on HEK293T cell membranes stably expressing mouse KOR. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [77] | ||||
| Indication | Pain | ||||
| Efficacy Data | Half Maximal Effect Concentration (EC50) |
0.7 ± 0.2 x 10-9 mol
|
|||
| MOA of PDC |
Here, we exploit the crystal structure of KOR bound to the dual KOR/delta-opioid receptor (DOR) epoxymorphinan opioid agonist MP11048 and use the Rosetta protein and peptide design software to computationally design peptide-small molecule conjugates targeting KOR. Utilizing the high affinity interaction of MP1104 with KOR as an anchor to initiate the design calculations, we seek to computationally design thioether cyclic peptides that interact with ECL2 and ECL3 of the receptor.
Click to Show/Hide
|
||||
| Description |
The TRUPATH screening platform has been developed as an alternative to cAMP second messenger assays to minimize signal overamplification35. Intriguingly, in contrast to U50,488 and MP1104, but similar to the mixed-action KOR agonist pentazocine, DNCP--NalA(1) exhibited KOR partial agonism at Gi2, Gi3, GoA, GoB and Ggastducin subtypes with Emax values ranging from 48% to 77%, whereas it elicited full agonism at Gi1 and Gz with Emax values of 81% and 101%, respectively. The highest potency in the picomolar range was observed at Gz subtype for DNCP--NalA(1), U50,488 and MP1104, while pentazocine revealed the least variation in potency and efficacy across the transducerome.
Click to Show/Hide
|
||||
| In Vivo Model | Experiments were performed in male SWISS mice. Radioligand binding (n=3) and functional cAMP assays (n=3-4) of DNCP-beta-NalA conjugates were performed on HEK293T cell membranes stably expressing mouse KOR. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [77] | ||||
| Indication | Pain | ||||
| Efficacy Data | Half Maximal Effect Concentration (EC50) |
0.9 ± 0.4 x 10-9 mol
|
|||
| MOA of PDC |
Here, we exploit the crystal structure of KOR bound to the dual KOR/delta-opioid receptor (DOR) epoxymorphinan opioid agonist MP11048 and use the Rosetta protein and peptide design software to computationally design peptide-small molecule conjugates targeting KOR. Utilizing the high affinity interaction of MP1104 with KOR as an anchor to initiate the design calculations, we seek to computationally design thioether cyclic peptides that interact with ECL2 and ECL3 of the receptor.
Click to Show/Hide
|
||||
| Description |
The TRUPATH screening platform has been developed as an alternative to cAMP second messenger assays to minimize signal overamplification35. Intriguingly, in contrast to U50,488 and MP1104, but similar to the mixed-action KOR agonist pentazocine, DNCP--NalA(1) exhibited KOR partial agonism at Gi2, Gi3, GoA, GoB and Ggastducin subtypes with Emax values ranging from 48% to 77%, whereas it elicited full agonism at Gi1 and Gz with Emax values of 81% and 101%, respectively. The highest potency in the picomolar range was observed at Gz subtype for DNCP--NalA(1), U50,488 and MP1104, while pentazocine revealed the least variation in potency and efficacy across the transducerome.
Click to Show/Hide
|
||||
| In Vivo Model | Experiments were performed in male SWISS mice. Radioligand binding (n=3) and functional cAMP assays (n=3-4) of DNCP-beta-NalA conjugates were performed on HEK293T cell membranes stably expressing mouse KOR. | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [77] | ||||
| Indication | Pain | ||||
| Efficacy Data | Half Maximal Effect Concentration (EC50) |
1.4 ± 0.3 x 10-9 mol
|
|||
| MOA of PDC |
Here, we exploit the crystal structure of KOR bound to the dual KOR/delta-opioid receptor (DOR) epoxymorphinan opioid agonist MP11048 and use the Rosetta protein and peptide design software to computationally design peptide-small molecule conjugates targeting KOR. Utilizing the high affinity interaction of MP1104 with KOR as an anchor to initiate the design calculations, we seek to computationally design thioether cyclic peptides that interact with ECL2 and ECL3 of the receptor.
Click to Show/Hide
|
||||
| Description |
The TRUPATH screening platform has been developed as an alternative to cAMP second messenger assays to minimize signal overamplification35. Intriguingly, in contrast to U50,488 and MP1104, but similar to the mixed-action KOR agonist pentazocine, DNCP--NalA(1) exhibited KOR partial agonism at Gi2, Gi3, GoA, GoB and Ggastducin subtypes with Emax values ranging from 48% to 77%, whereas it elicited full agonism at Gi1 and Gz with Emax values of 81% and 101%, respectively. The highest potency in the picomolar range was observed at Gz subtype for DNCP--NalA(1), U50,488 and MP1104, while pentazocine revealed the least variation in potency and efficacy across the transducerome.
Click to Show/Hide
|
||||
| In Vivo Model | Experiments were performed in male SWISS mice. Radioligand binding (n=3) and functional cAMP assays (n=3-4) of DNCP-beta-NalA conjugates were performed on HEK293T cell membranes stably expressing mouse KOR. | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [77] | ||||
| Indication | Pain | ||||
| Efficacy Data | Half Maximal Effect Concentration (EC50) |
1.7 ± 0.5 x 10-9 mol
|
|||
| MOA of PDC |
Here, we exploit the crystal structure of KOR bound to the dual KOR/delta-opioid receptor (DOR) epoxymorphinan opioid agonist MP11048 and use the Rosetta protein and peptide design software to computationally design peptide-small molecule conjugates targeting KOR. Utilizing the high affinity interaction of MP1104 with KOR as an anchor to initiate the design calculations, we seek to computationally design thioether cyclic peptides that interact with ECL2 and ECL3 of the receptor.
Click to Show/Hide
|
||||
| Description |
The TRUPATH screening platform has been developed as an alternative to cAMP second messenger assays to minimize signal overamplification35. Intriguingly, in contrast to U50,488 and MP1104, but similar to the mixed-action KOR agonist pentazocine, DNCP--NalA(1) exhibited KOR partial agonism at Gi2, Gi3, GoA, GoB and Ggastducin subtypes with Emax values ranging from 48% to 77%, whereas it elicited full agonism at Gi1 and Gz with Emax values of 81% and 101%, respectively. The highest potency in the picomolar range was observed at Gz subtype for DNCP--NalA(1), U50,488 and MP1104, while pentazocine revealed the least variation in potency and efficacy across the transducerome.
Click to Show/Hide
|
||||
| In Vivo Model | Experiments were performed in male SWISS mice. Radioligand binding (n=3) and functional cAMP assays (n=3-4) of DNCP-beta-NalA conjugates were performed on HEK293T cell membranes stably expressing mouse KOR. | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [77] | ||||
| Indication | Pain | ||||
| Efficacy Data | Half Maximal Effect Concentration (EC50) |
2.4 ± 0.9 x 10-9 mol
|
|||
| MOA of PDC |
Here, we exploit the crystal structure of KOR bound to the dual KOR/delta-opioid receptor (DOR) epoxymorphinan opioid agonist MP11048 and use the Rosetta protein and peptide design software to computationally design peptide-small molecule conjugates targeting KOR. Utilizing the high affinity interaction of MP1104 with KOR as an anchor to initiate the design calculations, we seek to computationally design thioether cyclic peptides that interact with ECL2 and ECL3 of the receptor.
Click to Show/Hide
|
||||
| Description |
The TRUPATH screening platform has been developed as an alternative to cAMP second messenger assays to minimize signal overamplification35. Intriguingly, in contrast to U50,488 and MP1104, but similar to the mixed-action KOR agonist pentazocine, DNCP--NalA(1) exhibited KOR partial agonism at Gi2, Gi3, GoA, GoB and Ggastducin subtypes with Emax values ranging from 48% to 77%, whereas it elicited full agonism at Gi1 and Gz with Emax values of 81% and 101%, respectively. The highest potency in the picomolar range was observed at Gz subtype for DNCP--NalA(1), U50,488 and MP1104, while pentazocine revealed the least variation in potency and efficacy across the transducerome.
Click to Show/Hide
|
||||
| In Vivo Model | Experiments were performed in male SWISS mice. Radioligand binding (n=3) and functional cAMP assays (n=3-4) of DNCP-beta-NalA conjugates were performed on HEK293T cell membranes stably expressing mouse KOR. | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [77] | ||||
| Indication | Pain | ||||
| Efficacy Data | Half Maximal Effect Concentration (EC50) |
3.1 ± 1.5 x 10-8 mol
|
|||
| MOA of PDC |
Here, we exploit the crystal structure of KOR bound to the dual KOR/delta-opioid receptor (DOR) epoxymorphinan opioid agonist MP11048 and use the Rosetta protein and peptide design software to computationally design peptide-small molecule conjugates targeting KOR. Utilizing the high affinity interaction of MP1104 with KOR as an anchor to initiate the design calculations, we seek to computationally design thioether cyclic peptides that interact with ECL2 and ECL3 of the receptor.
Click to Show/Hide
|
||||
| Description |
The TRUPATH screening platform has been developed as an alternative to cAMP second messenger assays to minimize signal overamplification35. Intriguingly, in contrast to U50,488 and MP1104, but similar to the mixed-action KOR agonist pentazocine, DNCP--NalA(1) exhibited KOR partial agonism at Gi2, Gi3, GoA, GoB and Ggastducin subtypes with Emax values ranging from 48% to 77%, whereas it elicited full agonism at Gi1 and Gz with Emax values of 81% and 101%, respectively. The highest potency in the picomolar range was observed at Gz subtype for DNCP--NalA(1), U50,488 and MP1104, while pentazocine revealed the least variation in potency and efficacy across the transducerome.
Click to Show/Hide
|
||||
| In Vivo Model | Experiments were performed in male SWISS mice. Radioligand binding (n=3) and functional cAMP assays (n=3-4) of DNCP-beta-NalA conjugates were performed on HEK293T cell membranes stably expressing mouse KOR. | ||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [77] | ||||
| Indication | Pain | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
31 ± 15 μM
|
|||
| Evaluation Method | [35S]GTPγS binding assay | ||||
| MOA of PDC |
Here, we exploit the crystal structure of KOR bound to the dual KOR/delta-opioid receptor (DOR) epoxymorphinan opioid agonist MP11048 and use the Rosetta protein and peptide design software to computationally design peptide-small molecule conjugates targeting KOR. Utilizing the high affinity interaction of MP1104 with KOR as an anchor to initiate the design calculations, we seek to computationally design thioether cyclic peptides that interact with ECL2 and ECL3 of the receptor.
Click to Show/Hide
|
||||
| Description |
Next, we determined the opioid receptor subtype selectivity profile of DNCP--NalA(1) in radioligand binding assays with membrane preparations from HEK293 cells stably expressing the mouse MOR and DOR, and CHO cells stably expressing human nociceptin (NOP) receptor. Herein, DNCP--NalA(1) bound to mouse MOR and DOR with Ki values of 5.4 and 318 nM, respectively, supporting an ~80-fold selectivity for KOR over DOR whereas it bound to the human NOP with a Ki value of ~1.3 μM, thus having an ~330-fold selectivity for KOR over NOP receptor. In the functional cAMP assay, DNCP--NalA(1) was inactive at both mouse MOR and DOR up to 10 μM. This was confirmed at the human MOR and DOR in the [35S]GTPγS binding assay. We next determined the mechanism of antagonism of DNCP--NalA(1) by measuring adenylyl cyclase-mediated cAMP inhibition and [35S]GTPγS binding at mouse and human MOR, respectively, using Schild regression analysis. The MOR expressed in HEK293 and CHO cells was activated by DAMGO in the absence and presence of increasing concentrations of DNCP--NalA(1). We observed a rightward shift of the concentration-response curves of DAMGO in cAMP and [35S]GTPγS binding assays. Schild analysis of DNCP--NalA(1) exhibited linear regression slopes of 0.9 and 1.5 and pA2 values of 9.1 and 7.9 in cAMP and [35S]GTPγS binding assays, respectively, which corresponds to an average functional affinity of 0.8 and 13 nM, respectively, thus demonstrating the competitive antagonism of the DNCP--NalA(1) at MOR.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | CHO cell | CVCL_0213 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [77] | ||||
| Indication | Pain | ||||
| Efficacy Data | Half Maximal Effect Concentration (EC50) |
2.0 ± 0.1 x 10-9 mol
|
|||
| Evaluation Method | Potency/efficacy cAMP assay) | ||||
| MOA of PDC |
Here, we exploit the crystal structure of KOR bound to the dual KOR/delta-opioid receptor (DOR) epoxymorphinan opioid agonist MP11048 and use the Rosetta protein and peptide design software to computationally design peptide-small molecule conjugates targeting KOR. Utilizing the high affinity interaction of MP1104 with KOR as an anchor to initiate the design calculations, we seek to computationally design thioether cyclic peptides that interact with ECL2 and ECL3 of the receptor.
Click to Show/Hide
|
||||
| Description |
We obtained four DNCP--NalA conjugates (1-4); the conjugation of DNCP (35) and DNCP (36) was unsuccessful. DNCP--NalA conjugates (1-4) were pharmacologically characterized via radioligand binding and functional cAMP assays to investigate their affinity, potency and efficacy at the mouse KOR. The DNCP--NalA conjugates exhibited affinities in the low nanomolar range, with DNCP--NalA(1) being the strongest binder at KOR with a Ki value of 3.9 nM, as compared to -NalA, which has an affinity Ki of 72 nM. All four DNCP--NalA conjugates were full agonists at KOR (Emax = 89-101%), while -NalA alone was only a partial agonist with an EC50 of 130 nM and Emax of 61%. The most potent conjugates were DNCP--NalA(1) and (4) with EC50 values of 2.0 nM and 1.0 nM, respectively. DNCP--NalA(2) and DNCP--NalA(3) showed agonist activities at KOR with EC50 values of 7.5 and 14 nM in cAMP assay, respectively, while their Ki values were 31 and 24 nM in radioligand binding assay, respectively. Receptor reserve may account for this discrepancy between potency and affinity values of DNCP--NalA(2) and DNCP--NalA(4) which can be observed in functional GPCR assays with opioid receptors30. The higher potency and efficacy of the conjugates at KOR compared to -NalA can be attributed to interactions of the macrocycles with the ECL2 region as described later.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | HEK-293T cell | CVCL_0063 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [77] | ||||
| Indication | Pain | ||||
| Efficacy Data | Half Maximal Effect Concentration (EC50) |
5.5 ± 1.7 x 10-9 mol
|
|||
| Evaluation Method | [35S]GTPγS binding assay | ||||
| MOA of PDC |
Here, we exploit the crystal structure of KOR bound to the dual KOR/delta-opioid receptor (DOR) epoxymorphinan opioid agonist MP11048 and use the Rosetta protein and peptide design software to computationally design peptide-small molecule conjugates targeting KOR. Utilizing the high affinity interaction of MP1104 with KOR as an anchor to initiate the design calculations, we seek to computationally design thioether cyclic peptides that interact with ECL2 and ECL3 of the receptor.
Click to Show/Hide
|
||||
| Description |
Next, we determined the opioid receptor subtype selectivity profile of DNCP--NalA(1) in radioligand binding assays with membrane preparations from HEK293 cells stably expressing the mouse MOR and DOR, and CHO cells stably expressing human nociceptin (NOP) receptor. Herein, DNCP--NalA(1) bound to mouse MOR and DOR with Ki values of 5.4 and 318 nM, respectively, supporting an ~80-fold selectivity for KOR over DOR whereas it bound to the human NOP with a Ki value of ~1.3 μM, thus having an ~330-fold selectivity for KOR over NOP receptor. In the functional cAMP assay, DNCP--NalA(1) was inactive at both mouse MOR and DOR up to 10 μM. This was confirmed at the human MOR and DOR in the [35S]GTPγS binding assay. We next determined the mechanism of antagonism of DNCP--NalA(1) by measuring adenylyl cyclase-mediated cAMP inhibition and [35S]GTPγS binding at mouse and human MOR, respectively, using Schild regression analysis. The MOR expressed in HEK293 and CHO cells was activated by DAMGO in the absence and presence of increasing concentrations of DNCP--NalA(1). We observed a rightward shift of the concentration-response curves of DAMGO in cAMP and [35S]GTPγS binding assays. Schild analysis of DNCP--NalA(1) exhibited linear regression slopes of 0.9 and 1.5 and pA2 values of 9.1 and 7.9 in cAMP and [35S]GTPγS binding assays, respectively, which corresponds to an average functional affinity of 0.8 and 13 nM, respectively, thus demonstrating the competitive antagonism of the DNCP--NalA(1) at MOR.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | CHO cell | CVCL_0213 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [77] | ||||
| Indication | Pain | ||||
| Efficacy Data | Half Maximal Effect Concentration (EC50) |
2.2 ± 1.3 x 10-8 mol
|
|||
| Evaluation Method | Potency/efficacy β-arrestin-2 recruitment assay | ||||
| MOA of PDC |
Here, we exploit the crystal structure of KOR bound to the dual KOR/delta-opioid receptor (DOR) epoxymorphinan opioid agonist MP11048 and use the Rosetta protein and peptide design software to computationally design peptide-small molecule conjugates targeting KOR. Utilizing the high affinity interaction of MP1104 with KOR as an anchor to initiate the design calculations, we seek to computationally design thioether cyclic peptides that interact with ECL2 and ECL3 of the receptor.
Click to Show/Hide
|
||||
| Description |
We obtained four DNCP--NalA conjugates (1-4); the conjugation of DNCP (35) and DNCP (36) was unsuccessful. DNCP--NalA conjugates (1-4) were pharmacologically characterized via radioligand binding and functional cAMP assays to investigate their affinity, potency and efficacy at the mouse KOR. The DNCP--NalA conjugates exhibited affinities in the low nanomolar range, with DNCP--NalA(1) being the strongest binder at KOR with a Ki value of 3.9 nM, as compared to -NalA, which has an affinity Ki of 72 nM. All four DNCP--NalA conjugates were full agonists at KOR (Emax = 89-101%), while -NalA alone was only a partial agonist with an EC50 of 130 nM and Emax of 61%. The most potent conjugates were DNCP--NalA(1) and (4) with EC50 values of 2.0 nM and 1.0 nM, respectively. DNCP--NalA(2) and DNCP--NalA(3) showed agonist activities at KOR with EC50 values of 7.5 and 14 nM in cAMP assay, respectively, while their Ki values were 31 and 24 nM in radioligand binding assay, respectively. Receptor reserve may account for this discrepancy between potency and affinity values of DNCP--NalA(2) and DNCP--NalA(4) which can be observed in functional GPCR assays with opioid receptors30. The higher potency and efficacy of the conjugates at KOR compared to -NalA can be attributed to interactions of the macrocycles with the ECL2 region as described later.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | HEK-293T cell | CVCL_0063 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [77] | ||||
| Indication | Pain | ||||
| Efficacy Data | Half Maximal Effect Concentration (EC50) |
2.8 ± 0.7 x 10-8 mol
|
|||
| Evaluation Method | Potency/efficacy β-arrestin-1 recruitment assay | ||||
| MOA of PDC |
Here, we exploit the crystal structure of KOR bound to the dual KOR/delta-opioid receptor (DOR) epoxymorphinan opioid agonist MP11048 and use the Rosetta protein and peptide design software to computationally design peptide-small molecule conjugates targeting KOR. Utilizing the high affinity interaction of MP1104 with KOR as an anchor to initiate the design calculations, we seek to computationally design thioether cyclic peptides that interact with ECL2 and ECL3 of the receptor.
Click to Show/Hide
|
||||
| Description |
We obtained four DNCP--NalA conjugates (1-4); the conjugation of DNCP (35) and DNCP (36) was unsuccessful. DNCP--NalA conjugates (1-4) were pharmacologically characterized via radioligand binding and functional cAMP assays to investigate their affinity, potency and efficacy at the mouse KOR. The DNCP--NalA conjugates exhibited affinities in the low nanomolar range, with DNCP--NalA(1) being the strongest binder at KOR with a Ki value of 3.9 nM, as compared to -NalA, which has an affinity Ki of 72 nM. All four DNCP--NalA conjugates were full agonists at KOR (Emax = 89-101%), while -NalA alone was only a partial agonist with an EC50 of 130 nM and Emax of 61%. The most potent conjugates were DNCP--NalA(1) and (4) with EC50 values of 2.0 nM and 1.0 nM, respectively. DNCP--NalA(2) and DNCP--NalA(3) showed agonist activities at KOR with EC50 values of 7.5 and 14 nM in cAMP assay, respectively, while their Ki values were 31 and 24 nM in radioligand binding assay, respectively. Receptor reserve may account for this discrepancy between potency and affinity values of DNCP--NalA(2) and DNCP--NalA(4) which can be observed in functional GPCR assays with opioid receptors30. The higher potency and efficacy of the conjugates at KOR compared to -NalA can be attributed to interactions of the macrocycles with the ECL2 region as described later.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | HEK-293T cell | CVCL_0063 | ||
Tripeptide 47 - Beta-carboline derivative conjugate 1 [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
7.46 ± 8.95%
|
|||
| Administration Time | 180min | ||||
| Administration Dosage | 0.05mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
8.23 ± 12.04%
|
|||
| Administration Time | 150min | ||||
| Administration Dosage | 0.05mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
13.42 ± 8.68%
|
|||
| Administration Time | 180min | ||||
| Administration Dosage | 0.10mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
17.97 ± 14.64%
|
|||
| Administration Time | 120min | ||||
| Administration Dosage | 0.05mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
18.86 ± 19.46%
|
|||
| Administration Time | 180min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
20.65 ± 18.94%
|
|||
| Administration Time | 90min | ||||
| Administration Dosage | 0.05mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
22.13 ± 14.89%
|
|||
| Administration Time | 150min | ||||
| Administration Dosage | 0.10mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 8 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
23.45 ± 15.31%
|
|||
| Administration Time | 60min | ||||
| Administration Dosage | 0.05mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 9 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
25.45 ± 15.37%
|
|||
| Administration Time | 120min | ||||
| Administration Dosage | 0.10mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 10 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
25.46 ± 12.12%
|
|||
| Administration Time | 150min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 11 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
29.22 ± 10.26%
|
|||
| Administration Time | 30min | ||||
| Administration Dosage | 0.05mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 12 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
32.73 ± 18.69%
|
|||
| Administration Time | 90min | ||||
| Administration Dosage | 0.10mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 13 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
34.55 ± 38.43%
|
|||
| Administration Time | 120min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 14 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
35.41 ± 13.56%
|
|||
| Administration Time | 30min | ||||
| Administration Dosage | 0.10mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 15 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
37.65 ± 28.53%
|
|||
| Administration Time | 60min | ||||
| Administration Dosage | 0.10mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 16 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
44.67 ± 41.08%
|
|||
| Administration Time | 90min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 17 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
48.25 ± 45.02%
|
|||
| Administration Time | 30min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 18 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
59.37 ± 37.02%
|
|||
| Administration Time | 60min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 19 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema weight |
1.29 ± 0.35 mg
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 20 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema weight |
1.65 ± 0.51 mg
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.1 mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 21 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema weight |
2.01 ± 0.48 mg
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.05 mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 22 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema inhibition rate |
38.70%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.05 mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 23 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema inhibition rate |
49.60%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.1 mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 24 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema inhibition rate |
60.70%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
Tripeptide 37 - Beta-carboline derivative conjugate 1 [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
9.06 ± 15.43%
|
|||
| Administration Time | 180min | ||||
| Administration Dosage | 0.05mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
11.55 ± 9.28%
|
|||
| Administration Time | 30min | ||||
| Administration Dosage | 0.05mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
14.38 ± 20.13%
|
|||
| Administration Time | 180min | ||||
| Administration Dosage | 0.10mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
15.43 ± 10.94%
|
|||
| Administration Time | 150min | ||||
| Administration Dosage | 0.05mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
18.99 ± 8.96%
|
|||
| Administration Time | 120min | ||||
| Administration Dosage | 0.05mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
21.39 ± 15.60%
|
|||
| Administration Time | 150min | ||||
| Administration Dosage | 0.10mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
23.08 ± 13.89%
|
|||
| Administration Time | 120min | ||||
| Administration Dosage | 0.10mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 8 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
23.58 ± 11.46%
|
|||
| Administration Time | 90min | ||||
| Administration Dosage | 0.05mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 9 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
25.69 ± 11.36%
|
|||
| Administration Time | 180min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 10 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
26.54 ± 10.33%
|
|||
| Administration Time | 60min | ||||
| Administration Dosage | 0.05mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 11 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
29.04 ± 22.58%
|
|||
| Administration Time | 150min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 12 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
32.32 ± 19.55%
|
|||
| Administration Time | 30min | ||||
| Administration Dosage | 0.10mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 13 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
34.48 ± 21.03%
|
|||
| Administration Time | 60min | ||||
| Administration Dosage | 0.10mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 14 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
38.33 ± 11.25%
|
|||
| Administration Time | 90min | ||||
| Administration Dosage | 0.10mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 15 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
41.28 ± 13.33%
|
|||
| Administration Time | 120min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 16 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
44.27 ± 42.46%
|
|||
| Administration Time | 30min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 17 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
52.22 ± 33.12%
|
|||
| Administration Time | 60min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 18 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
54.39 ± 23.67%
|
|||
| Administration Time | 90min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 19 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema weight |
1.47 ± 0.27 mg
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 20 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema weight |
1.79 ± 0.54 mg
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.1 mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 21 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema weight |
2.35 ± 0.43 mg
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.05 mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 22 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema inhibition rate |
28.30%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.05 mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 23 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema inhibition rate |
45.40%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.1 mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 24 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema inhibition rate |
55.20%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
Tripeptide 46 - Beta-carboline derivative conjugate 1 [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
10.36 ± 22.24%
|
|||
| Administration Time | 30min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
14.36 ± 28.12%
|
|||
| Administration Time | 180min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
22.37 ± 34.30%
|
|||
| Administration Time | 150min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
40.78 ± 32.03%
|
|||
| Administration Time | 60min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
46.45 ± 5.46%
|
|||
| Administration Time | 90min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
49.09 ± 29.03%
|
|||
| Administration Time | 120min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema weight |
2.05 ± 0.76 mg
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 8 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema inhibition rate |
37.50%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
Tripeptide 30 - Beta-carboline derivative conjugate 1 [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
12.25 ± 26.18%
|
|||
| Administration Time | 150min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
12.32 ± 14.94%
|
|||
| Administration Time | 180min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
17.23 ± 13.55%
|
|||
| Administration Time | 60min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
18.24 ± 11.56%
|
|||
| Administration Time | 30min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
29.25 ± 17.12%
|
|||
| Administration Time | 90min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
39.23 ± 17.12%
|
|||
| Administration Time | 120min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema weight |
2.35 ± 0.55 mg
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 8 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema inhibition rate |
28.40%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
Tripeptide 38 - Beta-carboline derivative conjugate 1 [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
13.29 ± 12.47%
|
|||
| Administration Time | 180min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
22.23 ± 14.66%
|
|||
| Administration Time | 90min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
24.33 ± 6.49%
|
|||
| Administration Time | 150min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
32.19 ± 14.78%
|
|||
| Administration Time | 60min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
37.25 ± 21.12%
|
|||
| Administration Time | 30min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
39.38 ± 13.48%
|
|||
| Administration Time | 120min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema weight |
1.39 ± 0.28 mg
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 8 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema inhibition rate |
57.60%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
Tripeptide 31 - Beta-carboline derivative conjugate 1 [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
13.54 ± 18.46%
|
|||
| Administration Time | 150min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
18.77 ± 13.45%
|
|||
| Administration Time | 180min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
22.17 ± 28.57%
|
|||
| Administration Time | 30min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
28.12 ± 19.81%
|
|||
| Administration Time | 60min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
34.55 ± 13.29%
|
|||
| Administration Time | 90min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
40.67 ± 33.57%
|
|||
| Administration Time | 120min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema weight |
1.97 ± 0.53 mg
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 8 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema inhibition rate |
40%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
Tripeptide 40 - Beta-carboline derivative conjugate 1 [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
14.23 ± 12.18%
|
|||
| Administration Time | 180min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
16.69 ± 10.33%
|
|||
| Administration Time | 150min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
28.55 ± 12.36%
|
|||
| Administration Time | 120min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
35.05 ± 29.27%
|
|||
| Administration Time | 30min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
35.19 ± 21.33%
|
|||
| Administration Time | 90min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
36.45 ± 27.36%
|
|||
| Administration Time | 60min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema weight |
1.59 ± 0.44 mg
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 8 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema inhibition rate |
51.50%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
Tripeptide 41 - Beta-carboline derivative conjugate 1 [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
15.25 ± 22.46%
|
|||
| Administration Time | 150min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
16.13 ± 13.39%
|
|||
| Administration Time | 180min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
27.32 ± 14.88%
|
|||
| Administration Time | 120min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
33.01 ± 38.16%
|
|||
| Administration Time | 30min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
38.01 ± 29.23%
|
|||
| Administration Time | 60min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
41.23 ± 44.46%
|
|||
| Administration Time | 90min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema weight |
1.43 ± 0.38 mg
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 8 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema inhibition rate |
56.40%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
Tripeptide 42 - Beta-carboline derivative conjugate 1 [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
15.32 ± 11.18%
|
|||
| Administration Time | 180min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
21.12 ± 14.45%
|
|||
| Administration Time | 150min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
31.34 ± 31.32%
|
|||
| Administration Time | 30min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
37.20 ± 15.13%
|
|||
| Administration Time | 60min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
40.56 ± 8.32%
|
|||
| Administration Time | 90min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
45.46 ± 23.54%
|
|||
| Administration Time | 120min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema weight |
1.70 ± 0.41 mg
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 8 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema inhibition rate |
48.20%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
Tripeptide 45 - Beta-carboline derivative conjugate 1 [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
15.68 ± 11.32%
|
|||
| Administration Time | 180min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
18.79 ± 15.74%
|
|||
| Administration Time | 120min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
21.35 ± 27.20%
|
|||
| Administration Time | 90min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
23.17 ± 25.32%
|
|||
| Administration Time | 30min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
25.05 ± 18.13%
|
|||
| Administration Time | 150min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
49.74 ± 35.11%
|
|||
| Administration Time | 60min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema weight |
1.73 ± 0.31 mg
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 8 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema inhibition rate |
47.30%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
Tripeptide 44 - Beta-carboline derivative conjugate 1 [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
16.03 ± 8.96%
|
|||
| Administration Time | 180min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
25.88 ± 11.77%
|
|||
| Administration Time | 150min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
35.79 ± 9.32%
|
|||
| Administration Time | 120min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
39.47 ± 25.44%
|
|||
| Administration Time | 30min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
45.77 ± 26.32%
|
|||
| Administration Time | 60min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
47.36 ± 28.21%
|
|||
| Administration Time | 90min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema weight |
1.85 ± 0.54 mg
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 8 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema inhibition rate |
43.60%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
Tripeptide 36 - Beta-carboline derivative conjugate 1 [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
18.32 ± 37.12%
|
|||
| Administration Time | 150min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
22.45 ± 43.22%
|
|||
| Administration Time | 180min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
32.33 ± 22.89%
|
|||
| Administration Time | 120min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
41.83 ± 39.35%
|
|||
| Administration Time | 30min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
47.33 ± 19.47%
|
|||
| Administration Time | 60min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
51.25 ± 11.84%
|
|||
| Administration Time | 90min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema weight |
1.89 ± 0.60 mg
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 8 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema inhibition rate |
42.40%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
Tripeptide 43 - Beta-carboline derivative conjugate 1 [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
19.22 ± 17.46%
|
|||
| Administration Time | 180min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
28.79 ± 10.13%
|
|||
| Administration Time | 150min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
37.58 ± 20.23%
|
|||
| Administration Time | 30min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
42.03 ± 6.41%
|
|||
| Administration Time | 120min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
46.48 ± 9.15%
|
|||
| Administration Time | 90min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
48.23 ± 14.23%
|
|||
| Administration Time | 60min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema weight |
1.55 ± 0.43 mg
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 8 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema weight |
1.81 ± 0.38 mg
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.1 mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 9 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema weight |
2.21 ± 0.47 mg
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.05 mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 10 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema inhibition rate |
32.60%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.05 mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 11 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema inhibition rate |
44.80%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.1 mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 12 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema inhibition rate |
52.70%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
Tripeptide 39 - Beta-carboline derivative conjugate 1 [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
19.25 ± 14.39%
|
|||
| Administration Time | 180min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
23.19 ± 15.36%
|
|||
| Administration Time | 150min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
30.75 ± 24.24%
|
|||
| Administration Time | 30min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
38.20 ± 32.09%
|
|||
| Administration Time | 60min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
40.36 ± 23.49%
|
|||
| Administration Time | 90min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
42.43 ± 27.22%
|
|||
| Administration Time | 120min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema weight |
1.62 ± 0.43 mg
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 8 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema inhibition rate |
50.60%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
Tripeptide 35 - Beta-carboline derivative conjugate 1 [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
19.45 ± 21.79%
|
|||
| Administration Time | 180min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
21.24 ± 15.74%
|
|||
| Administration Time | 30min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
22.12 ± 26.22%
|
|||
| Administration Time | 150min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
34.56 ± 35.77%
|
|||
| Administration Time | 90min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
39.25 ± 17.48%
|
|||
| Administration Time | 120min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
45.23 ± 11.14%
|
|||
| Administration Time | 60min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema weight |
1.45 ± 0.42 mg
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 8 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema inhibition rate |
55.80%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
Tripeptide 33 - Beta-carboline derivative conjugate 1 [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
20.94 ± 14.46%
|
|||
| Administration Time | 180min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
28.87 ± 21.93%
|
|||
| Administration Time | 150min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
30.25 ± 31.49%
|
|||
| Administration Time | 120min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
35.75 ± 30.35%
|
|||
| Administration Time | 30min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
41.18 ± 17.45%
|
|||
| Administration Time | 60min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
43.23 ± 32.49%
|
|||
| Administration Time | 90min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema weight |
1.61 ± 0.32 mg
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 8 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema weight |
2.20 ± 0.43 mg
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.1 mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 9 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema weight |
2.75 ± 0.66 mg
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.05 mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 10 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema inhibition rate |
16.20%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.05 mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 11 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema inhibition rate |
32.90%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.1 mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 12 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema inhibition rate |
50.90%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
Tripeptide 32 - Beta-carboline derivative conjugate [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
21.55 ± 17.29%
|
|||
| Administration Time | 180min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
29.38 ± 16.45%
|
|||
| Administration Time | 150min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
35.13 ± 22.25%
|
|||
| Administration Time | 60min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
37.12 ± 15.25%
|
|||
| Administration Time | 120min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
38.16 ± 19.26%
|
|||
| Administration Time | 30min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
46.08 ± 19.94%
|
|||
| Administration Time | 90min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema weight |
1.41 ± 0.49 mg
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 8 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema weight |
2.41 ± 0.58 mg
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.1 mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 9 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema weight |
2.91 ± 0.67 mg
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.05 mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 10 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema inhibition rate |
11.30%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.05 mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 11 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema inhibition rate |
26.50%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.1 mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 12 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema inhibition rate |
57%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
Tripeptide 34 - Beta-carboline derivative conjugate 1 [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
25.07 ± 11.21%
|
|||
| Administration Time | 180min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
26.47 ± 27.55%
|
|||
| Administration Time | 150min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
30.56 ± 21.46%
|
|||
| Administration Time | 30min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
38.45 ± 19.66%
|
|||
| Administration Time | 120min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
42.32 ± 16.41%
|
|||
| Administration Time | 60min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Pain threshold variation |
44.13 ± 28.22%
|
|||
| Administration Time | 90min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| Evaluation Method | Tail flick method | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were prescreened for analgesic activity at a dose of 0.15 mmol/kg. For each animal, pain thresholds were estimated at 30, 60, 120, 150 and 180 min following vehicle or drug administration. -Carboline-tripeptide conjugates exhibited moderate to good analgesic activity, with compounds 4a,e,k,l,o,p demonstrating especially good analgesic properties. All pain thresholds were increased after 30 min, yet maximal pain thresholds occurred at different time points for various synthetic derivatives. For example, the maximal pain thresholds for 4a,c,e,h,m were observed after 60 min, whereas for 4d,g,k,l,n,o,p this value was 90 min, and for 4b,f,i,q,r, 120 min post drug administration. The duration of the analgesic action of 4a,b,c,d,e,f,i,j was 150 min, while the same value for 4k,l,m,n,o,p was 180 min. We speculate that the physiological difference in analgesic activity is closely related to the tripeptide sequences of the -carbolines.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice. | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema weight |
1.78 ± 0.35 mg
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
| Experiment 8 Reporting the Activity Data of This PDC | [78] | ||||
| Indication | Mesenteric ischemia and reperfusion injury | ||||
| Efficacy Data | Edema inhibition rate |
45.70%
|
|||
| Administration Time | 30 min | ||||
| Administration Dosage | 0.15mmol/kg | ||||
| MOA of PDC |
It has been reported that some short peptides with certain sequences may be used as analgesic agents. In addition, our recent research findings indicated that some tripeptides with AA-Phe-Trp sequence present good analgesic activities. Since reactive oxygen species (ROS) and inflammatory leukocytes play an important role in the pathogenesis of I/R injury, antioxidant therapy and inhibition of post-ischemic neutrophil infiltration may have therapeutic efficacy during I/R injury. Ideally, I/R injury can be treated by a combination of the therapeutic agents. Generally, when two synergistic agents are administered individually but simultaneously, they will be transported to the site of action with different efficiencies. However, there are potential advantages in giving the co-administered agents as a single chemical entity. It is desirable to have the two synergistic agents reach a site simultaneously. Considering the antioxidant properties of β-carboline alkaloids coupled with the analgesic activity observed with AA-Phe-Trp tripeptides, we hypothesized that β-carboline and tripeptide (AA-Phe-Trp) conjugates might exhibit both enhanced antioxidant and analgesic activities. To test this hypothesis, we have synthesized a series of novel β-carboline-tripeptide (AA-Phe-Trp) conjugates, and evaluated their biological capacity to protect against mesenteric I/R injury.
Click to Show/Hide
|
||||
| Description |
All compounds were evaluated for anti-inflammatory capacity in a xylene-induced ear edema model. Briefly, this in vivo assay involves oral administration of test compounds in 0.5% carboxymethyl cellulose (CMC) suspension. Each compound was initially tested at 0.15 mmol/kg, and all revealed significant protection against xylene-induced inflammation, suggesting potent anti-inflammatory capacity. Since compounds (4a,e,k,o,p) exhibit good analgesic activities in the tail flick assay and superior anti-inflammatory activities in the xylene-induced ear edema model, these compounds were then examined in the same assay at lower doses to establish the detailed pharmacological activity profile. From Table 4, we noticed a dose-dependent anti-inflammatory response for 4a,e,k,o,p.
Click to Show/Hide
|
||||
| In Vivo Model | Male ICR mice xylene-induced ear edema model. | ||||
Tetrapeptide 8 - 1-(2,3-Dichlorophenyl)piperazine conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
3 ± 0.30 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
4 ± 0.26 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus infection | Staphylococcus | 1279 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
4 ± 0.41 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus flavus infection | Aspergillus flavus | 5059 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
5 ± 0.26 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
5 ± 0.05 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Xanthomonas oryzae infection | Xanthomonas oryzae | 347 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
5 ± 0.10 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
5 ± 0.47 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium oxysporum infection | Fusarium oxysporum | 5507 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
29 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Xanthomonas oryzae infection | Xanthomonas oryzae | 347 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
34 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
35 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
38 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus infection | Staphylococcus | 1279 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
41 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
43 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium oxysporum infection | Fusarium oxysporum | 5507 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
48 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus flavus infection | Aspergillus flavus | 5059 | ||
Pentapeptide 8 - 1-(2,3-Dichlorophenyl)piperazine conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
4 ± 0.11 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus infection | Staphylococcus | 1279 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
5 ± 0.10 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Xanthomonas oryzae infection | Xanthomonas oryzae | 347 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
5 ± 0.10 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
5 ± 0.05 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus flavus infection | Aspergillus flavus | 5059 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
5 ± 0.15 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium oxysporum infection | Fusarium oxysporum | 5507 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
6 ± 0.21 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
7 ± 0.20 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
27 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Xanthomonas oryzae infection | Xanthomonas oryzae | 347 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
31 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
32 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
35 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus infection | Staphylococcus | 1279 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
39 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
39 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium oxysporum infection | Fusarium oxysporum | 5507 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
42 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus flavus infection | Aspergillus flavus | 5059 | ||
Pentapeptide 7 - 1-(2,3-Dichlorophenyl)piperazine conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
9 ± 0.20 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus infection | Staphylococcus | 1279 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
10 ± 0.30 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
11 ± 0.20 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Xanthomonas oryzae infection | Xanthomonas oryzae | 347 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
11 ± 0.15 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus flavus infection | Aspergillus flavus | 5059 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
13 ± 0.26 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
15 ± 0.36 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
15 ± 0.10 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium oxysporum infection | Fusarium oxysporum | 5507 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
16 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Xanthomonas oryzae infection | Xanthomonas oryzae | 347 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
17 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
18 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus infection | Staphylococcus | 1279 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
18 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
20 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
21 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus flavus infection | Aspergillus flavus | 5059 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
21 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium oxysporum infection | Fusarium oxysporum | 5507 | ||
Tetrapeptide 9 - 1-(2,3-Dichlorophenyl)piperazine conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
11 ± 0.47 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus infection | Staphylococcus | 1279 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
12 ± 0.36 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
14 ± 0.30 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Xanthomonas oryzae infection | Xanthomonas oryzae | 347 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
14 ± 0.25 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus flavus infection | Aspergillus flavus | 5059 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
15 ± 0.47 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
17 ± 0.45 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium oxysporum infection | Fusarium oxysporum | 5507 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
18 ± 0.40 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
14 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Xanthomonas oryzae infection | Xanthomonas oryzae | 347 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
14 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
15 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus infection | Staphylococcus | 1279 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
16 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
17 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
17 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus flavus infection | Aspergillus flavus | 5059 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
19 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium oxysporum infection | Fusarium oxysporum | 5507 | ||
GDGVP - 3,4-Dihydro-4-oxo-2-quinazolinebutanoic acid conjugate 3 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Escherichia coli</i> infection | ||||
| Efficacy Data | Zone of inhibition |
13 ± 0.21 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Aspergillus niger</i> infection | ||||
| Efficacy Data | Zone of inhibition |
13 ± 0.34 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Klebsiella pneumoniae</i> infection | ||||
| Efficacy Data | Zone of inhibition |
14 ± 0.18 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Fusarium moniliforme</i> infection | ||||
| Efficacy Data | Zone of inhibition |
14 ± 0.48 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium moniliforme infection | Fusarium moniliforme | 117187 | ||
GDGVP - 3,4-Dihydro-4-oxo-2-quinazolinebutanoic acid conjugate 1 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Escherichia coli</i> infection | ||||
| Efficacy Data | Zone of inhibition |
13 ± 0.44 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Aspergillus niger</i> infection | ||||
| Efficacy Data | Zone of inhibition |
14 ± 0.14 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Klebsiella pneumoniae</i> infection | ||||
| Efficacy Data | Zone of inhibition |
15 ± 0.24 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Fusarium moniliforme</i> infection | ||||
| Efficacy Data | Zone of inhibition |
16 ± 0.45 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium moniliforme infection | Fusarium moniliforme | 117187 | ||
GDGVP - 3,4-Dihydro-4-oxo-2-quinazolinebutanoic acid conjugate 4 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Aspergillus niger</i> infection | ||||
| Efficacy Data | Zone of inhibition |
13 ± 0.17 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Escherichia coli</i> infection | ||||
| Efficacy Data | Zone of inhibition |
15 ± 0.27 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Fusarium moniliforme</i> infection | ||||
| Efficacy Data | Zone of inhibition |
15 ± 0.14 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium moniliforme infection | Fusarium moniliforme | 117187 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Klebsiella pneumoniae</i> infection | ||||
| Efficacy Data | Zone of inhibition |
16 ± 0.22 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
GTGVP - 3,4-Dihydro-4-oxo-2-quinazolinebutanoic acid conjugate 3 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Fusarium moniliforme</i> infection | ||||
| Efficacy Data | Zone of inhibition |
13 ± 0.28 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium moniliforme infection | Fusarium moniliforme | 117187 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Escherichia coli</i> infection | ||||
| Efficacy Data | Zone of inhibition |
14 ± 0.36 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Aspergillus niger</i> infection | ||||
| Efficacy Data | Zone of inhibition |
14 ± 0.39 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Klebsiella pneumoniae</i> infection | ||||
| Efficacy Data | Zone of inhibition |
16 ± 0.59 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
Tetrapeptide 10 - 1-(2,3-Dichlorophenyl)piperazine conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
14 ± 0.30 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
15 ± 0.43 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus infection | Staphylococcus | 1279 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
16 ± 0.25 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
16 ± 0.05 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Xanthomonas oryzae infection | Xanthomonas oryzae | 347 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
16 ± 0.34 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus flavus infection | Aspergillus flavus | 5059 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
19 ± 0.30 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium oxysporum infection | Fusarium oxysporum | 5507 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
22 ± 0.41 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
12 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Xanthomonas oryzae infection | Xanthomonas oryzae | 347 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
13 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
13 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus infection | Staphylococcus | 1279 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
14 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
15 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
16 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus flavus infection | Aspergillus flavus | 5059 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
18 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium oxysporum infection | Fusarium oxysporum | 5507 | ||
GTGVP - 3,4-Dihydro-4-oxo-2-quinazolinebutanoic acid conjugate 4 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Escherichia coli</i> infection | ||||
| Efficacy Data | Zone of inhibition |
15 ± 0.34 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Klebsiella pneumoniae</i> infection | ||||
| Efficacy Data | Zone of inhibition |
16 ± 0.74 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Fusarium moniliforme</i> infection | ||||
| Efficacy Data | Zone of inhibition |
16 ± 0.16 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium moniliforme infection | Fusarium moniliforme | 117187 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Aspergillus niger</i> infection | ||||
| Efficacy Data | Zone of inhibition |
17 ± 0.15 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
GDGVP - 3,4-Dihydro-4-oxo-2-quinazolinebutanoic acid conjugate 2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Escherichia coli</i> infection | ||||
| Efficacy Data | Zone of inhibition |
15 ± 0.28 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Aspergillus niger</i> infection | ||||
| Efficacy Data | Zone of inhibition |
15 ± 0.14 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Klebsiella pneumoniae</i> infection | ||||
| Efficacy Data | Zone of inhibition |
16 ± 0.24 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Fusarium moniliforme</i> infection | ||||
| Efficacy Data | Zone of inhibition |
16 ± 0.48 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium moniliforme infection | Fusarium moniliforme | 117187 | ||
GTGVP - 3,4-Dihydro-4-oxo-2-quinazolinebutanoic acid conjugate 1 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Aspergillus niger</i> infection | ||||
| Efficacy Data | Zone of inhibition |
15 ± 0.24 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Klebsiella pneumoniae</i> infection | ||||
| Efficacy Data | Zone of inhibition |
16 ± 0.46 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Fusarium moniliforme</i> infection | ||||
| Efficacy Data | Zone of inhibition |
16 ± 0.49 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium moniliforme infection | Fusarium moniliforme | 117187 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Escherichia coli</i> infection | ||||
| Efficacy Data | Zone of inhibition |
17 ± 0.22 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
GTGVP - 3,4-Dihydro-4-oxo-2-quinazolinebutanoic acid conjugate 2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Klebsiella pneumoniae</i> infection | ||||
| Efficacy Data | Zone of inhibition |
17 ± 0.61 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Aspergillus niger</i> infection | ||||
| Efficacy Data | Zone of inhibition |
17 ± 0.32 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Escherichia coli</i> infection | ||||
| Efficacy Data | Zone of inhibition |
18 ± 0.24 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Fusarium moniliforme</i> infection | ||||
| Efficacy Data | Zone of inhibition |
19 ± 0.49 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium moniliforme infection | Fusarium moniliforme | 117187 | ||
Elastin-base peptide 1 - 1-(2,3-Dichlorophenyl)piperazine conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
18 ± 0.32 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Xanthomonas oryzae infection | Xanthomonas oryzae | 347 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
20 ± 0.30 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
20 ± 0.40 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
20 ± 0.05 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus infection | Staphylococcus | 1279 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
26 ± 0.26 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus flavus infection | Aspergillus flavus | 5059 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
29 ± 0.10 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium oxysporum infection | Fusarium oxysporum | 5507 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Zone of inhibition |
37 ± 0.47 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| Evaluation Method | Agar well diffusion method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
3 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus flavus infection | Aspergillus flavus | 5059 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
4 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
5 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium oxysporum infection | Fusarium oxysporum | 5507 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
10 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
11 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Xanthomonas oryzae infection | Xanthomonas oryzae | 347 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
12 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [79] | ||||
| Indication | Bacterial infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
13 μg/mL
|
|||
| Administration Time | 16-18 h | ||||
| Evaluation Method | Microdilution method | ||||
| MOA of PDC |
Elastin protein-based polymers have their origin in repeating sequences of the mammalian elastic protein, elastin. The sequences of elastin peptides chosen are tetrapeptides, pentapeptides and tricosamers (30 amino acids) and also aromatic amino acids. These have been conjugated to 1-(2,3-dichlorophenyl)piperazine to study the effect of conjugation on the activity. The conjugates so obtained were characterized by physical and analytical techniques followed by the antimicrobial evaluation. The study revealed that all the conjugates have exhibited enhanced activity than the conventional drugs. Further, the conjugates of tricosamers have shown extraordinary activity against the fungal species with MIC value of 3-5 μg/ml which is five fold more potent than the antibiotic used.
Click to Show/Hide
|
||||
| Description |
Based on this observation we were very much projected towards the conjugation of elastin based peptides with varying chain length and hydrophobicity. We focused our attention on tetrapeptide elastin sequences with varying hydrophobicity. Among the analogs tested GGAP (29), GGIP (30) and GGFP (31), the latter has exhibited more activity i.e., GGFP with Phe unit has exerted nearly two times more potent activity than the antibiotics used where as the other two tetrapeptides conjugated heterocycle have shown comparatively less activity than GGFP which could be due to less hydrophobicity of amino acids present at the second position. Thus the order of activity among tetrapeptide conjugates is GGFP > GGIP > GGAP. In the light of the above findings, our subsequent goal was to incorporate GVGVP and GFGFP pentapeptide elastin sequences for conjugation. The pentapeptide conjugates have shown more than two fold potent activity compared to reference drugs used. Among these, GFGFP conjugate (33) has shown slight enhancement in the activity over GVGVP conjugate (32) which may be due to the presence of two more hydrophobic Phe units in 33. Inspection of the results further revealed that the conjugate 26 having single Phe residue has shown enhanced activity. On the other hand, retaining of only one Phe unit and increasing the peptide chain length has caused further increase in the activity (~two times) which is evident from the conjugate GGFP (31). Similarly, the remaining two tetrapeptide conjugates GGIP and GGAP have shown increased activity compared to Phe alone (26). Also, when the length of the peptide chain increased as well as two Phe residues were introduced (greater hydrophobicity) as in GFGFP (33) has exhibited two times greater activity than the standard drugs. This trend was followed in GVGVP conjugate (32) also. Hence, it can be inferred that as the length of the peptide chain as well as the hydrophobicity increases the activity also increases. Thus, the order of activity of the heterocyclic conjugated peptides is found to be GFGFP > GVGVP > GGFP > GGIP > GGAP > Phe > Trp > Tyr.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus infection | Staphylococcus | 1279 | ||
GDGVP - 3,4-Dihydro-4-oxo-2-quinazolinebutanoic acid conjugate 7 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Klebsiella pneumoniae</i> infection | ||||
| Efficacy Data | Zone of inhibition |
21 ± 0.45 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Aspergillus niger</i> infection | ||||
| Efficacy Data | Zone of inhibition |
22 ± 0.38 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Escherichia coli</i> infection | ||||
| Efficacy Data | Zone of inhibition |
23 ± 0.22 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Fusarium moniliforme</i> infection | ||||
| Efficacy Data | Zone of inhibition |
25 ± 0.42 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium moniliforme infection | Fusarium moniliforme | 117187 | ||
GDGVP - 3,4-Dihydro-4-oxo-2-quinazolinebutanoic acid conjugate 8 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Klebsiella pneumoniae</i> infection | ||||
| Efficacy Data | Zone of inhibition |
23 ± 0.41 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Escherichia coli</i> infection | ||||
| Efficacy Data | Zone of inhibition |
25 ± 0.21 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Fusarium moniliforme</i> infection | ||||
| Efficacy Data | Zone of inhibition |
26 ± 0.27 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium moniliforme infection | Fusarium moniliforme | 117187 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Aspergillus niger</i> infection | ||||
| Efficacy Data | Zone of inhibition |
27 ± 0.24 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
GDGVP - 3,4-Dihydro-4-oxo-2-quinazolinebutanoic acid conjugate 5 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Escherichia coli</i> infection | ||||
| Efficacy Data | Zone of inhibition |
23 ± 0.47 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Aspergillus niger</i> infection | ||||
| Efficacy Data | Zone of inhibition |
23 ± 0.37 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Fusarium moniliforme</i> infection | ||||
| Efficacy Data | Zone of inhibition |
26 ± 0.36 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium moniliforme infection | Fusarium moniliforme | 117187 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Klebsiella pneumoniae</i> infection | ||||
| Efficacy Data | Zone of inhibition |
27 ± 0.61 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
GWGVP - 3,4-Dihydro-4-oxo-2-quinazolinebutanoic acid conjugate 3 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Klebsiella pneumoniae</i> infection | ||||
| Efficacy Data | Zone of inhibition |
24 ± 0.36 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Escherichia coli</i> infection | ||||
| Efficacy Data | Zone of inhibition |
26 ± 0.31 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Aspergillus niger</i> infection | ||||
| Efficacy Data | Zone of inhibition |
27 ± 0.29 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Fusarium moniliforme</i> infection | ||||
| Efficacy Data | Zone of inhibition |
27 ± 0.37 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium moniliforme infection | Fusarium moniliforme | 117187 | ||
GYGVP - 3,4-Dihydro-4-oxo-2-quinazolinebutanoic acid conjugate 3 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Klebsiella pneumoniae</i> infection | ||||
| Efficacy Data | Zone of inhibition |
24 ± 0.56 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Aspergillus niger</i> infection | ||||
| Efficacy Data | Zone of inhibition |
25 ± 0.25 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Escherichia coli</i> infection | ||||
| Efficacy Data | Zone of inhibition |
26 ± 0.40 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Fusarium moniliforme</i> infection | ||||
| Efficacy Data | Zone of inhibition |
27 ± 0.36 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium moniliforme infection | Fusarium moniliforme | 117187 | ||
GWGVP - 3,4-Dihydro-4-oxo-2-quinazolinebutanoic acid conjugate 4 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Klebsiella pneumoniae</i> infection | ||||
| Efficacy Data | Zone of inhibition |
25 ± 0.81 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Escherichia coli</i> infection | ||||
| Efficacy Data | Zone of inhibition |
27 ± 0.33 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Aspergillus niger</i> infection | ||||
| Efficacy Data | Zone of inhibition |
30 ± 0.26 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Fusarium moniliforme</i> infection | ||||
| Efficacy Data | Zone of inhibition |
31 ± 0.36 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium moniliforme infection | Fusarium moniliforme | 117187 | ||
GYGVP - 3,4-Dihydro-4-oxo-2-quinazolinebutanoic acid conjugate 4 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Klebsiella pneumoniae</i> infection | ||||
| Efficacy Data | Zone of inhibition |
26 ± 0.60 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Escherichia coli</i> infection | ||||
| Efficacy Data | Zone of inhibition |
27 ± 0.35 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Aspergillus niger</i> infection | ||||
| Efficacy Data | Zone of inhibition |
29 ± 0.35 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Fusarium moniliforme</i> infection | ||||
| Efficacy Data | Zone of inhibition |
30 ± 0.14 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium moniliforme infection | Fusarium moniliforme | 117187 | ||
GYGVP - 3,4-Dihydro-4-oxo-2-quinazolinebutanoic acid conjugate 1 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Klebsiella pneumoniae</i> infection | ||||
| Efficacy Data | Zone of inhibition |
26 ± 0.12 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Escherichia coli</i> infection | ||||
| Efficacy Data | Zone of inhibition |
28 ± 0.42 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Aspergillus niger</i> infection | ||||
| Efficacy Data | Zone of inhibition |
28 ± 0.36 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Fusarium moniliforme</i> infection | ||||
| Efficacy Data | Zone of inhibition |
29 ± 0.29 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium moniliforme infection | Fusarium moniliforme | 117187 | ||
GDGVP - 3,4-Dihydro-4-oxo-2-quinazolinebutanoic acid conjugate 6 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Escherichia coli</i> infection | ||||
| Efficacy Data | Zone of inhibition |
26 ± 0.31 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Klebsiella pneumoniae</i> infection | ||||
| Efficacy Data | Zone of inhibition |
28 ± 0.22 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Aspergillus niger</i> infection | ||||
| Efficacy Data | Zone of inhibition |
28 ± 0.28 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Fusarium moniliforme</i> infection | ||||
| Efficacy Data | Zone of inhibition |
30 ± 0.54 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium moniliforme infection | Fusarium moniliforme | 117187 | ||
GYGVP - 3,4-Dihydro-4-oxo-2-quinazolinebutanoic acid conjugate 2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Klebsiella pneumoniae</i> infection | ||||
| Efficacy Data | Zone of inhibition |
28 ± 0.41 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Escherichia coli</i> infection | ||||
| Efficacy Data | Zone of inhibition |
29 ± 0.51 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Aspergillus niger</i> infection | ||||
| Efficacy Data | Zone of inhibition |
30 ± 0.34 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Fusarium moniliforme</i> infection | ||||
| Efficacy Data | Zone of inhibition |
31 ± 0.57 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium moniliforme infection | Fusarium moniliforme | 117187 | ||
GWGVP - 3,4-Dihydro-4-oxo-2-quinazolinebutanoic acid conjugate 1 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Klebsiella pneumoniae</i> infection | ||||
| Efficacy Data | Zone of inhibition |
28 ± 0.33 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Fusarium moniliforme</i> infection | ||||
| Efficacy Data | Zone of inhibition |
29 ± 0.30 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium moniliforme infection | Fusarium moniliforme | 117187 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Escherichia coli</i> infection | ||||
| Efficacy Data | Zone of inhibition |
30 ± 0.39 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Aspergillus niger</i> infection | ||||
| Efficacy Data | Zone of inhibition |
30 ± 0.33 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
GWGVP - 3,4-Dihydro-4-oxo-2-quinazolinebutanoic acid conjugate 2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Klebsiella pneumoniae</i> infection | ||||
| Efficacy Data | Zone of inhibition |
30 ± 0.90 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Klebsiella pneumoniae infection | Klebsiella pneumoniae | 573 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Escherichia coli</i> infection | ||||
| Efficacy Data | Zone of inhibition |
31 ± 0.61 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Escherichia coli infection | Escherichia coli | 562 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Aspergillus niger</i> infection | ||||
| Efficacy Data | Zone of inhibition |
32 ± 0.14 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Aspergillus niger infection | Aspergillus niger | 5061 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [80] | ||||
| Indication | <i>Fusarium moniliforme</i> infection | ||||
| Efficacy Data | Zone of inhibition |
32 ± 0.29 mm
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 30 μg/mL | ||||
| MOA of PDC |
We designed novel low charge, high hydrophobic pentapeptides with varying amino acids in the 2nd position and conjugated them to quinazolinone analogues to produce antimicrobial candidates. Preliminary structure-activity relationship revealed that constructs containing tryptophan, tyrosine, and phenylalanine amino acids were better antimicrobial agents than the standards used. Most of the candidates showed selective inhibitory power towards Gram-negative microorganisms. Most of the compounds could be considered as anti-Fusarium monoliforme since slightly superior results were observed for this species compared to A. niger. The alkyl chain length of the heterocyclic unit was found to be crucial for good activity. Finally, the presence of C-terminal polar group (COOH) seems to be favorable for antimicrobial results. Generating such hybrid compounds can be a promising approach to develop good desired biological activities.
Click to Show/Hide
|
||||
| Description |
The activity results showed that most of the compounds produced significant effects on the growth of the tested bacterial and fungal strains. The structure- antimicrobial activity relationship of the compounds revealed that quinazolinone precursors (I) and (II) conjugated with GWGVP ((XV), (XX), (XXV), and (XXX)), GYGVP ((XVI), (XXI), (XXVI), and (XXXI)), and GFGVP ((XXVII), (XXII), (XXVII), and (XXXII)) showed potent activities compared to conjugates of GDGVP ((XXVIII), (XXIII), (XXVIII), and (XXXIII)) and GTGVP ((XIX), XXIV), (XXIX), and (XXXIV)) and respective standard drugs. This could be due to aromaticity of the amino acids Trp, Tyr, and Phe, which are considered to play an important role in antimicrobial effects. It can be suggested that the presence of more hydrophobic units along with other non-polar amino acids provide amphipathicity to the compounds, which may help them in binding to the bacterial cell membranes followed by disruption. This becomes more evident with the loss of activity when the three aromatic and hydrophobic amino acids are substituted by more polar and charged Asp and Thr units. Among the above three hybrid analogues, tryptophan conjugates exhibited high potency, which may be due to the large aromaticity, hydrophobicity, light instability, and the ability to stabilize the amphiphilic structure necessary for antimicrobial activity. Further debenzylation of these compounds ((XXV), (XXVI), (XXVII), (XXX), (XXXI), and (XXXII)) resulted in more polar structures, which showed slight superior results than the benzylated analogues (XV), (XVI), (XVII), (XX), (XXI), and (XXII). This increased activity could be due to their higher polarity, which helps the molecules to establish a larger network for penetration through the cell membranes of microbes. Further, the increase in chain length of quinazolinones decreased the activity. Overall, the antimicrobial activity decreases in the order of X = Trp > Tyr > Phe > Thr > Asp.
Click to Show/Hide
|
||||
| In Vitro Model | Fusarium moniliforme infection | Fusarium moniliforme | 117187 | ||
MMC(TMZ)-TOC [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [76] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Survival rate |
80%
|
|||
| Administration Time | 4-6 h/day for 5 consecutive days | ||||
| Administration Dosage | 2 µmol/L | ||||
| Evaluation Method | Colony-forming assay | ||||
| MOA of PDC |
In this study, we hypothesized that the clinically utilized radiopharmaceutical 68Ga-DOTA-TOC could be converted to a radiolabeled peptide-drug conjugate (PDC) for selective delivery of TMZ to SSTR2-positive tumor cells. We used the intrinsic radiolabeling utility of the PDC to characterize receptor-binding properties, pharmacokinetics (PK), and tissue biodistribution in cells and animal studies. Receptor-targeted DNA-damaging properties and cytotoxic effects of the PDC were examined in different cell lines, and the effects of accumulated doses on MGMT content were evaluated in MGMT-positive cells.
Click to Show/Hide
|
||||
| Description |
TMZ is an alkylating agent that causes cell death through DNA-damaging mechanisms. To determine whether MMC(TMZ)-TOC induces the same cytotoxic effects through a receptor-dependent process, we first studied its DNA-damaging properties in IMR-32 cells that endogenously express SSTR2. We performed an alkaline comet assay to evaluate the ability of our drug conjugate to produce single- and double-stranded DNA breaks. As shown in Figure 3A,B, the targeted agent-induced DNA breaks that were similar to free TMZ, resulting in migrating DNA fragments (comet tail) from the nucleoid (comet head). The damaging effects of MMC(TMZ)-TOC were significantly reduced (P < 0.0001) when cells were preincubated with blocking doses of the high-affinity SSTR2 antagonist JR11, indicating receptor-dependent cytotoxicity. Next, we used a colony formation assay to assess the long-term survival and reproductive capability of the cells after treatment with the PDC. We treated BON1 (receptor-negative) and BON1-SSTR2 (receptor-positive) cells with 2 umol/L MMC(TMZ)-TOC or 10 umol/L TMZ for 5 consecutive days, seeded the cells at a low-density number, and allowed them to grow and form colonies for 14 days. The clonogenic survival data showed that MMC(TMZ)-TOC produced cytotoxic effects in the receptor-positive BON1-SSTR2 cells (P < 0.01) similar to clinically relevant doses of TMZ. Conversely, no cytotoxicity was observed in receptor-negative BON1 cells following treatment with the drug conjugate, further illustrating the receptor-dependent effects of the PDC.
Click to Show/Hide
|
||||
| In Vitro Model | SSR2 positive neuroendocrine tumour | BON1-SSTR2 cell | CVCL_3985 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [76] | ||||
| Indication | Neuroendocrine tumour | ||||
| Efficacy Data | Survival rate |
100%
|
|||
| Administration Time | 4-6 h/day for 5 consecutive days | ||||
| Administration Dosage | 2 µmol/L | ||||
| Evaluation Method | Colony-forming assay | ||||
| MOA of PDC |
In this study, we hypothesized that the clinically utilized radiopharmaceutical 68Ga-DOTA-TOC could be converted to a radiolabeled peptide-drug conjugate (PDC) for selective delivery of TMZ to SSTR2-positive tumor cells. We used the intrinsic radiolabeling utility of the PDC to characterize receptor-binding properties, pharmacokinetics (PK), and tissue biodistribution in cells and animal studies. Receptor-targeted DNA-damaging properties and cytotoxic effects of the PDC were examined in different cell lines, and the effects of accumulated doses on MGMT content were evaluated in MGMT-positive cells.
Click to Show/Hide
|
||||
| Description |
TMZ is an alkylating agent that causes cell death through DNA-damaging mechanisms. To determine whether MMC(TMZ)-TOC induces the same cytotoxic effects through a receptor-dependent process, we first studied its DNA-damaging properties in IMR-32 cells that endogenously express SSTR2. We performed an alkaline comet assay to evaluate the ability of our drug conjugate to produce single- and double-stranded DNA breaks. As shown in Figure 3A,B, the targeted agent-induced DNA breaks that were similar to free TMZ, resulting in migrating DNA fragments (comet tail) from the nucleoid (comet head). The damaging effects of MMC(TMZ)-TOC were significantly reduced (P < 0.0001) when cells were preincubated with blocking doses of the high-affinity SSTR2 antagonist JR11, indicating receptor-dependent cytotoxicity. Next, we used a colony formation assay to assess the long-term survival and reproductive capability of the cells after treatment with the PDC. We treated BON1 (receptor-negative) and BON1-SSTR2 (receptor-positive) cells with 2 umol/L MMC(TMZ)-TOC or 10 umol/L TMZ for 5 consecutive days, seeded the cells at a low-density number, and allowed them to grow and form colonies for 14 days. The clonogenic survival data showed that MMC(TMZ)-TOC produced cytotoxic effects in the receptor-positive BON1-SSTR2 cells (P < 0.01) similar to clinically relevant doses of TMZ. Conversely, no cytotoxicity was observed in receptor-negative BON1 cells following treatment with the drug conjugate, further illustrating the receptor-dependent effects of the PDC.
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic serotonin-producing neuroendocrine tumor | BON-1 cell | CVCL_3985 | ||
BA-NFs [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [81] | ||||
| Indication | DOX-induced cardiomyopathy | ||||
| Efficacy Data | SLC7A11/GAPDH increase rate |
83%
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 100 µM | ||||
| Evaluation Method | Western blotting assay | ||||
| MOA of PDC |
Based on the above background, we designed and synthesized supramolecular self-assembled nanofibers with AT1R-specific targeting moieties (Baicalin-FFYEEG-ARVYIHPF, BA-NFs). Baicalin (BA), with its capacity for ROS scavenging and ferroptosis inhibition, constitutes an ideal candidate for ameliorating DIC. BA was conjugated with the self-assembling peptide FF and targeting peptide ARVYIHPF to form a specific nanostructure to improve its aqueous solubility and incorporate a targeting functionality, thereby addressing its inherent limitations. Our results indicated BA-NFs can mitigate doxorubicin-induced cardiomyocyte ferroptosis and cardiac dysfunction. This remarkable therapeutic effect is primarily attributable to the targeting proficiency of BA-NFs for injured cardiomyocytes, culminating in superoxide and ROS scavenging, reduced iron deposition, alleviated lipid peroxidation, and enhanced SLC7A11 and GPX4 expression. Therefore, we propose that BA-NFs proffers an efficacious targeted drug delivery strategy for the amelioration of DIC.
Click to Show/Hide
|
||||
| Description |
The accumulation of iron is a characteristic feature of ferroptosis. Intracellular iron, especially labile ferrous iron, can react with oxidants to generate cytotoxic hydroxyl radicals via Fenton reactions, thereby promoting ferroptosis. FerroOrange staining revealed significantly increased intracellular Fe2+ levels in DOX-treated H9c2 cells compared to controls, indicating DOX induced iron dyshomeostasis, resulting in ferrous iron buildup and subsequent ferroptosis. BA-NFs alleviated the accumulation of intracellular ferrous ions, suppressing ferroptosis. Fe2+ accumulation may disrupt the balance between LPO and oxygen homeostasis. We observed that BA-NFs efficiently attenuated DOX-induced elevation in malondialdehyde (MDA), a lipid peroxidation end-product, while increased antioxidant glutathione (GSH) levels. These findings suggested that BA-NFs can prevent DOX-induced ferroptosis in cardiomyocytes. Furthermore, protein expression of SLC7A11 and GPX4, key components of the antioxidant defense system against ferroptosis, was examined. DOX significantly decreased SLC7A11 and GPX4 expression in H9c2 cells compared to controls, suggesting DOX disrupts cellular antioxidant systems leading to ferroptosis. BA and BA-NFs partially restored SLC7A11 and GPX4 expression versus DOX treatment, with BA-NFs exhibiting the most significant impact. Together, these results indicate BA-NFs specifically inhibits DOX-induced cardiomyocyte ferroptosis by reducing intracellular iron accumulation and enhancing antioxidant system gene expression. In addition, the cardiotoxic effects of DOX are mediated via excessive oxidative stress and induced mitochondrial damage, thereby activating intrinsic apoptosis as an additional pathway. Flow cytometric analysis using Annexin V-Fitc/PI staining revealed that BA-NFs potently attenuated DOX-induced cardiomyocyte apoptosis, with a more pronounced effect than BA alone. DOX enhanced the green fluorescent signal of cleaved caspase-3 (as a biomarker of apoptosis) in H9c2 cells, whereas BA-NFs significantly inhibited this upregulation. These results provided further evidence that BA-NFs serve as potential therapeutic approaches for DIC.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | H9c2 cell | CVCL_0286 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [81] | ||||
| Indication | DOX-induced cardiomyopathy | ||||
| Efficacy Data | MDA increase rate |
50%
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 100 µM | ||||
| Evaluation Method | Western blotting assay | ||||
| MOA of PDC |
Based on the above background, we designed and synthesized supramolecular self-assembled nanofibers with AT1R-specific targeting moieties (Baicalin-FFYEEG-ARVYIHPF, BA-NFs). Baicalin (BA), with its capacity for ROS scavenging and ferroptosis inhibition, constitutes an ideal candidate for ameliorating DIC. BA was conjugated with the self-assembling peptide FF and targeting peptide ARVYIHPF to form a specific nanostructure to improve its aqueous solubility and incorporate a targeting functionality, thereby addressing its inherent limitations. Our results indicated BA-NFs can mitigate doxorubicin-induced cardiomyocyte ferroptosis and cardiac dysfunction. This remarkable therapeutic effect is primarily attributable to the targeting proficiency of BA-NFs for injured cardiomyocytes, culminating in superoxide and ROS scavenging, reduced iron deposition, alleviated lipid peroxidation, and enhanced SLC7A11 and GPX4 expression. Therefore, we propose that BA-NFs proffers an efficacious targeted drug delivery strategy for the amelioration of DIC.
Click to Show/Hide
|
||||
| Description |
The accumulation of iron is a characteristic feature of ferroptosis. Intracellular iron, especially labile ferrous iron, can react with oxidants to generate cytotoxic hydroxyl radicals via Fenton reactions, thereby promoting ferroptosis. FerroOrange staining revealed significantly increased intracellular Fe2+ levels in DOX-treated H9c2 cells compared to controls, indicating DOX induced iron dyshomeostasis, resulting in ferrous iron buildup and subsequent ferroptosis. BA-NFs alleviated the accumulation of intracellular ferrous ions, suppressing ferroptosis. Fe2+ accumulation may disrupt the balance between LPO and oxygen homeostasis. We observed that BA-NFs efficiently attenuated DOX-induced elevation in malondialdehyde (MDA), a lipid peroxidation end-product, while increased antioxidant glutathione (GSH) levels. These findings suggested that BA-NFs can prevent DOX-induced ferroptosis in cardiomyocytes. Furthermore, protein expression of SLC7A11 and GPX4, key components of the antioxidant defense system against ferroptosis, was examined. DOX significantly decreased SLC7A11 and GPX4 expression in H9c2 cells compared to controls, suggesting DOX disrupts cellular antioxidant systems leading to ferroptosis. BA and BA-NFs partially restored SLC7A11 and GPX4 expression versus DOX treatment, with BA-NFs exhibiting the most significant impact. Together, these results indicate BA-NFs specifically inhibits DOX-induced cardiomyocyte ferroptosis by reducing intracellular iron accumulation and enhancing antioxidant system gene expression. In addition, the cardiotoxic effects of DOX are mediated via excessive oxidative stress and induced mitochondrial damage, thereby activating intrinsic apoptosis as an additional pathway. Flow cytometric analysis using Annexin V-Fitc/PI staining revealed that BA-NFs potently attenuated DOX-induced cardiomyocyte apoptosis, with a more pronounced effect than BA alone. DOX enhanced the green fluorescent signal of cleaved caspase-3 (as a biomarker of apoptosis) in H9c2 cells, whereas BA-NFs significantly inhibited this upregulation. These results provided further evidence that BA-NFs serve as potential therapeutic approaches for DIC.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | H9c2 cell | CVCL_0286 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [81] | ||||
| Indication | DOX-induced cardiomyopathy | ||||
| Efficacy Data | GSH/GSSG increase ratio |
85%
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 100 µM | ||||
| Evaluation Method | Western blotting assay | ||||
| MOA of PDC |
Based on the above background, we designed and synthesized supramolecular self-assembled nanofibers with AT1R-specific targeting moieties (Baicalin-FFYEEG-ARVYIHPF, BA-NFs). Baicalin (BA), with its capacity for ROS scavenging and ferroptosis inhibition, constitutes an ideal candidate for ameliorating DIC. BA was conjugated with the self-assembling peptide FF and targeting peptide ARVYIHPF to form a specific nanostructure to improve its aqueous solubility and incorporate a targeting functionality, thereby addressing its inherent limitations. Our results indicated BA-NFs can mitigate doxorubicin-induced cardiomyocyte ferroptosis and cardiac dysfunction. This remarkable therapeutic effect is primarily attributable to the targeting proficiency of BA-NFs for injured cardiomyocytes, culminating in superoxide and ROS scavenging, reduced iron deposition, alleviated lipid peroxidation, and enhanced SLC7A11 and GPX4 expression. Therefore, we propose that BA-NFs proffers an efficacious targeted drug delivery strategy for the amelioration of DIC.
Click to Show/Hide
|
||||
| Description |
The accumulation of iron is a characteristic feature of ferroptosis. Intracellular iron, especially labile ferrous iron, can react with oxidants to generate cytotoxic hydroxyl radicals via Fenton reactions, thereby promoting ferroptosis. FerroOrange staining revealed significantly increased intracellular Fe2+ levels in DOX-treated H9c2 cells compared to controls, indicating DOX induced iron dyshomeostasis, resulting in ferrous iron buildup and subsequent ferroptosis. BA-NFs alleviated the accumulation of intracellular ferrous ions, suppressing ferroptosis. Fe2+ accumulation may disrupt the balance between LPO and oxygen homeostasis. We observed that BA-NFs efficiently attenuated DOX-induced elevation in malondialdehyde (MDA), a lipid peroxidation end-product, while increased antioxidant glutathione (GSH) levels. These findings suggested that BA-NFs can prevent DOX-induced ferroptosis in cardiomyocytes. Furthermore, protein expression of SLC7A11 and GPX4, key components of the antioxidant defense system against ferroptosis, was examined. DOX significantly decreased SLC7A11 and GPX4 expression in H9c2 cells compared to controls, suggesting DOX disrupts cellular antioxidant systems leading to ferroptosis. BA and BA-NFs partially restored SLC7A11 and GPX4 expression versus DOX treatment, with BA-NFs exhibiting the most significant impact. Together, these results indicate BA-NFs specifically inhibits DOX-induced cardiomyocyte ferroptosis by reducing intracellular iron accumulation and enhancing antioxidant system gene expression. In addition, the cardiotoxic effects of DOX are mediated via excessive oxidative stress and induced mitochondrial damage, thereby activating intrinsic apoptosis as an additional pathway. Flow cytometric analysis using Annexin V-Fitc/PI staining revealed that BA-NFs potently attenuated DOX-induced cardiomyocyte apoptosis, with a more pronounced effect than BA alone. DOX enhanced the green fluorescent signal of cleaved caspase-3 (as a biomarker of apoptosis) in H9c2 cells, whereas BA-NFs significantly inhibited this upregulation. These results provided further evidence that BA-NFs serve as potential therapeutic approaches for DIC.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | H9c2 cell | CVCL_0286 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [81] | ||||
| Indication | DOX-induced cardiomyopathy | ||||
| Efficacy Data | GPX4/GAPDH increase ratio |
120%
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 100 µM | ||||
| Evaluation Method | Western blotting assay | ||||
| MOA of PDC |
Based on the above background, we designed and synthesized supramolecular self-assembled nanofibers with AT1R-specific targeting moieties (Baicalin-FFYEEG-ARVYIHPF, BA-NFs). Baicalin (BA), with its capacity for ROS scavenging and ferroptosis inhibition, constitutes an ideal candidate for ameliorating DIC. BA was conjugated with the self-assembling peptide FF and targeting peptide ARVYIHPF to form a specific nanostructure to improve its aqueous solubility and incorporate a targeting functionality, thereby addressing its inherent limitations. Our results indicated BA-NFs can mitigate doxorubicin-induced cardiomyocyte ferroptosis and cardiac dysfunction. This remarkable therapeutic effect is primarily attributable to the targeting proficiency of BA-NFs for injured cardiomyocytes, culminating in superoxide and ROS scavenging, reduced iron deposition, alleviated lipid peroxidation, and enhanced SLC7A11 and GPX4 expression. Therefore, we propose that BA-NFs proffers an efficacious targeted drug delivery strategy for the amelioration of DIC.
Click to Show/Hide
|
||||
| Description |
The accumulation of iron is a characteristic feature of ferroptosis. Intracellular iron, especially labile ferrous iron, can react with oxidants to generate cytotoxic hydroxyl radicals via Fenton reactions, thereby promoting ferroptosis. FerroOrange staining revealed significantly increased intracellular Fe2+ levels in DOX-treated H9c2 cells compared to controls, indicating DOX induced iron dyshomeostasis, resulting in ferrous iron buildup and subsequent ferroptosis. BA-NFs alleviated the accumulation of intracellular ferrous ions, suppressing ferroptosis. Fe2+ accumulation may disrupt the balance between LPO and oxygen homeostasis. We observed that BA-NFs efficiently attenuated DOX-induced elevation in malondialdehyde (MDA), a lipid peroxidation end-product, while increased antioxidant glutathione (GSH) levels. These findings suggested that BA-NFs can prevent DOX-induced ferroptosis in cardiomyocytes. Furthermore, protein expression of SLC7A11 and GPX4, key components of the antioxidant defense system against ferroptosis, was examined. DOX significantly decreased SLC7A11 and GPX4 expression in H9c2 cells compared to controls, suggesting DOX disrupts cellular antioxidant systems leading to ferroptosis. BA and BA-NFs partially restored SLC7A11 and GPX4 expression versus DOX treatment, with BA-NFs exhibiting the most significant impact. Together, these results indicate BA-NFs specifically inhibits DOX-induced cardiomyocyte ferroptosis by reducing intracellular iron accumulation and enhancing antioxidant system gene expression. In addition, the cardiotoxic effects of DOX are mediated via excessive oxidative stress and induced mitochondrial damage, thereby activating intrinsic apoptosis as an additional pathway. Flow cytometric analysis using Annexin V-Fitc/PI staining revealed that BA-NFs potently attenuated DOX-induced cardiomyocyte apoptosis, with a more pronounced effect than BA alone. DOX enhanced the green fluorescent signal of cleaved caspase-3 (as a biomarker of apoptosis) in H9c2 cells, whereas BA-NFs significantly inhibited this upregulation. These results provided further evidence that BA-NFs serve as potential therapeutic approaches for DIC.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | H9c2 cell | CVCL_0286 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [81] | ||||
| Indication | DOX-induced cardiomyopathy | ||||
| Efficacy Data | Fe2+ decrease ratio |
50%
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 100 µM | ||||
| Evaluation Method | Western blotting assay | ||||
| MOA of PDC |
Based on the above background, we designed and synthesized supramolecular self-assembled nanofibers with AT1R-specific targeting moieties (Baicalin-FFYEEG-ARVYIHPF, BA-NFs). Baicalin (BA), with its capacity for ROS scavenging and ferroptosis inhibition, constitutes an ideal candidate for ameliorating DIC. BA was conjugated with the self-assembling peptide FF and targeting peptide ARVYIHPF to form a specific nanostructure to improve its aqueous solubility and incorporate a targeting functionality, thereby addressing its inherent limitations. Our results indicated BA-NFs can mitigate doxorubicin-induced cardiomyocyte ferroptosis and cardiac dysfunction. This remarkable therapeutic effect is primarily attributable to the targeting proficiency of BA-NFs for injured cardiomyocytes, culminating in superoxide and ROS scavenging, reduced iron deposition, alleviated lipid peroxidation, and enhanced SLC7A11 and GPX4 expression. Therefore, we propose that BA-NFs proffers an efficacious targeted drug delivery strategy for the amelioration of DIC.
Click to Show/Hide
|
||||
| Description |
The accumulation of iron is a characteristic feature of ferroptosis. Intracellular iron, especially labile ferrous iron, can react with oxidants to generate cytotoxic hydroxyl radicals via Fenton reactions, thereby promoting ferroptosis. FerroOrange staining revealed significantly increased intracellular Fe2+ levels in DOX-treated H9c2 cells compared to controls, indicating DOX induced iron dyshomeostasis, resulting in ferrous iron buildup and subsequent ferroptosis. BA-NFs alleviated the accumulation of intracellular ferrous ions, suppressing ferroptosis. Fe2+ accumulation may disrupt the balance between LPO and oxygen homeostasis. We observed that BA-NFs efficiently attenuated DOX-induced elevation in malondialdehyde (MDA), a lipid peroxidation end-product, while increased antioxidant glutathione (GSH) levels. These findings suggested that BA-NFs can prevent DOX-induced ferroptosis in cardiomyocytes. Furthermore, protein expression of SLC7A11 and GPX4, key components of the antioxidant defense system against ferroptosis, was examined. DOX significantly decreased SLC7A11 and GPX4 expression in H9c2 cells compared to controls, suggesting DOX disrupts cellular antioxidant systems leading to ferroptosis. BA and BA-NFs partially restored SLC7A11 and GPX4 expression versus DOX treatment, with BA-NFs exhibiting the most significant impact. Together, these results indicate BA-NFs specifically inhibits DOX-induced cardiomyocyte ferroptosis by reducing intracellular iron accumulation and enhancing antioxidant system gene expression. In addition, the cardiotoxic effects of DOX are mediated via excessive oxidative stress and induced mitochondrial damage, thereby activating intrinsic apoptosis as an additional pathway. Flow cytometric analysis using Annexin V-Fitc/PI staining revealed that BA-NFs potently attenuated DOX-induced cardiomyocyte apoptosis, with a more pronounced effect than BA alone. DOX enhanced the green fluorescent signal of cleaved caspase-3 (as a biomarker of apoptosis) in H9c2 cells, whereas BA-NFs significantly inhibited this upregulation. These results provided further evidence that BA-NFs serve as potential therapeutic approaches for DIC.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | H9c2 cell | CVCL_0286 | ||
XRRR - Nalidixic acid conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [82] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 24 h | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
As expected, unconjugated NA showed limited activity toward MRSA and MSSA. Peptide vectors alone did not show significant toxicity to both MRSA and MSSA at all concentrations investigated. However, several of the peptide/drug conjugates, compounds 4 and 5 in particular, exhibited much lower IC50 and MIC values in both strains of the bacteria. A comparison of the hydrophobicities and molecular charges of the peptide/drug conjugates indicates that these two properties exert significant influence over the activities of these molecules. Hydrophobicity, measured by monitoring HPLC elution profiles, increased in the following order: 2 6 < 3 &tide; 4 < 5. The peptide conjugates that exhibited the highest potencies, compounds 4 and 5, had the highest levels of hydrophobicity. Compound 3, which had a similar level of hydrophobicity relative to these compounds, exhibited a lower level of toxicity, indicating that hydrophobicity alone does not control activity. This compound also carries a lower molecular charge, pointing to this parameter as another influencer of potency.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [82] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) | . Not detected | |||
| Administration Time | 24 h | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
As expected, unconjugated NA showed limited activity toward MRSA and MSSA. Peptide vectors alone did not show significant toxicity to both MRSA and MSSA at all concentrations investigated. However, several of the peptide/drug conjugates, compounds 4 and 5 in particular, exhibited much lower IC50 and MIC values in both strains of the bacteria. A comparison of the hydrophobicities and molecular charges of the peptide/drug conjugates indicates that these two properties exert significant influence over the activities of these molecules. Hydrophobicity, measured by monitoring HPLC elution profiles, increased in the following order: 2 6 < 3 &tide; 4 < 5. The peptide conjugates that exhibited the highest potencies, compounds 4 and 5, had the highest levels of hydrophobicity. Compound 3, which had a similar level of hydrophobicity relative to these compounds, exhibited a lower level of toxicity, indicating that hydrophobicity alone does not control activity. This compound also carries a lower molecular charge, pointing to this parameter as another influencer of potency.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [82] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 80 μM | |||
| Administration Time | 24 h | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
As expected, unconjugated NA showed limited activity toward MRSA and MSSA. Peptide vectors alone did not show significant toxicity to both MRSA and MSSA at all concentrations investigated. However, several of the peptide/drug conjugates, compounds 4 and 5 in particular, exhibited much lower IC50 and MIC values in both strains of the bacteria. A comparison of the hydrophobicities and molecular charges of the peptide/drug conjugates indicates that these two properties exert significant influence over the activities of these molecules. Hydrophobicity, measured by monitoring HPLC elution profiles, increased in the following order: 2 6 < 3 &tide; 4 < 5. The peptide conjugates that exhibited the highest potencies, compounds 4 and 5, had the highest levels of hydrophobicity. Compound 3, which had a similar level of hydrophobicity relative to these compounds, exhibited a lower level of toxicity, indicating that hydrophobicity alone does not control activity. This compound also carries a lower molecular charge, pointing to this parameter as another influencer of potency.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [82] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 80 μM | |||
| Administration Time | 24 h | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
As expected, unconjugated NA showed limited activity toward MRSA and MSSA. Peptide vectors alone did not show significant toxicity to both MRSA and MSSA at all concentrations investigated. However, several of the peptide/drug conjugates, compounds 4 and 5 in particular, exhibited much lower IC50 and MIC values in both strains of the bacteria. A comparison of the hydrophobicities and molecular charges of the peptide/drug conjugates indicates that these two properties exert significant influence over the activities of these molecules. Hydrophobicity, measured by monitoring HPLC elution profiles, increased in the following order: 2 6 < 3 &tide; 4 < 5. The peptide conjugates that exhibited the highest potencies, compounds 4 and 5, had the highest levels of hydrophobicity. Compound 3, which had a similar level of hydrophobicity relative to these compounds, exhibited a lower level of toxicity, indicating that hydrophobicity alone does not control activity. This compound also carries a lower molecular charge, pointing to this parameter as another influencer of potency.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [82] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 800 μM | |||
| Administration Time | 2 h | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
As expected, unconjugated NA showed limited activity toward MRSA and MSSA. Peptide vectors alone did not show significant toxicity to both MRSA and MSSA at all concentrations investigated. However, several of the peptide/drug conjugates, compounds 4 and 5 in particular, exhibited much lower IC50 and MIC values in both strains of the bacteria. A comparison of the hydrophobicities and molecular charges of the peptide/drug conjugates indicates that these two properties exert significant influence over the activities of these molecules. Hydrophobicity, measured by monitoring HPLC elution profiles, increased in the following order: 2 6 < 3 &tide; 4 < 5. The peptide conjugates that exhibited the highest potencies, compounds 4 and 5, had the highest levels of hydrophobicity. Compound 3, which had a similar level of hydrophobicity relative to these compounds, exhibited a lower level of toxicity, indicating that hydrophobicity alone does not control activity. This compound also carries a lower molecular charge, pointing to this parameter as another influencer of potency.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus infection strain | 1280 | ||
XRXRXRX - Nalidixic acid conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [82] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
13 μg/mL
|
|||
| Administration Time | 24 h | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
As expected, unconjugated NA showed limited activity toward MRSA and MSSA. Peptide vectors alone did not show significant toxicity to both MRSA and MSSA at all concentrations investigated. However, several of the peptide/drug conjugates, compounds 4 and 5 in particular, exhibited much lower IC50 and MIC values in both strains of the bacteria. A comparison of the hydrophobicities and molecular charges of the peptide/drug conjugates indicates that these two properties exert significant influence over the activities of these molecules. Hydrophobicity, measured by monitoring HPLC elution profiles, increased in the following order: 2 6 < 3 &tide; 4 < 5. The peptide conjugates that exhibited the highest potencies, compounds 4 and 5, had the highest levels of hydrophobicity. Compound 3, which had a similar level of hydrophobicity relative to these compounds, exhibited a lower level of toxicity, indicating that hydrophobicity alone does not control activity. This compound also carries a lower molecular charge, pointing to this parameter as another influencer of potency.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [82] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
13 μg/mL
|
|||
| Administration Time | 24 h | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
As expected, unconjugated NA showed limited activity toward MRSA and MSSA. Peptide vectors alone did not show significant toxicity to both MRSA and MSSA at all concentrations investigated. However, several of the peptide/drug conjugates, compounds 4 and 5 in particular, exhibited much lower IC50 and MIC values in both strains of the bacteria. A comparison of the hydrophobicities and molecular charges of the peptide/drug conjugates indicates that these two properties exert significant influence over the activities of these molecules. Hydrophobicity, measured by monitoring HPLC elution profiles, increased in the following order: 2 6 < 3 &tide; 4 < 5. The peptide conjugates that exhibited the highest potencies, compounds 4 and 5, had the highest levels of hydrophobicity. Compound 3, which had a similar level of hydrophobicity relative to these compounds, exhibited a lower level of toxicity, indicating that hydrophobicity alone does not control activity. This compound also carries a lower molecular charge, pointing to this parameter as another influencer of potency.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [82] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
6.0 ± 0.1 μM
|
|||
| Administration Time | 24 h | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
As expected, unconjugated NA showed limited activity toward MRSA and MSSA. Peptide vectors alone did not show significant toxicity to both MRSA and MSSA at all concentrations investigated. However, several of the peptide/drug conjugates, compounds 4 and 5 in particular, exhibited much lower IC50 and MIC values in both strains of the bacteria. A comparison of the hydrophobicities and molecular charges of the peptide/drug conjugates indicates that these two properties exert significant influence over the activities of these molecules. Hydrophobicity, measured by monitoring HPLC elution profiles, increased in the following order: 2 6 < 3 &tide; 4 < 5. The peptide conjugates that exhibited the highest potencies, compounds 4 and 5, had the highest levels of hydrophobicity. Compound 3, which had a similar level of hydrophobicity relative to these compounds, exhibited a lower level of toxicity, indicating that hydrophobicity alone does not control activity. This compound also carries a lower molecular charge, pointing to this parameter as another influencer of potency.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [82] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
7 ± 2 μM
|
|||
| Administration Time | 24 h | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
As expected, unconjugated NA showed limited activity toward MRSA and MSSA. Peptide vectors alone did not show significant toxicity to both MRSA and MSSA at all concentrations investigated. However, several of the peptide/drug conjugates, compounds 4 and 5 in particular, exhibited much lower IC50 and MIC values in both strains of the bacteria. A comparison of the hydrophobicities and molecular charges of the peptide/drug conjugates indicates that these two properties exert significant influence over the activities of these molecules. Hydrophobicity, measured by monitoring HPLC elution profiles, increased in the following order: 2 6 < 3 &tide; 4 < 5. The peptide conjugates that exhibited the highest potencies, compounds 4 and 5, had the highest levels of hydrophobicity. Compound 3, which had a similar level of hydrophobicity relative to these compounds, exhibited a lower level of toxicity, indicating that hydrophobicity alone does not control activity. This compound also carries a lower molecular charge, pointing to this parameter as another influencer of potency.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [82] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
25 μM
|
|||
| Administration Time | 2 h | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
As expected, unconjugated NA showed limited activity toward MRSA and MSSA. Peptide vectors alone did not show significant toxicity to both MRSA and MSSA at all concentrations investigated. However, several of the peptide/drug conjugates, compounds 4 and 5 in particular, exhibited much lower IC50 and MIC values in both strains of the bacteria. A comparison of the hydrophobicities and molecular charges of the peptide/drug conjugates indicates that these two properties exert significant influence over the activities of these molecules. Hydrophobicity, measured by monitoring HPLC elution profiles, increased in the following order: 2 6 < 3 &tide; 4 < 5. The peptide conjugates that exhibited the highest potencies, compounds 4 and 5, had the highest levels of hydrophobicity. Compound 3, which had a similar level of hydrophobicity relative to these compounds, exhibited a lower level of toxicity, indicating that hydrophobicity alone does not control activity. This compound also carries a lower molecular charge, pointing to this parameter as another influencer of potency.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus infection strain | 1280 | ||
RXRXRX - Nalidixic acid conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [82] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
18 μg/mL
|
|||
| Administration Time | 24 h | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
As expected, unconjugated NA showed limited activity toward MRSA and MSSA. Peptide vectors alone did not show significant toxicity to both MRSA and MSSA at all concentrations investigated. However, several of the peptide/drug conjugates, compounds 4 and 5 in particular, exhibited much lower IC50 and MIC values in both strains of the bacteria. A comparison of the hydrophobicities and molecular charges of the peptide/drug conjugates indicates that these two properties exert significant influence over the activities of these molecules. Hydrophobicity, measured by monitoring HPLC elution profiles, increased in the following order: 2 6 < 3 &tide; 4 < 5. The peptide conjugates that exhibited the highest potencies, compounds 4 and 5, had the highest levels of hydrophobicity. Compound 3, which had a similar level of hydrophobicity relative to these compounds, exhibited a lower level of toxicity, indicating that hydrophobicity alone does not control activity. This compound also carries a lower molecular charge, pointing to this parameter as another influencer of potency.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [82] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
18 μg/mL
|
|||
| Administration Time | 24 h | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
As expected, unconjugated NA showed limited activity toward MRSA and MSSA. Peptide vectors alone did not show significant toxicity to both MRSA and MSSA at all concentrations investigated. However, several of the peptide/drug conjugates, compounds 4 and 5 in particular, exhibited much lower IC50 and MIC values in both strains of the bacteria. A comparison of the hydrophobicities and molecular charges of the peptide/drug conjugates indicates that these two properties exert significant influence over the activities of these molecules. Hydrophobicity, measured by monitoring HPLC elution profiles, increased in the following order: 2 6 < 3 &tide; 4 < 5. The peptide conjugates that exhibited the highest potencies, compounds 4 and 5, had the highest levels of hydrophobicity. Compound 3, which had a similar level of hydrophobicity relative to these compounds, exhibited a lower level of toxicity, indicating that hydrophobicity alone does not control activity. This compound also carries a lower molecular charge, pointing to this parameter as another influencer of potency.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [82] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
6.0 ± 3.3 μM
|
|||
| Administration Time | 24 h | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
As expected, unconjugated NA showed limited activity toward MRSA and MSSA. Peptide vectors alone did not show significant toxicity to both MRSA and MSSA at all concentrations investigated. However, several of the peptide/drug conjugates, compounds 4 and 5 in particular, exhibited much lower IC50 and MIC values in both strains of the bacteria. A comparison of the hydrophobicities and molecular charges of the peptide/drug conjugates indicates that these two properties exert significant influence over the activities of these molecules. Hydrophobicity, measured by monitoring HPLC elution profiles, increased in the following order: 2 6 < 3 &tide; 4 < 5. The peptide conjugates that exhibited the highest potencies, compounds 4 and 5, had the highest levels of hydrophobicity. Compound 3, which had a similar level of hydrophobicity relative to these compounds, exhibited a lower level of toxicity, indicating that hydrophobicity alone does not control activity. This compound also carries a lower molecular charge, pointing to this parameter as another influencer of potency.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [82] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
7.0 ± 1.4 μM
|
|||
| Administration Time | 24 h | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
As expected, unconjugated NA showed limited activity toward MRSA and MSSA. Peptide vectors alone did not show significant toxicity to both MRSA and MSSA at all concentrations investigated. However, several of the peptide/drug conjugates, compounds 4 and 5 in particular, exhibited much lower IC50 and MIC values in both strains of the bacteria. A comparison of the hydrophobicities and molecular charges of the peptide/drug conjugates indicates that these two properties exert significant influence over the activities of these molecules. Hydrophobicity, measured by monitoring HPLC elution profiles, increased in the following order: 2 6 < 3 &tide; 4 < 5. The peptide conjugates that exhibited the highest potencies, compounds 4 and 5, had the highest levels of hydrophobicity. Compound 3, which had a similar level of hydrophobicity relative to these compounds, exhibited a lower level of toxicity, indicating that hydrophobicity alone does not control activity. This compound also carries a lower molecular charge, pointing to this parameter as another influencer of potency.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [82] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
25 μM
|
|||
| Administration Time | 2 h | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
As expected, unconjugated NA showed limited activity toward MRSA and MSSA. Peptide vectors alone did not show significant toxicity to both MRSA and MSSA at all concentrations investigated. However, several of the peptide/drug conjugates, compounds 4 and 5 in particular, exhibited much lower IC50 and MIC values in both strains of the bacteria. A comparison of the hydrophobicities and molecular charges of the peptide/drug conjugates indicates that these two properties exert significant influence over the activities of these molecules. Hydrophobicity, measured by monitoring HPLC elution profiles, increased in the following order: 2 6 < 3 &tide; 4 < 5. The peptide conjugates that exhibited the highest potencies, compounds 4 and 5, had the highest levels of hydrophobicity. Compound 3, which had a similar level of hydrophobicity relative to these compounds, exhibited a lower level of toxicity, indicating that hydrophobicity alone does not control activity. This compound also carries a lower molecular charge, pointing to this parameter as another influencer of potency.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus infection strain | 1280 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [82] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 80 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
To assess whether our most potent NA-peptide conjugates would possess a therapeutic window compatible with low levels of human toxicity, two types of human fibroblasts were treated with NA, compound 4, and the unconjugated peptide. The IC50 values were >10-fold higher for the fibroblasts versus the S. aureus strains tested. This trend indicates that the drug conjugates may possess a suitable therapeutic window for successful application as antibacterial agents.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human fibroblast strain 1 | Homo sapiens | ||
| Experiment 7 Reporting the Activity Data of This PDC | [82] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 80 μM | |||
| Administration Time | 24 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
To assess whether our most potent NA-peptide conjugates would possess a therapeutic window compatible with low levels of human toxicity, two types of human fibroblasts were treated with NA, compound 4, and the unconjugated peptide. The IC50 values were >10-fold higher for the fibroblasts versus the S. aureus strains tested. This trend indicates that the drug conjugates may possess a suitable therapeutic window for successful application as antibacterial agents.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human fibroblast strain 2 | Homo sapiens | ||
FRARXRARXRAR - Nalidixic acid conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [82] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
37 μg/mL
|
|||
| Administration Time | 24 h | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
As expected, unconjugated NA showed limited activity toward MRSA and MSSA. Peptide vectors alone did not show significant toxicity to both MRSA and MSSA at all concentrations investigated. However, several of the peptide/drug conjugates, compounds 4 and 5 in particular, exhibited much lower IC50 and MIC values in both strains of the bacteria. A comparison of the hydrophobicities and molecular charges of the peptide/drug conjugates indicates that these two properties exert significant influence over the activities of these molecules. Hydrophobicity, measured by monitoring HPLC elution profiles, increased in the following order: 2 6 < 3 &tide; 4 < 5. The peptide conjugates that exhibited the highest potencies, compounds 4 and 5, had the highest levels of hydrophobicity. Compound 3, which had a similar level of hydrophobicity relative to these compounds, exhibited a lower level of toxicity, indicating that hydrophobicity alone does not control activity. This compound also carries a lower molecular charge, pointing to this parameter as another influencer of potency.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [82] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
147 μg/mL
|
|||
| Administration Time | 24 h | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
As expected, unconjugated NA showed limited activity toward MRSA and MSSA. Peptide vectors alone did not show significant toxicity to both MRSA and MSSA at all concentrations investigated. However, several of the peptide/drug conjugates, compounds 4 and 5 in particular, exhibited much lower IC50 and MIC values in both strains of the bacteria. A comparison of the hydrophobicities and molecular charges of the peptide/drug conjugates indicates that these two properties exert significant influence over the activities of these molecules. Hydrophobicity, measured by monitoring HPLC elution profiles, increased in the following order: 2 6 < 3 &tide; 4 < 5. The peptide conjugates that exhibited the highest potencies, compounds 4 and 5, had the highest levels of hydrophobicity. Compound 3, which had a similar level of hydrophobicity relative to these compounds, exhibited a lower level of toxicity, indicating that hydrophobicity alone does not control activity. This compound also carries a lower molecular charge, pointing to this parameter as another influencer of potency.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [82] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
20 ± 2 μM
|
|||
| Administration Time | 24 h | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
As expected, unconjugated NA showed limited activity toward MRSA and MSSA. Peptide vectors alone did not show significant toxicity to both MRSA and MSSA at all concentrations investigated. However, several of the peptide/drug conjugates, compounds 4 and 5 in particular, exhibited much lower IC50 and MIC values in both strains of the bacteria. A comparison of the hydrophobicities and molecular charges of the peptide/drug conjugates indicates that these two properties exert significant influence over the activities of these molecules. Hydrophobicity, measured by monitoring HPLC elution profiles, increased in the following order: 2 6 < 3 &tide; 4 < 5. The peptide conjugates that exhibited the highest potencies, compounds 4 and 5, had the highest levels of hydrophobicity. Compound 3, which had a similar level of hydrophobicity relative to these compounds, exhibited a lower level of toxicity, indicating that hydrophobicity alone does not control activity. This compound also carries a lower molecular charge, pointing to this parameter as another influencer of potency.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [82] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
61 ± 2 μM
|
|||
| Administration Time | 24 h | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
As expected, unconjugated NA showed limited activity toward MRSA and MSSA. Peptide vectors alone did not show significant toxicity to both MRSA and MSSA at all concentrations investigated. However, several of the peptide/drug conjugates, compounds 4 and 5 in particular, exhibited much lower IC50 and MIC values in both strains of the bacteria. A comparison of the hydrophobicities and molecular charges of the peptide/drug conjugates indicates that these two properties exert significant influence over the activities of these molecules. Hydrophobicity, measured by monitoring HPLC elution profiles, increased in the following order: 2 6 < 3 &tide; 4 < 5. The peptide conjugates that exhibited the highest potencies, compounds 4 and 5, had the highest levels of hydrophobicity. Compound 3, which had a similar level of hydrophobicity relative to these compounds, exhibited a lower level of toxicity, indicating that hydrophobicity alone does not control activity. This compound also carries a lower molecular charge, pointing to this parameter as another influencer of potency.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [82] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
200 μM
|
|||
| Administration Time | 2 h | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
As expected, unconjugated NA showed limited activity toward MRSA and MSSA. Peptide vectors alone did not show significant toxicity to both MRSA and MSSA at all concentrations investigated. However, several of the peptide/drug conjugates, compounds 4 and 5 in particular, exhibited much lower IC50 and MIC values in both strains of the bacteria. A comparison of the hydrophobicities and molecular charges of the peptide/drug conjugates indicates that these two properties exert significant influence over the activities of these molecules. Hydrophobicity, measured by monitoring HPLC elution profiles, increased in the following order: 2 6 < 3 &tide; 4 < 5. The peptide conjugates that exhibited the highest potencies, compounds 4 and 5, had the highest levels of hydrophobicity. Compound 3, which had a similar level of hydrophobicity relative to these compounds, exhibited a lower level of toxicity, indicating that hydrophobicity alone does not control activity. This compound also carries a lower molecular charge, pointing to this parameter as another influencer of potency.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus infection strain | 1280 | ||
RXRX - Nalidixic acid conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [82] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
68 μg/mL
|
|||
| Administration Time | 24 h | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
As expected, unconjugated NA showed limited activity toward MRSA and MSSA. Peptide vectors alone did not show significant toxicity to both MRSA and MSSA at all concentrations investigated. However, several of the peptide/drug conjugates, compounds 4 and 5 in particular, exhibited much lower IC50 and MIC values in both strains of the bacteria. A comparison of the hydrophobicities and molecular charges of the peptide/drug conjugates indicates that these two properties exert significant influence over the activities of these molecules. Hydrophobicity, measured by monitoring HPLC elution profiles, increased in the following order: 2 6 < 3 &tide; 4 < 5. The peptide conjugates that exhibited the highest potencies, compounds 4 and 5, had the highest levels of hydrophobicity. Compound 3, which had a similar level of hydrophobicity relative to these compounds, exhibited a lower level of toxicity, indicating that hydrophobicity alone does not control activity. This compound also carries a lower molecular charge, pointing to this parameter as another influencer of potency.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [82] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Minimum inhibitory concentration (MIC) |
74 μg/mL
|
|||
| Administration Time | 24 h | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
As expected, unconjugated NA showed limited activity toward MRSA and MSSA. Peptide vectors alone did not show significant toxicity to both MRSA and MSSA at all concentrations investigated. However, several of the peptide/drug conjugates, compounds 4 and 5 in particular, exhibited much lower IC50 and MIC values in both strains of the bacteria. A comparison of the hydrophobicities and molecular charges of the peptide/drug conjugates indicates that these two properties exert significant influence over the activities of these molecules. Hydrophobicity, measured by monitoring HPLC elution profiles, increased in the following order: 2 6 < 3 &tide; 4 < 5. The peptide conjugates that exhibited the highest potencies, compounds 4 and 5, had the highest levels of hydrophobicity. Compound 3, which had a similar level of hydrophobicity relative to these compounds, exhibited a lower level of toxicity, indicating that hydrophobicity alone does not control activity. This compound also carries a lower molecular charge, pointing to this parameter as another influencer of potency.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [82] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
52 ± 1 μM
|
|||
| Administration Time | 24 h | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
As expected, unconjugated NA showed limited activity toward MRSA and MSSA. Peptide vectors alone did not show significant toxicity to both MRSA and MSSA at all concentrations investigated. However, several of the peptide/drug conjugates, compounds 4 and 5 in particular, exhibited much lower IC50 and MIC values in both strains of the bacteria. A comparison of the hydrophobicities and molecular charges of the peptide/drug conjugates indicates that these two properties exert significant influence over the activities of these molecules. Hydrophobicity, measured by monitoring HPLC elution profiles, increased in the following order: 2 6 < 3 &tide; 4 < 5. The peptide conjugates that exhibited the highest potencies, compounds 4 and 5, had the highest levels of hydrophobicity. Compound 3, which had a similar level of hydrophobicity relative to these compounds, exhibited a lower level of toxicity, indicating that hydrophobicity alone does not control activity. This compound also carries a lower molecular charge, pointing to this parameter as another influencer of potency.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [82] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
57 ± 0.8 μM
|
|||
| Administration Time | 24 h | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
As expected, unconjugated NA showed limited activity toward MRSA and MSSA. Peptide vectors alone did not show significant toxicity to both MRSA and MSSA at all concentrations investigated. However, several of the peptide/drug conjugates, compounds 4 and 5 in particular, exhibited much lower IC50 and MIC values in both strains of the bacteria. A comparison of the hydrophobicities and molecular charges of the peptide/drug conjugates indicates that these two properties exert significant influence over the activities of these molecules. Hydrophobicity, measured by monitoring HPLC elution profiles, increased in the following order: 2 6 < 3 &tide; 4 < 5. The peptide conjugates that exhibited the highest potencies, compounds 4 and 5, had the highest levels of hydrophobicity. Compound 3, which had a similar level of hydrophobicity relative to these compounds, exhibited a lower level of toxicity, indicating that hydrophobicity alone does not control activity. This compound also carries a lower molecular charge, pointing to this parameter as another influencer of potency.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus | 1280 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [82] | ||||
| Indication | <i>Staphylococcus aureus</i> infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
800 μM
|
|||
| Administration Time | 2 h | ||||
| MOA of PDC |
Nalidixic acid (NA) is a first-generation quinolone-based antibiotic that has a narrow spectrum and poor pharmacokinetics. Here, we describe a family of peptide-nalidixic acid conjugates featuring different levels of hydrophobicity and molecular charge prepared by solid-phase peptide synthesis that exhibit intriguing improvements in potency. In comparison to NA, which has a low level of potency in S. aureus, the NA peptide conjugates with optimized hydrophobicities and molecular charges exhibited significantly improved antibacterial activity. The most potent NA conjugate-featuring a peptide containing cyclohexylalanine and arginine-exhibited efficient bacterial uptake and, notably, specific inhibition of S. aureus DNA gyrase. A systematic study of peptide-NA conjugates revealed that a fine balance of cationic charge and hydrophobicity in an appendage anchored to the core of the drug is required to overcome the intrinsic resistance of S. aureus DNA gyrase toward this quinolone-based drug.
Click to Show/Hide
|
||||
| Description |
As expected, unconjugated NA showed limited activity toward MRSA and MSSA. Peptide vectors alone did not show significant toxicity to both MRSA and MSSA at all concentrations investigated. However, several of the peptide/drug conjugates, compounds 4 and 5 in particular, exhibited much lower IC50 and MIC values in both strains of the bacteria. A comparison of the hydrophobicities and molecular charges of the peptide/drug conjugates indicates that these two properties exert significant influence over the activities of these molecules. Hydrophobicity, measured by monitoring HPLC elution profiles, increased in the following order: 2 6 < 3 &tide; 4 < 5. The peptide conjugates that exhibited the highest potencies, compounds 4 and 5, had the highest levels of hydrophobicity. Compound 3, which had a similar level of hydrophobicity relative to these compounds, exhibited a lower level of toxicity, indicating that hydrophobicity alone does not control activity. This compound also carries a lower molecular charge, pointing to this parameter as another influencer of potency.
Click to Show/Hide
|
||||
| In Vitro Model | Staphylococcus aureus infection | Staphylococcus aureus infection strain | 1280 | ||
Dipeptide 23 - 4-Amino-7-chloroquinoline conjugate 8 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
0.83 μM
|
|||
| Administration Time | 48 h | ||||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
Activity against blood-stage CQ-resistant P. falciparum strain W2 was assessed as previously reported and given in detail in Experimental. Results demonstrated a complete lack of activity displayed by HECINs 9, correlating with their inability to inhibit heme polymerization. In turn, eleven out of the twelve HEDICINs 8 had IC50 values under 10 μM. Interestingly, three of the four most active HEDICIN blockers of heme polymerization (8b, 8j and 8l) were also among the four most active antiplasmodials, with IC50 below 2 μM. These results suggest that inhibition of heme polymerization is, at least in part, responsible for the anti-plasmodial activity of HEDICINs. An obvious exception to correlation between inhibition of heme polymerization and anti-plasmodial activity in HEDICINs is compound 8c; this bears a bulky electron-donating p-isopropyl group and did not inhibit heme polymerization in vitro, but displayed the highest anti-plasmodial activity. The inability of 8c to inhibit heme polymerization could be related to the bulkiness of the isopropyl group, but due to the higher hydrophobicity of this substituent, 8c was the most lipophilic HEDICIN assayed. Though we could not establish a full correlation between HEDICIN anti-plasmodial activity and estimated clogP values (not shown), the markedly higher lipophilicity of 8c, as compared to the other analogs, could promote a higher permeabilization of this compound into the infected RBC. Kirk and co-workers have demonstrated that P. falciparum parasites create new permeability pathways in host RBC, leading to increased permeability to small organic cations. In summary, though clean correlations could not be drawn between the anti-plasmodial activities displayed by the different HEDICINs (8) in vitro and molecular descriptors such as stereoelectronic factors (aryl substituents) or lipophilicity, it is demonstrated that these compounds displayed anti-plasmodial activity, whereas their HECIN counterparts (9), lacking the dipeptide spacer, did not. HEDICINs (8) inhibited heme polymerization in vitro, suggesting that this inhibitory activity is at least in part responsible for their anti-plasmodial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum | 5833 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 50 μM | |||
| Administration Time | 30 min | ||||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
The very different activities of compounds 8 and 9 suggest that the retro-enantio dipeptide spacer has a relevant role in determining anti-plasmodial activity. In view of this, we hypothesized that the anti-plasmodial activity of HEDICINs could also be partly due to falcipain inhibition. Therefore, both HEDICINs 8 and HECINs 9 were evaluated in vitro for inhibition of falcipains, using previously described methods, given in detail under Experimental. Only those compounds with IC50 < 50 μM against FP2 were assayed against FP3, as it has been established that FP2 has a larger catalytic cavity that accommodates a wider range of inhibitors than FP3. Consistent with this assumption, none of the compounds that inhibited FP2 in vitro displayed IC50 < 50 μM against FP3. Falcipain inhibition results contrasted with those for inhibition of heme polymerization or parasite development. HECINs 9 generally displayed more potent inhibition of falcipain than did HEDICINs 8. The ability of the test compounds to inhibit FP2 did not correlate with their anti-plasmodial activity. Although none of the HECINs 9 displayed anti-plasmodial activity, many inhibited FP2 and one of them, 9j, was actually the best FP2 inhibitor amongst the test compounds. Concerning HEDICINs, 8a, 8f and 8k, derived from cinnamic acid, m-fluorocinnamic acid, and m-nitrocinnamic acid, respectively, were the most active HEDICINs against FP2, with IC50 values of 19.7, 23.1 and 28.1 μM. Thus, stereoelectronic effects from aryl substituents did not correlate with inhibitory activities, as in HEDICINs 8 the most active compound was unsubstituted (8a), followed by the meta-fluorinated compound (8f); in turn, in HECINs 9, the meta-fluorinated derivative was inactive, whereas the most active of the set was 9j, which bears a meta-nitro substituent. The effect of amino acid configuration on HEDICIN activity was also assessed through synthesis and evaluation of 10a, the L-amino acid analog of 8a; interestingly, replacement of the D-amino acids by their natural L counterparts led to a clear decrease in both anti-plasmodial and falcipain-inhibitory activity. Therefore, amino acid configuration does influence compound behavior as either anti-plasmodial agent or falcipain inhibitor and, in the particular case of HEDICINs, data suggests that D-amino acids are preferable. Of note, the compound with highest anti-plasmodial activity, 8c, was completely devoid of inhibitory activity against FP2. This compound likely exerts its anti-plasmodial action by mechanisms other than inhibition of hemozoin formation or falcipain activity. Data on compound 8e reinforce the idea that falcipain or hemozoin inhibition are not the main mechanisms of action responsible for HEDICINs anti-plasmodial activity: 8e, bearing a p-amino substituent in the aryl ring, was the test compound which best reached our original goal of a dual-action inhibitor, by joining high hemozoin inhibitory activity with an IC50 20 μM against FP2; however, such was not translated into the highest anti-plasmodial activity being observed for 8e. Taken together, data from in vitro falcipain inhibition and parasite development assays suggest that the dipeptide spacer in HEDICINs 8 promotes uptake into infected RBCs. This hypothesis could explain why HEDICINs perform better than HECINs as antiplasmodials, despite the observation that HECINs were better falcipain-inhibitors than the HEDICINs. In addition, lipophilicity could have a role in compound uptake and anti-plasmodial action, as HEDICINs 8 are more lipophilic than HECINs 9 (due to the D-Leu and the D-hPhe hydrophobic side chains), and the most lipophilic compound, 8c, had the greatest anti-plasmodial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum strain W2 | 5833 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Haemozoin inhibition | < 50% | |||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
The capacity of test compounds 8 and 9 to inhibit heme polymerization in vitro was assessed by previously reported methods, given in detail under Experimental. The assays were run in 96-well microplates, where negative controls (water, DMSO) and positive controls (1 mM CQ) were included. Test compounds were assayed at 1 mM and data are given in Table 1. Interestingly, the dipeptide spacer was required to block heme polymerization, i.e., while HECINs 9 were not active, HEDICINs 8 displayed variable inhibitory efficiencies, with four out of the twelve compounds (8b, 8e, 8j and 8l) highly active (i.e., comparable to the reference drug, CQ). Though no clear trend could be established regarding stereoelectronic properties of the aryl substituent in compounds 8, it was clear that: (i) hydrogen (i.e., absence of a substituent) or halogens in the para position were detrimental for activity, whereas (ii) nitrogenated groups at either the ortho (8j, o-NO2) or the para (8e, p-NH2; 8l, p-NO2) position of the aryl ring was beneficial, but (iii) detrimental if placed in the meta position (8k, m-NO2). Furthermore, while small alkyl groups in para (8b, p-Me) were advantageous, bulkier groups as in 8c (p-iPr) led to complete loss of activity. Thus, replacement of CQ's aliphatic chain by an adequate dipeptidyl-cinnamoyl moiety as in 8b, 8e, 8j and 8l appears to preserve the parent drug's ability to inhibit hemozoin formation, suggesting that these novel compounds could be promising leads for new CQ surrogates.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum strain W2 | 5833 | ||
STRAP-4-MTX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [84] | ||||
| Indication | Cervical cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.01 ± 0.22 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Electrostatics modulates the interactions of CPPs with the corresponding cell surface. Therefore, for the design of the amino acid side chain sequences, we reverse-engineered the spatial electrostatic potential distribution of the previously designed peptides with cell penetration ability. The spatial electrostatic fingerprints of the earlier designed peptides were generated as described previously. The reverse-engineered sequences with a shorter (7-mer) chain length were compared to the previous designs through multiple iterations of amino acid sequences. Their respective electrostatic profiles were used for the design of four syndiotactic, cationic, amphipathic peptides with optimal sequence selection. These syndiotactic re-engineered amphipathic peptides (STRAPs) were tested for their ability to penetrate cells and deliver a small molecule (methotrexate). The amino acid constitutions of STRAP-1, STRAP-2, STRAP-3, and STRAP-4 are the same. However, STRAP-2 is the stereochemically reversed STRAP-1; similarly, STRAP-4 is the stereochemically reversed STRAP-3. The use of LDLD and DLDL stereochemical sequences in the design of STRAPs resulted in the differential electrostatic signatures for the same amino acid sequences. The designed peptides have a higher cellular uptake in TNBC cells (MDA-MB-231) than the standard control TAT peptide. They could penetrate cells by both active and passive processes, and their activity is not reduced in biological fluids (human plasma and bovine serum). Furthermore, the delivery of MTX as STRAP-MTX conjugates helped to overcome the drug resistance of the MDA-MB-231 cells under in vitro conditions. The delivery of the STARP-4-MTX conjugate in TNBC xenograft tumors was more effective in the reduction of both the tumor size and metastasis to the lungs, liver, spleen, and lymph nodes.
Click to Show/Hide
|
||||
| Description |
To deliver the functional molecule into the cells, we attached an anticancer drug, methotrexate (MTX), to the N-terminus of STRAPs. The peptides were tested for their capability to deliver the active drug molecule in breast and cervical cancer cells. MDA-MB-231 and HeLa cells were treated with varying concentrations of MTX and STRAP-MTX conjugates for 72 h. Cell viability was assessed using the MTT assay. The cytotoxicity of MTX increased when delivered as peptide-MTX conjugates to the cells. This resulted in an overall reduction in the inhibitory concentration cytotoxic to 50% of cells (IC50) for MTX. The MTX resistance for MDA-MB-231 cells is well-established. The IC50 for MTX in MDA-MB-231 cells was minimum when delivered as a STRAP-4-MTX conjugate. Similar lowering of IC50 values in HeLa cells was observed. We also treated HEK-293 cells with MTX-STRAP conjugates. The peptide-drug conjugates showed no significant toxicity to the HEK-293 cells under the tested conditions, thereby confirming the earlier reported observation of higher uptake rates in cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [84] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.34 ± 0.19 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Electrostatics modulates the interactions of CPPs with the corresponding cell surface. Therefore, for the design of the amino acid side chain sequences, we reverse-engineered the spatial electrostatic potential distribution of the previously designed peptides with cell penetration ability. The spatial electrostatic fingerprints of the earlier designed peptides were generated as described previously. The reverse-engineered sequences with a shorter (7-mer) chain length were compared to the previous designs through multiple iterations of amino acid sequences. Their respective electrostatic profiles were used for the design of four syndiotactic, cationic, amphipathic peptides with optimal sequence selection. These syndiotactic re-engineered amphipathic peptides (STRAPs) were tested for their ability to penetrate cells and deliver a small molecule (methotrexate). The amino acid constitutions of STRAP-1, STRAP-2, STRAP-3, and STRAP-4 are the same. However, STRAP-2 is the stereochemically reversed STRAP-1; similarly, STRAP-4 is the stereochemically reversed STRAP-3. The use of LDLD and DLDL stereochemical sequences in the design of STRAPs resulted in the differential electrostatic signatures for the same amino acid sequences. The designed peptides have a higher cellular uptake in TNBC cells (MDA-MB-231) than the standard control TAT peptide. They could penetrate cells by both active and passive processes, and their activity is not reduced in biological fluids (human plasma and bovine serum). Furthermore, the delivery of MTX as STRAP-MTX conjugates helped to overcome the drug resistance of the MDA-MB-231 cells under in vitro conditions. The delivery of the STARP-4-MTX conjugate in TNBC xenograft tumors was more effective in the reduction of both the tumor size and metastasis to the lungs, liver, spleen, and lymph nodes.
Click to Show/Hide
|
||||
| Description |
To deliver the functional molecule into the cells, we attached an anticancer drug, methotrexate (MTX), to the N-terminus of STRAPs. The peptides were tested for their capability to deliver the active drug molecule in breast and cervical cancer cells. MDA-MB-231 and HeLa cells were treated with varying concentrations of MTX and STRAP-MTX conjugates for 72 h. Cell viability was assessed using the MTT assay. The cytotoxicity of MTX increased when delivered as peptide-MTX conjugates to the cells. This resulted in an overall reduction in the inhibitory concentration cytotoxic to 50% of cells (IC50) for MTX. The MTX resistance for MDA-MB-231 cells is well-established. The IC50 for MTX in MDA-MB-231 cells was minimum when delivered as a STRAP-4-MTX conjugate. Similar lowering of IC50 values in HeLa cells was observed. We also treated HEK-293 cells with MTX-STRAP conjugates. The peptide-drug conjugates showed no significant toxicity to the HEK-293 cells under the tested conditions, thereby confirming the earlier reported observation of higher uptake rates in cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [84] | ||||
| Indication | Renal cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Electrostatics modulates the interactions of CPPs with the corresponding cell surface. Therefore, for the design of the amino acid side chain sequences, we reverse-engineered the spatial electrostatic potential distribution of the previously designed peptides with cell penetration ability. The spatial electrostatic fingerprints of the earlier designed peptides were generated as described previously. The reverse-engineered sequences with a shorter (7-mer) chain length were compared to the previous designs through multiple iterations of amino acid sequences. Their respective electrostatic profiles were used for the design of four syndiotactic, cationic, amphipathic peptides with optimal sequence selection. These syndiotactic re-engineered amphipathic peptides (STRAPs) were tested for their ability to penetrate cells and deliver a small molecule (methotrexate). The amino acid constitutions of STRAP-1, STRAP-2, STRAP-3, and STRAP-4 are the same. However, STRAP-2 is the stereochemically reversed STRAP-1; similarly, STRAP-4 is the stereochemically reversed STRAP-3. The use of LDLD and DLDL stereochemical sequences in the design of STRAPs resulted in the differential electrostatic signatures for the same amino acid sequences. The designed peptides have a higher cellular uptake in TNBC cells (MDA-MB-231) than the standard control TAT peptide. They could penetrate cells by both active and passive processes, and their activity is not reduced in biological fluids (human plasma and bovine serum). Furthermore, the delivery of MTX as STRAP-MTX conjugates helped to overcome the drug resistance of the MDA-MB-231 cells under in vitro conditions. The delivery of the STARP-4-MTX conjugate in TNBC xenograft tumors was more effective in the reduction of both the tumor size and metastasis to the lungs, liver, spleen, and lymph nodes.
Click to Show/Hide
|
||||
| Description |
To deliver the functional molecule into the cells, we attached an anticancer drug, methotrexate (MTX), to the N-terminus of STRAPs. The peptides were tested for their capability to deliver the active drug molecule in breast and cervical cancer cells. MDA-MB-231 and HeLa cells were treated with varying concentrations of MTX and STRAP-MTX conjugates for 72 h. Cell viability was assessed using the MTT assay. The cytotoxicity of MTX increased when delivered as peptide-MTX conjugates to the cells. This resulted in an overall reduction in the inhibitory concentration cytotoxic to 50% of cells (IC50) for MTX. The MTX resistance for MDA-MB-231 cells is well-established. The IC50 for MTX in MDA-MB-231 cells was minimum when delivered as a STRAP-4-MTX conjugate. Similar lowering of IC50 values in HeLa cells was observed. We also treated HEK-293 cells with MTX-STRAP conjugates. The peptide-drug conjugates showed no significant toxicity to the HEK-293 cells under the tested conditions, thereby confirming the earlier reported observation of higher uptake rates in cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | HEK293 cell | CVCL_0045 | ||
PP-P1 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [85] | ||||
| Indication | Zika virus infection | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.08 ± 0.14 µM
|
|||
| Administration Time | 1 h | ||||
| Evaluation Method | GraphPad Prism assay | ||||
| MOA of PDC |
Strategies for conjugating BBBpS to drug payloads have been actively explored over recent decades, with the number of new shuttles steadily increasing. Some recent entries, moreover, have shown to be capable not only of carrying drugs into but also removing toxins from the brain, preventing their accumulation. For our part, we have reported that peptide-porphyrin conjugates (PPCs), where a BBBpS and an antiviral porphyrin are covalently linked by an amide bond (amino and carboxyl groups in BBBpS and porphyrin, respectively) can successfully pass the BBB and act against brain-targeting viruses such as HIV. As for the BPB, the literature is scarce, with only a few described examples of peptides able to pass it. Herein, we report our results in developing new PPCs able to penetrate both BPB and BBB and act against ZIKV. The PPC production strategy, involving porphyrin (P), C- or N-terminal conjugation to a BBBpS, has been detailed in a recent publication and is illustrated in Scheme 1 and Scheme S1. In this paper we describe eight PPCs resulting from the combination of four BBBpS and two porphyrins, and their evaluation in terms of barrier crossing and anti-ZIKV activity. One of the conjugates, PP-P1, emerges as particularly effective against ZIKV, having also the ability to translocate across BPB and BBB.
Click to Show/Hide
|
||||
| Description |
The five PPCs that successfully translocated BBB and BPB were evaluated for ZIKV inactivation in vitro, using a plaque assay. Of them, two showed significant activity, namely MP-P5 (IC50 = 25.07 ± 0.05 μM, similar to activity against HIV) and PP-P1 (IC50 = 1.08 ± 0.14 μM). Additionally, a treatment assay performed with MP-P5 and PP-P1 revealed that both PPCs efficiently inhibit ZIKV replication when added 1 h and 7 h post-infection. As observed for HIV, non-conjugated porphyrins did not show activity against ZIKV, reinforcing the claim that BBBpS conjugation is not only critical for BBB/BPB translocation but also for antiviral activity. On the other hand, and somewhat unexpectedly, PP-P1, shown to be inactive in vitro against HIV, emerged as the most active anti-ZIKV conjugate. As described in the literature, the light-independent mechanism of action of porphyrins is based on a direct perturbation of the viral envelope. Porphyrins interact and accumulate on the envelope lipid membrane, causing a decrease in order and a consequent phase alteration that impairs viral entry processes. Since we ensured no-light conditions and no metal cations are coordinated to the porphyrin rings of the PPCsavoiding generation of reactive oxygen speciesthe antiviral light-independent mechanism is the only plausible one. Thus, one may confidently suggest that the PP-P1 specific anti-ZIKV activity is guided by preferential interaction with the ZIKV viral envelope and/or components.
Click to Show/Hide
|
||||
| In Vitro Model | ZIKV infection | Zika virus | 64320 | ||
| Half life period | 22.2 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [85] | ||||
| Indication | Human immunodeficiency virus infection | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Administration Time | 1 h | ||||
| Evaluation Method | GraphPad Prism assay | ||||
| MOA of PDC |
Strategies for conjugating BBBpS to drug payloads have been actively explored over recent decades, with the number of new shuttles steadily increasing. Some recent entries, moreover, have shown to be capable not only of carrying drugs into but also removing toxins from the brain, preventing their accumulation. For our part, we have reported that peptide-porphyrin conjugates (PPCs), where a BBBpS and an antiviral porphyrin are covalently linked by an amide bond (amino and carboxyl groups in BBBpS and porphyrin, respectively) can successfully pass the BBB and act against brain-targeting viruses such as HIV. As for the BPB, the literature is scarce, with only a few described examples of peptides able to pass it. Herein, we report our results in developing new PPCs able to penetrate both BPB and BBB and act against ZIKV. The PPC production strategy, involving porphyrin (P), C- or N-terminal conjugation to a BBBpS, has been detailed in a recent publication and is illustrated in Scheme 1 and Scheme S1. In this paper we describe eight PPCs resulting from the combination of four BBBpS and two porphyrins, and their evaluation in terms of barrier crossing and anti-ZIKV activity. One of the conjugates, PP-P1, emerges as particularly effective against ZIKV, having also the ability to translocate across BPB and BBB.
Click to Show/Hide
|
||||
| Description |
The five PPCs that successfully translocated BBB and BPB were evaluated for ZIKV inactivation in vitro, using a plaque assay. Of them, two showed significant activity, namely MP-P5 (IC50 = 25.07 ± 0.05 μM, similar to activity against HIV) and PP-P1 (IC50 = 1.08 ± 0.14 μM). Additionally, a treatment assay performed with MP-P5 and PP-P1 revealed that both PPCs efficiently inhibit ZIKV replication when added 1 h and 7 h post-infection. As observed for HIV, non-conjugated porphyrins did not show activity against ZIKV, reinforcing the claim that BBBpS conjugation is not only critical for BBB/BPB translocation but also for antiviral activity. On the other hand, and somewhat unexpectedly, PP-P1, shown to be inactive in vitro against HIV, emerged as the most active anti-ZIKV conjugate. As described in the literature, the light-independent mechanism of action of porphyrins is based on a direct perturbation of the viral envelope. Porphyrins interact and accumulate on the envelope lipid membrane, causing a decrease in order and a consequent phase alteration that impairs viral entry processes. Since we ensured no-light conditions and no metal cations are coordinated to the porphyrin rings of the PPCsavoiding generation of reactive oxygen speciesthe antiviral light-independent mechanism is the only plausible one. Thus, one may confidently suggest that the PP-P1 specific anti-ZIKV activity is guided by preferential interaction with the ZIKV viral envelope and/or components.
Click to Show/Hide
|
||||
| In Vitro Model | HIV Infection | Human immunodeficiency virus | 12721 | ||
| Half life period | 22.2 h | ||||
STRAP-3-MTX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [84] | ||||
| Indication | Cervical cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.22 ± 0.18 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Electrostatics modulates the interactions of CPPs with the corresponding cell surface. Therefore, for the design of the amino acid side chain sequences, we reverse-engineered the spatial electrostatic potential distribution of the previously designed peptides with cell penetration ability. The spatial electrostatic fingerprints of the earlier designed peptides were generated as described previously. The reverse-engineered sequences with a shorter (7-mer) chain length were compared to the previous designs through multiple iterations of amino acid sequences. Their respective electrostatic profiles were used for the design of four syndiotactic, cationic, amphipathic peptides with optimal sequence selection. These syndiotactic re-engineered amphipathic peptides (STRAPs) were tested for their ability to penetrate cells and deliver a small molecule (methotrexate). The amino acid constitutions of STRAP-1, STRAP-2, STRAP-3, and STRAP-4 are the same. However, STRAP-2 is the stereochemically reversed STRAP-1; similarly, STRAP-4 is the stereochemically reversed STRAP-3. The use of LDLD and DLDL stereochemical sequences in the design of STRAPs resulted in the differential electrostatic signatures for the same amino acid sequences. The designed peptides have a higher cellular uptake in TNBC cells (MDA-MB-231) than the standard control TAT peptide. They could penetrate cells by both active and passive processes, and their activity is not reduced in biological fluids (human plasma and bovine serum). Furthermore, the delivery of MTX as STRAP-MTX conjugates helped to overcome the drug resistance of the MDA-MB-231 cells under in vitro conditions. The delivery of the STARP-4-MTX conjugate in TNBC xenograft tumors was more effective in the reduction of both the tumor size and metastasis to the lungs, liver, spleen, and lymph nodes.
Click to Show/Hide
|
||||
| Description |
To deliver the functional molecule into the cells, we attached an anticancer drug, methotrexate (MTX), to the N-terminus of STRAPs. The peptides were tested for their capability to deliver the active drug molecule in breast and cervical cancer cells. MDA-MB-231 and HeLa cells were treated with varying concentrations of MTX and STRAP-MTX conjugates for 72 h. Cell viability was assessed using the MTT assay. The cytotoxicity of MTX increased when delivered as peptide-MTX conjugates to the cells. This resulted in an overall reduction in the inhibitory concentration cytotoxic to 50% of cells (IC50) for MTX. The MTX resistance for MDA-MB-231 cells is well-established. The IC50 for MTX in MDA-MB-231 cells was minimum when delivered as a STRAP-4-MTX conjugate. Similar lowering of IC50 values in HeLa cells was observed. We also treated HEK-293 cells with MTX-STRAP conjugates. The peptide-drug conjugates showed no significant toxicity to the HEK-293 cells under the tested conditions, thereby confirming the earlier reported observation of higher uptake rates in cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [84] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.79 ± 0.31 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Electrostatics modulates the interactions of CPPs with the corresponding cell surface. Therefore, for the design of the amino acid side chain sequences, we reverse-engineered the spatial electrostatic potential distribution of the previously designed peptides with cell penetration ability. The spatial electrostatic fingerprints of the earlier designed peptides were generated as described previously. The reverse-engineered sequences with a shorter (7-mer) chain length were compared to the previous designs through multiple iterations of amino acid sequences. Their respective electrostatic profiles were used for the design of four syndiotactic, cationic, amphipathic peptides with optimal sequence selection. These syndiotactic re-engineered amphipathic peptides (STRAPs) were tested for their ability to penetrate cells and deliver a small molecule (methotrexate). The amino acid constitutions of STRAP-1, STRAP-2, STRAP-3, and STRAP-4 are the same. However, STRAP-2 is the stereochemically reversed STRAP-1; similarly, STRAP-4 is the stereochemically reversed STRAP-3. The use of LDLD and DLDL stereochemical sequences in the design of STRAPs resulted in the differential electrostatic signatures for the same amino acid sequences. The designed peptides have a higher cellular uptake in TNBC cells (MDA-MB-231) than the standard control TAT peptide. They could penetrate cells by both active and passive processes, and their activity is not reduced in biological fluids (human plasma and bovine serum). Furthermore, the delivery of MTX as STRAP-MTX conjugates helped to overcome the drug resistance of the MDA-MB-231 cells under in vitro conditions. The delivery of the STARP-4-MTX conjugate in TNBC xenograft tumors was more effective in the reduction of both the tumor size and metastasis to the lungs, liver, spleen, and lymph nodes.
Click to Show/Hide
|
||||
| Description |
To deliver the functional molecule into the cells, we attached an anticancer drug, methotrexate (MTX), to the N-terminus of STRAPs. The peptides were tested for their capability to deliver the active drug molecule in breast and cervical cancer cells. MDA-MB-231 and HeLa cells were treated with varying concentrations of MTX and STRAP-MTX conjugates for 72 h. Cell viability was assessed using the MTT assay. The cytotoxicity of MTX increased when delivered as peptide-MTX conjugates to the cells. This resulted in an overall reduction in the inhibitory concentration cytotoxic to 50% of cells (IC50) for MTX. The MTX resistance for MDA-MB-231 cells is well-established. The IC50 for MTX in MDA-MB-231 cells was minimum when delivered as a STRAP-4-MTX conjugate. Similar lowering of IC50 values in HeLa cells was observed. We also treated HEK-293 cells with MTX-STRAP conjugates. The peptide-drug conjugates showed no significant toxicity to the HEK-293 cells under the tested conditions, thereby confirming the earlier reported observation of higher uptake rates in cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [84] | ||||
| Indication | Renal cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Electrostatics modulates the interactions of CPPs with the corresponding cell surface. Therefore, for the design of the amino acid side chain sequences, we reverse-engineered the spatial electrostatic potential distribution of the previously designed peptides with cell penetration ability. The spatial electrostatic fingerprints of the earlier designed peptides were generated as described previously. The reverse-engineered sequences with a shorter (7-mer) chain length were compared to the previous designs through multiple iterations of amino acid sequences. Their respective electrostatic profiles were used for the design of four syndiotactic, cationic, amphipathic peptides with optimal sequence selection. These syndiotactic re-engineered amphipathic peptides (STRAPs) were tested for their ability to penetrate cells and deliver a small molecule (methotrexate). The amino acid constitutions of STRAP-1, STRAP-2, STRAP-3, and STRAP-4 are the same. However, STRAP-2 is the stereochemically reversed STRAP-1; similarly, STRAP-4 is the stereochemically reversed STRAP-3. The use of LDLD and DLDL stereochemical sequences in the design of STRAPs resulted in the differential electrostatic signatures for the same amino acid sequences. The designed peptides have a higher cellular uptake in TNBC cells (MDA-MB-231) than the standard control TAT peptide. They could penetrate cells by both active and passive processes, and their activity is not reduced in biological fluids (human plasma and bovine serum). Furthermore, the delivery of MTX as STRAP-MTX conjugates helped to overcome the drug resistance of the MDA-MB-231 cells under in vitro conditions. The delivery of the STARP-4-MTX conjugate in TNBC xenograft tumors was more effective in the reduction of both the tumor size and metastasis to the lungs, liver, spleen, and lymph nodes.
Click to Show/Hide
|
||||
| Description |
To deliver the functional molecule into the cells, we attached an anticancer drug, methotrexate (MTX), to the N-terminus of STRAPs. The peptides were tested for their capability to deliver the active drug molecule in breast and cervical cancer cells. MDA-MB-231 and HeLa cells were treated with varying concentrations of MTX and STRAP-MTX conjugates for 72 h. Cell viability was assessed using the MTT assay. The cytotoxicity of MTX increased when delivered as peptide-MTX conjugates to the cells. This resulted in an overall reduction in the inhibitory concentration cytotoxic to 50% of cells (IC50) for MTX. The MTX resistance for MDA-MB-231 cells is well-established. The IC50 for MTX in MDA-MB-231 cells was minimum when delivered as a STRAP-4-MTX conjugate. Similar lowering of IC50 values in HeLa cells was observed. We also treated HEK-293 cells with MTX-STRAP conjugates. The peptide-drug conjugates showed no significant toxicity to the HEK-293 cells under the tested conditions, thereby confirming the earlier reported observation of higher uptake rates in cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | HEK293 cell | CVCL_0045 | ||
STRAP-1-MTX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [84] | ||||
| Indication | Cervical cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.63 ± 0.28 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Electrostatics modulates the interactions of CPPs with the corresponding cell surface. Therefore, for the design of the amino acid side chain sequences, we reverse-engineered the spatial electrostatic potential distribution of the previously designed peptides with cell penetration ability. The spatial electrostatic fingerprints of the earlier designed peptides were generated as described previously. The reverse-engineered sequences with a shorter (7-mer) chain length were compared to the previous designs through multiple iterations of amino acid sequences. Their respective electrostatic profiles were used for the design of four syndiotactic, cationic, amphipathic peptides with optimal sequence selection. These syndiotactic re-engineered amphipathic peptides (STRAPs) were tested for their ability to penetrate cells and deliver a small molecule (methotrexate). The amino acid constitutions of STRAP-1, STRAP-2, STRAP-3, and STRAP-4 are the same. However, STRAP-2 is the stereochemically reversed STRAP-1; similarly, STRAP-4 is the stereochemically reversed STRAP-3. The use of LDLD and DLDL stereochemical sequences in the design of STRAPs resulted in the differential electrostatic signatures for the same amino acid sequences. The designed peptides have a higher cellular uptake in TNBC cells (MDA-MB-231) than the standard control TAT peptide. They could penetrate cells by both active and passive processes, and their activity is not reduced in biological fluids (human plasma and bovine serum). Furthermore, the delivery of MTX as STRAP-MTX conjugates helped to overcome the drug resistance of the MDA-MB-231 cells under in vitro conditions. The delivery of the STARP-4-MTX conjugate in TNBC xenograft tumors was more effective in the reduction of both the tumor size and metastasis to the lungs, liver, spleen, and lymph nodes.
Click to Show/Hide
|
||||
| Description |
To deliver the functional molecule into the cells, we attached an anticancer drug, methotrexate (MTX), to the N-terminus of STRAPs. The peptides were tested for their capability to deliver the active drug molecule in breast and cervical cancer cells. MDA-MB-231 and HeLa cells were treated with varying concentrations of MTX and STRAP-MTX conjugates for 72 h. Cell viability was assessed using the MTT assay. The cytotoxicity of MTX increased when delivered as peptide-MTX conjugates to the cells. This resulted in an overall reduction in the inhibitory concentration cytotoxic to 50% of cells (IC50) for MTX. The MTX resistance for MDA-MB-231 cells is well-established. The IC50 for MTX in MDA-MB-231 cells was minimum when delivered as a STRAP-4-MTX conjugate. Similar lowering of IC50 values in HeLa cells was observed. We also treated HEK-293 cells with MTX-STRAP conjugates. The peptide-drug conjugates showed no significant toxicity to the HEK-293 cells under the tested conditions, thereby confirming the earlier reported observation of higher uptake rates in cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [84] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.26 ± 0.31 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Electrostatics modulates the interactions of CPPs with the corresponding cell surface. Therefore, for the design of the amino acid side chain sequences, we reverse-engineered the spatial electrostatic potential distribution of the previously designed peptides with cell penetration ability. The spatial electrostatic fingerprints of the earlier designed peptides were generated as described previously. The reverse-engineered sequences with a shorter (7-mer) chain length were compared to the previous designs through multiple iterations of amino acid sequences. Their respective electrostatic profiles were used for the design of four syndiotactic, cationic, amphipathic peptides with optimal sequence selection. These syndiotactic re-engineered amphipathic peptides (STRAPs) were tested for their ability to penetrate cells and deliver a small molecule (methotrexate). The amino acid constitutions of STRAP-1, STRAP-2, STRAP-3, and STRAP-4 are the same. However, STRAP-2 is the stereochemically reversed STRAP-1; similarly, STRAP-4 is the stereochemically reversed STRAP-3. The use of LDLD and DLDL stereochemical sequences in the design of STRAPs resulted in the differential electrostatic signatures for the same amino acid sequences. The designed peptides have a higher cellular uptake in TNBC cells (MDA-MB-231) than the standard control TAT peptide. They could penetrate cells by both active and passive processes, and their activity is not reduced in biological fluids (human plasma and bovine serum). Furthermore, the delivery of MTX as STRAP-MTX conjugates helped to overcome the drug resistance of the MDA-MB-231 cells under in vitro conditions. The delivery of the STARP-4-MTX conjugate in TNBC xenograft tumors was more effective in the reduction of both the tumor size and metastasis to the lungs, liver, spleen, and lymph nodes.
Click to Show/Hide
|
||||
| Description |
To deliver the functional molecule into the cells, we attached an anticancer drug, methotrexate (MTX), to the N-terminus of STRAPs. The peptides were tested for their capability to deliver the active drug molecule in breast and cervical cancer cells. MDA-MB-231 and HeLa cells were treated with varying concentrations of MTX and STRAP-MTX conjugates for 72 h. Cell viability was assessed using the MTT assay. The cytotoxicity of MTX increased when delivered as peptide-MTX conjugates to the cells. This resulted in an overall reduction in the inhibitory concentration cytotoxic to 50% of cells (IC50) for MTX. The MTX resistance for MDA-MB-231 cells is well-established. The IC50 for MTX in MDA-MB-231 cells was minimum when delivered as a STRAP-4-MTX conjugate. Similar lowering of IC50 values in HeLa cells was observed. We also treated HEK-293 cells with MTX-STRAP conjugates. The peptide-drug conjugates showed no significant toxicity to the HEK-293 cells under the tested conditions, thereby confirming the earlier reported observation of higher uptake rates in cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [84] | ||||
| Indication | Renal cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Electrostatics modulates the interactions of CPPs with the corresponding cell surface. Therefore, for the design of the amino acid side chain sequences, we reverse-engineered the spatial electrostatic potential distribution of the previously designed peptides with cell penetration ability. The spatial electrostatic fingerprints of the earlier designed peptides were generated as described previously. The reverse-engineered sequences with a shorter (7-mer) chain length were compared to the previous designs through multiple iterations of amino acid sequences. Their respective electrostatic profiles were used for the design of four syndiotactic, cationic, amphipathic peptides with optimal sequence selection. These syndiotactic re-engineered amphipathic peptides (STRAPs) were tested for their ability to penetrate cells and deliver a small molecule (methotrexate). The amino acid constitutions of STRAP-1, STRAP-2, STRAP-3, and STRAP-4 are the same. However, STRAP-2 is the stereochemically reversed STRAP-1; similarly, STRAP-4 is the stereochemically reversed STRAP-3. The use of LDLD and DLDL stereochemical sequences in the design of STRAPs resulted in the differential electrostatic signatures for the same amino acid sequences. The designed peptides have a higher cellular uptake in TNBC cells (MDA-MB-231) than the standard control TAT peptide. They could penetrate cells by both active and passive processes, and their activity is not reduced in biological fluids (human plasma and bovine serum). Furthermore, the delivery of MTX as STRAP-MTX conjugates helped to overcome the drug resistance of the MDA-MB-231 cells under in vitro conditions. The delivery of the STARP-4-MTX conjugate in TNBC xenograft tumors was more effective in the reduction of both the tumor size and metastasis to the lungs, liver, spleen, and lymph nodes.
Click to Show/Hide
|
||||
| Description |
To deliver the functional molecule into the cells, we attached an anticancer drug, methotrexate (MTX), to the N-terminus of STRAPs. The peptides were tested for their capability to deliver the active drug molecule in breast and cervical cancer cells. MDA-MB-231 and HeLa cells were treated with varying concentrations of MTX and STRAP-MTX conjugates for 72 h. Cell viability was assessed using the MTT assay. The cytotoxicity of MTX increased when delivered as peptide-MTX conjugates to the cells. This resulted in an overall reduction in the inhibitory concentration cytotoxic to 50% of cells (IC50) for MTX. The MTX resistance for MDA-MB-231 cells is well-established. The IC50 for MTX in MDA-MB-231 cells was minimum when delivered as a STRAP-4-MTX conjugate. Similar lowering of IC50 values in HeLa cells was observed. We also treated HEK-293 cells with MTX-STRAP conjugates. The peptide-drug conjugates showed no significant toxicity to the HEK-293 cells under the tested conditions, thereby confirming the earlier reported observation of higher uptake rates in cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | HEK293 cell | CVCL_0045 | ||
STRAP-2-MTX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [84] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.82 ± 0.24 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Electrostatics modulates the interactions of CPPs with the corresponding cell surface. Therefore, for the design of the amino acid side chain sequences, we reverse-engineered the spatial electrostatic potential distribution of the previously designed peptides with cell penetration ability. The spatial electrostatic fingerprints of the earlier designed peptides were generated as described previously. The reverse-engineered sequences with a shorter (7-mer) chain length were compared to the previous designs through multiple iterations of amino acid sequences. Their respective electrostatic profiles were used for the design of four syndiotactic, cationic, amphipathic peptides with optimal sequence selection. These syndiotactic re-engineered amphipathic peptides (STRAPs) were tested for their ability to penetrate cells and deliver a small molecule (methotrexate). The amino acid constitutions of STRAP-1, STRAP-2, STRAP-3, and STRAP-4 are the same. However, STRAP-2 is the stereochemically reversed STRAP-1; similarly, STRAP-4 is the stereochemically reversed STRAP-3. The use of LDLD and DLDL stereochemical sequences in the design of STRAPs resulted in the differential electrostatic signatures for the same amino acid sequences. The designed peptides have a higher cellular uptake in TNBC cells (MDA-MB-231) than the standard control TAT peptide. They could penetrate cells by both active and passive processes, and their activity is not reduced in biological fluids (human plasma and bovine serum). Furthermore, the delivery of MTX as STRAP-MTX conjugates helped to overcome the drug resistance of the MDA-MB-231 cells under in vitro conditions. The delivery of the STARP-4-MTX conjugate in TNBC xenograft tumors was more effective in the reduction of both the tumor size and metastasis to the lungs, liver, spleen, and lymph nodes.
Click to Show/Hide
|
||||
| Description |
To deliver the functional molecule into the cells, we attached an anticancer drug, methotrexate (MTX), to the N-terminus of STRAPs. The peptides were tested for their capability to deliver the active drug molecule in breast and cervical cancer cells. MDA-MB-231 and HeLa cells were treated with varying concentrations of MTX and STRAP-MTX conjugates for 72 h. Cell viability was assessed using the MTT assay. The cytotoxicity of MTX increased when delivered as peptide-MTX conjugates to the cells. This resulted in an overall reduction in the inhibitory concentration cytotoxic to 50% of cells (IC50) for MTX. The MTX resistance for MDA-MB-231 cells is well-established. The IC50 for MTX in MDA-MB-231 cells was minimum when delivered as a STRAP-4-MTX conjugate. Similar lowering of IC50 values in HeLa cells was observed. We also treated HEK-293 cells with MTX-STRAP conjugates. The peptide-drug conjugates showed no significant toxicity to the HEK-293 cells under the tested conditions, thereby confirming the earlier reported observation of higher uptake rates in cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [84] | ||||
| Indication | Cervical cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
11.8 ± 2.28 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Electrostatics modulates the interactions of CPPs with the corresponding cell surface. Therefore, for the design of the amino acid side chain sequences, we reverse-engineered the spatial electrostatic potential distribution of the previously designed peptides with cell penetration ability. The spatial electrostatic fingerprints of the earlier designed peptides were generated as described previously. The reverse-engineered sequences with a shorter (7-mer) chain length were compared to the previous designs through multiple iterations of amino acid sequences. Their respective electrostatic profiles were used for the design of four syndiotactic, cationic, amphipathic peptides with optimal sequence selection. These syndiotactic re-engineered amphipathic peptides (STRAPs) were tested for their ability to penetrate cells and deliver a small molecule (methotrexate). The amino acid constitutions of STRAP-1, STRAP-2, STRAP-3, and STRAP-4 are the same. However, STRAP-2 is the stereochemically reversed STRAP-1; similarly, STRAP-4 is the stereochemically reversed STRAP-3. The use of LDLD and DLDL stereochemical sequences in the design of STRAPs resulted in the differential electrostatic signatures for the same amino acid sequences. The designed peptides have a higher cellular uptake in TNBC cells (MDA-MB-231) than the standard control TAT peptide. They could penetrate cells by both active and passive processes, and their activity is not reduced in biological fluids (human plasma and bovine serum). Furthermore, the delivery of MTX as STRAP-MTX conjugates helped to overcome the drug resistance of the MDA-MB-231 cells under in vitro conditions. The delivery of the STARP-4-MTX conjugate in TNBC xenograft tumors was more effective in the reduction of both the tumor size and metastasis to the lungs, liver, spleen, and lymph nodes.
Click to Show/Hide
|
||||
| Description |
To deliver the functional molecule into the cells, we attached an anticancer drug, methotrexate (MTX), to the N-terminus of STRAPs. The peptides were tested for their capability to deliver the active drug molecule in breast and cervical cancer cells. MDA-MB-231 and HeLa cells were treated with varying concentrations of MTX and STRAP-MTX conjugates for 72 h. Cell viability was assessed using the MTT assay. The cytotoxicity of MTX increased when delivered as peptide-MTX conjugates to the cells. This resulted in an overall reduction in the inhibitory concentration cytotoxic to 50% of cells (IC50) for MTX. The MTX resistance for MDA-MB-231 cells is well-established. The IC50 for MTX in MDA-MB-231 cells was minimum when delivered as a STRAP-4-MTX conjugate. Similar lowering of IC50 values in HeLa cells was observed. We also treated HEK-293 cells with MTX-STRAP conjugates. The peptide-drug conjugates showed no significant toxicity to the HEK-293 cells under the tested conditions, thereby confirming the earlier reported observation of higher uptake rates in cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [84] | ||||
| Indication | Renal cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Electrostatics modulates the interactions of CPPs with the corresponding cell surface. Therefore, for the design of the amino acid side chain sequences, we reverse-engineered the spatial electrostatic potential distribution of the previously designed peptides with cell penetration ability. The spatial electrostatic fingerprints of the earlier designed peptides were generated as described previously. The reverse-engineered sequences with a shorter (7-mer) chain length were compared to the previous designs through multiple iterations of amino acid sequences. Their respective electrostatic profiles were used for the design of four syndiotactic, cationic, amphipathic peptides with optimal sequence selection. These syndiotactic re-engineered amphipathic peptides (STRAPs) were tested for their ability to penetrate cells and deliver a small molecule (methotrexate). The amino acid constitutions of STRAP-1, STRAP-2, STRAP-3, and STRAP-4 are the same. However, STRAP-2 is the stereochemically reversed STRAP-1; similarly, STRAP-4 is the stereochemically reversed STRAP-3. The use of LDLD and DLDL stereochemical sequences in the design of STRAPs resulted in the differential electrostatic signatures for the same amino acid sequences. The designed peptides have a higher cellular uptake in TNBC cells (MDA-MB-231) than the standard control TAT peptide. They could penetrate cells by both active and passive processes, and their activity is not reduced in biological fluids (human plasma and bovine serum). Furthermore, the delivery of MTX as STRAP-MTX conjugates helped to overcome the drug resistance of the MDA-MB-231 cells under in vitro conditions. The delivery of the STARP-4-MTX conjugate in TNBC xenograft tumors was more effective in the reduction of both the tumor size and metastasis to the lungs, liver, spleen, and lymph nodes.
Click to Show/Hide
|
||||
| Description |
To deliver the functional molecule into the cells, we attached an anticancer drug, methotrexate (MTX), to the N-terminus of STRAPs. The peptides were tested for their capability to deliver the active drug molecule in breast and cervical cancer cells. MDA-MB-231 and HeLa cells were treated with varying concentrations of MTX and STRAP-MTX conjugates for 72 h. Cell viability was assessed using the MTT assay. The cytotoxicity of MTX increased when delivered as peptide-MTX conjugates to the cells. This resulted in an overall reduction in the inhibitory concentration cytotoxic to 50% of cells (IC50) for MTX. The MTX resistance for MDA-MB-231 cells is well-established. The IC50 for MTX in MDA-MB-231 cells was minimum when delivered as a STRAP-4-MTX conjugate. Similar lowering of IC50 values in HeLa cells was observed. We also treated HEK-293 cells with MTX-STRAP conjugates. The peptide-drug conjugates showed no significant toxicity to the HEK-293 cells under the tested conditions, thereby confirming the earlier reported observation of higher uptake rates in cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | HEK293 cell | CVCL_0045 | ||
Dipeptide 23 - 4-Amino-7-chloroquinoline conjugate 9 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
1.96 μM
|
|||
| Administration Time | 48 h | ||||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
Activity against blood-stage CQ-resistant P. falciparum strain W2 was assessed as previously reported and given in detail in Experimental. Results demonstrated a complete lack of activity displayed by HECINs 9, correlating with their inability to inhibit heme polymerization. In turn, eleven out of the twelve HEDICINs 8 had IC50 values under 10 μM. Interestingly, three of the four most active HEDICIN blockers of heme polymerization (8b, 8j and 8l) were also among the four most active antiplasmodials, with IC50 below 2 μM. These results suggest that inhibition of heme polymerization is, at least in part, responsible for the anti-plasmodial activity of HEDICINs. An obvious exception to correlation between inhibition of heme polymerization and anti-plasmodial activity in HEDICINs is compound 8c; this bears a bulky electron-donating p-isopropyl group and did not inhibit heme polymerization in vitro, but displayed the highest anti-plasmodial activity. The inability of 8c to inhibit heme polymerization could be related to the bulkiness of the isopropyl group, but due to the higher hydrophobicity of this substituent, 8c was the most lipophilic HEDICIN assayed. Though we could not establish a full correlation between HEDICIN anti-plasmodial activity and estimated clogP values (not shown), the markedly higher lipophilicity of 8c, as compared to the other analogs, could promote a higher permeabilization of this compound into the infected RBC. Kirk and co-workers have demonstrated that P. falciparum parasites create new permeability pathways in host RBC, leading to increased permeability to small organic cations. In summary, though clean correlations could not be drawn between the anti-plasmodial activities displayed by the different HEDICINs (8) in vitro and molecular descriptors such as stereoelectronic factors (aryl substituents) or lipophilicity, it is demonstrated that these compounds displayed anti-plasmodial activity, whereas their HECIN counterparts (9), lacking the dipeptide spacer, did not. HEDICINs (8) inhibited heme polymerization in vitro, suggesting that this inhibitory activity is at least in part responsible for their anti-plasmodial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum | 5833 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 50 μM | |||
| Administration Time | 30 min | ||||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
The very different activities of compounds 8 and 9 suggest that the retro-enantio dipeptide spacer has a relevant role in determining anti-plasmodial activity. In view of this, we hypothesized that the anti-plasmodial activity of HEDICINs could also be partly due to falcipain inhibition. Therefore, both HEDICINs 8 and HECINs 9 were evaluated in vitro for inhibition of falcipains, using previously described methods, given in detail under Experimental. Only those compounds with IC50 < 50 μM against FP2 were assayed against FP3, as it has been established that FP2 has a larger catalytic cavity that accommodates a wider range of inhibitors than FP3. Consistent with this assumption, none of the compounds that inhibited FP2 in vitro displayed IC50 < 50 μM against FP3. Falcipain inhibition results contrasted with those for inhibition of heme polymerization or parasite development. HECINs 9 generally displayed more potent inhibition of falcipain than did HEDICINs 8. The ability of the test compounds to inhibit FP2 did not correlate with their anti-plasmodial activity. Although none of the HECINs 9 displayed anti-plasmodial activity, many inhibited FP2 and one of them, 9j, was actually the best FP2 inhibitor amongst the test compounds. Concerning HEDICINs, 8a, 8f and 8k, derived from cinnamic acid, m-fluorocinnamic acid, and m-nitrocinnamic acid, respectively, were the most active HEDICINs against FP2, with IC50 values of 19.7, 23.1 and 28.1 μM. Thus, stereoelectronic effects from aryl substituents did not correlate with inhibitory activities, as in HEDICINs 8 the most active compound was unsubstituted (8a), followed by the meta-fluorinated compound (8f); in turn, in HECINs 9, the meta-fluorinated derivative was inactive, whereas the most active of the set was 9j, which bears a meta-nitro substituent. The effect of amino acid configuration on HEDICIN activity was also assessed through synthesis and evaluation of 10a, the L-amino acid analog of 8a; interestingly, replacement of the D-amino acids by their natural L counterparts led to a clear decrease in both anti-plasmodial and falcipain-inhibitory activity. Therefore, amino acid configuration does influence compound behavior as either anti-plasmodial agent or falcipain inhibitor and, in the particular case of HEDICINs, data suggests that D-amino acids are preferable. Of note, the compound with highest anti-plasmodial activity, 8c, was completely devoid of inhibitory activity against FP2. This compound likely exerts its anti-plasmodial action by mechanisms other than inhibition of hemozoin formation or falcipain activity. Data on compound 8e reinforce the idea that falcipain or hemozoin inhibition are not the main mechanisms of action responsible for HEDICINs anti-plasmodial activity: 8e, bearing a p-amino substituent in the aryl ring, was the test compound which best reached our original goal of a dual-action inhibitor, by joining high hemozoin inhibitory activity with an IC50 20 μM against FP2; however, such was not translated into the highest anti-plasmodial activity being observed for 8e. Taken together, data from in vitro falcipain inhibition and parasite development assays suggest that the dipeptide spacer in HEDICINs 8 promotes uptake into infected RBCs. This hypothesis could explain why HEDICINs perform better than HECINs as antiplasmodials, despite the observation that HECINs were better falcipain-inhibitors than the HEDICINs. In addition, lipophilicity could have a role in compound uptake and anti-plasmodial action, as HEDICINs 8 are more lipophilic than HECINs 9 (due to the D-Leu and the D-hPhe hydrophobic side chains), and the most lipophilic compound, 8c, had the greatest anti-plasmodial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum strain W2 | 5833 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Haemozoin inhibition | ≥ 75% | |||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
The capacity of test compounds 8 and 9 to inhibit heme polymerization in vitro was assessed by previously reported methods, given in detail under Experimental. The assays were run in 96-well microplates, where negative controls (water, DMSO) and positive controls (1 mM CQ) were included. Test compounds were assayed at 1 mM and data are given in Table 1. Interestingly, the dipeptide spacer was required to block heme polymerization, i.e., while HECINs 9 were not active, HEDICINs 8 displayed variable inhibitory efficiencies, with four out of the twelve compounds (8b, 8e, 8j and 8l) highly active (i.e., comparable to the reference drug, CQ). Though no clear trend could be established regarding stereoelectronic properties of the aryl substituent in compounds 8, it was clear that: (i) hydrogen (i.e., absence of a substituent) or halogens in the para position were detrimental for activity, whereas (ii) nitrogenated groups at either the ortho (8j, o-NO2) or the para (8e, p-NH2; 8l, p-NO2) position of the aryl ring was beneficial, but (iii) detrimental if placed in the meta position (8k, m-NO2). Furthermore, while small alkyl groups in para (8b, p-Me) were advantageous, bulkier groups as in 8c (p-iPr) led to complete loss of activity. Thus, replacement of CQ's aliphatic chain by an adequate dipeptidyl-cinnamoyl moiety as in 8b, 8e, 8j and 8l appears to preserve the parent drug's ability to inhibit hemozoin formation, suggesting that these novel compounds could be promising leads for new CQ surrogates.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum strain W2 | 5833 | ||
Dipeptide 23 - 4-Amino-7-chloroquinoline conjugate 1 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
2.1 μM
|
|||
| Administration Time | 48 h | ||||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
Activity against blood-stage CQ-resistant P. falciparum strain W2 was assessed as previously reported and given in detail in Experimental. Results demonstrated a complete lack of activity displayed by HECINs 9, correlating with their inability to inhibit heme polymerization. In turn, eleven out of the twelve HEDICINs 8 had IC50 values under 10 μM. Interestingly, three of the four most active HEDICIN blockers of heme polymerization (8b, 8j and 8l) were also among the four most active antiplasmodials, with IC50 below 2 μM. These results suggest that inhibition of heme polymerization is, at least in part, responsible for the anti-plasmodial activity of HEDICINs. An obvious exception to correlation between inhibition of heme polymerization and anti-plasmodial activity in HEDICINs is compound 8c; this bears a bulky electron-donating p-isopropyl group and did not inhibit heme polymerization in vitro, but displayed the highest anti-plasmodial activity. The inability of 8c to inhibit heme polymerization could be related to the bulkiness of the isopropyl group, but due to the higher hydrophobicity of this substituent, 8c was the most lipophilic HEDICIN assayed. Though we could not establish a full correlation between HEDICIN anti-plasmodial activity and estimated clogP values (not shown), the markedly higher lipophilicity of 8c, as compared to the other analogs, could promote a higher permeabilization of this compound into the infected RBC. Kirk and co-workers have demonstrated that P. falciparum parasites create new permeability pathways in host RBC, leading to increased permeability to small organic cations. In summary, though clean correlations could not be drawn between the anti-plasmodial activities displayed by the different HEDICINs (8) in vitro and molecular descriptors such as stereoelectronic factors (aryl substituents) or lipophilicity, it is demonstrated that these compounds displayed anti-plasmodial activity, whereas their HECIN counterparts (9), lacking the dipeptide spacer, did not. HEDICINs (8) inhibited heme polymerization in vitro, suggesting that this inhibitory activity is at least in part responsible for their anti-plasmodial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum | 5833 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
28.1 μM
|
|||
| Administration Time | 30 min | ||||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
The very different activities of compounds 8 and 9 suggest that the retro-enantio dipeptide spacer has a relevant role in determining anti-plasmodial activity. In view of this, we hypothesized that the anti-plasmodial activity of HEDICINs could also be partly due to falcipain inhibition. Therefore, both HEDICINs 8 and HECINs 9 were evaluated in vitro for inhibition of falcipains, using previously described methods, given in detail under Experimental. Only those compounds with IC50 < 50 μM against FP2 were assayed against FP3, as it has been established that FP2 has a larger catalytic cavity that accommodates a wider range of inhibitors than FP3. Consistent with this assumption, none of the compounds that inhibited FP2 in vitro displayed IC50 < 50 μM against FP3. Falcipain inhibition results contrasted with those for inhibition of heme polymerization or parasite development. HECINs 9 generally displayed more potent inhibition of falcipain than did HEDICINs 8. The ability of the test compounds to inhibit FP2 did not correlate with their anti-plasmodial activity. Although none of the HECINs 9 displayed anti-plasmodial activity, many inhibited FP2 and one of them, 9j, was actually the best FP2 inhibitor amongst the test compounds. Concerning HEDICINs, 8a, 8f and 8k, derived from cinnamic acid, m-fluorocinnamic acid, and m-nitrocinnamic acid, respectively, were the most active HEDICINs against FP2, with IC50 values of 19.7, 23.1 and 28.1 μM. Thus, stereoelectronic effects from aryl substituents did not correlate with inhibitory activities, as in HEDICINs 8 the most active compound was unsubstituted (8a), followed by the meta-fluorinated compound (8f); in turn, in HECINs 9, the meta-fluorinated derivative was inactive, whereas the most active of the set was 9j, which bears a meta-nitro substituent. The effect of amino acid configuration on HEDICIN activity was also assessed through synthesis and evaluation of 10a, the L-amino acid analog of 8a; interestingly, replacement of the D-amino acids by their natural L counterparts led to a clear decrease in both anti-plasmodial and falcipain-inhibitory activity. Therefore, amino acid configuration does influence compound behavior as either anti-plasmodial agent or falcipain inhibitor and, in the particular case of HEDICINs, data suggests that D-amino acids are preferable. Of note, the compound with highest anti-plasmodial activity, 8c, was completely devoid of inhibitory activity against FP2. This compound likely exerts its anti-plasmodial action by mechanisms other than inhibition of hemozoin formation or falcipain activity. Data on compound 8e reinforce the idea that falcipain or hemozoin inhibition are not the main mechanisms of action responsible for HEDICINs anti-plasmodial activity: 8e, bearing a p-amino substituent in the aryl ring, was the test compound which best reached our original goal of a dual-action inhibitor, by joining high hemozoin inhibitory activity with an IC50 20 μM against FP2; however, such was not translated into the highest anti-plasmodial activity being observed for 8e. Taken together, data from in vitro falcipain inhibition and parasite development assays suggest that the dipeptide spacer in HEDICINs 8 promotes uptake into infected RBCs. This hypothesis could explain why HEDICINs perform better than HECINs as antiplasmodials, despite the observation that HECINs were better falcipain-inhibitors than the HEDICINs. In addition, lipophilicity could have a role in compound uptake and anti-plasmodial action, as HEDICINs 8 are more lipophilic than HECINs 9 (due to the D-Leu and the D-hPhe hydrophobic side chains), and the most lipophilic compound, 8c, had the greatest anti-plasmodial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum strain W2 | 5833 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 50 μM | |||
| Administration Time | 30 min | ||||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
The very different activities of compounds 8 and 9 suggest that the retro-enantio dipeptide spacer has a relevant role in determining anti-plasmodial activity. In view of this, we hypothesized that the anti-plasmodial activity of HEDICINs could also be partly due to falcipain inhibition. Therefore, both HEDICINs 8 and HECINs 9 were evaluated in vitro for inhibition of falcipains, using previously described methods, given in detail under Experimental. Only those compounds with IC50 < 50 μM against FP2 were assayed against FP3, as it has been established that FP2 has a larger catalytic cavity that accommodates a wider range of inhibitors than FP3. Consistent with this assumption, none of the compounds that inhibited FP2 in vitro displayed IC50 < 50 μM against FP3. Falcipain inhibition results contrasted with those for inhibition of heme polymerization or parasite development. HECINs 9 generally displayed more potent inhibition of falcipain than did HEDICINs 8. The ability of the test compounds to inhibit FP2 did not correlate with their anti-plasmodial activity. Although none of the HECINs 9 displayed anti-plasmodial activity, many inhibited FP2 and one of them, 9j, was actually the best FP2 inhibitor amongst the test compounds. Concerning HEDICINs, 8a, 8f and 8k, derived from cinnamic acid, m-fluorocinnamic acid, and m-nitrocinnamic acid, respectively, were the most active HEDICINs against FP2, with IC50 values of 19.7, 23.1 and 28.1 μM. Thus, stereoelectronic effects from aryl substituents did not correlate with inhibitory activities, as in HEDICINs 8 the most active compound was unsubstituted (8a), followed by the meta-fluorinated compound (8f); in turn, in HECINs 9, the meta-fluorinated derivative was inactive, whereas the most active of the set was 9j, which bears a meta-nitro substituent. The effect of amino acid configuration on HEDICIN activity was also assessed through synthesis and evaluation of 10a, the L-amino acid analog of 8a; interestingly, replacement of the D-amino acids by their natural L counterparts led to a clear decrease in both anti-plasmodial and falcipain-inhibitory activity. Therefore, amino acid configuration does influence compound behavior as either anti-plasmodial agent or falcipain inhibitor and, in the particular case of HEDICINs, data suggests that D-amino acids are preferable. Of note, the compound with highest anti-plasmodial activity, 8c, was completely devoid of inhibitory activity against FP2. This compound likely exerts its anti-plasmodial action by mechanisms other than inhibition of hemozoin formation or falcipain activity. Data on compound 8e reinforce the idea that falcipain or hemozoin inhibition are not the main mechanisms of action responsible for HEDICINs anti-plasmodial activity: 8e, bearing a p-amino substituent in the aryl ring, was the test compound which best reached our original goal of a dual-action inhibitor, by joining high hemozoin inhibitory activity with an IC50 20 μM against FP2; however, such was not translated into the highest anti-plasmodial activity being observed for 8e. Taken together, data from in vitro falcipain inhibition and parasite development assays suggest that the dipeptide spacer in HEDICINs 8 promotes uptake into infected RBCs. This hypothesis could explain why HEDICINs perform better than HECINs as antiplasmodials, despite the observation that HECINs were better falcipain-inhibitors than the HEDICINs. In addition, lipophilicity could have a role in compound uptake and anti-plasmodial action, as HEDICINs 8 are more lipophilic than HECINs 9 (due to the D-Leu and the D-hPhe hydrophobic side chains), and the most lipophilic compound, 8c, had the greatest anti-plasmodial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum strain W2 | 5833 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Haemozoin inhibition | < 50% | |||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
The capacity of test compounds 8 and 9 to inhibit heme polymerization in vitro was assessed by previously reported methods, given in detail under Experimental. The assays were run in 96-well microplates, where negative controls (water, DMSO) and positive controls (1 mM CQ) were included. Test compounds were assayed at 1 mM and data are given in Table 1. Interestingly, the dipeptide spacer was required to block heme polymerization, i.e., while HECINs 9 were not active, HEDICINs 8 displayed variable inhibitory efficiencies, with four out of the twelve compounds (8b, 8e, 8j and 8l) highly active (i.e., comparable to the reference drug, CQ). Though no clear trend could be established regarding stereoelectronic properties of the aryl substituent in compounds 8, it was clear that: (i) hydrogen (i.e., absence of a substituent) or halogens in the para position were detrimental for activity, whereas (ii) nitrogenated groups at either the ortho (8j, o-NO2) or the para (8e, p-NH2; 8l, p-NO2) position of the aryl ring was beneficial, but (iii) detrimental if placed in the meta position (8k, m-NO2). Furthermore, while small alkyl groups in para (8b, p-Me) were advantageous, bulkier groups as in 8c (p-iPr) led to complete loss of activity. Thus, replacement of CQ's aliphatic chain by an adequate dipeptidyl-cinnamoyl moiety as in 8b, 8e, 8j and 8l appears to preserve the parent drug's ability to inhibit hemozoin formation, suggesting that these novel compounds could be promising leads for new CQ surrogates.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum strain W2 | 5833 | ||
Dipeptide 23 - 4-Amino-7-chloroquinoline conjugate 4 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
2.55 μM
|
|||
| Administration Time | 48 h | ||||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
Activity against blood-stage CQ-resistant P. falciparum strain W2 was assessed as previously reported and given in detail in Experimental. Results demonstrated a complete lack of activity displayed by HECINs 9, correlating with their inability to inhibit heme polymerization. In turn, eleven out of the twelve HEDICINs 8 had IC50 values under 10 μM. Interestingly, three of the four most active HEDICIN blockers of heme polymerization (8b, 8j and 8l) were also among the four most active antiplasmodials, with IC50 below 2 μM. These results suggest that inhibition of heme polymerization is, at least in part, responsible for the anti-plasmodial activity of HEDICINs. An obvious exception to correlation between inhibition of heme polymerization and anti-plasmodial activity in HEDICINs is compound 8c; this bears a bulky electron-donating p-isopropyl group and did not inhibit heme polymerization in vitro, but displayed the highest anti-plasmodial activity. The inability of 8c to inhibit heme polymerization could be related to the bulkiness of the isopropyl group, but due to the higher hydrophobicity of this substituent, 8c was the most lipophilic HEDICIN assayed. Though we could not establish a full correlation between HEDICIN anti-plasmodial activity and estimated clogP values (not shown), the markedly higher lipophilicity of 8c, as compared to the other analogs, could promote a higher permeabilization of this compound into the infected RBC. Kirk and co-workers have demonstrated that P. falciparum parasites create new permeability pathways in host RBC, leading to increased permeability to small organic cations. In summary, though clean correlations could not be drawn between the anti-plasmodial activities displayed by the different HEDICINs (8) in vitro and molecular descriptors such as stereoelectronic factors (aryl substituents) or lipophilicity, it is demonstrated that these compounds displayed anti-plasmodial activity, whereas their HECIN counterparts (9), lacking the dipeptide spacer, did not. HEDICINs (8) inhibited heme polymerization in vitro, suggesting that this inhibitory activity is at least in part responsible for their anti-plasmodial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum | 5833 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
48.3 μM
|
|||
| Administration Time | 30 min | ||||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
The very different activities of compounds 8 and 9 suggest that the retro-enantio dipeptide spacer has a relevant role in determining anti-plasmodial activity. In view of this, we hypothesized that the anti-plasmodial activity of HEDICINs could also be partly due to falcipain inhibition. Therefore, both HEDICINs 8 and HECINs 9 were evaluated in vitro for inhibition of falcipains, using previously described methods, given in detail under Experimental. Only those compounds with IC50 < 50 μM against FP2 were assayed against FP3, as it has been established that FP2 has a larger catalytic cavity that accommodates a wider range of inhibitors than FP3. Consistent with this assumption, none of the compounds that inhibited FP2 in vitro displayed IC50 < 50 μM against FP3. Falcipain inhibition results contrasted with those for inhibition of heme polymerization or parasite development. HECINs 9 generally displayed more potent inhibition of falcipain than did HEDICINs 8. The ability of the test compounds to inhibit FP2 did not correlate with their anti-plasmodial activity. Although none of the HECINs 9 displayed anti-plasmodial activity, many inhibited FP2 and one of them, 9j, was actually the best FP2 inhibitor amongst the test compounds. Concerning HEDICINs, 8a, 8f and 8k, derived from cinnamic acid, m-fluorocinnamic acid, and m-nitrocinnamic acid, respectively, were the most active HEDICINs against FP2, with IC50 values of 19.7, 23.1 and 28.1 μM. Thus, stereoelectronic effects from aryl substituents did not correlate with inhibitory activities, as in HEDICINs 8 the most active compound was unsubstituted (8a), followed by the meta-fluorinated compound (8f); in turn, in HECINs 9, the meta-fluorinated derivative was inactive, whereas the most active of the set was 9j, which bears a meta-nitro substituent. The effect of amino acid configuration on HEDICIN activity was also assessed through synthesis and evaluation of 10a, the L-amino acid analog of 8a; interestingly, replacement of the D-amino acids by their natural L counterparts led to a clear decrease in both anti-plasmodial and falcipain-inhibitory activity. Therefore, amino acid configuration does influence compound behavior as either anti-plasmodial agent or falcipain inhibitor and, in the particular case of HEDICINs, data suggests that D-amino acids are preferable. Of note, the compound with highest anti-plasmodial activity, 8c, was completely devoid of inhibitory activity against FP2. This compound likely exerts its anti-plasmodial action by mechanisms other than inhibition of hemozoin formation or falcipain activity. Data on compound 8e reinforce the idea that falcipain or hemozoin inhibition are not the main mechanisms of action responsible for HEDICINs anti-plasmodial activity: 8e, bearing a p-amino substituent in the aryl ring, was the test compound which best reached our original goal of a dual-action inhibitor, by joining high hemozoin inhibitory activity with an IC50 20 μM against FP2; however, such was not translated into the highest anti-plasmodial activity being observed for 8e. Taken together, data from in vitro falcipain inhibition and parasite development assays suggest that the dipeptide spacer in HEDICINs 8 promotes uptake into infected RBCs. This hypothesis could explain why HEDICINs perform better than HECINs as antiplasmodials, despite the observation that HECINs were better falcipain-inhibitors than the HEDICINs. In addition, lipophilicity could have a role in compound uptake and anti-plasmodial action, as HEDICINs 8 are more lipophilic than HECINs 9 (due to the D-Leu and the D-hPhe hydrophobic side chains), and the most lipophilic compound, 8c, had the greatest anti-plasmodial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum strain W2 | 5833 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 50 μM | |||
| Administration Time | 30 min | ||||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
The very different activities of compounds 8 and 9 suggest that the retro-enantio dipeptide spacer has a relevant role in determining anti-plasmodial activity. In view of this, we hypothesized that the anti-plasmodial activity of HEDICINs could also be partly due to falcipain inhibition. Therefore, both HEDICINs 8 and HECINs 9 were evaluated in vitro for inhibition of falcipains, using previously described methods, given in detail under Experimental. Only those compounds with IC50 < 50 μM against FP2 were assayed against FP3, as it has been established that FP2 has a larger catalytic cavity that accommodates a wider range of inhibitors than FP3. Consistent with this assumption, none of the compounds that inhibited FP2 in vitro displayed IC50 < 50 μM against FP3. Falcipain inhibition results contrasted with those for inhibition of heme polymerization or parasite development. HECINs 9 generally displayed more potent inhibition of falcipain than did HEDICINs 8. The ability of the test compounds to inhibit FP2 did not correlate with their anti-plasmodial activity. Although none of the HECINs 9 displayed anti-plasmodial activity, many inhibited FP2 and one of them, 9j, was actually the best FP2 inhibitor amongst the test compounds. Concerning HEDICINs, 8a, 8f and 8k, derived from cinnamic acid, m-fluorocinnamic acid, and m-nitrocinnamic acid, respectively, were the most active HEDICINs against FP2, with IC50 values of 19.7, 23.1 and 28.1 μM. Thus, stereoelectronic effects from aryl substituents did not correlate with inhibitory activities, as in HEDICINs 8 the most active compound was unsubstituted (8a), followed by the meta-fluorinated compound (8f); in turn, in HECINs 9, the meta-fluorinated derivative was inactive, whereas the most active of the set was 9j, which bears a meta-nitro substituent. The effect of amino acid configuration on HEDICIN activity was also assessed through synthesis and evaluation of 10a, the L-amino acid analog of 8a; interestingly, replacement of the D-amino acids by their natural L counterparts led to a clear decrease in both anti-plasmodial and falcipain-inhibitory activity. Therefore, amino acid configuration does influence compound behavior as either anti-plasmodial agent or falcipain inhibitor and, in the particular case of HEDICINs, data suggests that D-amino acids are preferable. Of note, the compound with highest anti-plasmodial activity, 8c, was completely devoid of inhibitory activity against FP2. This compound likely exerts its anti-plasmodial action by mechanisms other than inhibition of hemozoin formation or falcipain activity. Data on compound 8e reinforce the idea that falcipain or hemozoin inhibition are not the main mechanisms of action responsible for HEDICINs anti-plasmodial activity: 8e, bearing a p-amino substituent in the aryl ring, was the test compound which best reached our original goal of a dual-action inhibitor, by joining high hemozoin inhibitory activity with an IC50 20 μM against FP2; however, such was not translated into the highest anti-plasmodial activity being observed for 8e. Taken together, data from in vitro falcipain inhibition and parasite development assays suggest that the dipeptide spacer in HEDICINs 8 promotes uptake into infected RBCs. This hypothesis could explain why HEDICINs perform better than HECINs as antiplasmodials, despite the observation that HECINs were better falcipain-inhibitors than the HEDICINs. In addition, lipophilicity could have a role in compound uptake and anti-plasmodial action, as HEDICINs 8 are more lipophilic than HECINs 9 (due to the D-Leu and the D-hPhe hydrophobic side chains), and the most lipophilic compound, 8c, had the greatest anti-plasmodial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum strain W2 | 5833 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Haemozoin inhibition | < 50% | |||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
The capacity of test compounds 8 and 9 to inhibit heme polymerization in vitro was assessed by previously reported methods, given in detail under Experimental. The assays were run in 96-well microplates, where negative controls (water, DMSO) and positive controls (1 mM CQ) were included. Test compounds were assayed at 1 mM and data are given in Table 1. Interestingly, the dipeptide spacer was required to block heme polymerization, i.e., while HECINs 9 were not active, HEDICINs 8 displayed variable inhibitory efficiencies, with four out of the twelve compounds (8b, 8e, 8j and 8l) highly active (i.e., comparable to the reference drug, CQ). Though no clear trend could be established regarding stereoelectronic properties of the aryl substituent in compounds 8, it was clear that: (i) hydrogen (i.e., absence of a substituent) or halogens in the para position were detrimental for activity, whereas (ii) nitrogenated groups at either the ortho (8j, o-NO2) or the para (8e, p-NH2; 8l, p-NO2) position of the aryl ring was beneficial, but (iii) detrimental if placed in the meta position (8k, m-NO2). Furthermore, while small alkyl groups in para (8b, p-Me) were advantageous, bulkier groups as in 8c (p-iPr) led to complete loss of activity. Thus, replacement of CQ's aliphatic chain by an adequate dipeptidyl-cinnamoyl moiety as in 8b, 8e, 8j and 8l appears to preserve the parent drug's ability to inhibit hemozoin formation, suggesting that these novel compounds could be promising leads for new CQ surrogates.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum strain W2 | 5833 | ||
Dipeptide 23 - 4-Amino-7-chloroquinoline conjugate 5 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
2.89 μM
|
|||
| Administration Time | 48 h | ||||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
Activity against blood-stage CQ-resistant P. falciparum strain W2 was assessed as previously reported and given in detail in Experimental. Results demonstrated a complete lack of activity displayed by HECINs 9, correlating with their inability to inhibit heme polymerization. In turn, eleven out of the twelve HEDICINs 8 had IC50 values under 10 μM. Interestingly, three of the four most active HEDICIN blockers of heme polymerization (8b, 8j and 8l) were also among the four most active antiplasmodials, with IC50 below 2 μM. These results suggest that inhibition of heme polymerization is, at least in part, responsible for the anti-plasmodial activity of HEDICINs. An obvious exception to correlation between inhibition of heme polymerization and anti-plasmodial activity in HEDICINs is compound 8c; this bears a bulky electron-donating p-isopropyl group and did not inhibit heme polymerization in vitro, but displayed the highest anti-plasmodial activity. The inability of 8c to inhibit heme polymerization could be related to the bulkiness of the isopropyl group, but due to the higher hydrophobicity of this substituent, 8c was the most lipophilic HEDICIN assayed. Though we could not establish a full correlation between HEDICIN anti-plasmodial activity and estimated clogP values (not shown), the markedly higher lipophilicity of 8c, as compared to the other analogs, could promote a higher permeabilization of this compound into the infected RBC. Kirk and co-workers have demonstrated that P. falciparum parasites create new permeability pathways in host RBC, leading to increased permeability to small organic cations. In summary, though clean correlations could not be drawn between the anti-plasmodial activities displayed by the different HEDICINs (8) in vitro and molecular descriptors such as stereoelectronic factors (aryl substituents) or lipophilicity, it is demonstrated that these compounds displayed anti-plasmodial activity, whereas their HECIN counterparts (9), lacking the dipeptide spacer, did not. HEDICINs (8) inhibited heme polymerization in vitro, suggesting that this inhibitory activity is at least in part responsible for their anti-plasmodial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum | 5833 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 50 μM | |||
| Administration Time | 30 min | ||||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
The very different activities of compounds 8 and 9 suggest that the retro-enantio dipeptide spacer has a relevant role in determining anti-plasmodial activity. In view of this, we hypothesized that the anti-plasmodial activity of HEDICINs could also be partly due to falcipain inhibition. Therefore, both HEDICINs 8 and HECINs 9 were evaluated in vitro for inhibition of falcipains, using previously described methods, given in detail under Experimental. Only those compounds with IC50 < 50 μM against FP2 were assayed against FP3, as it has been established that FP2 has a larger catalytic cavity that accommodates a wider range of inhibitors than FP3. Consistent with this assumption, none of the compounds that inhibited FP2 in vitro displayed IC50 < 50 μM against FP3. Falcipain inhibition results contrasted with those for inhibition of heme polymerization or parasite development. HECINs 9 generally displayed more potent inhibition of falcipain than did HEDICINs 8. The ability of the test compounds to inhibit FP2 did not correlate with their anti-plasmodial activity. Although none of the HECINs 9 displayed anti-plasmodial activity, many inhibited FP2 and one of them, 9j, was actually the best FP2 inhibitor amongst the test compounds. Concerning HEDICINs, 8a, 8f and 8k, derived from cinnamic acid, m-fluorocinnamic acid, and m-nitrocinnamic acid, respectively, were the most active HEDICINs against FP2, with IC50 values of 19.7, 23.1 and 28.1 μM. Thus, stereoelectronic effects from aryl substituents did not correlate with inhibitory activities, as in HEDICINs 8 the most active compound was unsubstituted (8a), followed by the meta-fluorinated compound (8f); in turn, in HECINs 9, the meta-fluorinated derivative was inactive, whereas the most active of the set was 9j, which bears a meta-nitro substituent. The effect of amino acid configuration on HEDICIN activity was also assessed through synthesis and evaluation of 10a, the L-amino acid analog of 8a; interestingly, replacement of the D-amino acids by their natural L counterparts led to a clear decrease in both anti-plasmodial and falcipain-inhibitory activity. Therefore, amino acid configuration does influence compound behavior as either anti-plasmodial agent or falcipain inhibitor and, in the particular case of HEDICINs, data suggests that D-amino acids are preferable. Of note, the compound with highest anti-plasmodial activity, 8c, was completely devoid of inhibitory activity against FP2. This compound likely exerts its anti-plasmodial action by mechanisms other than inhibition of hemozoin formation or falcipain activity. Data on compound 8e reinforce the idea that falcipain or hemozoin inhibition are not the main mechanisms of action responsible for HEDICINs anti-plasmodial activity: 8e, bearing a p-amino substituent in the aryl ring, was the test compound which best reached our original goal of a dual-action inhibitor, by joining high hemozoin inhibitory activity with an IC50 20 μM against FP2; however, such was not translated into the highest anti-plasmodial activity being observed for 8e. Taken together, data from in vitro falcipain inhibition and parasite development assays suggest that the dipeptide spacer in HEDICINs 8 promotes uptake into infected RBCs. This hypothesis could explain why HEDICINs perform better than HECINs as antiplasmodials, despite the observation that HECINs were better falcipain-inhibitors than the HEDICINs. In addition, lipophilicity could have a role in compound uptake and anti-plasmodial action, as HEDICINs 8 are more lipophilic than HECINs 9 (due to the D-Leu and the D-hPhe hydrophobic side chains), and the most lipophilic compound, 8c, had the greatest anti-plasmodial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum strain W2 | 5833 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Haemozoin inhibition | < 50% | |||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
The capacity of test compounds 8 and 9 to inhibit heme polymerization in vitro was assessed by previously reported methods, given in detail under Experimental. The assays were run in 96-well microplates, where negative controls (water, DMSO) and positive controls (1 mM CQ) were included. Test compounds were assayed at 1 mM and data are given in Table 1. Interestingly, the dipeptide spacer was required to block heme polymerization, i.e., while HECINs 9 were not active, HEDICINs 8 displayed variable inhibitory efficiencies, with four out of the twelve compounds (8b, 8e, 8j and 8l) highly active (i.e., comparable to the reference drug, CQ). Though no clear trend could be established regarding stereoelectronic properties of the aryl substituent in compounds 8, it was clear that: (i) hydrogen (i.e., absence of a substituent) or halogens in the para position were detrimental for activity, whereas (ii) nitrogenated groups at either the ortho (8j, o-NO2) or the para (8e, p-NH2; 8l, p-NO2) position of the aryl ring was beneficial, but (iii) detrimental if placed in the meta position (8k, m-NO2). Furthermore, while small alkyl groups in para (8b, p-Me) were advantageous, bulkier groups as in 8c (p-iPr) led to complete loss of activity. Thus, replacement of CQ's aliphatic chain by an adequate dipeptidyl-cinnamoyl moiety as in 8b, 8e, 8j and 8l appears to preserve the parent drug's ability to inhibit hemozoin formation, suggesting that these novel compounds could be promising leads for new CQ surrogates.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum strain W2 | 5833 | ||
PP-P5 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [85] | ||||
| Indication | Human immunodeficiency virus infection | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.02 ± 0.35 µM
|
|||
| Administration Time | 1 h | ||||
| Evaluation Method | GraphPad Prism assay | ||||
| MOA of PDC |
Strategies for conjugating BBBpS to drug payloads have been actively explored over recent decades, with the number of new shuttles steadily increasing. Some recent entries, moreover, have shown to be capable not only of carrying drugs into but also removing toxins from the brain, preventing their accumulation. For our part, we have reported that peptide-porphyrin conjugates (PPCs), where a BBBpS and an antiviral porphyrin are covalently linked by an amide bond (amino and carboxyl groups in BBBpS and porphyrin, respectively) can successfully pass the BBB and act against brain-targeting viruses such as HIV. As for the BPB, the literature is scarce, with only a few described examples of peptides able to pass it. Herein, we report our results in developing new PPCs able to penetrate both BPB and BBB and act against ZIKV. The PPC production strategy, involving porphyrin (P), C- or N-terminal conjugation to a BBBpS, has been detailed in a recent publication and is illustrated in Scheme 1 and Scheme S1. In this paper we describe eight PPCs resulting from the combination of four BBBpS and two porphyrins, and their evaluation in terms of barrier crossing and anti-ZIKV activity. One of the conjugates, PP-P1, emerges as particularly effective against ZIKV, having also the ability to translocate across BPB and BBB.
Click to Show/Hide
|
||||
| Description |
The five PPCs that successfully translocated BBB and BPB were evaluated for ZIKV inactivation in vitro, using a plaque assay. Of them, two showed significant activity, namely MP-P5 (IC50 = 25.07 ± 0.05 μM, similar to activity against HIV) and PP-P1 (IC50 = 1.08 ± 0.14 μM). Additionally, a treatment assay performed with MP-P5 and PP-P1 revealed that both PPCs efficiently inhibit ZIKV replication when added 1 h and 7 h post-infection. As observed for HIV, non-conjugated porphyrins did not show activity against ZIKV, reinforcing the claim that BBBpS conjugation is not only critical for BBB/BPB translocation but also for antiviral activity. On the other hand, and somewhat unexpectedly, PP-P1, shown to be inactive in vitro against HIV, emerged as the most active anti-ZIKV conjugate. As described in the literature, the light-independent mechanism of action of porphyrins is based on a direct perturbation of the viral envelope. Porphyrins interact and accumulate on the envelope lipid membrane, causing a decrease in order and a consequent phase alteration that impairs viral entry processes. Since we ensured no-light conditions and no metal cations are coordinated to the porphyrin rings of the PPCsavoiding generation of reactive oxygen speciesthe antiviral light-independent mechanism is the only plausible one. Thus, one may confidently suggest that the PP-P1 specific anti-ZIKV activity is guided by preferential interaction with the ZIKV viral envelope and/or components.
Click to Show/Hide
|
||||
| In Vitro Model | HIV Infection | Human immunodeficiency virus | 12721 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [85] | ||||
| Indication | Zika virus infection | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Administration Time | 1 h | ||||
| Evaluation Method | GraphPad Prism assay | ||||
| MOA of PDC |
Strategies for conjugating BBBpS to drug payloads have been actively explored over recent decades, with the number of new shuttles steadily increasing. Some recent entries, moreover, have shown to be capable not only of carrying drugs into but also removing toxins from the brain, preventing their accumulation. For our part, we have reported that peptide-porphyrin conjugates (PPCs), where a BBBpS and an antiviral porphyrin are covalently linked by an amide bond (amino and carboxyl groups in BBBpS and porphyrin, respectively) can successfully pass the BBB and act against brain-targeting viruses such as HIV. As for the BPB, the literature is scarce, with only a few described examples of peptides able to pass it. Herein, we report our results in developing new PPCs able to penetrate both BPB and BBB and act against ZIKV. The PPC production strategy, involving porphyrin (P), C- or N-terminal conjugation to a BBBpS, has been detailed in a recent publication and is illustrated in Scheme 1 and Scheme S1. In this paper we describe eight PPCs resulting from the combination of four BBBpS and two porphyrins, and their evaluation in terms of barrier crossing and anti-ZIKV activity. One of the conjugates, PP-P1, emerges as particularly effective against ZIKV, having also the ability to translocate across BPB and BBB.
Click to Show/Hide
|
||||
| Description |
The five PPCs that successfully translocated BBB and BPB were evaluated for ZIKV inactivation in vitro, using a plaque assay. Of them, two showed significant activity, namely MP-P5 (IC50 = 25.07 ± 0.05 μM, similar to activity against HIV) and PP-P1 (IC50 = 1.08 ± 0.14 μM). Additionally, a treatment assay performed with MP-P5 and PP-P1 revealed that both PPCs efficiently inhibit ZIKV replication when added 1 h and 7 h post-infection. As observed for HIV, non-conjugated porphyrins did not show activity against ZIKV, reinforcing the claim that BBBpS conjugation is not only critical for BBB/BPB translocation but also for antiviral activity. On the other hand, and somewhat unexpectedly, PP-P1, shown to be inactive in vitro against HIV, emerged as the most active anti-ZIKV conjugate. As described in the literature, the light-independent mechanism of action of porphyrins is based on a direct perturbation of the viral envelope. Porphyrins interact and accumulate on the envelope lipid membrane, causing a decrease in order and a consequent phase alteration that impairs viral entry processes. Since we ensured no-light conditions and no metal cations are coordinated to the porphyrin rings of the PPCsavoiding generation of reactive oxygen speciesthe antiviral light-independent mechanism is the only plausible one. Thus, one may confidently suggest that the PP-P1 specific anti-ZIKV activity is guided by preferential interaction with the ZIKV viral envelope and/or components.
Click to Show/Hide
|
||||
| In Vitro Model | ZIKV infection | Zika virus | 64320 | ||
Dipeptide 23 - 4-Amino-7-chloroquinoline conjugate 12 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
3.28 μM
|
|||
| Administration Time | 48 h | ||||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
Activity against blood-stage CQ-resistant P. falciparum strain W2 was assessed as previously reported and given in detail in Experimental. Results demonstrated a complete lack of activity displayed by HECINs 9, correlating with their inability to inhibit heme polymerization. In turn, eleven out of the twelve HEDICINs 8 had IC50 values under 10 μM. Interestingly, three of the four most active HEDICIN blockers of heme polymerization (8b, 8j and 8l) were also among the four most active antiplasmodials, with IC50 below 2 μM. These results suggest that inhibition of heme polymerization is, at least in part, responsible for the anti-plasmodial activity of HEDICINs. An obvious exception to correlation between inhibition of heme polymerization and anti-plasmodial activity in HEDICINs is compound 8c; this bears a bulky electron-donating p-isopropyl group and did not inhibit heme polymerization in vitro, but displayed the highest anti-plasmodial activity. The inability of 8c to inhibit heme polymerization could be related to the bulkiness of the isopropyl group, but due to the higher hydrophobicity of this substituent, 8c was the most lipophilic HEDICIN assayed. Though we could not establish a full correlation between HEDICIN anti-plasmodial activity and estimated clogP values (not shown), the markedly higher lipophilicity of 8c, as compared to the other analogs, could promote a higher permeabilization of this compound into the infected RBC. Kirk and co-workers have demonstrated that P. falciparum parasites create new permeability pathways in host RBC, leading to increased permeability to small organic cations. In summary, though clean correlations could not be drawn between the anti-plasmodial activities displayed by the different HEDICINs (8) in vitro and molecular descriptors such as stereoelectronic factors (aryl substituents) or lipophilicity, it is demonstrated that these compounds displayed anti-plasmodial activity, whereas their HECIN counterparts (9), lacking the dipeptide spacer, did not. HEDICINs (8) inhibited heme polymerization in vitro, suggesting that this inhibitory activity is at least in part responsible for their anti-plasmodial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum | 5833 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
20.3 μM
|
|||
| Administration Time | 30 min | ||||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
The very different activities of compounds 8 and 9 suggest that the retro-enantio dipeptide spacer has a relevant role in determining anti-plasmodial activity. In view of this, we hypothesized that the anti-plasmodial activity of HEDICINs could also be partly due to falcipain inhibition. Therefore, both HEDICINs 8 and HECINs 9 were evaluated in vitro for inhibition of falcipains, using previously described methods, given in detail under Experimental. Only those compounds with IC50 < 50 μM against FP2 were assayed against FP3, as it has been established that FP2 has a larger catalytic cavity that accommodates a wider range of inhibitors than FP3. Consistent with this assumption, none of the compounds that inhibited FP2 in vitro displayed IC50 < 50 μM against FP3. Falcipain inhibition results contrasted with those for inhibition of heme polymerization or parasite development. HECINs 9 generally displayed more potent inhibition of falcipain than did HEDICINs 8. The ability of the test compounds to inhibit FP2 did not correlate with their anti-plasmodial activity. Although none of the HECINs 9 displayed anti-plasmodial activity, many inhibited FP2 and one of them, 9j, was actually the best FP2 inhibitor amongst the test compounds. Concerning HEDICINs, 8a, 8f and 8k, derived from cinnamic acid, m-fluorocinnamic acid, and m-nitrocinnamic acid, respectively, were the most active HEDICINs against FP2, with IC50 values of 19.7, 23.1 and 28.1 μM. Thus, stereoelectronic effects from aryl substituents did not correlate with inhibitory activities, as in HEDICINs 8 the most active compound was unsubstituted (8a), followed by the meta-fluorinated compound (8f); in turn, in HECINs 9, the meta-fluorinated derivative was inactive, whereas the most active of the set was 9j, which bears a meta-nitro substituent. The effect of amino acid configuration on HEDICIN activity was also assessed through synthesis and evaluation of 10a, the L-amino acid analog of 8a; interestingly, replacement of the D-amino acids by their natural L counterparts led to a clear decrease in both anti-plasmodial and falcipain-inhibitory activity. Therefore, amino acid configuration does influence compound behavior as either anti-plasmodial agent or falcipain inhibitor and, in the particular case of HEDICINs, data suggests that D-amino acids are preferable. Of note, the compound with highest anti-plasmodial activity, 8c, was completely devoid of inhibitory activity against FP2. This compound likely exerts its anti-plasmodial action by mechanisms other than inhibition of hemozoin formation or falcipain activity. Data on compound 8e reinforce the idea that falcipain or hemozoin inhibition are not the main mechanisms of action responsible for HEDICINs anti-plasmodial activity: 8e, bearing a p-amino substituent in the aryl ring, was the test compound which best reached our original goal of a dual-action inhibitor, by joining high hemozoin inhibitory activity with an IC50 20 μM against FP2; however, such was not translated into the highest anti-plasmodial activity being observed for 8e. Taken together, data from in vitro falcipain inhibition and parasite development assays suggest that the dipeptide spacer in HEDICINs 8 promotes uptake into infected RBCs. This hypothesis could explain why HEDICINs perform better than HECINs as antiplasmodials, despite the observation that HECINs were better falcipain-inhibitors than the HEDICINs. In addition, lipophilicity could have a role in compound uptake and anti-plasmodial action, as HEDICINs 8 are more lipophilic than HECINs 9 (due to the D-Leu and the D-hPhe hydrophobic side chains), and the most lipophilic compound, 8c, had the greatest anti-plasmodial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum strain W2 | 5833 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 50 μM | |||
| Administration Time | 30 min | ||||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
The very different activities of compounds 8 and 9 suggest that the retro-enantio dipeptide spacer has a relevant role in determining anti-plasmodial activity. In view of this, we hypothesized that the anti-plasmodial activity of HEDICINs could also be partly due to falcipain inhibition. Therefore, both HEDICINs 8 and HECINs 9 were evaluated in vitro for inhibition of falcipains, using previously described methods, given in detail under Experimental. Only those compounds with IC50 < 50 μM against FP2 were assayed against FP3, as it has been established that FP2 has a larger catalytic cavity that accommodates a wider range of inhibitors than FP3. Consistent with this assumption, none of the compounds that inhibited FP2 in vitro displayed IC50 < 50 μM against FP3. Falcipain inhibition results contrasted with those for inhibition of heme polymerization or parasite development. HECINs 9 generally displayed more potent inhibition of falcipain than did HEDICINs 8. The ability of the test compounds to inhibit FP2 did not correlate with their anti-plasmodial activity. Although none of the HECINs 9 displayed anti-plasmodial activity, many inhibited FP2 and one of them, 9j, was actually the best FP2 inhibitor amongst the test compounds. Concerning HEDICINs, 8a, 8f and 8k, derived from cinnamic acid, m-fluorocinnamic acid, and m-nitrocinnamic acid, respectively, were the most active HEDICINs against FP2, with IC50 values of 19.7, 23.1 and 28.1 μM. Thus, stereoelectronic effects from aryl substituents did not correlate with inhibitory activities, as in HEDICINs 8 the most active compound was unsubstituted (8a), followed by the meta-fluorinated compound (8f); in turn, in HECINs 9, the meta-fluorinated derivative was inactive, whereas the most active of the set was 9j, which bears a meta-nitro substituent. The effect of amino acid configuration on HEDICIN activity was also assessed through synthesis and evaluation of 10a, the L-amino acid analog of 8a; interestingly, replacement of the D-amino acids by their natural L counterparts led to a clear decrease in both anti-plasmodial and falcipain-inhibitory activity. Therefore, amino acid configuration does influence compound behavior as either anti-plasmodial agent or falcipain inhibitor and, in the particular case of HEDICINs, data suggests that D-amino acids are preferable. Of note, the compound with highest anti-plasmodial activity, 8c, was completely devoid of inhibitory activity against FP2. This compound likely exerts its anti-plasmodial action by mechanisms other than inhibition of hemozoin formation or falcipain activity. Data on compound 8e reinforce the idea that falcipain or hemozoin inhibition are not the main mechanisms of action responsible for HEDICINs anti-plasmodial activity: 8e, bearing a p-amino substituent in the aryl ring, was the test compound which best reached our original goal of a dual-action inhibitor, by joining high hemozoin inhibitory activity with an IC50 20 μM against FP2; however, such was not translated into the highest anti-plasmodial activity being observed for 8e. Taken together, data from in vitro falcipain inhibition and parasite development assays suggest that the dipeptide spacer in HEDICINs 8 promotes uptake into infected RBCs. This hypothesis could explain why HEDICINs perform better than HECINs as antiplasmodials, despite the observation that HECINs were better falcipain-inhibitors than the HEDICINs. In addition, lipophilicity could have a role in compound uptake and anti-plasmodial action, as HEDICINs 8 are more lipophilic than HECINs 9 (due to the D-Leu and the D-hPhe hydrophobic side chains), and the most lipophilic compound, 8c, had the greatest anti-plasmodial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum strain W2 | 5833 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Haemozoin inhibition | ≥ 75% | |||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
The capacity of test compounds 8 and 9 to inhibit heme polymerization in vitro was assessed by previously reported methods, given in detail under Experimental. The assays were run in 96-well microplates, where negative controls (water, DMSO) and positive controls (1 mM CQ) were included. Test compounds were assayed at 1 mM and data are given in Table 1. Interestingly, the dipeptide spacer was required to block heme polymerization, i.e., while HECINs 9 were not active, HEDICINs 8 displayed variable inhibitory efficiencies, with four out of the twelve compounds (8b, 8e, 8j and 8l) highly active (i.e., comparable to the reference drug, CQ). Though no clear trend could be established regarding stereoelectronic properties of the aryl substituent in compounds 8, it was clear that: (i) hydrogen (i.e., absence of a substituent) or halogens in the para position were detrimental for activity, whereas (ii) nitrogenated groups at either the ortho (8j, o-NO2) or the para (8e, p-NH2; 8l, p-NO2) position of the aryl ring was beneficial, but (iii) detrimental if placed in the meta position (8k, m-NO2). Furthermore, while small alkyl groups in para (8b, p-Me) were advantageous, bulkier groups as in 8c (p-iPr) led to complete loss of activity. Thus, replacement of CQ's aliphatic chain by an adequate dipeptidyl-cinnamoyl moiety as in 8b, 8e, 8j and 8l appears to preserve the parent drug's ability to inhibit hemozoin formation, suggesting that these novel compounds could be promising leads for new CQ surrogates.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum strain W2 | 5833 | ||
Ac-EMC [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [86] | ||||
| Indication | SARS-CoV infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
3.43 ± 0.54 μM
|
|||
| MOA of PDC |
Peptide-drug conjugates (PDCs) are a class of novel molecules widely designed and synthesized for delivering drug payloads. In this work, we designed a novel PDC in which the GRL0617 was linked to the sulfonium-tethered peptides derived from PLpro-specific substrate LRGG. This conjugate could covalently label PLpro active site C111. Two conjugates EM-C and EC-M showed a promising in vitro IC50 of 7.40 ± 0.37 and 8.63 ± 0.55 μM, respectively. Both conjugates could effectively inhibit anti-ISGylation activities of PLpro in cells, and there is low toxicity of PDC EC-M and EM-C in different cells.
Click to Show/Hide
|
||||
| Description |
The enzymatic activities of SARS-CoV-2 PLpro were tested using the fluorogenic peptide substrate LRGG-AMC. The sulfonium-tethered peptides without GRL0617 could not efficiently inhibit SARS-CoV-2 PLpro, while the sulfonium-tethered peptide conjugate GRL0617 has a better inhibition ability. Specifically, IC50 values of EM-C and EC-M were 7.40 ± 0.37 and 8.63 ± 0.55 μM, respectively. Enzymatic activities of PLpro were inhibited by different PDCs or GRL0617. (A) Enzymatic activity of SARS-CoV-2 PLpro was inhibited by different PDCs or GRL0617. The activity assay was performed using the peptide LRGG-ACC as a substrate. Error bars represent standard errors of mean of at least three independent measurements. (B) IC50 of different PDCs or GRL0617 in SARS-CoV-2 PLpro. IC50 of EM-C was 7.40 ± 0.37 μM. IC50 of EC-M was 8.63 ± 0.55 μM. IC50 of ELRGG was 15.38 ± 1.63 μM. IC50 of GRL0617 was 2.64 ± 0.34 μM. (C) Enzymatic activity of SARS-CoV PLpro was inhibited by different PDCs or GRL0617. (D) Enzyme activity of MERS PLpro was inhibited by different PDCs or GRL0617. Also, we tested the peptide inhibition ability to SARS-CoV PLpro and MERS PLpro. There is a high sequence identity (83%) between the SARS-CoV-2 PLpro and SARS-CoV PLpro. The IC50 value of EM-C against SARS-CoV PLpro was 3.43 ± 0.54 μM, close to GRL0617 (IC50 = 2.60 ± 0.05 μM). Its linear analogue showed weaker inhibition. The GRL0617, EC-M, EM-C, and ELRGG could not inhibit MERS PLpro. Both EM-C and EC-M showed better inhibition for SARS-CoV PLpro than SARS-CoV-2 PLpro.
Click to Show/Hide
|
||||
| In Vitro Model | COVID-19 | SARS-CoV-2 | 2697049 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [86] | ||||
| Indication | SARS-CoV infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
7.40±0.37 μM
|
|||
| MOA of PDC |
Peptide-drug conjugates (PDCs) are a class of novel molecules widely designed and synthesized for delivering drug payloads. In this work, we designed a novel PDC in which the GRL0617 was linked to the sulfonium-tethered peptides derived from PLpro-specific substrate LRGG. This conjugate could covalently label PLpro active site C111. Two conjugates EM-C and EC-M showed a promising in vitro IC50 of 7.40 ± 0.37 and 8.63 ± 0.55 μM, respectively. Both conjugates could effectively inhibit anti-ISGylation activities of PLpro in cells, and there is low toxicity of PDC EC-M and EM-C in different cells.
Click to Show/Hide
|
||||
| Description |
The enzymatic activities of SARS-CoV-2 PLpro were tested using the fluorogenic peptide substrate LRGG-AMC. The sulfonium-tethered peptides without GRL0617 could not efficiently inhibit SARS-CoV-2 PLpro, while the sulfonium-tethered peptide conjugate GRL0617 has a better inhibition ability. Specifically, IC50 values of EM-C and EC-M were 7.40 ± 0.37 and 8.63 ± 0.55 μM, respectively. Enzymatic activities of PLpro were inhibited by different PDCs or GRL0617. (A) Enzymatic activity of SARS-CoV-2 PLpro was inhibited by different PDCs or GRL0617. The activity assay was performed using the peptide LRGG-ACC as a substrate. Error bars represent standard errors of mean of at least three independent measurements. (B) IC50 of different PDCs or GRL0617 in SARS-CoV-2 PLpro. IC50 of EM-C was 7.40 ± 0.37 μM. IC50 of EC-M was 8.63 ± 0.55 μM. IC50 of ELRGG was 15.38 ± 1.63 μM. IC50 of GRL0617 was 2.64 ± 0.34 μM. (C) Enzymatic activity of SARS-CoV PLpro was inhibited by different PDCs or GRL0617. (D) Enzyme activity of MERS PLpro was inhibited by different PDCs or GRL0617. Also, we tested the peptide inhibition ability to SARS-CoV PLpro and MERS PLpro. There is a high sequence identity (83%) between the SARS-CoV-2 PLpro and SARS-CoV PLpro. The IC50 value of EM-C against SARS-CoV PLpro was 3.43 ± 0.54 μM, close to GRL0617 (IC50 = 2.60 ± 0.05 μM). Its linear analogue showed weaker inhibition. The GRL0617, EC-M, EM-C, and ELRGG could not inhibit MERS PLpro. Both EM-C and EC-M showed better inhibition for SARS-CoV PLpro than SARS-CoV-2 PLpro.
Click to Show/Hide
|
||||
| In Vitro Model | COVID-19 | SARS-CoV-2 | 2697049 | ||
PDC-5c [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [87] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.8 (5.4 ± 0.1) μM
|
|||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [87] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4.0 (5.4 ± 0.1) μM
|
|||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-468 cell | CVCL_0419 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [87] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 10 µM | |||
| In Vitro Model | Invasive breast carcinoma | T-47D cell | CVCL_0553 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [87] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
2.8 (8.5 ± 0.1) nM
|
|||
| In Vitro Model | Normal | COS-7 cell | CVCL_0224 | ||
Dipeptide 23 - 4-Amino-7-chloroquinoline conjugate 10 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
4.89 μM
|
|||
| Administration Time | 48 h | ||||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
Activity against blood-stage CQ-resistant P. falciparum strain W2 was assessed as previously reported and given in detail in Experimental. Results demonstrated a complete lack of activity displayed by HECINs 9, correlating with their inability to inhibit heme polymerization. In turn, eleven out of the twelve HEDICINs 8 had IC50 values under 10 μM. Interestingly, three of the four most active HEDICIN blockers of heme polymerization (8b, 8j and 8l) were also among the four most active antiplasmodials, with IC50 below 2 μM. These results suggest that inhibition of heme polymerization is, at least in part, responsible for the anti-plasmodial activity of HEDICINs. An obvious exception to correlation between inhibition of heme polymerization and anti-plasmodial activity in HEDICINs is compound 8c; this bears a bulky electron-donating p-isopropyl group and did not inhibit heme polymerization in vitro, but displayed the highest anti-plasmodial activity. The inability of 8c to inhibit heme polymerization could be related to the bulkiness of the isopropyl group, but due to the higher hydrophobicity of this substituent, 8c was the most lipophilic HEDICIN assayed. Though we could not establish a full correlation between HEDICIN anti-plasmodial activity and estimated clogP values (not shown), the markedly higher lipophilicity of 8c, as compared to the other analogs, could promote a higher permeabilization of this compound into the infected RBC. Kirk and co-workers have demonstrated that P. falciparum parasites create new permeability pathways in host RBC, leading to increased permeability to small organic cations. In summary, though clean correlations could not be drawn between the anti-plasmodial activities displayed by the different HEDICINs (8) in vitro and molecular descriptors such as stereoelectronic factors (aryl substituents) or lipophilicity, it is demonstrated that these compounds displayed anti-plasmodial activity, whereas their HECIN counterparts (9), lacking the dipeptide spacer, did not. HEDICINs (8) inhibited heme polymerization in vitro, suggesting that this inhibitory activity is at least in part responsible for their anti-plasmodial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum | 5833 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
19.6 μM
|
|||
| Administration Time | 30 min | ||||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
The very different activities of compounds 8 and 9 suggest that the retro-enantio dipeptide spacer has a relevant role in determining anti-plasmodial activity. In view of this, we hypothesized that the anti-plasmodial activity of HEDICINs could also be partly due to falcipain inhibition. Therefore, both HEDICINs 8 and HECINs 9 were evaluated in vitro for inhibition of falcipains, using previously described methods, given in detail under Experimental. Only those compounds with IC50 < 50 μM against FP2 were assayed against FP3, as it has been established that FP2 has a larger catalytic cavity that accommodates a wider range of inhibitors than FP3. Consistent with this assumption, none of the compounds that inhibited FP2 in vitro displayed IC50 < 50 μM against FP3. Falcipain inhibition results contrasted with those for inhibition of heme polymerization or parasite development. HECINs 9 generally displayed more potent inhibition of falcipain than did HEDICINs 8. The ability of the test compounds to inhibit FP2 did not correlate with their anti-plasmodial activity. Although none of the HECINs 9 displayed anti-plasmodial activity, many inhibited FP2 and one of them, 9j, was actually the best FP2 inhibitor amongst the test compounds. Concerning HEDICINs, 8a, 8f and 8k, derived from cinnamic acid, m-fluorocinnamic acid, and m-nitrocinnamic acid, respectively, were the most active HEDICINs against FP2, with IC50 values of 19.7, 23.1 and 28.1 μM. Thus, stereoelectronic effects from aryl substituents did not correlate with inhibitory activities, as in HEDICINs 8 the most active compound was unsubstituted (8a), followed by the meta-fluorinated compound (8f); in turn, in HECINs 9, the meta-fluorinated derivative was inactive, whereas the most active of the set was 9j, which bears a meta-nitro substituent. The effect of amino acid configuration on HEDICIN activity was also assessed through synthesis and evaluation of 10a, the L-amino acid analog of 8a; interestingly, replacement of the D-amino acids by their natural L counterparts led to a clear decrease in both anti-plasmodial and falcipain-inhibitory activity. Therefore, amino acid configuration does influence compound behavior as either anti-plasmodial agent or falcipain inhibitor and, in the particular case of HEDICINs, data suggests that D-amino acids are preferable. Of note, the compound with highest anti-plasmodial activity, 8c, was completely devoid of inhibitory activity against FP2. This compound likely exerts its anti-plasmodial action by mechanisms other than inhibition of hemozoin formation or falcipain activity. Data on compound 8e reinforce the idea that falcipain or hemozoin inhibition are not the main mechanisms of action responsible for HEDICINs anti-plasmodial activity: 8e, bearing a p-amino substituent in the aryl ring, was the test compound which best reached our original goal of a dual-action inhibitor, by joining high hemozoin inhibitory activity with an IC50 20 μM against FP2; however, such was not translated into the highest anti-plasmodial activity being observed for 8e. Taken together, data from in vitro falcipain inhibition and parasite development assays suggest that the dipeptide spacer in HEDICINs 8 promotes uptake into infected RBCs. This hypothesis could explain why HEDICINs perform better than HECINs as antiplasmodials, despite the observation that HECINs were better falcipain-inhibitors than the HEDICINs. In addition, lipophilicity could have a role in compound uptake and anti-plasmodial action, as HEDICINs 8 are more lipophilic than HECINs 9 (due to the D-Leu and the D-hPhe hydrophobic side chains), and the most lipophilic compound, 8c, had the greatest anti-plasmodial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum strain W2 | 5833 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 50 μM | |||
| Administration Time | 30 min | ||||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
The very different activities of compounds 8 and 9 suggest that the retro-enantio dipeptide spacer has a relevant role in determining anti-plasmodial activity. In view of this, we hypothesized that the anti-plasmodial activity of HEDICINs could also be partly due to falcipain inhibition. Therefore, both HEDICINs 8 and HECINs 9 were evaluated in vitro for inhibition of falcipains, using previously described methods, given in detail under Experimental. Only those compounds with IC50 < 50 μM against FP2 were assayed against FP3, as it has been established that FP2 has a larger catalytic cavity that accommodates a wider range of inhibitors than FP3. Consistent with this assumption, none of the compounds that inhibited FP2 in vitro displayed IC50 < 50 μM against FP3. Falcipain inhibition results contrasted with those for inhibition of heme polymerization or parasite development. HECINs 9 generally displayed more potent inhibition of falcipain than did HEDICINs 8. The ability of the test compounds to inhibit FP2 did not correlate with their anti-plasmodial activity. Although none of the HECINs 9 displayed anti-plasmodial activity, many inhibited FP2 and one of them, 9j, was actually the best FP2 inhibitor amongst the test compounds. Concerning HEDICINs, 8a, 8f and 8k, derived from cinnamic acid, m-fluorocinnamic acid, and m-nitrocinnamic acid, respectively, were the most active HEDICINs against FP2, with IC50 values of 19.7, 23.1 and 28.1 μM. Thus, stereoelectronic effects from aryl substituents did not correlate with inhibitory activities, as in HEDICINs 8 the most active compound was unsubstituted (8a), followed by the meta-fluorinated compound (8f); in turn, in HECINs 9, the meta-fluorinated derivative was inactive, whereas the most active of the set was 9j, which bears a meta-nitro substituent. The effect of amino acid configuration on HEDICIN activity was also assessed through synthesis and evaluation of 10a, the L-amino acid analog of 8a; interestingly, replacement of the D-amino acids by their natural L counterparts led to a clear decrease in both anti-plasmodial and falcipain-inhibitory activity. Therefore, amino acid configuration does influence compound behavior as either anti-plasmodial agent or falcipain inhibitor and, in the particular case of HEDICINs, data suggests that D-amino acids are preferable. Of note, the compound with highest anti-plasmodial activity, 8c, was completely devoid of inhibitory activity against FP2. This compound likely exerts its anti-plasmodial action by mechanisms other than inhibition of hemozoin formation or falcipain activity. Data on compound 8e reinforce the idea that falcipain or hemozoin inhibition are not the main mechanisms of action responsible for HEDICINs anti-plasmodial activity: 8e, bearing a p-amino substituent in the aryl ring, was the test compound which best reached our original goal of a dual-action inhibitor, by joining high hemozoin inhibitory activity with an IC50 20 μM against FP2; however, such was not translated into the highest anti-plasmodial activity being observed for 8e. Taken together, data from in vitro falcipain inhibition and parasite development assays suggest that the dipeptide spacer in HEDICINs 8 promotes uptake into infected RBCs. This hypothesis could explain why HEDICINs perform better than HECINs as antiplasmodials, despite the observation that HECINs were better falcipain-inhibitors than the HEDICINs. In addition, lipophilicity could have a role in compound uptake and anti-plasmodial action, as HEDICINs 8 are more lipophilic than HECINs 9 (due to the D-Leu and the D-hPhe hydrophobic side chains), and the most lipophilic compound, 8c, had the greatest anti-plasmodial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum strain W2 | 5833 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Haemozoin inhibition | < 50% | |||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
The capacity of test compounds 8 and 9 to inhibit heme polymerization in vitro was assessed by previously reported methods, given in detail under Experimental. The assays were run in 96-well microplates, where negative controls (water, DMSO) and positive controls (1 mM CQ) were included. Test compounds were assayed at 1 mM and data are given in Table 1. Interestingly, the dipeptide spacer was required to block heme polymerization, i.e., while HECINs 9 were not active, HEDICINs 8 displayed variable inhibitory efficiencies, with four out of the twelve compounds (8b, 8e, 8j and 8l) highly active (i.e., comparable to the reference drug, CQ). Though no clear trend could be established regarding stereoelectronic properties of the aryl substituent in compounds 8, it was clear that: (i) hydrogen (i.e., absence of a substituent) or halogens in the para position were detrimental for activity, whereas (ii) nitrogenated groups at either the ortho (8j, o-NO2) or the para (8e, p-NH2; 8l, p-NO2) position of the aryl ring was beneficial, but (iii) detrimental if placed in the meta position (8k, m-NO2). Furthermore, while small alkyl groups in para (8b, p-Me) were advantageous, bulkier groups as in 8c (p-iPr) led to complete loss of activity. Thus, replacement of CQ's aliphatic chain by an adequate dipeptidyl-cinnamoyl moiety as in 8b, 8e, 8j and 8l appears to preserve the parent drug's ability to inhibit hemozoin formation, suggesting that these novel compounds could be promising leads for new CQ surrogates.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum strain W2 | 5833 | ||
PP-P6 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [85] | ||||
| Indication | Human immunodeficiency virus infection | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
5.28 ± 1.39 µM
|
|||
| Administration Time | 1 h | ||||
| Evaluation Method | GraphPad Prism assay | ||||
| MOA of PDC |
Strategies for conjugating BBBpS to drug payloads have been actively explored over recent decades, with the number of new shuttles steadily increasing. Some recent entries, moreover, have shown to be capable not only of carrying drugs into but also removing toxins from the brain, preventing their accumulation. For our part, we have reported that peptide-porphyrin conjugates (PPCs), where a BBBpS and an antiviral porphyrin are covalently linked by an amide bond (amino and carboxyl groups in BBBpS and porphyrin, respectively) can successfully pass the BBB and act against brain-targeting viruses such as HIV. As for the BPB, the literature is scarce, with only a few described examples of peptides able to pass it. Herein, we report our results in developing new PPCs able to penetrate both BPB and BBB and act against ZIKV. The PPC production strategy, involving porphyrin (P), C- or N-terminal conjugation to a BBBpS, has been detailed in a recent publication and is illustrated in Scheme 1 and Scheme S1. In this paper we describe eight PPCs resulting from the combination of four BBBpS and two porphyrins, and their evaluation in terms of barrier crossing and anti-ZIKV activity. One of the conjugates, PP-P1, emerges as particularly effective against ZIKV, having also the ability to translocate across BPB and BBB.
Click to Show/Hide
|
||||
| Description |
The five PPCs that successfully translocated BBB and BPB were evaluated for ZIKV inactivation in vitro, using a plaque assay. Of them, two showed significant activity, namely MP-P5 (IC50 = 25.07 ± 0.05 μM, similar to activity against HIV) and PP-P1 (IC50 = 1.08 ± 0.14 μM). Additionally, a treatment assay performed with MP-P5 and PP-P1 revealed that both PPCs efficiently inhibit ZIKV replication when added 1 h and 7 h post-infection. As observed for HIV, non-conjugated porphyrins did not show activity against ZIKV, reinforcing the claim that BBBpS conjugation is not only critical for BBB/BPB translocation but also for antiviral activity. On the other hand, and somewhat unexpectedly, PP-P1, shown to be inactive in vitro against HIV, emerged as the most active anti-ZIKV conjugate. As described in the literature, the light-independent mechanism of action of porphyrins is based on a direct perturbation of the viral envelope. Porphyrins interact and accumulate on the envelope lipid membrane, causing a decrease in order and a consequent phase alteration that impairs viral entry processes. Since we ensured no-light conditions and no metal cations are coordinated to the porphyrin rings of the PPCsavoiding generation of reactive oxygen speciesthe antiviral light-independent mechanism is the only plausible one. Thus, one may confidently suggest that the PP-P1 specific anti-ZIKV activity is guided by preferential interaction with the ZIKV viral envelope and/or components.
Click to Show/Hide
|
||||
| In Vitro Model | HIV Infection | Human immunodeficiency virus | 12721 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [85] | ||||
| Indication | Zika virus infection | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Administration Time | 1 h | ||||
| Evaluation Method | GraphPad Prism assay | ||||
| MOA of PDC |
Strategies for conjugating BBBpS to drug payloads have been actively explored over recent decades, with the number of new shuttles steadily increasing. Some recent entries, moreover, have shown to be capable not only of carrying drugs into but also removing toxins from the brain, preventing their accumulation. For our part, we have reported that peptide-porphyrin conjugates (PPCs), where a BBBpS and an antiviral porphyrin are covalently linked by an amide bond (amino and carboxyl groups in BBBpS and porphyrin, respectively) can successfully pass the BBB and act against brain-targeting viruses such as HIV. As for the BPB, the literature is scarce, with only a few described examples of peptides able to pass it. Herein, we report our results in developing new PPCs able to penetrate both BPB and BBB and act against ZIKV. The PPC production strategy, involving porphyrin (P), C- or N-terminal conjugation to a BBBpS, has been detailed in a recent publication and is illustrated in Scheme 1 and Scheme S1. In this paper we describe eight PPCs resulting from the combination of four BBBpS and two porphyrins, and their evaluation in terms of barrier crossing and anti-ZIKV activity. One of the conjugates, PP-P1, emerges as particularly effective against ZIKV, having also the ability to translocate across BPB and BBB.
Click to Show/Hide
|
||||
| Description |
The five PPCs that successfully translocated BBB and BPB were evaluated for ZIKV inactivation in vitro, using a plaque assay. Of them, two showed significant activity, namely MP-P5 (IC50 = 25.07 ± 0.05 μM, similar to activity against HIV) and PP-P1 (IC50 = 1.08 ± 0.14 μM). Additionally, a treatment assay performed with MP-P5 and PP-P1 revealed that both PPCs efficiently inhibit ZIKV replication when added 1 h and 7 h post-infection. As observed for HIV, non-conjugated porphyrins did not show activity against ZIKV, reinforcing the claim that BBBpS conjugation is not only critical for BBB/BPB translocation but also for antiviral activity. On the other hand, and somewhat unexpectedly, PP-P1, shown to be inactive in vitro against HIV, emerged as the most active anti-ZIKV conjugate. As described in the literature, the light-independent mechanism of action of porphyrins is based on a direct perturbation of the viral envelope. Porphyrins interact and accumulate on the envelope lipid membrane, causing a decrease in order and a consequent phase alteration that impairs viral entry processes. Since we ensured no-light conditions and no metal cations are coordinated to the porphyrin rings of the PPCsavoiding generation of reactive oxygen speciesthe antiviral light-independent mechanism is the only plausible one. Thus, one may confidently suggest that the PP-P1 specific anti-ZIKV activity is guided by preferential interaction with the ZIKV viral envelope and/or components.
Click to Show/Hide
|
||||
| In Vitro Model | ZIKV infection | Zika virus | 64320 | ||
Dipeptide 23 - 4-Amino-7-chloroquinoline conjugate 6 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
5.43 μM
|
|||
| Administration Time | 48 h | ||||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
Activity against blood-stage CQ-resistant P. falciparum strain W2 was assessed as previously reported and given in detail in Experimental. Results demonstrated a complete lack of activity displayed by HECINs 9, correlating with their inability to inhibit heme polymerization. In turn, eleven out of the twelve HEDICINs 8 had IC50 values under 10 μM. Interestingly, three of the four most active HEDICIN blockers of heme polymerization (8b, 8j and 8l) were also among the four most active antiplasmodials, with IC50 below 2 μM. These results suggest that inhibition of heme polymerization is, at least in part, responsible for the anti-plasmodial activity of HEDICINs. An obvious exception to correlation between inhibition of heme polymerization and anti-plasmodial activity in HEDICINs is compound 8c; this bears a bulky electron-donating p-isopropyl group and did not inhibit heme polymerization in vitro, but displayed the highest anti-plasmodial activity. The inability of 8c to inhibit heme polymerization could be related to the bulkiness of the isopropyl group, but due to the higher hydrophobicity of this substituent, 8c was the most lipophilic HEDICIN assayed. Though we could not establish a full correlation between HEDICIN anti-plasmodial activity and estimated clogP values (not shown), the markedly higher lipophilicity of 8c, as compared to the other analogs, could promote a higher permeabilization of this compound into the infected RBC. Kirk and co-workers have demonstrated that P. falciparum parasites create new permeability pathways in host RBC, leading to increased permeability to small organic cations. In summary, though clean correlations could not be drawn between the anti-plasmodial activities displayed by the different HEDICINs (8) in vitro and molecular descriptors such as stereoelectronic factors (aryl substituents) or lipophilicity, it is demonstrated that these compounds displayed anti-plasmodial activity, whereas their HECIN counterparts (9), lacking the dipeptide spacer, did not. HEDICINs (8) inhibited heme polymerization in vitro, suggesting that this inhibitory activity is at least in part responsible for their anti-plasmodial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum | 5833 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
23.1 μM
|
|||
| Administration Time | 30 min | ||||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
The very different activities of compounds 8 and 9 suggest that the retro-enantio dipeptide spacer has a relevant role in determining anti-plasmodial activity. In view of this, we hypothesized that the anti-plasmodial activity of HEDICINs could also be partly due to falcipain inhibition. Therefore, both HEDICINs 8 and HECINs 9 were evaluated in vitro for inhibition of falcipains, using previously described methods, given in detail under Experimental. Only those compounds with IC50 < 50 μM against FP2 were assayed against FP3, as it has been established that FP2 has a larger catalytic cavity that accommodates a wider range of inhibitors than FP3. Consistent with this assumption, none of the compounds that inhibited FP2 in vitro displayed IC50 < 50 μM against FP3. Falcipain inhibition results contrasted with those for inhibition of heme polymerization or parasite development. HECINs 9 generally displayed more potent inhibition of falcipain than did HEDICINs 8. The ability of the test compounds to inhibit FP2 did not correlate with their anti-plasmodial activity. Although none of the HECINs 9 displayed anti-plasmodial activity, many inhibited FP2 and one of them, 9j, was actually the best FP2 inhibitor amongst the test compounds. Concerning HEDICINs, 8a, 8f and 8k, derived from cinnamic acid, m-fluorocinnamic acid, and m-nitrocinnamic acid, respectively, were the most active HEDICINs against FP2, with IC50 values of 19.7, 23.1 and 28.1 μM. Thus, stereoelectronic effects from aryl substituents did not correlate with inhibitory activities, as in HEDICINs 8 the most active compound was unsubstituted (8a), followed by the meta-fluorinated compound (8f); in turn, in HECINs 9, the meta-fluorinated derivative was inactive, whereas the most active of the set was 9j, which bears a meta-nitro substituent. The effect of amino acid configuration on HEDICIN activity was also assessed through synthesis and evaluation of 10a, the L-amino acid analog of 8a; interestingly, replacement of the D-amino acids by their natural L counterparts led to a clear decrease in both anti-plasmodial and falcipain-inhibitory activity. Therefore, amino acid configuration does influence compound behavior as either anti-plasmodial agent or falcipain inhibitor and, in the particular case of HEDICINs, data suggests that D-amino acids are preferable. Of note, the compound with highest anti-plasmodial activity, 8c, was completely devoid of inhibitory activity against FP2. This compound likely exerts its anti-plasmodial action by mechanisms other than inhibition of hemozoin formation or falcipain activity. Data on compound 8e reinforce the idea that falcipain or hemozoin inhibition are not the main mechanisms of action responsible for HEDICINs anti-plasmodial activity: 8e, bearing a p-amino substituent in the aryl ring, was the test compound which best reached our original goal of a dual-action inhibitor, by joining high hemozoin inhibitory activity with an IC50 20 μM against FP2; however, such was not translated into the highest anti-plasmodial activity being observed for 8e. Taken together, data from in vitro falcipain inhibition and parasite development assays suggest that the dipeptide spacer in HEDICINs 8 promotes uptake into infected RBCs. This hypothesis could explain why HEDICINs perform better than HECINs as antiplasmodials, despite the observation that HECINs were better falcipain-inhibitors than the HEDICINs. In addition, lipophilicity could have a role in compound uptake and anti-plasmodial action, as HEDICINs 8 are more lipophilic than HECINs 9 (due to the D-Leu and the D-hPhe hydrophobic side chains), and the most lipophilic compound, 8c, had the greatest anti-plasmodial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum strain W2 | 5833 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 50 μM | |||
| Administration Time | 30 min | ||||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
The very different activities of compounds 8 and 9 suggest that the retro-enantio dipeptide spacer has a relevant role in determining anti-plasmodial activity. In view of this, we hypothesized that the anti-plasmodial activity of HEDICINs could also be partly due to falcipain inhibition. Therefore, both HEDICINs 8 and HECINs 9 were evaluated in vitro for inhibition of falcipains, using previously described methods, given in detail under Experimental. Only those compounds with IC50 < 50 μM against FP2 were assayed against FP3, as it has been established that FP2 has a larger catalytic cavity that accommodates a wider range of inhibitors than FP3. Consistent with this assumption, none of the compounds that inhibited FP2 in vitro displayed IC50 < 50 μM against FP3. Falcipain inhibition results contrasted with those for inhibition of heme polymerization or parasite development. HECINs 9 generally displayed more potent inhibition of falcipain than did HEDICINs 8. The ability of the test compounds to inhibit FP2 did not correlate with their anti-plasmodial activity. Although none of the HECINs 9 displayed anti-plasmodial activity, many inhibited FP2 and one of them, 9j, was actually the best FP2 inhibitor amongst the test compounds. Concerning HEDICINs, 8a, 8f and 8k, derived from cinnamic acid, m-fluorocinnamic acid, and m-nitrocinnamic acid, respectively, were the most active HEDICINs against FP2, with IC50 values of 19.7, 23.1 and 28.1 μM. Thus, stereoelectronic effects from aryl substituents did not correlate with inhibitory activities, as in HEDICINs 8 the most active compound was unsubstituted (8a), followed by the meta-fluorinated compound (8f); in turn, in HECINs 9, the meta-fluorinated derivative was inactive, whereas the most active of the set was 9j, which bears a meta-nitro substituent. The effect of amino acid configuration on HEDICIN activity was also assessed through synthesis and evaluation of 10a, the L-amino acid analog of 8a; interestingly, replacement of the D-amino acids by their natural L counterparts led to a clear decrease in both anti-plasmodial and falcipain-inhibitory activity. Therefore, amino acid configuration does influence compound behavior as either anti-plasmodial agent or falcipain inhibitor and, in the particular case of HEDICINs, data suggests that D-amino acids are preferable. Of note, the compound with highest anti-plasmodial activity, 8c, was completely devoid of inhibitory activity against FP2. This compound likely exerts its anti-plasmodial action by mechanisms other than inhibition of hemozoin formation or falcipain activity. Data on compound 8e reinforce the idea that falcipain or hemozoin inhibition are not the main mechanisms of action responsible for HEDICINs anti-plasmodial activity: 8e, bearing a p-amino substituent in the aryl ring, was the test compound which best reached our original goal of a dual-action inhibitor, by joining high hemozoin inhibitory activity with an IC50 20 μM against FP2; however, such was not translated into the highest anti-plasmodial activity being observed for 8e. Taken together, data from in vitro falcipain inhibition and parasite development assays suggest that the dipeptide spacer in HEDICINs 8 promotes uptake into infected RBCs. This hypothesis could explain why HEDICINs perform better than HECINs as antiplasmodials, despite the observation that HECINs were better falcipain-inhibitors than the HEDICINs. In addition, lipophilicity could have a role in compound uptake and anti-plasmodial action, as HEDICINs 8 are more lipophilic than HECINs 9 (due to the D-Leu and the D-hPhe hydrophobic side chains), and the most lipophilic compound, 8c, had the greatest anti-plasmodial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum strain W2 | 5833 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Haemozoin inhibition | < 50% | |||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
The capacity of test compounds 8 and 9 to inhibit heme polymerization in vitro was assessed by previously reported methods, given in detail under Experimental. The assays were run in 96-well microplates, where negative controls (water, DMSO) and positive controls (1 mM CQ) were included. Test compounds were assayed at 1 mM and data are given in Table 1. Interestingly, the dipeptide spacer was required to block heme polymerization, i.e., while HECINs 9 were not active, HEDICINs 8 displayed variable inhibitory efficiencies, with four out of the twelve compounds (8b, 8e, 8j and 8l) highly active (i.e., comparable to the reference drug, CQ). Though no clear trend could be established regarding stereoelectronic properties of the aryl substituent in compounds 8, it was clear that: (i) hydrogen (i.e., absence of a substituent) or halogens in the para position were detrimental for activity, whereas (ii) nitrogenated groups at either the ortho (8j, o-NO2) or the para (8e, p-NH2; 8l, p-NO2) position of the aryl ring was beneficial, but (iii) detrimental if placed in the meta position (8k, m-NO2). Furthermore, while small alkyl groups in para (8b, p-Me) were advantageous, bulkier groups as in 8c (p-iPr) led to complete loss of activity. Thus, replacement of CQ's aliphatic chain by an adequate dipeptidyl-cinnamoyl moiety as in 8b, 8e, 8j and 8l appears to preserve the parent drug's ability to inhibit hemozoin formation, suggesting that these novel compounds could be promising leads for new CQ surrogates.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum strain W2 | 5833 | ||
Ac-ECM [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [86] | ||||
| Indication | SARS-CoV infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
8.63 ± 0.55 μM
|
|||
| MOA of PDC |
Peptide-drug conjugates (PDCs) are a class of novel molecules widely designed and synthesized for delivering drug payloads. In this work, we designed a novel PDC in which the GRL0617 was linked to the sulfonium-tethered peptides derived from PLpro-specific substrate LRGG. This conjugate could covalently label PLpro active site C111. Two conjugates EM-C and EC-M showed a promising in vitro IC50 of 7.40 ± 0.37 and 8.63 ± 0.55 μM, respectively. Both conjugates could effectively inhibit anti-ISGylation activities of PLpro in cells, and there is low toxicity of PDC EC-M and EM-C in different cells.
Click to Show/Hide
|
||||
| Description |
The enzymatic activities of SARS-CoV-2 PLpro were tested using the fluorogenic peptide substrate LRGG-AMC. The sulfonium-tethered peptides without GRL0617 could not efficiently inhibit SARS-CoV-2 PLpro, while the sulfonium-tethered peptide conjugate GRL0617 has a better inhibition ability. Specifically, IC50 values of EM-C and EC-M were 7.40 ± 0.37 and 8.63 ± 0.55 μM, respectively. Enzymatic activities of PLpro were inhibited by different PDCs or GRL0617. (A) Enzymatic activity of SARS-CoV-2 PLpro was inhibited by different PDCs or GRL0617. The activity assay was performed using the peptide LRGG-ACC as a substrate. Error bars represent standard errors of mean of at least three independent measurements. (B) IC50 of different PDCs or GRL0617 in SARS-CoV-2 PLpro. IC50 of EM-C was 7.40 ± 0.37 μM. IC50 of EC-M was 8.63 ± 0.55 μM. IC50 of ELRGG was 15.38 ± 1.63 μM. IC50 of GRL0617 was 2.64 ± 0.34 μM. (C) Enzymatic activity of SARS-CoV PLpro was inhibited by different PDCs or GRL0617. (D) Enzyme activity of MERS PLpro was inhibited by different PDCs or GRL0617. Also, we tested the peptide inhibition ability to SARS-CoV PLpro and MERS PLpro. There is a high sequence identity (83%) between the SARS-CoV-2 PLpro and SARS-CoV PLpro. The IC50 value of EM-C against SARS-CoV PLpro was 3.43 ± 0.54 μM, close to GRL0617 (IC50 = 2.60 ± 0.05 μM). Its linear analogue showed weaker inhibition. The GRL0617, EC-M, EM-C, and ELRGG could not inhibit MERS PLpro. Both EM-C and EC-M showed better inhibition for SARS-CoV PLpro than SARS-CoV-2 PLpro.
Click to Show/Hide
|
||||
| In Vitro Model | COVID-19 | SARS-CoV-2 | 2697049 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [86] | ||||
| Indication | SARS-CoV infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
16.38±0.81 μM
|
|||
| MOA of PDC |
Peptide-drug conjugates (PDCs) are a class of novel molecules widely designed and synthesized for delivering drug payloads. In this work, we designed a novel PDC in which the GRL0617 was linked to the sulfonium-tethered peptides derived from PLpro-specific substrate LRGG. This conjugate could covalently label PLpro active site C111. Two conjugates EM-C and EC-M showed a promising in vitro IC50 of 7.40 ± 0.37 and 8.63 ± 0.55 μM, respectively. Both conjugates could effectively inhibit anti-ISGylation activities of PLpro in cells, and there is low toxicity of PDC EC-M and EM-C in different cells.
Click to Show/Hide
|
||||
| Description |
The enzymatic activities of SARS-CoV-2 PLpro were tested using the fluorogenic peptide substrate LRGG-AMC. The sulfonium-tethered peptides without GRL0617 could not efficiently inhibit SARS-CoV-2 PLpro, while the sulfonium-tethered peptide conjugate GRL0617 has a better inhibition ability. Specifically, IC50 values of EM-C and EC-M were 7.40 ± 0.37 and 8.63 ± 0.55 μM, respectively. Enzymatic activities of PLpro were inhibited by different PDCs or GRL0617. (A) Enzymatic activity of SARS-CoV-2 PLpro was inhibited by different PDCs or GRL0617. The activity assay was performed using the peptide LRGG-ACC as a substrate. Error bars represent standard errors of mean of at least three independent measurements. (B) IC50 of different PDCs or GRL0617 in SARS-CoV-2 PLpro. IC50 of EM-C was 7.40 ± 0.37 μM. IC50 of EC-M was 8.63 ± 0.55 μM. IC50 of ELRGG was 15.38 ± 1.63 μM. IC50 of GRL0617 was 2.64 ± 0.34 μM. (C) Enzymatic activity of SARS-CoV PLpro was inhibited by different PDCs or GRL0617. (D) Enzyme activity of MERS PLpro was inhibited by different PDCs or GRL0617. Also, we tested the peptide inhibition ability to SARS-CoV PLpro and MERS PLpro. There is a high sequence identity (83%) between the SARS-CoV-2 PLpro and SARS-CoV PLpro. The IC50 value of EM-C against SARS-CoV PLpro was 3.43 ± 0.54 μM, close to GRL0617 (IC50 = 2.60 ± 0.05 μM). Its linear analogue showed weaker inhibition. The GRL0617, EC-M, EM-C, and ELRGG could not inhibit MERS PLpro. Both EM-C and EC-M showed better inhibition for SARS-CoV PLpro than SARS-CoV-2 PLpro.
Click to Show/Hide
|
||||
| In Vitro Model | COVID-19 | SARS-CoV-2 | 2697049 | ||
Dipeptide 23 - 4-Amino-7-chloroquinoline conjugate 7 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
10.8 μM
|
|||
| Administration Time | 48 h | ||||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
Activity against blood-stage CQ-resistant P. falciparum strain W2 was assessed as previously reported and given in detail in Experimental. Results demonstrated a complete lack of activity displayed by HECINs 9, correlating with their inability to inhibit heme polymerization. In turn, eleven out of the twelve HEDICINs 8 had IC50 values under 10 μM. Interestingly, three of the four most active HEDICIN blockers of heme polymerization (8b, 8j and 8l) were also among the four most active antiplasmodials, with IC50 below 2 μM. These results suggest that inhibition of heme polymerization is, at least in part, responsible for the anti-plasmodial activity of HEDICINs. An obvious exception to correlation between inhibition of heme polymerization and anti-plasmodial activity in HEDICINs is compound 8c; this bears a bulky electron-donating p-isopropyl group and did not inhibit heme polymerization in vitro, but displayed the highest anti-plasmodial activity. The inability of 8c to inhibit heme polymerization could be related to the bulkiness of the isopropyl group, but due to the higher hydrophobicity of this substituent, 8c was the most lipophilic HEDICIN assayed. Though we could not establish a full correlation between HEDICIN anti-plasmodial activity and estimated clogP values (not shown), the markedly higher lipophilicity of 8c, as compared to the other analogs, could promote a higher permeabilization of this compound into the infected RBC. Kirk and co-workers have demonstrated that P. falciparum parasites create new permeability pathways in host RBC, leading to increased permeability to small organic cations. In summary, though clean correlations could not be drawn between the anti-plasmodial activities displayed by the different HEDICINs (8) in vitro and molecular descriptors such as stereoelectronic factors (aryl substituents) or lipophilicity, it is demonstrated that these compounds displayed anti-plasmodial activity, whereas their HECIN counterparts (9), lacking the dipeptide spacer, did not. HEDICINs (8) inhibited heme polymerization in vitro, suggesting that this inhibitory activity is at least in part responsible for their anti-plasmodial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum | 5833 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 50 μM | |||
| Administration Time | 30 min | ||||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
The very different activities of compounds 8 and 9 suggest that the retro-enantio dipeptide spacer has a relevant role in determining anti-plasmodial activity. In view of this, we hypothesized that the anti-plasmodial activity of HEDICINs could also be partly due to falcipain inhibition. Therefore, both HEDICINs 8 and HECINs 9 were evaluated in vitro for inhibition of falcipains, using previously described methods, given in detail under Experimental. Only those compounds with IC50 < 50 μM against FP2 were assayed against FP3, as it has been established that FP2 has a larger catalytic cavity that accommodates a wider range of inhibitors than FP3. Consistent with this assumption, none of the compounds that inhibited FP2 in vitro displayed IC50 < 50 μM against FP3. Falcipain inhibition results contrasted with those for inhibition of heme polymerization or parasite development. HECINs 9 generally displayed more potent inhibition of falcipain than did HEDICINs 8. The ability of the test compounds to inhibit FP2 did not correlate with their anti-plasmodial activity. Although none of the HECINs 9 displayed anti-plasmodial activity, many inhibited FP2 and one of them, 9j, was actually the best FP2 inhibitor amongst the test compounds. Concerning HEDICINs, 8a, 8f and 8k, derived from cinnamic acid, m-fluorocinnamic acid, and m-nitrocinnamic acid, respectively, were the most active HEDICINs against FP2, with IC50 values of 19.7, 23.1 and 28.1 μM. Thus, stereoelectronic effects from aryl substituents did not correlate with inhibitory activities, as in HEDICINs 8 the most active compound was unsubstituted (8a), followed by the meta-fluorinated compound (8f); in turn, in HECINs 9, the meta-fluorinated derivative was inactive, whereas the most active of the set was 9j, which bears a meta-nitro substituent. The effect of amino acid configuration on HEDICIN activity was also assessed through synthesis and evaluation of 10a, the L-amino acid analog of 8a; interestingly, replacement of the D-amino acids by their natural L counterparts led to a clear decrease in both anti-plasmodial and falcipain-inhibitory activity. Therefore, amino acid configuration does influence compound behavior as either anti-plasmodial agent or falcipain inhibitor and, in the particular case of HEDICINs, data suggests that D-amino acids are preferable. Of note, the compound with highest anti-plasmodial activity, 8c, was completely devoid of inhibitory activity against FP2. This compound likely exerts its anti-plasmodial action by mechanisms other than inhibition of hemozoin formation or falcipain activity. Data on compound 8e reinforce the idea that falcipain or hemozoin inhibition are not the main mechanisms of action responsible for HEDICINs anti-plasmodial activity: 8e, bearing a p-amino substituent in the aryl ring, was the test compound which best reached our original goal of a dual-action inhibitor, by joining high hemozoin inhibitory activity with an IC50 20 μM against FP2; however, such was not translated into the highest anti-plasmodial activity being observed for 8e. Taken together, data from in vitro falcipain inhibition and parasite development assays suggest that the dipeptide spacer in HEDICINs 8 promotes uptake into infected RBCs. This hypothesis could explain why HEDICINs perform better than HECINs as antiplasmodials, despite the observation that HECINs were better falcipain-inhibitors than the HEDICINs. In addition, lipophilicity could have a role in compound uptake and anti-plasmodial action, as HEDICINs 8 are more lipophilic than HECINs 9 (due to the D-Leu and the D-hPhe hydrophobic side chains), and the most lipophilic compound, 8c, had the greatest anti-plasmodial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum strain W2 | 5833 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [83] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Haemozoin inhibition |
50%-75%
|
|||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
The capacity of test compounds 8 and 9 to inhibit heme polymerization in vitro was assessed by previously reported methods, given in detail under Experimental. The assays were run in 96-well microplates, where negative controls (water, DMSO) and positive controls (1 mM CQ) were included. Test compounds were assayed at 1 mM and data are given in Table 1. Interestingly, the dipeptide spacer was required to block heme polymerization, i.e., while HECINs 9 were not active, HEDICINs 8 displayed variable inhibitory efficiencies, with four out of the twelve compounds (8b, 8e, 8j and 8l) highly active (i.e., comparable to the reference drug, CQ). Though no clear trend could be established regarding stereoelectronic properties of the aryl substituent in compounds 8, it was clear that: (i) hydrogen (i.e., absence of a substituent) or halogens in the para position were detrimental for activity, whereas (ii) nitrogenated groups at either the ortho (8j, o-NO2) or the para (8e, p-NH2; 8l, p-NO2) position of the aryl ring was beneficial, but (iii) detrimental if placed in the meta position (8k, m-NO2). Furthermore, while small alkyl groups in para (8b, p-Me) were advantageous, bulkier groups as in 8c (p-iPr) led to complete loss of activity. Thus, replacement of CQ's aliphatic chain by an adequate dipeptidyl-cinnamoyl moiety as in 8b, 8e, 8j and 8l appears to preserve the parent drug's ability to inhibit hemozoin formation, suggesting that these novel compounds could be promising leads for new CQ surrogates.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum strain W2 | 5833 | ||
Ac-ELRGG [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [86] | ||||
| Indication | SARS-CoV infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
15.38±1.63 μM
|
|||
| MOA of PDC |
Peptide-drug conjugates (PDCs) are a class of novel molecules widely designed and synthesized for delivering drug payloads. In this work, we designed a novel PDC in which the GRL0617 was linked to the sulfonium-tethered peptides derived from PLpro-specific substrate LRGG. This conjugate could covalently label PLpro active site C111. Two conjugates EM-C and EC-M showed a promising in vitro IC50 of 7.40 ± 0.37 and 8.63 ± 0.55 μM, respectively. Both conjugates could effectively inhibit anti-ISGylation activities of PLpro in cells, and there is low toxicity of PDC EC-M and EM-C in different cells.
Click to Show/Hide
|
||||
| Description |
The enzymatic activities of SARS-CoV-2 PLpro were tested using the fluorogenic peptide substrate LRGG-AMC. The sulfonium-tethered peptides without GRL0617 could not efficiently inhibit SARS-CoV-2 PLpro, while the sulfonium-tethered peptide conjugate GRL0617 has a better inhibition ability. Specifically, IC50 values of EM-C and EC-M were 7.40 ± 0.37 and 8.63 ± 0.55 μM, respectively. Enzymatic activities of PLpro were inhibited by different PDCs or GRL0617. (A) Enzymatic activity of SARS-CoV-2 PLpro was inhibited by different PDCs or GRL0617. The activity assay was performed using the peptide LRGG-ACC as a substrate. Error bars represent standard errors of mean of at least three independent measurements. (B) IC50 of different PDCs or GRL0617 in SARS-CoV-2 PLpro. IC50 of EM-C was 7.40 ± 0.37 μM. IC50 of EC-M was 8.63 ± 0.55 μM. IC50 of ELRGG was 15.38 ± 1.63 μM. IC50 of GRL0617 was 2.64 ± 0.34 μM. (C) Enzymatic activity of SARS-CoV PLpro was inhibited by different PDCs or GRL0617. (D) Enzyme activity of MERS PLpro was inhibited by different PDCs or GRL0617. Also, we tested the peptide inhibition ability to SARS-CoV PLpro and MERS PLpro. There is a high sequence identity (83%) between the SARS-CoV-2 PLpro and SARS-CoV PLpro. The IC50 value of EM-C against SARS-CoV PLpro was 3.43 ± 0.54 μM, close to GRL0617 (IC50 = 2.60 ± 0.05 μM). Its linear analogue showed weaker inhibition. The GRL0617, EC-M, EM-C, and ELRGG could not inhibit MERS PLpro. Both EM-C and EC-M showed better inhibition for SARS-CoV PLpro than SARS-CoV-2 PLpro.
Click to Show/Hide
|
||||
| In Vitro Model | COVID-19 | SARS-CoV-2 | 2697049 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [86] | ||||
| Indication | SARS-CoV infection | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
31.75 ±1.55 μM
|
|||
| MOA of PDC |
Peptide-drug conjugates (PDCs) are a class of novel molecules widely designed and synthesized for delivering drug payloads. In this work, we designed a novel PDC in which the GRL0617 was linked to the sulfonium-tethered peptides derived from PLpro-specific substrate LRGG. This conjugate could covalently label PLpro active site C111. Two conjugates EM-C and EC-M showed a promising in vitro IC50 of 7.40 ± 0.37 and 8.63 ± 0.55 μM, respectively. Both conjugates could effectively inhibit anti-ISGylation activities of PLpro in cells, and there is low toxicity of PDC EC-M and EM-C in different cells.
Click to Show/Hide
|
||||
| Description |
The enzymatic activities of SARS-CoV-2 PLpro were tested using the fluorogenic peptide substrate LRGG-AMC. The sulfonium-tethered peptides without GRL0617 could not efficiently inhibit SARS-CoV-2 PLpro, while the sulfonium-tethered peptide conjugate GRL0617 has a better inhibition ability. Specifically, IC50 values of EM-C and EC-M were 7.40 ± 0.37 and 8.63 ± 0.55 μM, respectively. Enzymatic activities of PLpro were inhibited by different PDCs or GRL0617. (A) Enzymatic activity of SARS-CoV-2 PLpro was inhibited by different PDCs or GRL0617. The activity assay was performed using the peptide LRGG-ACC as a substrate. Error bars represent standard errors of mean of at least three independent measurements. (B) IC50 of different PDCs or GRL0617 in SARS-CoV-2 PLpro. IC50 of EM-C was 7.40 ± 0.37 μM. IC50 of EC-M was 8.63 ± 0.55 μM. IC50 of ELRGG was 15.38 ± 1.63 μM. IC50 of GRL0617 was 2.64 ± 0.34 μM. (C) Enzymatic activity of SARS-CoV PLpro was inhibited by different PDCs or GRL0617. (D) Enzyme activity of MERS PLpro was inhibited by different PDCs or GRL0617. Also, we tested the peptide inhibition ability to SARS-CoV PLpro and MERS PLpro. There is a high sequence identity (83%) between the SARS-CoV-2 PLpro and SARS-CoV PLpro. The IC50 value of EM-C against SARS-CoV PLpro was 3.43 ± 0.54 μM, close to GRL0617 (IC50 = 2.60 ± 0.05 μM). Its linear analogue showed weaker inhibition. The GRL0617, EC-M, EM-C, and ELRGG could not inhibit MERS PLpro. Both EM-C and EC-M showed better inhibition for SARS-CoV PLpro than SARS-CoV-2 PLpro.
Click to Show/Hide
|
||||
| In Vitro Model | COVID-19 | SARS-CoV-2 | 2697049 | ||
PDC-5a [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [87] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
18 (4.8 ± 0.1) μM
|
|||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-468 cell | CVCL_0419 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [87] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 25 µM | |||
| In Vitro Model | Invasive breast carcinoma | T-47D cell | CVCL_0553 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [87] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 25 µM | |||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [87] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
2.9 (8.5 ± 0.1) nM
|
|||
| In Vitro Model | Normal | COS-7 cell | CVCL_0224 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [87] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
209 (6.7 ± 0.1) nM
|
|||
| In Vitro Model | Normal | COS-7 cell | CVCL_0224 | ||
CRB-FFFK-cyclen [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [88] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
23.8 μM
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The DNA-alkylating agent chlorambucil (CRB) belongs to aryl nitrogen mustard antitumor drugs, and has been widely used for treating different types of cancerous diseases. However, the clinical application of CRB is severely restricted by its poor aqueous solubility, lack of targeting, short degradation half-life and severe side effects. Macrocyclic polyamines have many applications in medicine, industry and other fields, owing to their chemical and biological properties. Some of them, such as cyclen and cyclam, could be protonated below physiological pH (7.4), and the lower the pH, the higher the degree of protonation. It is reported that the pH of the tumor environment is lower than the physiological pH, which is beneficial to the protonation of macrocyclic polyamines. Modification of macrocyclic polyamine to self-assembling peptide-drug amphiphiles can increase the cell accumulation of the hydrogel, because the cationic hydrogel has a high affinity to a negatively charged cell membrane and nucleus. Therefore, a macrocyclic polyamine containing peptide hydrogel could serve as a suitable delivery system to improve the pharmacokinetic properties of CRB, achieving improved delivery efficacy and enhanced antitumor activity without severe side effects. Herein, we report a self-assembling peptide-based cationic supramolecular nanomedicine bearing the small molecule agent CRB and macrocyclic polyamine cyclen. We found that the CRB-FFFK-cyclen conjugate could readily transform into a hydrogel through a heating-cooling process, and the resulting hydrogel could significantly improve drug stability, cellular uptake and, antitumor activity.
Click to Show/Hide
|
||||
| Description |
The IC50 values of CRB-FFFK-cyclen nanofiber against A549 cells, HeLa cells and MCF-7 cells were 23.8, 50.2, and 127.4 μM, respectively.
|
||||
| In Vitro Model | Lung adenocarcinoma | A-549 cell | CVCL_0023 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [88] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
50.2 μM
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The DNA-alkylating agent chlorambucil (CRB) belongs to aryl nitrogen mustard antitumor drugs, and has been widely used for treating different types of cancerous diseases. However, the clinical application of CRB is severely restricted by its poor aqueous solubility, lack of targeting, short degradation half-life and severe side effects. Macrocyclic polyamines have many applications in medicine, industry and other fields, owing to their chemical and biological properties. Some of them, such as cyclen and cyclam, could be protonated below physiological pH (7.4), and the lower the pH, the higher the degree of protonation. It is reported that the pH of the tumor environment is lower than the physiological pH, which is beneficial to the protonation of macrocyclic polyamines. Modification of macrocyclic polyamine to self-assembling peptide-drug amphiphiles can increase the cell accumulation of the hydrogel, because the cationic hydrogel has a high affinity to a negatively charged cell membrane and nucleus. Therefore, a macrocyclic polyamine containing peptide hydrogel could serve as a suitable delivery system to improve the pharmacokinetic properties of CRB, achieving improved delivery efficacy and enhanced antitumor activity without severe side effects. Herein, we report a self-assembling peptide-based cationic supramolecular nanomedicine bearing the small molecule agent CRB and macrocyclic polyamine cyclen. We found that the CRB-FFFK-cyclen conjugate could readily transform into a hydrogel through a heating-cooling process, and the resulting hydrogel could significantly improve drug stability, cellular uptake and, antitumor activity.
Click to Show/Hide
|
||||
| Description |
The IC50 values of CRB-FFFK-cyclen nanofiber against A549 cells, HeLa cells and MCF-7 cells were 23.8, 50.2, and 127.4 μM, respectively.
|
||||
| In Vitro Model | Human papillomavirus-related cervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [88] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
127.4 μM
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The DNA-alkylating agent chlorambucil (CRB) belongs to aryl nitrogen mustard antitumor drugs, and has been widely used for treating different types of cancerous diseases. However, the clinical application of CRB is severely restricted by its poor aqueous solubility, lack of targeting, short degradation half-life and severe side effects. Macrocyclic polyamines have many applications in medicine, industry and other fields, owing to their chemical and biological properties. Some of them, such as cyclen and cyclam, could be protonated below physiological pH (7.4), and the lower the pH, the higher the degree of protonation. It is reported that the pH of the tumor environment is lower than the physiological pH, which is beneficial to the protonation of macrocyclic polyamines. Modification of macrocyclic polyamine to self-assembling peptide-drug amphiphiles can increase the cell accumulation of the hydrogel, because the cationic hydrogel has a high affinity to a negatively charged cell membrane and nucleus. Therefore, a macrocyclic polyamine containing peptide hydrogel could serve as a suitable delivery system to improve the pharmacokinetic properties of CRB, achieving improved delivery efficacy and enhanced antitumor activity without severe side effects. Herein, we report a self-assembling peptide-based cationic supramolecular nanomedicine bearing the small molecule agent CRB and macrocyclic polyamine cyclen. We found that the CRB-FFFK-cyclen conjugate could readily transform into a hydrogel through a heating-cooling process, and the resulting hydrogel could significantly improve drug stability, cellular uptake and, antitumor activity.
Click to Show/Hide
|
||||
| Description |
The IC50 values of CRB-FFFK-cyclen nanofiber against A549 cells, HeLa cells and MCF-7 cells were 23.8, 50.2, and 127.4 μM, respectively.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [88] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability |
15.00%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 500 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The DNA-alkylating agent chlorambucil (CRB) belongs to aryl nitrogen mustard antitumor drugs, and has been widely used for treating different types of cancerous diseases. However, the clinical application of CRB is severely restricted by its poor aqueous solubility, lack of targeting, short degradation half-life and severe side effects. Macrocyclic polyamines have many applications in medicine, industry and other fields, owing to their chemical and biological properties. Some of them, such as cyclen and cyclam, could be protonated below physiological pH (7.4), and the lower the pH, the higher the degree of protonation. It is reported that the pH of the tumor environment is lower than the physiological pH, which is beneficial to the protonation of macrocyclic polyamines. Modification of macrocyclic polyamine to self-assembling peptide-drug amphiphiles can increase the cell accumulation of the hydrogel, because the cationic hydrogel has a high affinity to a negatively charged cell membrane and nucleus. Therefore, a macrocyclic polyamine containing peptide hydrogel could serve as a suitable delivery system to improve the pharmacokinetic properties of CRB, achieving improved delivery efficacy and enhanced antitumor activity without severe side effects. Herein, we report a self-assembling peptide-based cationic supramolecular nanomedicine bearing the small molecule agent CRB and macrocyclic polyamine cyclen. We found that the CRB-FFFK-cyclen conjugate could readily transform into a hydrogel through a heating-cooling process, and the resulting hydrogel could significantly improve drug stability, cellular uptake and, antitumor activity.
Click to Show/Hide
|
||||
| Description |
CRB and CRB-FFFK-cyclen hydrogel both exhibited broad-spectrum anticancer activities against different types of cancer cells in a dose-dependent manner.
|
||||
| In Vitro Model | Lung adenocarcinoma | A-549 cell | CVCL_0023 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [88] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability |
15.00%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 500 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The DNA-alkylating agent chlorambucil (CRB) belongs to aryl nitrogen mustard antitumor drugs, and has been widely used for treating different types of cancerous diseases. However, the clinical application of CRB is severely restricted by its poor aqueous solubility, lack of targeting, short degradation half-life and severe side effects. Macrocyclic polyamines have many applications in medicine, industry and other fields, owing to their chemical and biological properties. Some of them, such as cyclen and cyclam, could be protonated below physiological pH (7.4), and the lower the pH, the higher the degree of protonation. It is reported that the pH of the tumor environment is lower than the physiological pH, which is beneficial to the protonation of macrocyclic polyamines. Modification of macrocyclic polyamine to self-assembling peptide-drug amphiphiles can increase the cell accumulation of the hydrogel, because the cationic hydrogel has a high affinity to a negatively charged cell membrane and nucleus. Therefore, a macrocyclic polyamine containing peptide hydrogel could serve as a suitable delivery system to improve the pharmacokinetic properties of CRB, achieving improved delivery efficacy and enhanced antitumor activity without severe side effects. Herein, we report a self-assembling peptide-based cationic supramolecular nanomedicine bearing the small molecule agent CRB and macrocyclic polyamine cyclen. We found that the CRB-FFFK-cyclen conjugate could readily transform into a hydrogel through a heating-cooling process, and the resulting hydrogel could significantly improve drug stability, cellular uptake and, antitumor activity.
Click to Show/Hide
|
||||
| Description |
CRB and CRB-FFFK-cyclen hydrogel both exhibited broad-spectrum anticancer activities against different types of cancer cells in a dose-dependent manner.
|
||||
| In Vitro Model | Human papillomavirus-related cervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [88] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability |
20.00%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 250 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The DNA-alkylating agent chlorambucil (CRB) belongs to aryl nitrogen mustard antitumor drugs, and has been widely used for treating different types of cancerous diseases. However, the clinical application of CRB is severely restricted by its poor aqueous solubility, lack of targeting, short degradation half-life and severe side effects. Macrocyclic polyamines have many applications in medicine, industry and other fields, owing to their chemical and biological properties. Some of them, such as cyclen and cyclam, could be protonated below physiological pH (7.4), and the lower the pH, the higher the degree of protonation. It is reported that the pH of the tumor environment is lower than the physiological pH, which is beneficial to the protonation of macrocyclic polyamines. Modification of macrocyclic polyamine to self-assembling peptide-drug amphiphiles can increase the cell accumulation of the hydrogel, because the cationic hydrogel has a high affinity to a negatively charged cell membrane and nucleus. Therefore, a macrocyclic polyamine containing peptide hydrogel could serve as a suitable delivery system to improve the pharmacokinetic properties of CRB, achieving improved delivery efficacy and enhanced antitumor activity without severe side effects. Herein, we report a self-assembling peptide-based cationic supramolecular nanomedicine bearing the small molecule agent CRB and macrocyclic polyamine cyclen. We found that the CRB-FFFK-cyclen conjugate could readily transform into a hydrogel through a heating-cooling process, and the resulting hydrogel could significantly improve drug stability, cellular uptake and, antitumor activity.
Click to Show/Hide
|
||||
| Description |
CRB and CRB-FFFK-cyclen hydrogel both exhibited broad-spectrum anticancer activities against different types of cancer cells in a dose-dependent manner.
|
||||
| In Vitro Model | Lung adenocarcinoma | A-549 cell | CVCL_0023 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [88] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability |
20.00%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 250 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The DNA-alkylating agent chlorambucil (CRB) belongs to aryl nitrogen mustard antitumor drugs, and has been widely used for treating different types of cancerous diseases. However, the clinical application of CRB is severely restricted by its poor aqueous solubility, lack of targeting, short degradation half-life and severe side effects. Macrocyclic polyamines have many applications in medicine, industry and other fields, owing to their chemical and biological properties. Some of them, such as cyclen and cyclam, could be protonated below physiological pH (7.4), and the lower the pH, the higher the degree of protonation. It is reported that the pH of the tumor environment is lower than the physiological pH, which is beneficial to the protonation of macrocyclic polyamines. Modification of macrocyclic polyamine to self-assembling peptide-drug amphiphiles can increase the cell accumulation of the hydrogel, because the cationic hydrogel has a high affinity to a negatively charged cell membrane and nucleus. Therefore, a macrocyclic polyamine containing peptide hydrogel could serve as a suitable delivery system to improve the pharmacokinetic properties of CRB, achieving improved delivery efficacy and enhanced antitumor activity without severe side effects. Herein, we report a self-assembling peptide-based cationic supramolecular nanomedicine bearing the small molecule agent CRB and macrocyclic polyamine cyclen. We found that the CRB-FFFK-cyclen conjugate could readily transform into a hydrogel through a heating-cooling process, and the resulting hydrogel could significantly improve drug stability, cellular uptake and, antitumor activity.
Click to Show/Hide
|
||||
| Description |
CRB and CRB-FFFK-cyclen hydrogel both exhibited broad-spectrum anticancer activities against different types of cancer cells in a dose-dependent manner.
|
||||
| In Vitro Model | Human papillomavirus-related cervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [88] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability |
20.00%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 500 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The DNA-alkylating agent chlorambucil (CRB) belongs to aryl nitrogen mustard antitumor drugs, and has been widely used for treating different types of cancerous diseases. However, the clinical application of CRB is severely restricted by its poor aqueous solubility, lack of targeting, short degradation half-life and severe side effects. Macrocyclic polyamines have many applications in medicine, industry and other fields, owing to their chemical and biological properties. Some of them, such as cyclen and cyclam, could be protonated below physiological pH (7.4), and the lower the pH, the higher the degree of protonation. It is reported that the pH of the tumor environment is lower than the physiological pH, which is beneficial to the protonation of macrocyclic polyamines. Modification of macrocyclic polyamine to self-assembling peptide-drug amphiphiles can increase the cell accumulation of the hydrogel, because the cationic hydrogel has a high affinity to a negatively charged cell membrane and nucleus. Therefore, a macrocyclic polyamine containing peptide hydrogel could serve as a suitable delivery system to improve the pharmacokinetic properties of CRB, achieving improved delivery efficacy and enhanced antitumor activity without severe side effects. Herein, we report a self-assembling peptide-based cationic supramolecular nanomedicine bearing the small molecule agent CRB and macrocyclic polyamine cyclen. We found that the CRB-FFFK-cyclen conjugate could readily transform into a hydrogel through a heating-cooling process, and the resulting hydrogel could significantly improve drug stability, cellular uptake and, antitumor activity.
Click to Show/Hide
|
||||
| Description |
CRB and CRB-FFFK-cyclen hydrogel both exhibited broad-spectrum anticancer activities against different types of cancer cells in a dose-dependent manner.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [88] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability |
28.00%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 125 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The DNA-alkylating agent chlorambucil (CRB) belongs to aryl nitrogen mustard antitumor drugs, and has been widely used for treating different types of cancerous diseases. However, the clinical application of CRB is severely restricted by its poor aqueous solubility, lack of targeting, short degradation half-life and severe side effects. Macrocyclic polyamines have many applications in medicine, industry and other fields, owing to their chemical and biological properties. Some of them, such as cyclen and cyclam, could be protonated below physiological pH (7.4), and the lower the pH, the higher the degree of protonation. It is reported that the pH of the tumor environment is lower than the physiological pH, which is beneficial to the protonation of macrocyclic polyamines. Modification of macrocyclic polyamine to self-assembling peptide-drug amphiphiles can increase the cell accumulation of the hydrogel, because the cationic hydrogel has a high affinity to a negatively charged cell membrane and nucleus. Therefore, a macrocyclic polyamine containing peptide hydrogel could serve as a suitable delivery system to improve the pharmacokinetic properties of CRB, achieving improved delivery efficacy and enhanced antitumor activity without severe side effects. Herein, we report a self-assembling peptide-based cationic supramolecular nanomedicine bearing the small molecule agent CRB and macrocyclic polyamine cyclen. We found that the CRB-FFFK-cyclen conjugate could readily transform into a hydrogel through a heating-cooling process, and the resulting hydrogel could significantly improve drug stability, cellular uptake and, antitumor activity.
Click to Show/Hide
|
||||
| Description |
CRB and CRB-FFFK-cyclen hydrogel both exhibited broad-spectrum anticancer activities against different types of cancer cells in a dose-dependent manner.
|
||||
| In Vitro Model | Lung adenocarcinoma | A-549 cell | CVCL_0023 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [88] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability |
30.00%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 62.5 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The DNA-alkylating agent chlorambucil (CRB) belongs to aryl nitrogen mustard antitumor drugs, and has been widely used for treating different types of cancerous diseases. However, the clinical application of CRB is severely restricted by its poor aqueous solubility, lack of targeting, short degradation half-life and severe side effects. Macrocyclic polyamines have many applications in medicine, industry and other fields, owing to their chemical and biological properties. Some of them, such as cyclen and cyclam, could be protonated below physiological pH (7.4), and the lower the pH, the higher the degree of protonation. It is reported that the pH of the tumor environment is lower than the physiological pH, which is beneficial to the protonation of macrocyclic polyamines. Modification of macrocyclic polyamine to self-assembling peptide-drug amphiphiles can increase the cell accumulation of the hydrogel, because the cationic hydrogel has a high affinity to a negatively charged cell membrane and nucleus. Therefore, a macrocyclic polyamine containing peptide hydrogel could serve as a suitable delivery system to improve the pharmacokinetic properties of CRB, achieving improved delivery efficacy and enhanced antitumor activity without severe side effects. Herein, we report a self-assembling peptide-based cationic supramolecular nanomedicine bearing the small molecule agent CRB and macrocyclic polyamine cyclen. We found that the CRB-FFFK-cyclen conjugate could readily transform into a hydrogel through a heating-cooling process, and the resulting hydrogel could significantly improve drug stability, cellular uptake and, antitumor activity.
Click to Show/Hide
|
||||
| Description |
CRB and CRB-FFFK-cyclen hydrogel both exhibited broad-spectrum anticancer activities against different types of cancer cells in a dose-dependent manner.
|
||||
| In Vitro Model | Lung adenocarcinoma | A-549 cell | CVCL_0023 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [88] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability |
30.00%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 125 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The DNA-alkylating agent chlorambucil (CRB) belongs to aryl nitrogen mustard antitumor drugs, and has been widely used for treating different types of cancerous diseases. However, the clinical application of CRB is severely restricted by its poor aqueous solubility, lack of targeting, short degradation half-life and severe side effects. Macrocyclic polyamines have many applications in medicine, industry and other fields, owing to their chemical and biological properties. Some of them, such as cyclen and cyclam, could be protonated below physiological pH (7.4), and the lower the pH, the higher the degree of protonation. It is reported that the pH of the tumor environment is lower than the physiological pH, which is beneficial to the protonation of macrocyclic polyamines. Modification of macrocyclic polyamine to self-assembling peptide-drug amphiphiles can increase the cell accumulation of the hydrogel, because the cationic hydrogel has a high affinity to a negatively charged cell membrane and nucleus. Therefore, a macrocyclic polyamine containing peptide hydrogel could serve as a suitable delivery system to improve the pharmacokinetic properties of CRB, achieving improved delivery efficacy and enhanced antitumor activity without severe side effects. Herein, we report a self-assembling peptide-based cationic supramolecular nanomedicine bearing the small molecule agent CRB and macrocyclic polyamine cyclen. We found that the CRB-FFFK-cyclen conjugate could readily transform into a hydrogel through a heating-cooling process, and the resulting hydrogel could significantly improve drug stability, cellular uptake and, antitumor activity.
Click to Show/Hide
|
||||
| Description |
CRB and CRB-FFFK-cyclen hydrogel both exhibited broad-spectrum anticancer activities against different types of cancer cells in a dose-dependent manner.
|
||||
| In Vitro Model | Human papillomavirus-related cervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [88] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability |
35.00%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 31.3 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The DNA-alkylating agent chlorambucil (CRB) belongs to aryl nitrogen mustard antitumor drugs, and has been widely used for treating different types of cancerous diseases. However, the clinical application of CRB is severely restricted by its poor aqueous solubility, lack of targeting, short degradation half-life and severe side effects. Macrocyclic polyamines have many applications in medicine, industry and other fields, owing to their chemical and biological properties. Some of them, such as cyclen and cyclam, could be protonated below physiological pH (7.4), and the lower the pH, the higher the degree of protonation. It is reported that the pH of the tumor environment is lower than the physiological pH, which is beneficial to the protonation of macrocyclic polyamines. Modification of macrocyclic polyamine to self-assembling peptide-drug amphiphiles can increase the cell accumulation of the hydrogel, because the cationic hydrogel has a high affinity to a negatively charged cell membrane and nucleus. Therefore, a macrocyclic polyamine containing peptide hydrogel could serve as a suitable delivery system to improve the pharmacokinetic properties of CRB, achieving improved delivery efficacy and enhanced antitumor activity without severe side effects. Herein, we report a self-assembling peptide-based cationic supramolecular nanomedicine bearing the small molecule agent CRB and macrocyclic polyamine cyclen. We found that the CRB-FFFK-cyclen conjugate could readily transform into a hydrogel through a heating-cooling process, and the resulting hydrogel could significantly improve drug stability, cellular uptake and, antitumor activity.
Click to Show/Hide
|
||||
| Description |
CRB and CRB-FFFK-cyclen hydrogel both exhibited broad-spectrum anticancer activities against different types of cancer cells in a dose-dependent manner.
|
||||
| In Vitro Model | Lung adenocarcinoma | A-549 cell | CVCL_0023 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [88] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability |
40.00%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 250 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The DNA-alkylating agent chlorambucil (CRB) belongs to aryl nitrogen mustard antitumor drugs, and has been widely used for treating different types of cancerous diseases. However, the clinical application of CRB is severely restricted by its poor aqueous solubility, lack of targeting, short degradation half-life and severe side effects. Macrocyclic polyamines have many applications in medicine, industry and other fields, owing to their chemical and biological properties. Some of them, such as cyclen and cyclam, could be protonated below physiological pH (7.4), and the lower the pH, the higher the degree of protonation. It is reported that the pH of the tumor environment is lower than the physiological pH, which is beneficial to the protonation of macrocyclic polyamines. Modification of macrocyclic polyamine to self-assembling peptide-drug amphiphiles can increase the cell accumulation of the hydrogel, because the cationic hydrogel has a high affinity to a negatively charged cell membrane and nucleus. Therefore, a macrocyclic polyamine containing peptide hydrogel could serve as a suitable delivery system to improve the pharmacokinetic properties of CRB, achieving improved delivery efficacy and enhanced antitumor activity without severe side effects. Herein, we report a self-assembling peptide-based cationic supramolecular nanomedicine bearing the small molecule agent CRB and macrocyclic polyamine cyclen. We found that the CRB-FFFK-cyclen conjugate could readily transform into a hydrogel through a heating-cooling process, and the resulting hydrogel could significantly improve drug stability, cellular uptake and, antitumor activity.
Click to Show/Hide
|
||||
| Description |
CRB and CRB-FFFK-cyclen hydrogel both exhibited broad-spectrum anticancer activities against different types of cancer cells in a dose-dependent manner.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [88] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability |
50.00%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 125 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The DNA-alkylating agent chlorambucil (CRB) belongs to aryl nitrogen mustard antitumor drugs, and has been widely used for treating different types of cancerous diseases. However, the clinical application of CRB is severely restricted by its poor aqueous solubility, lack of targeting, short degradation half-life and severe side effects. Macrocyclic polyamines have many applications in medicine, industry and other fields, owing to their chemical and biological properties. Some of them, such as cyclen and cyclam, could be protonated below physiological pH (7.4), and the lower the pH, the higher the degree of protonation. It is reported that the pH of the tumor environment is lower than the physiological pH, which is beneficial to the protonation of macrocyclic polyamines. Modification of macrocyclic polyamine to self-assembling peptide-drug amphiphiles can increase the cell accumulation of the hydrogel, because the cationic hydrogel has a high affinity to a negatively charged cell membrane and nucleus. Therefore, a macrocyclic polyamine containing peptide hydrogel could serve as a suitable delivery system to improve the pharmacokinetic properties of CRB, achieving improved delivery efficacy and enhanced antitumor activity without severe side effects. Herein, we report a self-assembling peptide-based cationic supramolecular nanomedicine bearing the small molecule agent CRB and macrocyclic polyamine cyclen. We found that the CRB-FFFK-cyclen conjugate could readily transform into a hydrogel through a heating-cooling process, and the resulting hydrogel could significantly improve drug stability, cellular uptake and, antitumor activity.
Click to Show/Hide
|
||||
| Description |
CRB and CRB-FFFK-cyclen hydrogel both exhibited broad-spectrum anticancer activities against different types of cancer cells in a dose-dependent manner.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [88] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability |
55.00%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 62.5 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The DNA-alkylating agent chlorambucil (CRB) belongs to aryl nitrogen mustard antitumor drugs, and has been widely used for treating different types of cancerous diseases. However, the clinical application of CRB is severely restricted by its poor aqueous solubility, lack of targeting, short degradation half-life and severe side effects. Macrocyclic polyamines have many applications in medicine, industry and other fields, owing to their chemical and biological properties. Some of them, such as cyclen and cyclam, could be protonated below physiological pH (7.4), and the lower the pH, the higher the degree of protonation. It is reported that the pH of the tumor environment is lower than the physiological pH, which is beneficial to the protonation of macrocyclic polyamines. Modification of macrocyclic polyamine to self-assembling peptide-drug amphiphiles can increase the cell accumulation of the hydrogel, because the cationic hydrogel has a high affinity to a negatively charged cell membrane and nucleus. Therefore, a macrocyclic polyamine containing peptide hydrogel could serve as a suitable delivery system to improve the pharmacokinetic properties of CRB, achieving improved delivery efficacy and enhanced antitumor activity without severe side effects. Herein, we report a self-assembling peptide-based cationic supramolecular nanomedicine bearing the small molecule agent CRB and macrocyclic polyamine cyclen. We found that the CRB-FFFK-cyclen conjugate could readily transform into a hydrogel through a heating-cooling process, and the resulting hydrogel could significantly improve drug stability, cellular uptake and, antitumor activity.
Click to Show/Hide
|
||||
| Description |
CRB and CRB-FFFK-cyclen hydrogel both exhibited broad-spectrum anticancer activities against different types of cancer cells in a dose-dependent manner.
|
||||
| In Vitro Model | Human papillomavirus-related cervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [88] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability |
60.00%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 15.6 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The DNA-alkylating agent chlorambucil (CRB) belongs to aryl nitrogen mustard antitumor drugs, and has been widely used for treating different types of cancerous diseases. However, the clinical application of CRB is severely restricted by its poor aqueous solubility, lack of targeting, short degradation half-life and severe side effects. Macrocyclic polyamines have many applications in medicine, industry and other fields, owing to their chemical and biological properties. Some of them, such as cyclen and cyclam, could be protonated below physiological pH (7.4), and the lower the pH, the higher the degree of protonation. It is reported that the pH of the tumor environment is lower than the physiological pH, which is beneficial to the protonation of macrocyclic polyamines. Modification of macrocyclic polyamine to self-assembling peptide-drug amphiphiles can increase the cell accumulation of the hydrogel, because the cationic hydrogel has a high affinity to a negatively charged cell membrane and nucleus. Therefore, a macrocyclic polyamine containing peptide hydrogel could serve as a suitable delivery system to improve the pharmacokinetic properties of CRB, achieving improved delivery efficacy and enhanced antitumor activity without severe side effects. Herein, we report a self-assembling peptide-based cationic supramolecular nanomedicine bearing the small molecule agent CRB and macrocyclic polyamine cyclen. We found that the CRB-FFFK-cyclen conjugate could readily transform into a hydrogel through a heating-cooling process, and the resulting hydrogel could significantly improve drug stability, cellular uptake and, antitumor activity.
Click to Show/Hide
|
||||
| Description |
CRB and CRB-FFFK-cyclen hydrogel both exhibited broad-spectrum anticancer activities against different types of cancer cells in a dose-dependent manner.
|
||||
| In Vitro Model | Lung adenocarcinoma | A-549 cell | CVCL_0023 | ||
| Experiment 17 Reporting the Activity Data of This PDC | [88] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability |
60.00%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 31.3 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The DNA-alkylating agent chlorambucil (CRB) belongs to aryl nitrogen mustard antitumor drugs, and has been widely used for treating different types of cancerous diseases. However, the clinical application of CRB is severely restricted by its poor aqueous solubility, lack of targeting, short degradation half-life and severe side effects. Macrocyclic polyamines have many applications in medicine, industry and other fields, owing to their chemical and biological properties. Some of them, such as cyclen and cyclam, could be protonated below physiological pH (7.4), and the lower the pH, the higher the degree of protonation. It is reported that the pH of the tumor environment is lower than the physiological pH, which is beneficial to the protonation of macrocyclic polyamines. Modification of macrocyclic polyamine to self-assembling peptide-drug amphiphiles can increase the cell accumulation of the hydrogel, because the cationic hydrogel has a high affinity to a negatively charged cell membrane and nucleus. Therefore, a macrocyclic polyamine containing peptide hydrogel could serve as a suitable delivery system to improve the pharmacokinetic properties of CRB, achieving improved delivery efficacy and enhanced antitumor activity without severe side effects. Herein, we report a self-assembling peptide-based cationic supramolecular nanomedicine bearing the small molecule agent CRB and macrocyclic polyamine cyclen. We found that the CRB-FFFK-cyclen conjugate could readily transform into a hydrogel through a heating-cooling process, and the resulting hydrogel could significantly improve drug stability, cellular uptake and, antitumor activity.
Click to Show/Hide
|
||||
| Description |
CRB and CRB-FFFK-cyclen hydrogel both exhibited broad-spectrum anticancer activities against different types of cancer cells in a dose-dependent manner.
|
||||
| In Vitro Model | Human papillomavirus-related cervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 18 Reporting the Activity Data of This PDC | [88] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability |
62.00%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 15.6 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The DNA-alkylating agent chlorambucil (CRB) belongs to aryl nitrogen mustard antitumor drugs, and has been widely used for treating different types of cancerous diseases. However, the clinical application of CRB is severely restricted by its poor aqueous solubility, lack of targeting, short degradation half-life and severe side effects. Macrocyclic polyamines have many applications in medicine, industry and other fields, owing to their chemical and biological properties. Some of them, such as cyclen and cyclam, could be protonated below physiological pH (7.4), and the lower the pH, the higher the degree of protonation. It is reported that the pH of the tumor environment is lower than the physiological pH, which is beneficial to the protonation of macrocyclic polyamines. Modification of macrocyclic polyamine to self-assembling peptide-drug amphiphiles can increase the cell accumulation of the hydrogel, because the cationic hydrogel has a high affinity to a negatively charged cell membrane and nucleus. Therefore, a macrocyclic polyamine containing peptide hydrogel could serve as a suitable delivery system to improve the pharmacokinetic properties of CRB, achieving improved delivery efficacy and enhanced antitumor activity without severe side effects. Herein, we report a self-assembling peptide-based cationic supramolecular nanomedicine bearing the small molecule agent CRB and macrocyclic polyamine cyclen. We found that the CRB-FFFK-cyclen conjugate could readily transform into a hydrogel through a heating-cooling process, and the resulting hydrogel could significantly improve drug stability, cellular uptake and, antitumor activity.
Click to Show/Hide
|
||||
| Description |
CRB and CRB-FFFK-cyclen hydrogel both exhibited broad-spectrum anticancer activities against different types of cancer cells in a dose-dependent manner.
|
||||
| In Vitro Model | Human papillomavirus-related cervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 19 Reporting the Activity Data of This PDC | [88] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability |
64.00%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 7.8 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The DNA-alkylating agent chlorambucil (CRB) belongs to aryl nitrogen mustard antitumor drugs, and has been widely used for treating different types of cancerous diseases. However, the clinical application of CRB is severely restricted by its poor aqueous solubility, lack of targeting, short degradation half-life and severe side effects. Macrocyclic polyamines have many applications in medicine, industry and other fields, owing to their chemical and biological properties. Some of them, such as cyclen and cyclam, could be protonated below physiological pH (7.4), and the lower the pH, the higher the degree of protonation. It is reported that the pH of the tumor environment is lower than the physiological pH, which is beneficial to the protonation of macrocyclic polyamines. Modification of macrocyclic polyamine to self-assembling peptide-drug amphiphiles can increase the cell accumulation of the hydrogel, because the cationic hydrogel has a high affinity to a negatively charged cell membrane and nucleus. Therefore, a macrocyclic polyamine containing peptide hydrogel could serve as a suitable delivery system to improve the pharmacokinetic properties of CRB, achieving improved delivery efficacy and enhanced antitumor activity without severe side effects. Herein, we report a self-assembling peptide-based cationic supramolecular nanomedicine bearing the small molecule agent CRB and macrocyclic polyamine cyclen. We found that the CRB-FFFK-cyclen conjugate could readily transform into a hydrogel through a heating-cooling process, and the resulting hydrogel could significantly improve drug stability, cellular uptake and, antitumor activity.
Click to Show/Hide
|
||||
| Description |
CRB and CRB-FFFK-cyclen hydrogel both exhibited broad-spectrum anticancer activities against different types of cancer cells in a dose-dependent manner.
|
||||
| In Vitro Model | Human papillomavirus-related cervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 20 Reporting the Activity Data of This PDC | [88] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability |
70.00%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 62.5 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The DNA-alkylating agent chlorambucil (CRB) belongs to aryl nitrogen mustard antitumor drugs, and has been widely used for treating different types of cancerous diseases. However, the clinical application of CRB is severely restricted by its poor aqueous solubility, lack of targeting, short degradation half-life and severe side effects. Macrocyclic polyamines have many applications in medicine, industry and other fields, owing to their chemical and biological properties. Some of them, such as cyclen and cyclam, could be protonated below physiological pH (7.4), and the lower the pH, the higher the degree of protonation. It is reported that the pH of the tumor environment is lower than the physiological pH, which is beneficial to the protonation of macrocyclic polyamines. Modification of macrocyclic polyamine to self-assembling peptide-drug amphiphiles can increase the cell accumulation of the hydrogel, because the cationic hydrogel has a high affinity to a negatively charged cell membrane and nucleus. Therefore, a macrocyclic polyamine containing peptide hydrogel could serve as a suitable delivery system to improve the pharmacokinetic properties of CRB, achieving improved delivery efficacy and enhanced antitumor activity without severe side effects. Herein, we report a self-assembling peptide-based cationic supramolecular nanomedicine bearing the small molecule agent CRB and macrocyclic polyamine cyclen. We found that the CRB-FFFK-cyclen conjugate could readily transform into a hydrogel through a heating-cooling process, and the resulting hydrogel could significantly improve drug stability, cellular uptake and, antitumor activity.
Click to Show/Hide
|
||||
| Description |
CRB and CRB-FFFK-cyclen hydrogel both exhibited broad-spectrum anticancer activities against different types of cancer cells in a dose-dependent manner.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 21 Reporting the Activity Data of This PDC | [88] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability |
78.00%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 7.8 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The DNA-alkylating agent chlorambucil (CRB) belongs to aryl nitrogen mustard antitumor drugs, and has been widely used for treating different types of cancerous diseases. However, the clinical application of CRB is severely restricted by its poor aqueous solubility, lack of targeting, short degradation half-life and severe side effects. Macrocyclic polyamines have many applications in medicine, industry and other fields, owing to their chemical and biological properties. Some of them, such as cyclen and cyclam, could be protonated below physiological pH (7.4), and the lower the pH, the higher the degree of protonation. It is reported that the pH of the tumor environment is lower than the physiological pH, which is beneficial to the protonation of macrocyclic polyamines. Modification of macrocyclic polyamine to self-assembling peptide-drug amphiphiles can increase the cell accumulation of the hydrogel, because the cationic hydrogel has a high affinity to a negatively charged cell membrane and nucleus. Therefore, a macrocyclic polyamine containing peptide hydrogel could serve as a suitable delivery system to improve the pharmacokinetic properties of CRB, achieving improved delivery efficacy and enhanced antitumor activity without severe side effects. Herein, we report a self-assembling peptide-based cationic supramolecular nanomedicine bearing the small molecule agent CRB and macrocyclic polyamine cyclen. We found that the CRB-FFFK-cyclen conjugate could readily transform into a hydrogel through a heating-cooling process, and the resulting hydrogel could significantly improve drug stability, cellular uptake and, antitumor activity.
Click to Show/Hide
|
||||
| Description |
CRB and CRB-FFFK-cyclen hydrogel both exhibited broad-spectrum anticancer activities against different types of cancer cells in a dose-dependent manner.
|
||||
| In Vitro Model | Lung adenocarcinoma | A-549 cell | CVCL_0023 | ||
| Experiment 22 Reporting the Activity Data of This PDC | [88] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability |
78.00%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 31.3 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The DNA-alkylating agent chlorambucil (CRB) belongs to aryl nitrogen mustard antitumor drugs, and has been widely used for treating different types of cancerous diseases. However, the clinical application of CRB is severely restricted by its poor aqueous solubility, lack of targeting, short degradation half-life and severe side effects. Macrocyclic polyamines have many applications in medicine, industry and other fields, owing to their chemical and biological properties. Some of them, such as cyclen and cyclam, could be protonated below physiological pH (7.4), and the lower the pH, the higher the degree of protonation. It is reported that the pH of the tumor environment is lower than the physiological pH, which is beneficial to the protonation of macrocyclic polyamines. Modification of macrocyclic polyamine to self-assembling peptide-drug amphiphiles can increase the cell accumulation of the hydrogel, because the cationic hydrogel has a high affinity to a negatively charged cell membrane and nucleus. Therefore, a macrocyclic polyamine containing peptide hydrogel could serve as a suitable delivery system to improve the pharmacokinetic properties of CRB, achieving improved delivery efficacy and enhanced antitumor activity without severe side effects. Herein, we report a self-assembling peptide-based cationic supramolecular nanomedicine bearing the small molecule agent CRB and macrocyclic polyamine cyclen. We found that the CRB-FFFK-cyclen conjugate could readily transform into a hydrogel through a heating-cooling process, and the resulting hydrogel could significantly improve drug stability, cellular uptake and, antitumor activity.
Click to Show/Hide
|
||||
| Description |
CRB and CRB-FFFK-cyclen hydrogel both exhibited broad-spectrum anticancer activities against different types of cancer cells in a dose-dependent manner.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 23 Reporting the Activity Data of This PDC | [88] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability |
80.00%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 15.6 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The DNA-alkylating agent chlorambucil (CRB) belongs to aryl nitrogen mustard antitumor drugs, and has been widely used for treating different types of cancerous diseases. However, the clinical application of CRB is severely restricted by its poor aqueous solubility, lack of targeting, short degradation half-life and severe side effects. Macrocyclic polyamines have many applications in medicine, industry and other fields, owing to their chemical and biological properties. Some of them, such as cyclen and cyclam, could be protonated below physiological pH (7.4), and the lower the pH, the higher the degree of protonation. It is reported that the pH of the tumor environment is lower than the physiological pH, which is beneficial to the protonation of macrocyclic polyamines. Modification of macrocyclic polyamine to self-assembling peptide-drug amphiphiles can increase the cell accumulation of the hydrogel, because the cationic hydrogel has a high affinity to a negatively charged cell membrane and nucleus. Therefore, a macrocyclic polyamine containing peptide hydrogel could serve as a suitable delivery system to improve the pharmacokinetic properties of CRB, achieving improved delivery efficacy and enhanced antitumor activity without severe side effects. Herein, we report a self-assembling peptide-based cationic supramolecular nanomedicine bearing the small molecule agent CRB and macrocyclic polyamine cyclen. We found that the CRB-FFFK-cyclen conjugate could readily transform into a hydrogel through a heating-cooling process, and the resulting hydrogel could significantly improve drug stability, cellular uptake and, antitumor activity.
Click to Show/Hide
|
||||
| Description |
CRB and CRB-FFFK-cyclen hydrogel both exhibited broad-spectrum anticancer activities against different types of cancer cells in a dose-dependent manner.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 24 Reporting the Activity Data of This PDC | [88] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability |
83.00%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 7.8 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The DNA-alkylating agent chlorambucil (CRB) belongs to aryl nitrogen mustard antitumor drugs, and has been widely used for treating different types of cancerous diseases. However, the clinical application of CRB is severely restricted by its poor aqueous solubility, lack of targeting, short degradation half-life and severe side effects. Macrocyclic polyamines have many applications in medicine, industry and other fields, owing to their chemical and biological properties. Some of them, such as cyclen and cyclam, could be protonated below physiological pH (7.4), and the lower the pH, the higher the degree of protonation. It is reported that the pH of the tumor environment is lower than the physiological pH, which is beneficial to the protonation of macrocyclic polyamines. Modification of macrocyclic polyamine to self-assembling peptide-drug amphiphiles can increase the cell accumulation of the hydrogel, because the cationic hydrogel has a high affinity to a negatively charged cell membrane and nucleus. Therefore, a macrocyclic polyamine containing peptide hydrogel could serve as a suitable delivery system to improve the pharmacokinetic properties of CRB, achieving improved delivery efficacy and enhanced antitumor activity without severe side effects. Herein, we report a self-assembling peptide-based cationic supramolecular nanomedicine bearing the small molecule agent CRB and macrocyclic polyamine cyclen. We found that the CRB-FFFK-cyclen conjugate could readily transform into a hydrogel through a heating-cooling process, and the resulting hydrogel could significantly improve drug stability, cellular uptake and, antitumor activity.
Click to Show/Hide
|
||||
| Description |
CRB and CRB-FFFK-cyclen hydrogel both exhibited broad-spectrum anticancer activities against different types of cancer cells in a dose-dependent manner.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
MP-P5 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [85] | ||||
| Indication | Zika virus infection | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
25.07 ± 0.05 µM
|
|||
| Administration Time | 1 h | ||||
| Evaluation Method | GraphPad Prism assay | ||||
| MOA of PDC |
Strategies for conjugating BBBpS to drug payloads have been actively explored over recent decades, with the number of new shuttles steadily increasing. Some recent entries, moreover, have shown to be capable not only of carrying drugs into but also removing toxins from the brain, preventing their accumulation. For our part, we have reported that peptide-porphyrin conjugates (PPCs), where a BBBpS and an antiviral porphyrin are covalently linked by an amide bond (amino and carboxyl groups in BBBpS and porphyrin, respectively) can successfully pass the BBB and act against brain-targeting viruses such as HIV. As for the BPB, the literature is scarce, with only a few described examples of peptides able to pass it. Herein, we report our results in developing new PPCs able to penetrate both BPB and BBB and act against ZIKV. The PPC production strategy, involving porphyrin (P), C- or N-terminal conjugation to a BBBpS, has been detailed in a recent publication and is illustrated in Scheme 1 and Scheme S1. In this paper we describe eight PPCs resulting from the combination of four BBBpS and two porphyrins, and their evaluation in terms of barrier crossing and anti-ZIKV activity. One of the conjugates, PP-P1, emerges as particularly effective against ZIKV, having also the ability to translocate across BPB and BBB.
Click to Show/Hide
|
||||
| Description |
The five PPCs that successfully translocated BBB and BPB were evaluated for ZIKV inactivation in vitro, using a plaque assay. Of them, two showed significant activity, namely MP-P5 (IC50 = 25.07 ± 0.05 μM, similar to activity against HIV) and PP-P1 (IC50 = 1.08 ± 0.14 μM). Additionally, a treatment assay performed with MP-P5 and PP-P1 revealed that both PPCs efficiently inhibit ZIKV replication when added 1 h and 7 h post-infection. As observed for HIV, non-conjugated porphyrins did not show activity against ZIKV, reinforcing the claim that BBBpS conjugation is not only critical for BBB/BPB translocation but also for antiviral activity. On the other hand, and somewhat unexpectedly, PP-P1, shown to be inactive in vitro against HIV, emerged as the most active anti-ZIKV conjugate. As described in the literature, the light-independent mechanism of action of porphyrins is based on a direct perturbation of the viral envelope. Porphyrins interact and accumulate on the envelope lipid membrane, causing a decrease in order and a consequent phase alteration that impairs viral entry processes. Since we ensured no-light conditions and no metal cations are coordinated to the porphyrin rings of the PPCsavoiding generation of reactive oxygen speciesthe antiviral light-independent mechanism is the only plausible one. Thus, one may confidently suggest that the PP-P1 specific anti-ZIKV activity is guided by preferential interaction with the ZIKV viral envelope and/or components.
Click to Show/Hide
|
||||
| In Vitro Model | ZIKV infection | Zika virus | 64320 | ||
| Half life period | 21.1 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [85] | ||||
| Indication | Human immunodeficiency virus infection | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
33.1 ± 1.38 µM
|
|||
| Administration Time | 1 h | ||||
| Evaluation Method | GraphPad Prism assay | ||||
| MOA of PDC |
Strategies for conjugating BBBpS to drug payloads have been actively explored over recent decades, with the number of new shuttles steadily increasing. Some recent entries, moreover, have shown to be capable not only of carrying drugs into but also removing toxins from the brain, preventing their accumulation. For our part, we have reported that peptide-porphyrin conjugates (PPCs), where a BBBpS and an antiviral porphyrin are covalently linked by an amide bond (amino and carboxyl groups in BBBpS and porphyrin, respectively) can successfully pass the BBB and act against brain-targeting viruses such as HIV. As for the BPB, the literature is scarce, with only a few described examples of peptides able to pass it. Herein, we report our results in developing new PPCs able to penetrate both BPB and BBB and act against ZIKV. The PPC production strategy, involving porphyrin (P), C- or N-terminal conjugation to a BBBpS, has been detailed in a recent publication and is illustrated in Scheme 1 and Scheme S1. In this paper we describe eight PPCs resulting from the combination of four BBBpS and two porphyrins, and their evaluation in terms of barrier crossing and anti-ZIKV activity. One of the conjugates, PP-P1, emerges as particularly effective against ZIKV, having also the ability to translocate across BPB and BBB.
Click to Show/Hide
|
||||
| Description |
The five PPCs that successfully translocated BBB and BPB were evaluated for ZIKV inactivation in vitro, using a plaque assay. Of them, two showed significant activity, namely MP-P5 (IC50 = 25.07 ± 0.05 μM, similar to activity against HIV) and PP-P1 (IC50 = 1.08 ± 0.14 μM). Additionally, a treatment assay performed with MP-P5 and PP-P1 revealed that both PPCs efficiently inhibit ZIKV replication when added 1 h and 7 h post-infection. As observed for HIV, non-conjugated porphyrins did not show activity against ZIKV, reinforcing the claim that BBBpS conjugation is not only critical for BBB/BPB translocation but also for antiviral activity. On the other hand, and somewhat unexpectedly, PP-P1, shown to be inactive in vitro against HIV, emerged as the most active anti-ZIKV conjugate. As described in the literature, the light-independent mechanism of action of porphyrins is based on a direct perturbation of the viral envelope. Porphyrins interact and accumulate on the envelope lipid membrane, causing a decrease in order and a consequent phase alteration that impairs viral entry processes. Since we ensured no-light conditions and no metal cations are coordinated to the porphyrin rings of the PPCsavoiding generation of reactive oxygen speciesthe antiviral light-independent mechanism is the only plausible one. Thus, one may confidently suggest that the PP-P1 specific anti-ZIKV activity is guided by preferential interaction with the ZIKV viral envelope and/or components.
Click to Show/Hide
|
||||
| In Vitro Model | HIV Infection | Human immunodeficiency virus | 12721 | ||
| Half life period | 21.1 h | ||||
MP-P1 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [85] | ||||
| Indication | Zika virus infection | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Administration Time | 1 h | ||||
| Evaluation Method | GraphPad Prism assay | ||||
| MOA of PDC |
Strategies for conjugating BBBpS to drug payloads have been actively explored over recent decades, with the number of new shuttles steadily increasing. Some recent entries, moreover, have shown to be capable not only of carrying drugs into but also removing toxins from the brain, preventing their accumulation. For our part, we have reported that peptide-porphyrin conjugates (PPCs), where a BBBpS and an antiviral porphyrin are covalently linked by an amide bond (amino and carboxyl groups in BBBpS and porphyrin, respectively) can successfully pass the BBB and act against brain-targeting viruses such as HIV. As for the BPB, the literature is scarce, with only a few described examples of peptides able to pass it. Herein, we report our results in developing new PPCs able to penetrate both BPB and BBB and act against ZIKV. The PPC production strategy, involving porphyrin (P), C- or N-terminal conjugation to a BBBpS, has been detailed in a recent publication and is illustrated in Scheme 1 and Scheme S1. In this paper we describe eight PPCs resulting from the combination of four BBBpS and two porphyrins, and their evaluation in terms of barrier crossing and anti-ZIKV activity. One of the conjugates, PP-P1, emerges as particularly effective against ZIKV, having also the ability to translocate across BPB and BBB.
Click to Show/Hide
|
||||
| Description |
The five PPCs that successfully translocated BBB and BPB were evaluated for ZIKV inactivation in vitro, using a plaque assay. Of them, two showed significant activity, namely MP-P5 (IC50 = 25.07 ± 0.05 μM, similar to activity against HIV) and PP-P1 (IC50 = 1.08 ± 0.14 μM). Additionally, a treatment assay performed with MP-P5 and PP-P1 revealed that both PPCs efficiently inhibit ZIKV replication when added 1 h and 7 h post-infection. As observed for HIV, non-conjugated porphyrins did not show activity against ZIKV, reinforcing the claim that BBBpS conjugation is not only critical for BBB/BPB translocation but also for antiviral activity. On the other hand, and somewhat unexpectedly, PP-P1, shown to be inactive in vitro against HIV, emerged as the most active anti-ZIKV conjugate. As described in the literature, the light-independent mechanism of action of porphyrins is based on a direct perturbation of the viral envelope. Porphyrins interact and accumulate on the envelope lipid membrane, causing a decrease in order and a consequent phase alteration that impairs viral entry processes. Since we ensured no-light conditions and no metal cations are coordinated to the porphyrin rings of the PPCsavoiding generation of reactive oxygen speciesthe antiviral light-independent mechanism is the only plausible one. Thus, one may confidently suggest that the PP-P1 specific anti-ZIKV activity is guided by preferential interaction with the ZIKV viral envelope and/or components.
Click to Show/Hide
|
||||
| In Vitro Model | ZIKV infection | Zika virus | 64320 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [85] | ||||
| Indication | Human immunodeficiency virus infection | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Administration Time | 1 h | ||||
| Evaluation Method | GraphPad Prism assay | ||||
| MOA of PDC |
Strategies for conjugating BBBpS to drug payloads have been actively explored over recent decades, with the number of new shuttles steadily increasing. Some recent entries, moreover, have shown to be capable not only of carrying drugs into but also removing toxins from the brain, preventing their accumulation. For our part, we have reported that peptide-porphyrin conjugates (PPCs), where a BBBpS and an antiviral porphyrin are covalently linked by an amide bond (amino and carboxyl groups in BBBpS and porphyrin, respectively) can successfully pass the BBB and act against brain-targeting viruses such as HIV. As for the BPB, the literature is scarce, with only a few described examples of peptides able to pass it. Herein, we report our results in developing new PPCs able to penetrate both BPB and BBB and act against ZIKV. The PPC production strategy, involving porphyrin (P), C- or N-terminal conjugation to a BBBpS, has been detailed in a recent publication and is illustrated in Scheme 1 and Scheme S1. In this paper we describe eight PPCs resulting from the combination of four BBBpS and two porphyrins, and their evaluation in terms of barrier crossing and anti-ZIKV activity. One of the conjugates, PP-P1, emerges as particularly effective against ZIKV, having also the ability to translocate across BPB and BBB.
Click to Show/Hide
|
||||
| Description |
The five PPCs that successfully translocated BBB and BPB were evaluated for ZIKV inactivation in vitro, using a plaque assay. Of them, two showed significant activity, namely MP-P5 (IC50 = 25.07 ± 0.05 μM, similar to activity against HIV) and PP-P1 (IC50 = 1.08 ± 0.14 μM). Additionally, a treatment assay performed with MP-P5 and PP-P1 revealed that both PPCs efficiently inhibit ZIKV replication when added 1 h and 7 h post-infection. As observed for HIV, non-conjugated porphyrins did not show activity against ZIKV, reinforcing the claim that BBBpS conjugation is not only critical for BBB/BPB translocation but also for antiviral activity. On the other hand, and somewhat unexpectedly, PP-P1, shown to be inactive in vitro against HIV, emerged as the most active anti-ZIKV conjugate. As described in the literature, the light-independent mechanism of action of porphyrins is based on a direct perturbation of the viral envelope. Porphyrins interact and accumulate on the envelope lipid membrane, causing a decrease in order and a consequent phase alteration that impairs viral entry processes. Since we ensured no-light conditions and no metal cations are coordinated to the porphyrin rings of the PPCsavoiding generation of reactive oxygen speciesthe antiviral light-independent mechanism is the only plausible one. Thus, one may confidently suggest that the PP-P1 specific anti-ZIKV activity is guided by preferential interaction with the ZIKV viral envelope and/or components.
Click to Show/Hide
|
||||
| In Vitro Model | HIV Infection | Human immunodeficiency virus | 12721 | ||
MP-P2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [85] | ||||
| Indication | Zika virus infection | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Administration Time | 1 h | ||||
| Evaluation Method | GraphPad Prism assay | ||||
| MOA of PDC |
Strategies for conjugating BBBpS to drug payloads have been actively explored over recent decades, with the number of new shuttles steadily increasing. Some recent entries, moreover, have shown to be capable not only of carrying drugs into but also removing toxins from the brain, preventing their accumulation. For our part, we have reported that peptide-porphyrin conjugates (PPCs), where a BBBpS and an antiviral porphyrin are covalently linked by an amide bond (amino and carboxyl groups in BBBpS and porphyrin, respectively) can successfully pass the BBB and act against brain-targeting viruses such as HIV. As for the BPB, the literature is scarce, with only a few described examples of peptides able to pass it. Herein, we report our results in developing new PPCs able to penetrate both BPB and BBB and act against ZIKV. The PPC production strategy, involving porphyrin (P), C- or N-terminal conjugation to a BBBpS, has been detailed in a recent publication and is illustrated in Scheme 1 and Scheme S1. In this paper we describe eight PPCs resulting from the combination of four BBBpS and two porphyrins, and their evaluation in terms of barrier crossing and anti-ZIKV activity. One of the conjugates, PP-P1, emerges as particularly effective against ZIKV, having also the ability to translocate across BPB and BBB.
Click to Show/Hide
|
||||
| Description |
The five PPCs that successfully translocated BBB and BPB were evaluated for ZIKV inactivation in vitro, using a plaque assay. Of them, two showed significant activity, namely MP-P5 (IC50 = 25.07 ± 0.05 μM, similar to activity against HIV) and PP-P1 (IC50 = 1.08 ± 0.14 μM). Additionally, a treatment assay performed with MP-P5 and PP-P1 revealed that both PPCs efficiently inhibit ZIKV replication when added 1 h and 7 h post-infection. As observed for HIV, non-conjugated porphyrins did not show activity against ZIKV, reinforcing the claim that BBBpS conjugation is not only critical for BBB/BPB translocation but also for antiviral activity. On the other hand, and somewhat unexpectedly, PP-P1, shown to be inactive in vitro against HIV, emerged as the most active anti-ZIKV conjugate. As described in the literature, the light-independent mechanism of action of porphyrins is based on a direct perturbation of the viral envelope. Porphyrins interact and accumulate on the envelope lipid membrane, causing a decrease in order and a consequent phase alteration that impairs viral entry processes. Since we ensured no-light conditions and no metal cations are coordinated to the porphyrin rings of the PPCsavoiding generation of reactive oxygen speciesthe antiviral light-independent mechanism is the only plausible one. Thus, one may confidently suggest that the PP-P1 specific anti-ZIKV activity is guided by preferential interaction with the ZIKV viral envelope and/or components.
Click to Show/Hide
|
||||
| In Vitro Model | ZIKV infection | Zika virus | 64320 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [85] | ||||
| Indication | Human immunodeficiency virus infection | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Administration Time | 1 h | ||||
| Evaluation Method | GraphPad Prism assay | ||||
| MOA of PDC |
Strategies for conjugating BBBpS to drug payloads have been actively explored over recent decades, with the number of new shuttles steadily increasing. Some recent entries, moreover, have shown to be capable not only of carrying drugs into but also removing toxins from the brain, preventing their accumulation. For our part, we have reported that peptide-porphyrin conjugates (PPCs), where a BBBpS and an antiviral porphyrin are covalently linked by an amide bond (amino and carboxyl groups in BBBpS and porphyrin, respectively) can successfully pass the BBB and act against brain-targeting viruses such as HIV. As for the BPB, the literature is scarce, with only a few described examples of peptides able to pass it. Herein, we report our results in developing new PPCs able to penetrate both BPB and BBB and act against ZIKV. The PPC production strategy, involving porphyrin (P), C- or N-terminal conjugation to a BBBpS, has been detailed in a recent publication and is illustrated in Scheme 1 and Scheme S1. In this paper we describe eight PPCs resulting from the combination of four BBBpS and two porphyrins, and their evaluation in terms of barrier crossing and anti-ZIKV activity. One of the conjugates, PP-P1, emerges as particularly effective against ZIKV, having also the ability to translocate across BPB and BBB.
Click to Show/Hide
|
||||
| Description |
The five PPCs that successfully translocated BBB and BPB were evaluated for ZIKV inactivation in vitro, using a plaque assay. Of them, two showed significant activity, namely MP-P5 (IC50 = 25.07 ± 0.05 μM, similar to activity against HIV) and PP-P1 (IC50 = 1.08 ± 0.14 μM). Additionally, a treatment assay performed with MP-P5 and PP-P1 revealed that both PPCs efficiently inhibit ZIKV replication when added 1 h and 7 h post-infection. As observed for HIV, non-conjugated porphyrins did not show activity against ZIKV, reinforcing the claim that BBBpS conjugation is not only critical for BBB/BPB translocation but also for antiviral activity. On the other hand, and somewhat unexpectedly, PP-P1, shown to be inactive in vitro against HIV, emerged as the most active anti-ZIKV conjugate. As described in the literature, the light-independent mechanism of action of porphyrins is based on a direct perturbation of the viral envelope. Porphyrins interact and accumulate on the envelope lipid membrane, causing a decrease in order and a consequent phase alteration that impairs viral entry processes. Since we ensured no-light conditions and no metal cations are coordinated to the porphyrin rings of the PPCsavoiding generation of reactive oxygen speciesthe antiviral light-independent mechanism is the only plausible one. Thus, one may confidently suggest that the PP-P1 specific anti-ZIKV activity is guided by preferential interaction with the ZIKV viral envelope and/or components.
Click to Show/Hide
|
||||
| In Vitro Model | HIV Infection | Human immunodeficiency virus | 12721 | ||
MP-P6 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [85] | ||||
| Indication | Zika virus infection | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Administration Time | 1 h | ||||
| Evaluation Method | GraphPad Prism assay | ||||
| MOA of PDC |
Strategies for conjugating BBBpS to drug payloads have been actively explored over recent decades, with the number of new shuttles steadily increasing. Some recent entries, moreover, have shown to be capable not only of carrying drugs into but also removing toxins from the brain, preventing their accumulation. For our part, we have reported that peptide-porphyrin conjugates (PPCs), where a BBBpS and an antiviral porphyrin are covalently linked by an amide bond (amino and carboxyl groups in BBBpS and porphyrin, respectively) can successfully pass the BBB and act against brain-targeting viruses such as HIV. As for the BPB, the literature is scarce, with only a few described examples of peptides able to pass it. Herein, we report our results in developing new PPCs able to penetrate both BPB and BBB and act against ZIKV. The PPC production strategy, involving porphyrin (P), C- or N-terminal conjugation to a BBBpS, has been detailed in a recent publication and is illustrated in Scheme 1 and Scheme S1. In this paper we describe eight PPCs resulting from the combination of four BBBpS and two porphyrins, and their evaluation in terms of barrier crossing and anti-ZIKV activity. One of the conjugates, PP-P1, emerges as particularly effective against ZIKV, having also the ability to translocate across BPB and BBB.
Click to Show/Hide
|
||||
| Description |
The five PPCs that successfully translocated BBB and BPB were evaluated for ZIKV inactivation in vitro, using a plaque assay. Of them, two showed significant activity, namely MP-P5 (IC50 = 25.07 ± 0.05 μM, similar to activity against HIV) and PP-P1 (IC50 = 1.08 ± 0.14 μM). Additionally, a treatment assay performed with MP-P5 and PP-P1 revealed that both PPCs efficiently inhibit ZIKV replication when added 1 h and 7 h post-infection. As observed for HIV, non-conjugated porphyrins did not show activity against ZIKV, reinforcing the claim that BBBpS conjugation is not only critical for BBB/BPB translocation but also for antiviral activity. On the other hand, and somewhat unexpectedly, PP-P1, shown to be inactive in vitro against HIV, emerged as the most active anti-ZIKV conjugate. As described in the literature, the light-independent mechanism of action of porphyrins is based on a direct perturbation of the viral envelope. Porphyrins interact and accumulate on the envelope lipid membrane, causing a decrease in order and a consequent phase alteration that impairs viral entry processes. Since we ensured no-light conditions and no metal cations are coordinated to the porphyrin rings of the PPCsavoiding generation of reactive oxygen speciesthe antiviral light-independent mechanism is the only plausible one. Thus, one may confidently suggest that the PP-P1 specific anti-ZIKV activity is guided by preferential interaction with the ZIKV viral envelope and/or components.
Click to Show/Hide
|
||||
| In Vitro Model | ZIKV infection | Zika virus | 64320 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [85] | ||||
| Indication | Human immunodeficiency virus infection | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Administration Time | 1 h | ||||
| Evaluation Method | GraphPad Prism assay | ||||
| MOA of PDC |
Strategies for conjugating BBBpS to drug payloads have been actively explored over recent decades, with the number of new shuttles steadily increasing. Some recent entries, moreover, have shown to be capable not only of carrying drugs into but also removing toxins from the brain, preventing their accumulation. For our part, we have reported that peptide-porphyrin conjugates (PPCs), where a BBBpS and an antiviral porphyrin are covalently linked by an amide bond (amino and carboxyl groups in BBBpS and porphyrin, respectively) can successfully pass the BBB and act against brain-targeting viruses such as HIV. As for the BPB, the literature is scarce, with only a few described examples of peptides able to pass it. Herein, we report our results in developing new PPCs able to penetrate both BPB and BBB and act against ZIKV. The PPC production strategy, involving porphyrin (P), C- or N-terminal conjugation to a BBBpS, has been detailed in a recent publication and is illustrated in Scheme 1 and Scheme S1. In this paper we describe eight PPCs resulting from the combination of four BBBpS and two porphyrins, and their evaluation in terms of barrier crossing and anti-ZIKV activity. One of the conjugates, PP-P1, emerges as particularly effective against ZIKV, having also the ability to translocate across BPB and BBB.
Click to Show/Hide
|
||||
| Description |
The five PPCs that successfully translocated BBB and BPB were evaluated for ZIKV inactivation in vitro, using a plaque assay. Of them, two showed significant activity, namely MP-P5 (IC50 = 25.07 ± 0.05 μM, similar to activity against HIV) and PP-P1 (IC50 = 1.08 ± 0.14 μM). Additionally, a treatment assay performed with MP-P5 and PP-P1 revealed that both PPCs efficiently inhibit ZIKV replication when added 1 h and 7 h post-infection. As observed for HIV, non-conjugated porphyrins did not show activity against ZIKV, reinforcing the claim that BBBpS conjugation is not only critical for BBB/BPB translocation but also for antiviral activity. On the other hand, and somewhat unexpectedly, PP-P1, shown to be inactive in vitro against HIV, emerged as the most active anti-ZIKV conjugate. As described in the literature, the light-independent mechanism of action of porphyrins is based on a direct perturbation of the viral envelope. Porphyrins interact and accumulate on the envelope lipid membrane, causing a decrease in order and a consequent phase alteration that impairs viral entry processes. Since we ensured no-light conditions and no metal cations are coordinated to the porphyrin rings of the PPCsavoiding generation of reactive oxygen speciesthe antiviral light-independent mechanism is the only plausible one. Thus, one may confidently suggest that the PP-P1 specific anti-ZIKV activity is guided by preferential interaction with the ZIKV viral envelope and/or components.
Click to Show/Hide
|
||||
| In Vitro Model | HIV Infection | Human immunodeficiency virus | 12721 | ||
PP-P2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [85] | ||||
| Indication | Zika virus infection | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Administration Time | 1 h | ||||
| Evaluation Method | GraphPad Prism assay | ||||
| MOA of PDC |
Strategies for conjugating BBBpS to drug payloads have been actively explored over recent decades, with the number of new shuttles steadily increasing. Some recent entries, moreover, have shown to be capable not only of carrying drugs into but also removing toxins from the brain, preventing their accumulation. For our part, we have reported that peptide-porphyrin conjugates (PPCs), where a BBBpS and an antiviral porphyrin are covalently linked by an amide bond (amino and carboxyl groups in BBBpS and porphyrin, respectively) can successfully pass the BBB and act against brain-targeting viruses such as HIV. As for the BPB, the literature is scarce, with only a few described examples of peptides able to pass it. Herein, we report our results in developing new PPCs able to penetrate both BPB and BBB and act against ZIKV. The PPC production strategy, involving porphyrin (P), C- or N-terminal conjugation to a BBBpS, has been detailed in a recent publication and is illustrated in Scheme 1 and Scheme S1. In this paper we describe eight PPCs resulting from the combination of four BBBpS and two porphyrins, and their evaluation in terms of barrier crossing and anti-ZIKV activity. One of the conjugates, PP-P1, emerges as particularly effective against ZIKV, having also the ability to translocate across BPB and BBB.
Click to Show/Hide
|
||||
| Description |
The five PPCs that successfully translocated BBB and BPB were evaluated for ZIKV inactivation in vitro, using a plaque assay. Of them, two showed significant activity, namely MP-P5 (IC50 = 25.07 ± 0.05 μM, similar to activity against HIV) and PP-P1 (IC50 = 1.08 ± 0.14 μM). Additionally, a treatment assay performed with MP-P5 and PP-P1 revealed that both PPCs efficiently inhibit ZIKV replication when added 1 h and 7 h post-infection. As observed for HIV, non-conjugated porphyrins did not show activity against ZIKV, reinforcing the claim that BBBpS conjugation is not only critical for BBB/BPB translocation but also for antiviral activity. On the other hand, and somewhat unexpectedly, PP-P1, shown to be inactive in vitro against HIV, emerged as the most active anti-ZIKV conjugate. As described in the literature, the light-independent mechanism of action of porphyrins is based on a direct perturbation of the viral envelope. Porphyrins interact and accumulate on the envelope lipid membrane, causing a decrease in order and a consequent phase alteration that impairs viral entry processes. Since we ensured no-light conditions and no metal cations are coordinated to the porphyrin rings of the PPCsavoiding generation of reactive oxygen speciesthe antiviral light-independent mechanism is the only plausible one. Thus, one may confidently suggest that the PP-P1 specific anti-ZIKV activity is guided by preferential interaction with the ZIKV viral envelope and/or components.
Click to Show/Hide
|
||||
| In Vitro Model | ZIKV infection | Zika virus | 64320 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [85] | ||||
| Indication | Human immunodeficiency virus infection | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Administration Time | 1 h | ||||
| Evaluation Method | GraphPad Prism assay | ||||
| MOA of PDC |
Strategies for conjugating BBBpS to drug payloads have been actively explored over recent decades, with the number of new shuttles steadily increasing. Some recent entries, moreover, have shown to be capable not only of carrying drugs into but also removing toxins from the brain, preventing their accumulation. For our part, we have reported that peptide-porphyrin conjugates (PPCs), where a BBBpS and an antiviral porphyrin are covalently linked by an amide bond (amino and carboxyl groups in BBBpS and porphyrin, respectively) can successfully pass the BBB and act against brain-targeting viruses such as HIV. As for the BPB, the literature is scarce, with only a few described examples of peptides able to pass it. Herein, we report our results in developing new PPCs able to penetrate both BPB and BBB and act against ZIKV. The PPC production strategy, involving porphyrin (P), C- or N-terminal conjugation to a BBBpS, has been detailed in a recent publication and is illustrated in Scheme 1 and Scheme S1. In this paper we describe eight PPCs resulting from the combination of four BBBpS and two porphyrins, and their evaluation in terms of barrier crossing and anti-ZIKV activity. One of the conjugates, PP-P1, emerges as particularly effective against ZIKV, having also the ability to translocate across BPB and BBB.
Click to Show/Hide
|
||||
| Description |
The five PPCs that successfully translocated BBB and BPB were evaluated for ZIKV inactivation in vitro, using a plaque assay. Of them, two showed significant activity, namely MP-P5 (IC50 = 25.07 ± 0.05 μM, similar to activity against HIV) and PP-P1 (IC50 = 1.08 ± 0.14 μM). Additionally, a treatment assay performed with MP-P5 and PP-P1 revealed that both PPCs efficiently inhibit ZIKV replication when added 1 h and 7 h post-infection. As observed for HIV, non-conjugated porphyrins did not show activity against ZIKV, reinforcing the claim that BBBpS conjugation is not only critical for BBB/BPB translocation but also for antiviral activity. On the other hand, and somewhat unexpectedly, PP-P1, shown to be inactive in vitro against HIV, emerged as the most active anti-ZIKV conjugate. As described in the literature, the light-independent mechanism of action of porphyrins is based on a direct perturbation of the viral envelope. Porphyrins interact and accumulate on the envelope lipid membrane, causing a decrease in order and a consequent phase alteration that impairs viral entry processes. Since we ensured no-light conditions and no metal cations are coordinated to the porphyrin rings of the PPCsavoiding generation of reactive oxygen speciesthe antiviral light-independent mechanism is the only plausible one. Thus, one may confidently suggest that the PP-P1 specific anti-ZIKV activity is guided by preferential interaction with the ZIKV viral envelope and/or components.
Click to Show/Hide
|
||||
| In Vitro Model | HIV Infection | Human immunodeficiency virus | 12721 | ||
RR-BA [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [89] | ||||
| Indication | Colorectal cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
149.5 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | Ez Cytox cell viability assay kit | ||||
| MOA of PDC |
Here, we developed a cathepsin B inhibitor using a peptide and bile acid for anticancer therapy. Among the peptide sequences that cathepsin B can recognize, positively charged Arg-Arg (RR) was used as a cathepsin-B-specific peptide without any linkers. A hydrophobic bile acid (BA) was coupled via amide linkages to further enhance the inhibitory effects of the peptides. Out of several types of bile acids, ursodeoxycholic acid (UDCA), a safe drug approved by the US Food and Drug Administration (FDA), was used for this study. The nano-sized Arg-Arg and BA conjugate (RR-BA) can be self-assembled in an aqueous solution, forming nanoparticles due to the strong positive charge and the hydrophobicity of bile acids. This pharmaceutical substance has shown promising results in mouse colorectal cancer (CT26) cell treatment and animal experiments. This type of pharmaceutical therapy could be a potential option for cancer treatment.
Click to Show/Hide
|
||||
| Description |
Cell viability and corresponding IC50 values of CT26 tumor cells in the concentration range of 0.1-1000 μM RR-BA, RR peptides, or BA were measured after 24 h and 48 h incubation. The 24 h cytotoxicity IC50 value of RR peptide and BA is NA and RR-BA is 375.5 μM. The 48 h cytotoxicity IC50 value of RR peptide and BA is NA and RR-BA is 149.5 μM. *** Denotes statistically significant differences in a comparison of RR-BA with RR peptides and BA (*** p < 0.001). Results represent means ± S.D.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | CT26 cell | CVCL_7254 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [89] | ||||
| Indication | Colorectal cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
375.5 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | Ez Cytox cell viability assay kit assay | ||||
| MOA of PDC |
Here, we developed a cathepsin B inhibitor using a peptide and bile acid for anticancer therapy. Among the peptide sequences that cathepsin B can recognize, positively charged Arg-Arg (RR) was used as a cathepsin-B-specific peptide without any linkers. A hydrophobic bile acid (BA) was coupled via amide linkages to further enhance the inhibitory effects of the peptides. Out of several types of bile acids, ursodeoxycholic acid (UDCA), a safe drug approved by the US Food and Drug Administration (FDA), was used for this study. The nano-sized Arg-Arg and BA conjugate (RR-BA) can be self-assembled in an aqueous solution, forming nanoparticles due to the strong positive charge and the hydrophobicity of bile acids. This pharmaceutical substance has shown promising results in mouse colorectal cancer (CT26) cell treatment and animal experiments. This type of pharmaceutical therapy could be a potential option for cancer treatment.
Click to Show/Hide
|
||||
| Description |
Cell viability and corresponding IC50 values of CT26 tumor cells in the concentration range of 0.1-1000 μM RR-BA, RR peptides, or BA were measured after 24 h and 48 h incubation. The 24 h cytotoxicity IC50 value of RR peptide and BA is NA and RR-BA is 375.5 μM. The 48 h cytotoxicity IC50 value of RR peptide and BA is NA and RR-BA is 149.5 μM. *** Denotes statistically significant differences in a comparison of RR-BA with RR peptides and BA (*** p < 0.001). Results represent means ± S.D.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | CT26 cell | CVCL_7254 | ||
De Novo Cyclic Peptide (DNCP)-β-naloxamine (NalA) conjugates 4 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [77] | ||||
| Indication | Pain | ||||
| Efficacy Data | Half Maximal Effect Concentration (EC50) |
1.0 ± 0.1 x 10-9 mol
|
|||
| Evaluation Method | Potency/efficacy cAMP assay | ||||
| MOA of PDC |
Here, we exploit the crystal structure of KOR bound to the dual KOR/delta-opioid receptor (DOR) epoxymorphinan opioid agonist MP11048 and use the Rosetta protein and peptide design software to computationally design peptide-small molecule conjugates targeting KOR. Utilizing the high affinity interaction of MP1104 with KOR as an anchor to initiate the design calculations, we seek to computationally design thioether cyclic peptides that interact with ECL2 and ECL3 of the receptor.
Click to Show/Hide
|
||||
| Description |
We obtained four DNCP--NalA conjugates (1-4); the conjugation of DNCP (35) and DNCP (36) was unsuccessful. DNCP--NalA conjugates (1-4) were pharmacologically characterized via radioligand binding and functional cAMP assays to investigate their affinity, potency and efficacy at the mouse KOR. The DNCP--NalA conjugates exhibited affinities in the low nanomolar range, with DNCP--NalA(1) being the strongest binder at KOR with a Ki value of 3.9 nM, as compared to -NalA, which has an affinity Ki of 72 nM. All four DNCP--NalA conjugates were full agonists at KOR (Emax = 89-101%), while -NalA alone was only a partial agonist with an EC50 of 130 nM and Emax of 61%. The most potent conjugates were DNCP--NalA(1) and (4) with EC50 values of 2.0 nM and 1.0 nM, respectively. DNCP--NalA(2) and DNCP--NalA(3) showed agonist activities at KOR with EC50 values of 7.5 and 14 nM in cAMP assay, respectively, while their Ki values were 31 and 24 nM in radioligand binding assay, respectively. Receptor reserve may account for this discrepancy between potency and affinity values of DNCP--NalA(2) and DNCP--NalA(4) which can be observed in functional GPCR assays with opioid receptors30. The higher potency and efficacy of the conjugates at KOR compared to -NalA can be attributed to interactions of the macrocycles with the ECL2 region as described later.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | HEK-293T cell | CVCL_0063 | ||
De Novo Cyclic Peptide (DNCP)-β-naloxamine (NalA) conjugates 2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [77] | ||||
| Indication | Pain | ||||
| Efficacy Data | Half Maximal Effect Concentration (EC50) |
7.5 ± 0.6 x 10-9 mol
|
|||
| Evaluation Method | Potency/efficacy cAMP assay | ||||
| MOA of PDC |
Here, we exploit the crystal structure of KOR bound to the dual KOR/delta-opioid receptor (DOR) epoxymorphinan opioid agonist MP11048 and use the Rosetta protein and peptide design software to computationally design peptide-small molecule conjugates targeting KOR. Utilizing the high affinity interaction of MP1104 with KOR as an anchor to initiate the design calculations, we seek to computationally design thioether cyclic peptides that interact with ECL2 and ECL3 of the receptor.
Click to Show/Hide
|
||||
| Description |
We obtained four DNCP--NalA conjugates (1-4); the conjugation of DNCP (35) and DNCP (36) was unsuccessful. DNCP--NalA conjugates (1-4) were pharmacologically characterized via radioligand binding and functional cAMP assays to investigate their affinity, potency and efficacy at the mouse KOR. The DNCP--NalA conjugates exhibited affinities in the low nanomolar range, with DNCP--NalA(1) being the strongest binder at KOR with a Ki value of 3.9 nM, as compared to -NalA, which has an affinity Ki of 72 nM. All four DNCP--NalA conjugates were full agonists at KOR (Emax = 89-101%), while -NalA alone was only a partial agonist with an EC50 of 130 nM and Emax of 61%. The most potent conjugates were DNCP--NalA(1) and (4) with EC50 values of 2.0 nM and 1.0 nM, respectively. DNCP--NalA(2) and DNCP--NalA(3) showed agonist activities at KOR with EC50 values of 7.5 and 14 nM in cAMP assay, respectively, while their Ki values were 31 and 24 nM in radioligand binding assay, respectively. Receptor reserve may account for this discrepancy between potency and affinity values of DNCP--NalA(2) and DNCP--NalA(4) which can be observed in functional GPCR assays with opioid receptors30. The higher potency and efficacy of the conjugates at KOR compared to -NalA can be attributed to interactions of the macrocycles with the ECL2 region as described later.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | HEK-293T cell | CVCL_0063 | ||
De Novo Cyclic Peptide (DNCP)-β-naloxamine (NalA) conjugates 3 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [77] | ||||
| Indication | Pain | ||||
| Efficacy Data | Half Maximal Effect Concentration (EC50) |
1.4 ± 0.7 x 10-8 mol
|
|||
| Evaluation Method | Potency/efficacy cAMP assay | ||||
| MOA of PDC |
Here, we exploit the crystal structure of KOR bound to the dual KOR/delta-opioid receptor (DOR) epoxymorphinan opioid agonist MP11048 and use the Rosetta protein and peptide design software to computationally design peptide-small molecule conjugates targeting KOR. Utilizing the high affinity interaction of MP1104 with KOR as an anchor to initiate the design calculations, we seek to computationally design thioether cyclic peptides that interact with ECL2 and ECL3 of the receptor.
Click to Show/Hide
|
||||
| Description |
We obtained four DNCP--NalA conjugates (1-4); the conjugation of DNCP (35) and DNCP (36) was unsuccessful. DNCP--NalA conjugates (1-4) were pharmacologically characterized via radioligand binding and functional cAMP assays to investigate their affinity, potency and efficacy at the mouse KOR. The DNCP--NalA conjugates exhibited affinities in the low nanomolar range, with DNCP--NalA(1) being the strongest binder at KOR with a Ki value of 3.9 nM, as compared to -NalA, which has an affinity Ki of 72 nM. All four DNCP--NalA conjugates were full agonists at KOR (Emax = 89-101%), while -NalA alone was only a partial agonist with an EC50 of 130 nM and Emax of 61%. The most potent conjugates were DNCP--NalA(1) and (4) with EC50 values of 2.0 nM and 1.0 nM, respectively. DNCP--NalA(2) and DNCP--NalA(3) showed agonist activities at KOR with EC50 values of 7.5 and 14 nM in cAMP assay, respectively, while their Ki values were 31 and 24 nM in radioligand binding assay, respectively. Receptor reserve may account for this discrepancy between potency and affinity values of DNCP--NalA(2) and DNCP--NalA(4) which can be observed in functional GPCR assays with opioid receptors30. The higher potency and efficacy of the conjugates at KOR compared to -NalA can be attributed to interactions of the macrocycles with the ECL2 region as described later.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | HEK-293T cell | CVCL_0063 | ||
99mTc-HYNIC-CLB-c(NGR) [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [90] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Growth inhibition rate |
30%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 1 µM | ||||
| Evaluation Method | MTT assay | ||||
| Description |
The exposure of cells to PDC resulted in significant growth inhibition at all the concentrations as compared to the drug or the peptide alone. It was observed that nearly 25-fold excess concentration of the drug is required to achieve similar cytotoxicity as obtained on exposure to PDC. The RGD peptide-CLB conjugate has also been reported to show higher growth inhibition in B16F10 cells as compared to the drug or peptide alone.24 The enhanced cytotoxic effect of PDC even at lower levels indicates clear advantage in reducing the systemic exposure and side effects of the drug.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [90] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Growth inhibition rate |
33%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 5 µM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Although RNT with 177Lu-DOTATATE/PSMA is known as a novel and effective therapy option for cancer that significantly improves the quality of life and survival of patients, it may have acute or chronic side effects. Therefore, any method that can ameliorate these side effects is useful in the RNT process. For this purpose, a few clinical studies have reported that antioxidants as free radical scavengers such as amifostine and vitamins C and E can reduce radioiodine-related side effects, particularly in salivary glands in thyroid cancer patients.
Click to Show/Hide
|
||||
| Description |
The exposure of cells to PDC resulted in significant growth inhibition at all the concentrations as compared to the drug or the peptide alone. It was observed that nearly 25-fold excess concentration of the drug is required to achieve similar cytotoxicity as obtained on exposure to PDC. The RGD peptide-CLB conjugate has also been reported to show higher growth inhibition in B16F10 cells as compared to the drug or peptide alone.24 The enhanced cytotoxic effect of PDC even at lower levels indicates clear advantage in reducing the systemic exposure and side effects of the drug.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [90] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Growth inhibition rate |
40%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 10 µM | ||||
| Evaluation Method | MTT assay | ||||
| Description |
The exposure of cells to PDC resulted in significant growth inhibition at all the concentrations as compared to the drug or the peptide alone. It was observed that nearly 25-fold excess concentration of the drug is required to achieve similar cytotoxicity as obtained on exposure to PDC. The RGD peptide-CLB conjugate has also been reported to show higher growth inhibition in B16F10 cells as compared to the drug or peptide alone.24 The enhanced cytotoxic effect of PDC even at lower levels indicates clear advantage in reducing the systemic exposure and side effects of the drug.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [90] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Growth inhibition rate |
55%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 25 µM | ||||
| Evaluation Method | MTT assay | ||||
| Description |
The exposure of cells to PDC resulted in significant growth inhibition at all the concentrations as compared to the drug or the peptide alone. It was observed that nearly 25-fold excess concentration of the drug is required to achieve similar cytotoxicity as obtained on exposure to PDC. The RGD peptide-CLB conjugate has also been reported to show higher growth inhibition in B16F10 cells as compared to the drug or peptide alone.24 The enhanced cytotoxic effect of PDC even at lower levels indicates clear advantage in reducing the systemic exposure and side effects of the drug.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [90] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Growth inhibition rate |
75%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 50 µM | ||||
| Evaluation Method | MTT assay | ||||
| Description |
The exposure of cells to PDC resulted in significant growth inhibition at all the concentrations as compared to the drug or the peptide alone. It was observed that nearly 25-fold excess concentration of the drug is required to achieve similar cytotoxicity as obtained on exposure to PDC. The RGD peptide-CLB conjugate has also been reported to show higher growth inhibition in B16F10 cells as compared to the drug or peptide alone.24 The enhanced cytotoxic effect of PDC even at lower levels indicates clear advantage in reducing the systemic exposure and side effects of the drug.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
PDC-CPT1 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [91] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
38%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 5 µM | ||||
| Evaluation Method | MTT assay | ||||
| Description |
Antiproliferative results showed that PTX1 inhibited cell proliferation by 18.7%. The anti-proliferative activity of CPT1 was diminished by 1.9-fold as compared to CPT whereas the activity of CPT2 was comparable to CPT, since CPT2 reduced the cell viability to 61%.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
Linear arginineglycineaspartic acid (RGD) - Naproxen conjugate 3 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [92] | ||||
| Indication | Invasive breast carcinoma | ||||
| Efficacy Data | Cell inhibition rate |
0%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 100 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The two tripeptide sequences, arginine-glycine-aspartic acid (RGD) and asparagine-glycine-arginine (NGR) motifs have been identified based on phage display studies and they have been used widely in the field of targeted drug delivery. RGD is a well-known peptide sequence for targeting integrin receptors and can bind to avβ3 and avβ5 integrin receptors subunits, which are overexpressed in the angiogenesis process of cancer cells. Because av integrin is overexpressed on the surface of cancer cells, an integrin ligand can be used as a targeting system for cancer treatment. RGD peptide conjugated with cytotoxic agents (RGD-drug conjugates) is likely to exhibit a tumor-targeting and thus antiangiogenic synergetic effect. During the last few years, a number of RGD-cytotoxic drugs have been developed and showed promising activities in vitro and in vivo. Conjugation of camptothecin with RGD is an example for improving the therapeutic index of an antitumoral drug.[20c] Synthesis of dimeric RGD peptide-paclitaxel conjugate is another successful example of targeted drug delivery. Other motif that has been used for tumor targeting is NGR tripeptide. This sequence can bind to CD13 that is specially overexpressed in tumor blood vessels and is involved in angiogenesis, invasion, and metastasis. Because RGD is a peptide tag which targets the process of angiogenesis and NGR also targets tumor blood vessels, we decided to synthesize the conjugated forms of two famous NSAIDs, naproxen, and ibuprofen, with these two tripeptides. To investigate the impact of possible steric hindrance due to the attachment of the drug to the peptide, a linear six-carbon segment (hexanoic acid) was also used as a spacer.
Click to Show/Hide
|
||||
| Description |
NGR conjugate forms of both ibuprofen and naproxen showed improved activity when they were tested against SKOV-3 cell line which is positive for APN/CD13. Interestingly, both ibuprofen and naproxen show increased activity against this cell line when a six-carbon spacer is used for their conjugation to NGR. This is probably due to the less steric hindrance for NGR interaction with its binding protein on the cell surface. Ibuprofen-spacer-NGR and naproxen-spacer-NGR showed the same pattern of increased activity against HT-1080 tumor cells which this cell line show high expression of CD13. Surprisingly, NGR conjugates of both drugs without spacer did not show improved activity compared to the nonconjugated forms against this cell line. Therefore, it could be speculated that HT-1080 cells are more sensitive to the steric hindrance for interaction between NGR and its binding protein. None of the conjugates of ibuprofen and naproxen with or without spacer showed significantly improved activity against A2780 (as a tumor cell with normal RGD-binding protein) and OVCAR3. Therefore, it could be inferred again that the RGD motif is not qualified as a targeting tool for ibuprofen and naproxen.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [92] | ||||
| Efficacy Data | Cell inhibition rate |
0%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 100 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The two tripeptide sequences, arginine-glycine-aspartic acid (RGD) and asparagine-glycine-arginine (NGR) motifs have been identified based on phage display studies and they have been used widely in the field of targeted drug delivery. RGD is a well-known peptide sequence for targeting integrin receptors and can bind to avβ3 and avβ5 integrin receptors subunits, which are overexpressed in the angiogenesis process of cancer cells. Because av integrin is overexpressed on the surface of cancer cells, an integrin ligand can be used as a targeting system for cancer treatment. RGD peptide conjugated with cytotoxic agents (RGD-drug conjugates) is likely to exhibit a tumor-targeting and thus antiangiogenic synergetic effect. During the last few years, a number of RGD-cytotoxic drugs have been developed and showed promising activities in vitro and in vivo. Conjugation of camptothecin with RGD is an example for improving the therapeutic index of an antitumoral drug.[20c] Synthesis of dimeric RGD peptide-paclitaxel conjugate is another successful example of targeted drug delivery. Other motif that has been used for tumor targeting is NGR tripeptide. This sequence can bind to CD13 that is specially overexpressed in tumor blood vessels and is involved in angiogenesis, invasion, and metastasis. Because RGD is a peptide tag which targets the process of angiogenesis and NGR also targets tumor blood vessels, we decided to synthesize the conjugated forms of two famous NSAIDs, naproxen, and ibuprofen, with these two tripeptides. To investigate the impact of possible steric hindrance due to the attachment of the drug to the peptide, a linear six-carbon segment (hexanoic acid) was also used as a spacer.
Click to Show/Hide
|
||||
| Description |
NGR conjugate forms of both ibuprofen and naproxen showed improved activity when they were tested against SKOV-3 cell line which is positive for APN/CD13. Interestingly, both ibuprofen and naproxen show increased activity against this cell line when a six-carbon spacer is used for their conjugation to NGR. This is probably due to the less steric hindrance for NGR interaction with its binding protein on the cell surface. Ibuprofen-spacer-NGR and naproxen-spacer-NGR showed the same pattern of increased activity against HT-1080 tumor cells which this cell line show high expression of CD13. Surprisingly, NGR conjugates of both drugs without spacer did not show improved activity compared to the nonconjugated forms against this cell line. Therefore, it could be speculated that HT-1080 cells are more sensitive to the steric hindrance for interaction between NGR and its binding protein. None of the conjugates of ibuprofen and naproxen with or without spacer showed significantly improved activity against A2780 (as a tumor cell with normal RGD-binding protein) and OVCAR3. Therefore, it could be inferred again that the RGD motif is not qualified as a targeting tool for ibuprofen and naproxen.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human fibroblast cells | Homo sapiens | ||
| Experiment 3 Reporting the Activity Data of This PDC | [92] | ||||
| Indication | Ovarian endometrioid adenocarcinoma | ||||
| Efficacy Data | Cell inhibition rate |
28 ± 2.3%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 100 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The two tripeptide sequences, arginine-glycine-aspartic acid (RGD) and asparagine-glycine-arginine (NGR) motifs have been identified based on phage display studies and they have been used widely in the field of targeted drug delivery. RGD is a well-known peptide sequence for targeting integrin receptors and can bind to avβ3 and avβ5 integrin receptors subunits, which are overexpressed in the angiogenesis process of cancer cells. Because av integrin is overexpressed on the surface of cancer cells, an integrin ligand can be used as a targeting system for cancer treatment. RGD peptide conjugated with cytotoxic agents (RGD-drug conjugates) is likely to exhibit a tumor-targeting and thus antiangiogenic synergetic effect. During the last few years, a number of RGD-cytotoxic drugs have been developed and showed promising activities in vitro and in vivo. Conjugation of camptothecin with RGD is an example for improving the therapeutic index of an antitumoral drug.[20c] Synthesis of dimeric RGD peptide-paclitaxel conjugate is another successful example of targeted drug delivery. Other motif that has been used for tumor targeting is NGR tripeptide. This sequence can bind to CD13 that is specially overexpressed in tumor blood vessels and is involved in angiogenesis, invasion, and metastasis. Because RGD is a peptide tag which targets the process of angiogenesis and NGR also targets tumor blood vessels, we decided to synthesize the conjugated forms of two famous NSAIDs, naproxen, and ibuprofen, with these two tripeptides. To investigate the impact of possible steric hindrance due to the attachment of the drug to the peptide, a linear six-carbon segment (hexanoic acid) was also used as a spacer.
Click to Show/Hide
|
||||
| Description |
NGR conjugate forms of both ibuprofen and naproxen showed improved activity when they were tested against SKOV-3 cell line which is positive for APN/CD13. Interestingly, both ibuprofen and naproxen show increased activity against this cell line when a six-carbon spacer is used for their conjugation to NGR. This is probably due to the less steric hindrance for NGR interaction with its binding protein on the cell surface. Ibuprofen-spacer-NGR and naproxen-spacer-NGR showed the same pattern of increased activity against HT-1080 tumor cells which this cell line show high expression of CD13. Surprisingly, NGR conjugates of both drugs without spacer did not show improved activity compared to the nonconjugated forms against this cell line. Therefore, it could be speculated that HT-1080 cells are more sensitive to the steric hindrance for interaction between NGR and its binding protein. None of the conjugates of ibuprofen and naproxen with or without spacer showed significantly improved activity against A2780 (as a tumor cell with normal RGD-binding protein) and OVCAR3. Therefore, it could be inferred again that the RGD motif is not qualified as a targeting tool for ibuprofen and naproxen.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [92] | ||||
| Indication | Ovarian serous adenocarcinoma | ||||
| Efficacy Data | Cell inhibition rate |
41 ± 2.0%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 100 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The two tripeptide sequences, arginine-glycine-aspartic acid (RGD) and asparagine-glycine-arginine (NGR) motifs have been identified based on phage display studies and they have been used widely in the field of targeted drug delivery. RGD is a well-known peptide sequence for targeting integrin receptors and can bind to avβ3 and avβ5 integrin receptors subunits, which are overexpressed in the angiogenesis process of cancer cells. Because av integrin is overexpressed on the surface of cancer cells, an integrin ligand can be used as a targeting system for cancer treatment. RGD peptide conjugated with cytotoxic agents (RGD-drug conjugates) is likely to exhibit a tumor-targeting and thus antiangiogenic synergetic effect. During the last few years, a number of RGD-cytotoxic drugs have been developed and showed promising activities in vitro and in vivo. Conjugation of camptothecin with RGD is an example for improving the therapeutic index of an antitumoral drug.[20c] Synthesis of dimeric RGD peptide-paclitaxel conjugate is another successful example of targeted drug delivery. Other motif that has been used for tumor targeting is NGR tripeptide. This sequence can bind to CD13 that is specially overexpressed in tumor blood vessels and is involved in angiogenesis, invasion, and metastasis. Because RGD is a peptide tag which targets the process of angiogenesis and NGR also targets tumor blood vessels, we decided to synthesize the conjugated forms of two famous NSAIDs, naproxen, and ibuprofen, with these two tripeptides. To investigate the impact of possible steric hindrance due to the attachment of the drug to the peptide, a linear six-carbon segment (hexanoic acid) was also used as a spacer.
Click to Show/Hide
|
||||
| Description |
NGR conjugate forms of both ibuprofen and naproxen showed improved activity when they were tested against SKOV-3 cell line which is positive for APN/CD13. Interestingly, both ibuprofen and naproxen show increased activity against this cell line when a six-carbon spacer is used for their conjugation to NGR. This is probably due to the less steric hindrance for NGR interaction with its binding protein on the cell surface. Ibuprofen-spacer-NGR and naproxen-spacer-NGR showed the same pattern of increased activity against HT-1080 tumor cells which this cell line show high expression of CD13. Surprisingly, NGR conjugates of both drugs without spacer did not show improved activity compared to the nonconjugated forms against this cell line. Therefore, it could be speculated that HT-1080 cells are more sensitive to the steric hindrance for interaction between NGR and its binding protein. None of the conjugates of ibuprofen and naproxen with or without spacer showed significantly improved activity against A2780 (as a tumor cell with normal RGD-binding protein) and OVCAR3. Therefore, it could be inferred again that the RGD motif is not qualified as a targeting tool for ibuprofen and naproxen.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous adenocarcinoma | OVCAR-3 cell | CVCL_0465 | ||
Linear asparagineglycinearginine (NGR) 1 - Naproxen conjugate 1 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [92] | ||||
| Indication | Invasive breast carcinoma | ||||
| Efficacy Data | Cell inhibition rate |
0%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 100 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The two tripeptide sequences, arginine-glycine-aspartic acid (RGD) and asparagine-glycine-arginine (NGR) motifs have been identified based on phage display studies and they have been used widely in the field of targeted drug delivery. RGD is a well-known peptide sequence for targeting integrin receptors and can bind to avβ3 and avβ5 integrin receptors subunits, which are overexpressed in the angiogenesis process of cancer cells. Because av integrin is overexpressed on the surface of cancer cells, an integrin ligand can be used as a targeting system for cancer treatment. RGD peptide conjugated with cytotoxic agents (RGD-drug conjugates) is likely to exhibit a tumor-targeting and thus antiangiogenic synergetic effect. During the last few years, a number of RGD-cytotoxic drugs have been developed and showed promising activities in vitro and in vivo. Conjugation of camptothecin with RGD is an example for improving the therapeutic index of an antitumoral drug.[20c] Synthesis of dimeric RGD peptide-paclitaxel conjugate is another successful example of targeted drug delivery. Other motif that has been used for tumor targeting is NGR tripeptide. This sequence can bind to CD13 that is specially overexpressed in tumor blood vessels and is involved in angiogenesis, invasion, and metastasis. Because RGD is a peptide tag which targets the process of angiogenesis and NGR also targets tumor blood vessels, we decided to synthesize the conjugated forms of two famous NSAIDs, naproxen, and ibuprofen, with these two tripeptides. To investigate the impact of possible steric hindrance due to the attachment of the drug to the peptide, a linear six-carbon segment (hexanoic acid) was also used as a spacer.
Click to Show/Hide
|
||||
| Description |
NGR conjugate forms of both ibuprofen and naproxen showed improved activity when they were tested against SKOV-3 cell line which is positive for APN/CD13. Interestingly, both ibuprofen and naproxen show increased activity against this cell line when a six-carbon spacer is used for their conjugation to NGR. This is probably due to the less steric hindrance for NGR interaction with its binding protein on the cell surface. Ibuprofen-spacer-NGR and naproxen-spacer-NGR showed the same pattern of increased activity against HT-1080 tumor cells which this cell line show high expression of CD13. Surprisingly, NGR conjugates of both drugs without spacer did not show improved activity compared to the nonconjugated forms against this cell line. Therefore, it could be speculated that HT-1080 cells are more sensitive to the steric hindrance for interaction between NGR and its binding protein. None of the conjugates of ibuprofen and naproxen with or without spacer showed significantly improved activity against A2780 (as a tumor cell with normal RGD-binding protein) and OVCAR3. Therefore, it could be inferred again that the RGD motif is not qualified as a targeting tool for ibuprofen and naproxen.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [92] | ||||
| Efficacy Data | Cell inhibition rate |
0%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 100 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The two tripeptide sequences, arginine-glycine-aspartic acid (RGD) and asparagine-glycine-arginine (NGR) motifs have been identified based on phage display studies and they have been used widely in the field of targeted drug delivery. RGD is a well-known peptide sequence for targeting integrin receptors and can bind to avβ3 and avβ5 integrin receptors subunits, which are overexpressed in the angiogenesis process of cancer cells. Because av integrin is overexpressed on the surface of cancer cells, an integrin ligand can be used as a targeting system for cancer treatment. RGD peptide conjugated with cytotoxic agents (RGD-drug conjugates) is likely to exhibit a tumor-targeting and thus antiangiogenic synergetic effect. During the last few years, a number of RGD-cytotoxic drugs have been developed and showed promising activities in vitro and in vivo. Conjugation of camptothecin with RGD is an example for improving the therapeutic index of an antitumoral drug.[20c] Synthesis of dimeric RGD peptide-paclitaxel conjugate is another successful example of targeted drug delivery. Other motif that has been used for tumor targeting is NGR tripeptide. This sequence can bind to CD13 that is specially overexpressed in tumor blood vessels and is involved in angiogenesis, invasion, and metastasis. Because RGD is a peptide tag which targets the process of angiogenesis and NGR also targets tumor blood vessels, we decided to synthesize the conjugated forms of two famous NSAIDs, naproxen, and ibuprofen, with these two tripeptides. To investigate the impact of possible steric hindrance due to the attachment of the drug to the peptide, a linear six-carbon segment (hexanoic acid) was also used as a spacer.
Click to Show/Hide
|
||||
| Description |
NGR conjugate forms of both ibuprofen and naproxen showed improved activity when they were tested against SKOV-3 cell line which is positive for APN/CD13. Interestingly, both ibuprofen and naproxen show increased activity against this cell line when a six-carbon spacer is used for their conjugation to NGR. This is probably due to the less steric hindrance for NGR interaction with its binding protein on the cell surface. Ibuprofen-spacer-NGR and naproxen-spacer-NGR showed the same pattern of increased activity against HT-1080 tumor cells which this cell line show high expression of CD13. Surprisingly, NGR conjugates of both drugs without spacer did not show improved activity compared to the nonconjugated forms against this cell line. Therefore, it could be speculated that HT-1080 cells are more sensitive to the steric hindrance for interaction between NGR and its binding protein. None of the conjugates of ibuprofen and naproxen with or without spacer showed significantly improved activity against A2780 (as a tumor cell with normal RGD-binding protein) and OVCAR3. Therefore, it could be inferred again that the RGD motif is not qualified as a targeting tool for ibuprofen and naproxen.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human fibroblast cells | Homo sapiens | ||
| Experiment 3 Reporting the Activity Data of This PDC | [92] | ||||
| Indication | Fibrosarcoma | ||||
| Efficacy Data | Cell inhibition rate |
32 ± 4.1%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 100 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The two tripeptide sequences, arginine-glycine-aspartic acid (RGD) and asparagine-glycine-arginine (NGR) motifs have been identified based on phage display studies and they have been used widely in the field of targeted drug delivery. RGD is a well-known peptide sequence for targeting integrin receptors and can bind to avβ3 and avβ5 integrin receptors subunits, which are overexpressed in the angiogenesis process of cancer cells. Because av integrin is overexpressed on the surface of cancer cells, an integrin ligand can be used as a targeting system for cancer treatment. RGD peptide conjugated with cytotoxic agents (RGD-drug conjugates) is likely to exhibit a tumor-targeting and thus antiangiogenic synergetic effect. During the last few years, a number of RGD-cytotoxic drugs have been developed and showed promising activities in vitro and in vivo. Conjugation of camptothecin with RGD is an example for improving the therapeutic index of an antitumoral drug.[20c] Synthesis of dimeric RGD peptide-paclitaxel conjugate is another successful example of targeted drug delivery. Other motif that has been used for tumor targeting is NGR tripeptide. This sequence can bind to CD13 that is specially overexpressed in tumor blood vessels and is involved in angiogenesis, invasion, and metastasis. Because RGD is a peptide tag which targets the process of angiogenesis and NGR also targets tumor blood vessels, we decided to synthesize the conjugated forms of two famous NSAIDs, naproxen, and ibuprofen, with these two tripeptides. To investigate the impact of possible steric hindrance due to the attachment of the drug to the peptide, a linear six-carbon segment (hexanoic acid) was also used as a spacer.
Click to Show/Hide
|
||||
| Description |
NGR conjugate forms of both ibuprofen and naproxen showed improved activity when they were tested against SKOV-3 cell line which is positive for APN/CD13. Interestingly, both ibuprofen and naproxen show increased activity against this cell line when a six-carbon spacer is used for their conjugation to NGR. This is probably due to the less steric hindrance for NGR interaction with its binding protein on the cell surface. Ibuprofen-spacer-NGR and naproxen-spacer-NGR showed the same pattern of increased activity against HT-1080 tumor cells which this cell line show high expression of CD13. Surprisingly, NGR conjugates of both drugs without spacer did not show improved activity compared to the nonconjugated forms against this cell line. Therefore, it could be speculated that HT-1080 cells are more sensitive to the steric hindrance for interaction between NGR and its binding protein. None of the conjugates of ibuprofen and naproxen with or without spacer showed significantly improved activity against A2780 (as a tumor cell with normal RGD-binding protein) and OVCAR3. Therefore, it could be inferred again that the RGD motif is not qualified as a targeting tool for ibuprofen and naproxen.
Click to Show/Hide
|
||||
| In Vitro Model | Fibrosarcoma | HT-1080 cell | CVCL_0317 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [92] | ||||
| Indication | Ovarian endometrioid adenocarcinoma | ||||
| Efficacy Data | Cell inhibition rate |
48 ± 1.1%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 100 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The two tripeptide sequences, arginine-glycine-aspartic acid (RGD) and asparagine-glycine-arginine (NGR) motifs have been identified based on phage display studies and they have been used widely in the field of targeted drug delivery. RGD is a well-known peptide sequence for targeting integrin receptors and can bind to avβ3 and avβ5 integrin receptors subunits, which are overexpressed in the angiogenesis process of cancer cells. Because av integrin is overexpressed on the surface of cancer cells, an integrin ligand can be used as a targeting system for cancer treatment. RGD peptide conjugated with cytotoxic agents (RGD-drug conjugates) is likely to exhibit a tumor-targeting and thus antiangiogenic synergetic effect. During the last few years, a number of RGD-cytotoxic drugs have been developed and showed promising activities in vitro and in vivo. Conjugation of camptothecin with RGD is an example for improving the therapeutic index of an antitumoral drug.[20c] Synthesis of dimeric RGD peptide-paclitaxel conjugate is another successful example of targeted drug delivery. Other motif that has been used for tumor targeting is NGR tripeptide. This sequence can bind to CD13 that is specially overexpressed in tumor blood vessels and is involved in angiogenesis, invasion, and metastasis. Because RGD is a peptide tag which targets the process of angiogenesis and NGR also targets tumor blood vessels, we decided to synthesize the conjugated forms of two famous NSAIDs, naproxen, and ibuprofen, with these two tripeptides. To investigate the impact of possible steric hindrance due to the attachment of the drug to the peptide, a linear six-carbon segment (hexanoic acid) was also used as a spacer.
Click to Show/Hide
|
||||
| Description |
NGR conjugate forms of both ibuprofen and naproxen showed improved activity when they were tested against SKOV-3 cell line which is positive for APN/CD13. Interestingly, both ibuprofen and naproxen show increased activity against this cell line when a six-carbon spacer is used for their conjugation to NGR. This is probably due to the less steric hindrance for NGR interaction with its binding protein on the cell surface. Ibuprofen-spacer-NGR and naproxen-spacer-NGR showed the same pattern of increased activity against HT-1080 tumor cells which this cell line show high expression of CD13. Surprisingly, NGR conjugates of both drugs without spacer did not show improved activity compared to the nonconjugated forms against this cell line. Therefore, it could be speculated that HT-1080 cells are more sensitive to the steric hindrance for interaction between NGR and its binding protein. None of the conjugates of ibuprofen and naproxen with or without spacer showed significantly improved activity against A2780 (as a tumor cell with normal RGD-binding protein) and OVCAR3. Therefore, it could be inferred again that the RGD motif is not qualified as a targeting tool for ibuprofen and naproxen.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [92] | ||||
| Indication | Ovarian serous cystadenocarcinoma | ||||
| Efficacy Data | Cell inhibition rate |
53 ± 1.4%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 100 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The two tripeptide sequences, arginine-glycine-aspartic acid (RGD) and asparagine-glycine-arginine (NGR) motifs have been identified based on phage display studies and they have been used widely in the field of targeted drug delivery. RGD is a well-known peptide sequence for targeting integrin receptors and can bind to avβ3 and avβ5 integrin receptors subunits, which are overexpressed in the angiogenesis process of cancer cells. Because av integrin is overexpressed on the surface of cancer cells, an integrin ligand can be used as a targeting system for cancer treatment. RGD peptide conjugated with cytotoxic agents (RGD-drug conjugates) is likely to exhibit a tumor-targeting and thus antiangiogenic synergetic effect. During the last few years, a number of RGD-cytotoxic drugs have been developed and showed promising activities in vitro and in vivo. Conjugation of camptothecin with RGD is an example for improving the therapeutic index of an antitumoral drug.[20c] Synthesis of dimeric RGD peptide-paclitaxel conjugate is another successful example of targeted drug delivery. Other motif that has been used for tumor targeting is NGR tripeptide. This sequence can bind to CD13 that is specially overexpressed in tumor blood vessels and is involved in angiogenesis, invasion, and metastasis. Because RGD is a peptide tag which targets the process of angiogenesis and NGR also targets tumor blood vessels, we decided to synthesize the conjugated forms of two famous NSAIDs, naproxen, and ibuprofen, with these two tripeptides. To investigate the impact of possible steric hindrance due to the attachment of the drug to the peptide, a linear six-carbon segment (hexanoic acid) was also used as a spacer.
Click to Show/Hide
|
||||
| Description |
NGR conjugate forms of both ibuprofen and naproxen showed improved activity when they were tested against SKOV-3 cell line which is positive for APN/CD13. Interestingly, both ibuprofen and naproxen show increased activity against this cell line when a six-carbon spacer is used for their conjugation to NGR. This is probably due to the less steric hindrance for NGR interaction with its binding protein on the cell surface. Ibuprofen-spacer-NGR and naproxen-spacer-NGR showed the same pattern of increased activity against HT-1080 tumor cells which this cell line show high expression of CD13. Surprisingly, NGR conjugates of both drugs without spacer did not show improved activity compared to the nonconjugated forms against this cell line. Therefore, it could be speculated that HT-1080 cells are more sensitive to the steric hindrance for interaction between NGR and its binding protein. None of the conjugates of ibuprofen and naproxen with or without spacer showed significantly improved activity against A2780 (as a tumor cell with normal RGD-binding protein) and OVCAR3. Therefore, it could be inferred again that the RGD motif is not qualified as a targeting tool for ibuprofen and naproxen.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
Linear arginineglycineaspartic acid (RGD) - Ibuprofen conjugate 1 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [92] | ||||
| Indication | Invasive breast carcinoma | ||||
| Efficacy Data | Cell inhibition rate |
0%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 100 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The two tripeptide sequences, arginine-glycine-aspartic acid (RGD) and asparagine-glycine-arginine (NGR) motifs have been identified based on phage display studies and they have been used widely in the field of targeted drug delivery. RGD is a well-known peptide sequence for targeting integrin receptors and can bind to avβ3 and avβ5 integrin receptors subunits, which are overexpressed in the angiogenesis process of cancer cells. Because av integrin is overexpressed on the surface of cancer cells, an integrin ligand can be used as a targeting system for cancer treatment. RGD peptide conjugated with cytotoxic agents (RGD-drug conjugates) is likely to exhibit a tumor-targeting and thus antiangiogenic synergetic effect. During the last few years, a number of RGD-cytotoxic drugs have been developed and showed promising activities in vitro and in vivo. Conjugation of camptothecin with RGD is an example for improving the therapeutic index of an antitumoral drug.[20c] Synthesis of dimeric RGD peptide-paclitaxel conjugate is another successful example of targeted drug delivery. Other motif that has been used for tumor targeting is NGR tripeptide. This sequence can bind to CD13 that is specially overexpressed in tumor blood vessels and is involved in angiogenesis, invasion, and metastasis. Because RGD is a peptide tag which targets the process of angiogenesis and NGR also targets tumor blood vessels, we decided to synthesize the conjugated forms of two famous NSAIDs, naproxen, and ibuprofen, with these two tripeptides. To investigate the impact of possible steric hindrance due to the attachment of the drug to the peptide, a linear six-carbon segment (hexanoic acid) was also used as a spacer.
Click to Show/Hide
|
||||
| Description |
NGR conjugate forms of both ibuprofen and naproxen showed improved activity when they were tested against SKOV-3 cell line which is positive for APN/CD13. Interestingly, both ibuprofen and naproxen show increased activity against this cell line when a six-carbon spacer is used for their conjugation to NGR. This is probably due to the less steric hindrance for NGR interaction with its binding protein on the cell surface. Ibuprofen-spacer-NGR and naproxen-spacer-NGR showed the same pattern of increased activity against HT-1080 tumor cells which this cell line show high expression of CD13. Surprisingly, NGR conjugates of both drugs without spacer did not show improved activity compared to the nonconjugated forms against this cell line. Therefore, it could be speculated that HT-1080 cells are more sensitive to the steric hindrance for interaction between NGR and its binding protein. None of the conjugates of ibuprofen and naproxen with or without spacer showed significantly improved activity against A2780 (as a tumor cell with normal RGD-binding protein) and OVCAR3. Therefore, it could be inferred again that the RGD motif is not qualified as a targeting tool for ibuprofen and naproxen.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [92] | ||||
| Efficacy Data | Cell inhibition rate |
0%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 100 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The two tripeptide sequences, arginine-glycine-aspartic acid (RGD) and asparagine-glycine-arginine (NGR) motifs have been identified based on phage display studies and they have been used widely in the field of targeted drug delivery. RGD is a well-known peptide sequence for targeting integrin receptors and can bind to avβ3 and avβ5 integrin receptors subunits, which are overexpressed in the angiogenesis process of cancer cells. Because av integrin is overexpressed on the surface of cancer cells, an integrin ligand can be used as a targeting system for cancer treatment. RGD peptide conjugated with cytotoxic agents (RGD-drug conjugates) is likely to exhibit a tumor-targeting and thus antiangiogenic synergetic effect. During the last few years, a number of RGD-cytotoxic drugs have been developed and showed promising activities in vitro and in vivo. Conjugation of camptothecin with RGD is an example for improving the therapeutic index of an antitumoral drug.[20c] Synthesis of dimeric RGD peptide-paclitaxel conjugate is another successful example of targeted drug delivery. Other motif that has been used for tumor targeting is NGR tripeptide. This sequence can bind to CD13 that is specially overexpressed in tumor blood vessels and is involved in angiogenesis, invasion, and metastasis. Because RGD is a peptide tag which targets the process of angiogenesis and NGR also targets tumor blood vessels, we decided to synthesize the conjugated forms of two famous NSAIDs, naproxen, and ibuprofen, with these two tripeptides. To investigate the impact of possible steric hindrance due to the attachment of the drug to the peptide, a linear six-carbon segment (hexanoic acid) was also used as a spacer.
Click to Show/Hide
|
||||
| Description |
NGR conjugate forms of both ibuprofen and naproxen showed improved activity when they were tested against SKOV-3 cell line which is positive for APN/CD13. Interestingly, both ibuprofen and naproxen show increased activity against this cell line when a six-carbon spacer is used for their conjugation to NGR. This is probably due to the less steric hindrance for NGR interaction with its binding protein on the cell surface. Ibuprofen-spacer-NGR and naproxen-spacer-NGR showed the same pattern of increased activity against HT-1080 tumor cells which this cell line show high expression of CD13. Surprisingly, NGR conjugates of both drugs without spacer did not show improved activity compared to the nonconjugated forms against this cell line. Therefore, it could be speculated that HT-1080 cells are more sensitive to the steric hindrance for interaction between NGR and its binding protein. None of the conjugates of ibuprofen and naproxen with or without spacer showed significantly improved activity against A2780 (as a tumor cell with normal RGD-binding protein) and OVCAR3. Therefore, it could be inferred again that the RGD motif is not qualified as a targeting tool for ibuprofen and naproxen.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human fibroblast cells | Homo sapiens | ||
| Experiment 3 Reporting the Activity Data of This PDC | [92] | ||||
| Indication | Ovarian serous adenocarcinoma | ||||
| Efficacy Data | Cell inhibition rate |
11 ± 4.1%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 100 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The two tripeptide sequences, arginine-glycine-aspartic acid (RGD) and asparagine-glycine-arginine (NGR) motifs have been identified based on phage display studies and they have been used widely in the field of targeted drug delivery. RGD is a well-known peptide sequence for targeting integrin receptors and can bind to avβ3 and avβ5 integrin receptors subunits, which are overexpressed in the angiogenesis process of cancer cells. Because av integrin is overexpressed on the surface of cancer cells, an integrin ligand can be used as a targeting system for cancer treatment. RGD peptide conjugated with cytotoxic agents (RGD-drug conjugates) is likely to exhibit a tumor-targeting and thus antiangiogenic synergetic effect. During the last few years, a number of RGD-cytotoxic drugs have been developed and showed promising activities in vitro and in vivo. Conjugation of camptothecin with RGD is an example for improving the therapeutic index of an antitumoral drug.[20c] Synthesis of dimeric RGD peptide-paclitaxel conjugate is another successful example of targeted drug delivery. Other motif that has been used for tumor targeting is NGR tripeptide. This sequence can bind to CD13 that is specially overexpressed in tumor blood vessels and is involved in angiogenesis, invasion, and metastasis. Because RGD is a peptide tag which targets the process of angiogenesis and NGR also targets tumor blood vessels, we decided to synthesize the conjugated forms of two famous NSAIDs, naproxen, and ibuprofen, with these two tripeptides. To investigate the impact of possible steric hindrance due to the attachment of the drug to the peptide, a linear six-carbon segment (hexanoic acid) was also used as a spacer.
Click to Show/Hide
|
||||
| Description |
NGR conjugate forms of both ibuprofen and naproxen showed improved activity when they were tested against SKOV-3 cell line which is positive for APN/CD13. Interestingly, both ibuprofen and naproxen show increased activity against this cell line when a six-carbon spacer is used for their conjugation to NGR. This is probably due to the less steric hindrance for NGR interaction with its binding protein on the cell surface. Ibuprofen-spacer-NGR and naproxen-spacer-NGR showed the same pattern of increased activity against HT-1080 tumor cells which this cell line show high expression of CD13. Surprisingly, NGR conjugates of both drugs without spacer did not show improved activity compared to the nonconjugated forms against this cell line. Therefore, it could be speculated that HT-1080 cells are more sensitive to the steric hindrance for interaction between NGR and its binding protein. None of the conjugates of ibuprofen and naproxen with or without spacer showed significantly improved activity against A2780 (as a tumor cell with normal RGD-binding protein) and OVCAR3. Therefore, it could be inferred again that the RGD motif is not qualified as a targeting tool for ibuprofen and naproxen.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous adenocarcinoma | OVCAR-3 cell | CVCL_0465 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [92] | ||||
| Indication | Ovarian endometrioid adenocarcinoma | ||||
| Efficacy Data | Cell inhibition rate |
45 ± 2.0%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 100 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The two tripeptide sequences, arginine-glycine-aspartic acid (RGD) and asparagine-glycine-arginine (NGR) motifs have been identified based on phage display studies and they have been used widely in the field of targeted drug delivery. RGD is a well-known peptide sequence for targeting integrin receptors and can bind to avβ3 and avβ5 integrin receptors subunits, which are overexpressed in the angiogenesis process of cancer cells. Because av integrin is overexpressed on the surface of cancer cells, an integrin ligand can be used as a targeting system for cancer treatment. RGD peptide conjugated with cytotoxic agents (RGD-drug conjugates) is likely to exhibit a tumor-targeting and thus antiangiogenic synergetic effect. During the last few years, a number of RGD-cytotoxic drugs have been developed and showed promising activities in vitro and in vivo. Conjugation of camptothecin with RGD is an example for improving the therapeutic index of an antitumoral drug.[20c] Synthesis of dimeric RGD peptide-paclitaxel conjugate is another successful example of targeted drug delivery. Other motif that has been used for tumor targeting is NGR tripeptide. This sequence can bind to CD13 that is specially overexpressed in tumor blood vessels and is involved in angiogenesis, invasion, and metastasis. Because RGD is a peptide tag which targets the process of angiogenesis and NGR also targets tumor blood vessels, we decided to synthesize the conjugated forms of two famous NSAIDs, naproxen, and ibuprofen, with these two tripeptides. To investigate the impact of possible steric hindrance due to the attachment of the drug to the peptide, a linear six-carbon segment (hexanoic acid) was also used as a spacer.
Click to Show/Hide
|
||||
| Description |
NGR conjugate forms of both ibuprofen and naproxen showed improved activity when they were tested against SKOV-3 cell line which is positive for APN/CD13. Interestingly, both ibuprofen and naproxen show increased activity against this cell line when a six-carbon spacer is used for their conjugation to NGR. This is probably due to the less steric hindrance for NGR interaction with its binding protein on the cell surface. Ibuprofen-spacer-NGR and naproxen-spacer-NGR showed the same pattern of increased activity against HT-1080 tumor cells which this cell line show high expression of CD13. Surprisingly, NGR conjugates of both drugs without spacer did not show improved activity compared to the nonconjugated forms against this cell line. Therefore, it could be speculated that HT-1080 cells are more sensitive to the steric hindrance for interaction between NGR and its binding protein. None of the conjugates of ibuprofen and naproxen with or without spacer showed significantly improved activity against A2780 (as a tumor cell with normal RGD-binding protein) and OVCAR3. Therefore, it could be inferred again that the RGD motif is not qualified as a targeting tool for ibuprofen and naproxen.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
Linear asparagineglycinearginine (NGR) 2 - Ibuprofen conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [92] | ||||
| Indication | Invasive breast carcinoma | ||||
| Efficacy Data | Cell inhibition rate |
0%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 100 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The two tripeptide sequences, arginine-glycine-aspartic acid (RGD) and asparagine-glycine-arginine (NGR) motifs have been identified based on phage display studies and they have been used widely in the field of targeted drug delivery. RGD is a well-known peptide sequence for targeting integrin receptors and can bind to avβ3 and avβ5 integrin receptors subunits, which are overexpressed in the angiogenesis process of cancer cells. Because av integrin is overexpressed on the surface of cancer cells, an integrin ligand can be used as a targeting system for cancer treatment. RGD peptide conjugated with cytotoxic agents (RGD-drug conjugates) is likely to exhibit a tumor-targeting and thus antiangiogenic synergetic effect. During the last few years, a number of RGD-cytotoxic drugs have been developed and showed promising activities in vitro and in vivo. Conjugation of camptothecin with RGD is an example for improving the therapeutic index of an antitumoral drug.[20c] Synthesis of dimeric RGD peptide-paclitaxel conjugate is another successful example of targeted drug delivery. Other motif that has been used for tumor targeting is NGR tripeptide. This sequence can bind to CD13 that is specially overexpressed in tumor blood vessels and is involved in angiogenesis, invasion, and metastasis. Because RGD is a peptide tag which targets the process of angiogenesis and NGR also targets tumor blood vessels, we decided to synthesize the conjugated forms of two famous NSAIDs, naproxen, and ibuprofen, with these two tripeptides. To investigate the impact of possible steric hindrance due to the attachment of the drug to the peptide, a linear six-carbon segment (hexanoic acid) was also used as a spacer.
Click to Show/Hide
|
||||
| Description |
NGR conjugate forms of both ibuprofen and naproxen showed improved activity when they were tested against SKOV-3 cell line which is positive for APN/CD13. Interestingly, both ibuprofen and naproxen show increased activity against this cell line when a six-carbon spacer is used for their conjugation to NGR. This is probably due to the less steric hindrance for NGR interaction with its binding protein on the cell surface. Ibuprofen-spacer-NGR and naproxen-spacer-NGR showed the same pattern of increased activity against HT-1080 tumor cells which this cell line show high expression of CD13. Surprisingly, NGR conjugates of both drugs without spacer did not show improved activity compared to the nonconjugated forms against this cell line. Therefore, it could be speculated that HT-1080 cells are more sensitive to the steric hindrance for interaction between NGR and its binding protein. None of the conjugates of ibuprofen and naproxen with or without spacer showed significantly improved activity against A2780 (as a tumor cell with normal RGD-binding protein) and OVCAR3. Therefore, it could be inferred again that the RGD motif is not qualified as a targeting tool for ibuprofen and naproxen.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [92] | ||||
| Efficacy Data | Cell inhibition rate |
0%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 100 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The two tripeptide sequences, arginine-glycine-aspartic acid (RGD) and asparagine-glycine-arginine (NGR) motifs have been identified based on phage display studies and they have been used widely in the field of targeted drug delivery. RGD is a well-known peptide sequence for targeting integrin receptors and can bind to avβ3 and avβ5 integrin receptors subunits, which are overexpressed in the angiogenesis process of cancer cells. Because av integrin is overexpressed on the surface of cancer cells, an integrin ligand can be used as a targeting system for cancer treatment. RGD peptide conjugated with cytotoxic agents (RGD-drug conjugates) is likely to exhibit a tumor-targeting and thus antiangiogenic synergetic effect. During the last few years, a number of RGD-cytotoxic drugs have been developed and showed promising activities in vitro and in vivo. Conjugation of camptothecin with RGD is an example for improving the therapeutic index of an antitumoral drug.[20c] Synthesis of dimeric RGD peptide-paclitaxel conjugate is another successful example of targeted drug delivery. Other motif that has been used for tumor targeting is NGR tripeptide. This sequence can bind to CD13 that is specially overexpressed in tumor blood vessels and is involved in angiogenesis, invasion, and metastasis. Because RGD is a peptide tag which targets the process of angiogenesis and NGR also targets tumor blood vessels, we decided to synthesize the conjugated forms of two famous NSAIDs, naproxen, and ibuprofen, with these two tripeptides. To investigate the impact of possible steric hindrance due to the attachment of the drug to the peptide, a linear six-carbon segment (hexanoic acid) was also used as a spacer.
Click to Show/Hide
|
||||
| Description |
NGR conjugate forms of both ibuprofen and naproxen showed improved activity when they were tested against SKOV-3 cell line which is positive for APN/CD13. Interestingly, both ibuprofen and naproxen show increased activity against this cell line when a six-carbon spacer is used for their conjugation to NGR. This is probably due to the less steric hindrance for NGR interaction with its binding protein on the cell surface. Ibuprofen-spacer-NGR and naproxen-spacer-NGR showed the same pattern of increased activity against HT-1080 tumor cells which this cell line show high expression of CD13. Surprisingly, NGR conjugates of both drugs without spacer did not show improved activity compared to the nonconjugated forms against this cell line. Therefore, it could be speculated that HT-1080 cells are more sensitive to the steric hindrance for interaction between NGR and its binding protein. None of the conjugates of ibuprofen and naproxen with or without spacer showed significantly improved activity against A2780 (as a tumor cell with normal RGD-binding protein) and OVCAR3. Therefore, it could be inferred again that the RGD motif is not qualified as a targeting tool for ibuprofen and naproxen.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human fibroblast cells | Homo sapiens | ||
| Experiment 3 Reporting the Activity Data of This PDC | [92] | ||||
| Indication | Fibrosarcoma | ||||
| Efficacy Data | Cell inhibition rate |
36 ± 3.2%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 100 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The two tripeptide sequences, arginine-glycine-aspartic acid (RGD) and asparagine-glycine-arginine (NGR) motifs have been identified based on phage display studies and they have been used widely in the field of targeted drug delivery. RGD is a well-known peptide sequence for targeting integrin receptors and can bind to avβ3 and avβ5 integrin receptors subunits, which are overexpressed in the angiogenesis process of cancer cells. Because av integrin is overexpressed on the surface of cancer cells, an integrin ligand can be used as a targeting system for cancer treatment. RGD peptide conjugated with cytotoxic agents (RGD-drug conjugates) is likely to exhibit a tumor-targeting and thus antiangiogenic synergetic effect. During the last few years, a number of RGD-cytotoxic drugs have been developed and showed promising activities in vitro and in vivo. Conjugation of camptothecin with RGD is an example for improving the therapeutic index of an antitumoral drug.[20c] Synthesis of dimeric RGD peptide-paclitaxel conjugate is another successful example of targeted drug delivery. Other motif that has been used for tumor targeting is NGR tripeptide. This sequence can bind to CD13 that is specially overexpressed in tumor blood vessels and is involved in angiogenesis, invasion, and metastasis. Because RGD is a peptide tag which targets the process of angiogenesis and NGR also targets tumor blood vessels, we decided to synthesize the conjugated forms of two famous NSAIDs, naproxen, and ibuprofen, with these two tripeptides. To investigate the impact of possible steric hindrance due to the attachment of the drug to the peptide, a linear six-carbon segment (hexanoic acid) was also used as a spacer.
Click to Show/Hide
|
||||
| Description |
NGR conjugate forms of both ibuprofen and naproxen showed improved activity when they were tested against SKOV-3 cell line which is positive for APN/CD13. Interestingly, both ibuprofen and naproxen show increased activity against this cell line when a six-carbon spacer is used for their conjugation to NGR. This is probably due to the less steric hindrance for NGR interaction with its binding protein on the cell surface. Ibuprofen-spacer-NGR and naproxen-spacer-NGR showed the same pattern of increased activity against HT-1080 tumor cells which this cell line show high expression of CD13. Surprisingly, NGR conjugates of both drugs without spacer did not show improved activity compared to the nonconjugated forms against this cell line. Therefore, it could be speculated that HT-1080 cells are more sensitive to the steric hindrance for interaction between NGR and its binding protein. None of the conjugates of ibuprofen and naproxen with or without spacer showed significantly improved activity against A2780 (as a tumor cell with normal RGD-binding protein) and OVCAR3. Therefore, it could be inferred again that the RGD motif is not qualified as a targeting tool for ibuprofen and naproxen.
Click to Show/Hide
|
||||
| In Vitro Model | Fibrosarcoma | HT-1080 cell | CVCL_0317 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [92] | ||||
| Indication | Ovarian endometrioid adenocarcinoma | ||||
| Efficacy Data | Cell inhibition rate |
44 ± 2.4%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 100 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The two tripeptide sequences, arginine-glycine-aspartic acid (RGD) and asparagine-glycine-arginine (NGR) motifs have been identified based on phage display studies and they have been used widely in the field of targeted drug delivery. RGD is a well-known peptide sequence for targeting integrin receptors and can bind to avβ3 and avβ5 integrin receptors subunits, which are overexpressed in the angiogenesis process of cancer cells. Because av integrin is overexpressed on the surface of cancer cells, an integrin ligand can be used as a targeting system for cancer treatment. RGD peptide conjugated with cytotoxic agents (RGD-drug conjugates) is likely to exhibit a tumor-targeting and thus antiangiogenic synergetic effect. During the last few years, a number of RGD-cytotoxic drugs have been developed and showed promising activities in vitro and in vivo. Conjugation of camptothecin with RGD is an example for improving the therapeutic index of an antitumoral drug.[20c] Synthesis of dimeric RGD peptide-paclitaxel conjugate is another successful example of targeted drug delivery. Other motif that has been used for tumor targeting is NGR tripeptide. This sequence can bind to CD13 that is specially overexpressed in tumor blood vessels and is involved in angiogenesis, invasion, and metastasis. Because RGD is a peptide tag which targets the process of angiogenesis and NGR also targets tumor blood vessels, we decided to synthesize the conjugated forms of two famous NSAIDs, naproxen, and ibuprofen, with these two tripeptides. To investigate the impact of possible steric hindrance due to the attachment of the drug to the peptide, a linear six-carbon segment (hexanoic acid) was also used as a spacer.
Click to Show/Hide
|
||||
| Description |
NGR conjugate forms of both ibuprofen and naproxen showed improved activity when they were tested against SKOV-3 cell line which is positive for APN/CD13. Interestingly, both ibuprofen and naproxen show increased activity against this cell line when a six-carbon spacer is used for their conjugation to NGR. This is probably due to the less steric hindrance for NGR interaction with its binding protein on the cell surface. Ibuprofen-spacer-NGR and naproxen-spacer-NGR showed the same pattern of increased activity against HT-1080 tumor cells which this cell line show high expression of CD13. Surprisingly, NGR conjugates of both drugs without spacer did not show improved activity compared to the nonconjugated forms against this cell line. Therefore, it could be speculated that HT-1080 cells are more sensitive to the steric hindrance for interaction between NGR and its binding protein. None of the conjugates of ibuprofen and naproxen with or without spacer showed significantly improved activity against A2780 (as a tumor cell with normal RGD-binding protein) and OVCAR3. Therefore, it could be inferred again that the RGD motif is not qualified as a targeting tool for ibuprofen and naproxen.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [92] | ||||
| Indication | Ovarian serous cystadenocarcinoma | ||||
| Efficacy Data | Cell inhibition rate |
46 ± 0.7%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 100 μM | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The two tripeptide sequences, arginine-glycine-aspartic acid (RGD) and asparagine-glycine-arginine (NGR) motifs have been identified based on phage display studies and they have been used widely in the field of targeted drug delivery. RGD is a well-known peptide sequence for targeting integrin receptors and can bind to avβ3 and avβ5 integrin receptors subunits, which are overexpressed in the angiogenesis process of cancer cells. Because av integrin is overexpressed on the surface of cancer cells, an integrin ligand can be used as a targeting system for cancer treatment. RGD peptide conjugated with cytotoxic agents (RGD-drug conjugates) is likely to exhibit a tumor-targeting and thus antiangiogenic synergetic effect. During the last few years, a number of RGD-cytotoxic drugs have been developed and showed promising activities in vitro and in vivo. Conjugation of camptothecin with RGD is an example for improving the therapeutic index of an antitumoral drug.[20c] Synthesis of dimeric RGD peptide-paclitaxel conjugate is another successful example of targeted drug delivery. Other motif that has been used for tumor targeting is NGR tripeptide. This sequence can bind to CD13 that is specially overexpressed in tumor blood vessels and is involved in angiogenesis, invasion, and metastasis. Because RGD is a peptide tag which targets the process of angiogenesis and NGR also targets tumor blood vessels, we decided to synthesize the conjugated forms of two famous NSAIDs, naproxen, and ibuprofen, with these two tripeptides. To investigate the impact of possible steric hindrance due to the attachment of the drug to the peptide, a linear six-carbon segment (hexanoic acid) was also used as a spacer.
Click to Show/Hide
|
||||
| Description |
NGR conjugate forms of both ibuprofen and naproxen showed improved activity when they were tested against SKOV-3 cell line which is positive for APN/CD13. Interestingly, both ibuprofen and naproxen show increased activity against this cell line when a six-carbon spacer is used for their conjugation to NGR. This is probably due to the less steric hindrance for NGR interaction with its binding protein on the cell surface. Ibuprofen-spacer-NGR and naproxen-spacer-NGR showed the same pattern of increased activity against HT-1080 tumor cells which this cell line show high expression of CD13. Surprisingly, NGR conjugates of both drugs without spacer did not show improved activity compared to the nonconjugated forms against this cell line. Therefore, it could be speculated that HT-1080 cells are more sensitive to the steric hindrance for interaction between NGR and its binding protein. None of the conjugates of ibuprofen and naproxen with or without spacer showed significantly improved activity against A2780 (as a tumor cell with normal RGD-binding protein) and OVCAR3. Therefore, it could be inferred again that the RGD motif is not qualified as a targeting tool for ibuprofen and naproxen.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
P6-melphalan [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [93] | ||||
| Indication | Tumor | ||||
| Efficacy Data | A20 growth inhibition |
25.00%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 50 µM | ||||
| Evaluation Method | XTT assay | ||||
| Description |
Conjugation of the drugs to P4 affected their efficacy toward A20 cells. For chlorambucil and melphalan, conjugation reduced the cytotoxic effect and this was significant for chlorambucil at 25 μM (p = 0.0013). On the other hand, conjugation significantly improved the cytotoxic effect of bendamustine at 25 (p = 0.043) and 50 μM (p = 0.048). The efficacies of all P6-conjugates were significantly lower than those of P4-conjugates at concentrations above 10 μM.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse reticulum cell sarcoma | A20 cell | CVCL_1940 | ||
P4-Melph-PEG-AuNP [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [93] | ||||
| Indication | Tumor | ||||
| Efficacy Data | A20 growth inhibition |
45.00%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 50 µM | ||||
| Evaluation Method | XTT assay | ||||
| Description |
All three P4-PDC-coated gold nanoparticles pre-incubated for 24 or 48 h induced statistically similar cytotoxicity in A20 to that induced by freshly prepared PDC4 and to coated particles without pre-incubation (the latter data not shown).
|
||||
| In Vitro Model | Mouse reticulum cell sarcoma | A20 cell | CVCL_1940 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [93] | ||||
| Indication | Tumor | ||||
| Efficacy Data | A20 growth inhibition |
78.00%
|
|||
| Administration Time | 48 h | ||||
| Administration Dosage | 50 µM | ||||
| Evaluation Method | XTT assay | ||||
| MOA of PDC |
Vitamin C as a water-soluble vitamin is the reduced form of ascorbic acid. No significant adverse effect of taking high doses of vitamin C (over 2000 mg/day) has been reported due to the water-soluble feature of vitamin C. Vitamin C directly reacts with hydroxy, alkoxyl, and lipid peroxyl radicals and converts them to alcohol, water, and hydroperoxide lipid, respectively. It has been shown that taking vitamin C before radioiodine therapy can ameliorate the oxidative stress effect of radioiodine. The radioprotective effects of vitamin C are mainly due to its free radical scavenging activity.
Click to Show/Hide
|
||||
| Description |
All three P4-PDC-coated gold nanoparticles pre-incubated for 24 or 48 h induced statistically similar cytotoxicity in A20 to that induced by freshly prepared PDC4 and to coated particles without pre-incubation (the latter data not shown).
|
||||
| In Vitro Model | Mouse reticulum cell sarcoma | A20 cell | CVCL_1940 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [93] | ||||
| Indication | Tumor | ||||
| Efficacy Data | A20 growth inhibition |
80.00%
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 50 µM | ||||
| Evaluation Method | XTT assay | ||||
| Description |
All three P4-PDC-coated gold nanoparticles pre-incubated for 24 or 48 h induced statistically similar cytotoxicity in A20 to that induced by freshly prepared PDC4 and to coated particles without pre-incubation (the latter data not shown).
|
||||
| In Vitro Model | Mouse reticulum cell sarcoma | A20 cell | CVCL_1940 | ||
P4-melphalan [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [93] | ||||
| Indication | Tumor | ||||
| Efficacy Data | A20 growth inhibition |
75.00%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 50 µM | ||||
| Evaluation Method | XTT assay | ||||
| Description |
Conjugation of the drugs to P4 affected their efficacy toward A20 cells. For chlorambucil and melphalan, conjugation reduced the cytotoxic effect and this was significant for chlorambucil at 25 μM (p = 0.0013). On the other hand, conjugation significantly improved the cytotoxic effect of bendamustine at 25 (p = 0.043) and 50 μM (p = 0.048). The efficacies of all P6-conjugates were significantly lower than those of P4-conjugates at concentrations above 10 μM.
Click to Show/Hide
|
||||
| In Vitro Model | Mouse reticulum cell sarcoma | A20 cell | CVCL_1940 | ||
Mipsagargin [Discontinued]
Identified from the Human Clinical Data
| Experiment 1 Reporting the Activity Data of This PDC | [94] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Time to disease progression |
134 Day
|
|||
| Patients Enrolled |
Patients with hepatocellular carcinoma who progressed on or after treatment with sorafenib or intolerant of sorafenib.
|
||||
| Administration Time | Days 1, 2, and 3 of a 28-day cycle | ||||
| Administration Dosage | 40 mg/m2 | ||||
| Description |
The median TTP was 134.0 days, median PFS was 129.0 days, and median OS was 205.0 days.
|
||||
| Experiment 2 Reporting the Activity Data of This PDC | [95] | ||||
| Indication | Non-small cell lung cancer | ||||
| Efficacy Data | Progression-free survival (PFS) |
52 day
|
|||
| Patients Enrolled |
Patients with non-small cell lung cancer.
|
||||
| Administration Time | Day 1/2/3 | ||||
| Administration Dosage | Dose level ( mg/m2) 10 | ||||
| Description |
Among the 42 patients considered to be evaluable for efficacy, there were no observations of objective response. However, 12 (28.6%) of patients had disease stabilisation. Overall, PFS was a median of 52.0 days (n1/442; censored 8; Q1, Q3: 50.0, 112.0). However, among the 12 patients with SD as best response, progression-free survival (PFS) was considerably higher: 129 days with 8 patients censored at the date of last study visit (Table 5A). Among the 16 efficacy-evaluable patients enrolled in part 2, 5 patients presented to the study with a diagnosis of hepatocellular carcinoma and had progressed on or were intolerant of sorafenib. In these 5 patients, prolonged disease stabilisation with an average PFS of 166 days (6 months) was observed (Table 5B); in patients with HCC who received more than 2 cycles of treatment, the PFS was 209 days, approaching 7.5 months
Click to Show/Hide
|
||||
| Experiment 3 Reporting the Activity Data of This PDC | [95] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Progression-free survival (PFS) |
57 day
|
|||
| Patients Enrolled |
Patients with pancreas cancer.
|
||||
| Administration Time | Day 1/2/3 | ||||
| Administration Dosage | Dose level ( mg/m2) 40 | ||||
| Description |
Among the 42 patients considered to be evaluable for efficacy, there were no observations of objective response. However, 12 (28.6%) of patients had disease stabilisation. Overall, PFS was a median of 52.0 days (n1/442; censored 8; Q1, Q3: 50.0, 112.0). However, among the 12 patients with SD as best response, progression-free survival (PFS) was considerably higher: 129 days with 8 patients censored at the date of last study visit (Table 5A). Among the 16 efficacy-evaluable patients enrolled in part 2, 5 patients presented to the study with a diagnosis of hepatocellular carcinoma and had progressed on or were intolerant of sorafenib. In these 5 patients, prolonged disease stabilisation with an average PFS of 166 days (6 months) was observed (Table 5B); in patients with HCC who received more than 2 cycles of treatment, the PFS was 209 days, approaching 7.5 months
Click to Show/Hide
|
||||
| Experiment 4 Reporting the Activity Data of This PDC | [95] | ||||
| Indication | Adenocarcinoma | ||||
| Efficacy Data | Progression-free survival (PFS) |
63 day
|
|||
| Patients Enrolled |
Patients with adenocarcinoma cancer.
|
||||
| Administration Time | Day 1/2/3 | ||||
| Administration Dosage | Dose level ( mg/m2) 66.8 | ||||
| Description |
Among the 42 patients considered to be evaluable for efficacy, there were no observations of objective response. However, 12 (28.6%) of patients had disease stabilisation. Overall, PFS was a median of 52.0 days (n1/442; censored 8; Q1, Q3: 50.0, 112.0). However, among the 12 patients with SD as best response, progression-free survival (PFS) was considerably higher: 129 days with 8 patients censored at the date of last study visit (Table 5A). Among the 16 efficacy-evaluable patients enrolled in part 2, 5 patients presented to the study with a diagnosis of hepatocellular carcinoma and had progressed on or were intolerant of sorafenib. In these 5 patients, prolonged disease stabilisation with an average PFS of 166 days (6 months) was observed (Table 5B); in patients with HCC who received more than 2 cycles of treatment, the PFS was 209 days, approaching 7.5 months
Click to Show/Hide
|
||||
| Experiment 5 Reporting the Activity Data of This PDC | [95] | ||||
| Indication | Advanced solid tumour | ||||
| Efficacy Data | Progression-free survival (PFS) |
77 day
|
|||
| Patients Enrolled |
Patients with endometrial cancer.
|
||||
| Administration Time | Day 1/2/3 | ||||
| Administration Dosage | Dose level ( mg/m2) 40/66.8/66.8 | ||||
| Description |
Among the 42 patients considered to be evaluable for efficacy, there were no observations of objective response. However, 12 (28.6%) of patients had disease stabilisation. Overall, PFS was a median of 52.0 days (n1/442; censored 8; Q1, Q3: 50.0, 112.0). However, among the 12 patients with SD as best response, progression-free survival (PFS) was considerably higher: 129 days with 8 patients censored at the date of last study visit (Table 5A). Among the 16 efficacy-evaluable patients enrolled in part 2, 5 patients presented to the study with a diagnosis of hepatocellular carcinoma and had progressed on or were intolerant of sorafenib. In these 5 patients, prolonged disease stabilisation with an average PFS of 166 days (6 months) was observed (Table 5B); in patients with HCC who received more than 2 cycles of treatment, the PFS was 209 days, approaching 7.5 months
Click to Show/Hide
|
||||
| Experiment 6 Reporting the Activity Data of This PDC | [95] | ||||
| Indication | Cholangiocarcinoma | ||||
| Efficacy Data | Progression-free survival (PFS) |
89 day
|
|||
| Patients Enrolled |
Patients with cholangiocarcinoma cancer.
|
||||
| Administration Time | Day 1/2/3 | ||||
| Administration Dosage | Dose level ( mg/m2) 2.5 | ||||
| Description |
Among the 42 patients considered to be evaluable for efficacy, there were no observations of objective response. However, 12 (28.6%) of patients had disease stabilisation. Overall, PFS was a median of 52.0 days (n1/442; censored 8; Q1, Q3: 50.0, 112.0). However, among the 12 patients with SD as best response, progression-free survival (PFS) was considerably higher: 129 days with 8 patients censored at the date of last study visit (Table 5A). Among the 16 efficacy-evaluable patients enrolled in part 2, 5 patients presented to the study with a diagnosis of hepatocellular carcinoma and had progressed on or were intolerant of sorafenib. In these 5 patients, prolonged disease stabilisation with an average PFS of 166 days (6 months) was observed (Table 5B); in patients with HCC who received more than 2 cycles of treatment, the PFS was 209 days, approaching 7.5 months
Click to Show/Hide
|
||||
| Experiment 7 Reporting the Activity Data of This PDC | [95] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Progression-free survival (PFS) |
112 day
|
|||
| Patients Enrolled |
Patients with prostate cancer.
|
||||
| Administration Time | Day 1/2/3 | ||||
| Administration Dosage | Dose level ( mg/m2) 1.2 | ||||
| Description |
Among the 42 patients considered to be evaluable for efficacy, there were no observations of objective response. However, 12 (28.6%) of patients had disease stabilisation. Overall, PFS was a median of 52.0 days (n1/442; censored 8; Q1, Q3: 50.0, 112.0). However, among the 12 patients with SD as best response, progression-free survival (PFS) was considerably higher: 129 days with 8 patients censored at the date of last study visit (Table 5A). Among the 16 efficacy-evaluable patients enrolled in part 2, 5 patients presented to the study with a diagnosis of hepatocellular carcinoma and had progressed on or were intolerant of sorafenib. In these 5 patients, prolonged disease stabilisation with an average PFS of 166 days (6 months) was observed (Table 5B); in patients with HCC who received more than 2 cycles of treatment, the PFS was 209 days, approaching 7.5 months
Click to Show/Hide
|
||||
| Experiment 8 Reporting the Activity Data of This PDC | [95] | ||||
| Indication | Colorectal cancer | ||||
| Efficacy Data | Progression-free survival (PFS) |
112 day
|
|||
| Patients Enrolled |
Patients with colorectal cancer.
|
||||
| Administration Time | Day 1/2/3 | ||||
| Administration Dosage | Dose level ( mg/m2) 5 | ||||
| Description |
Among the 42 patients considered to be evaluable for efficacy, there were no observations of objective response. However, 12 (28.6%) of patients had disease stabilisation. Overall, PFS was a median of 52.0 days (n1/442; censored 8; Q1, Q3: 50.0, 112.0). However, among the 12 patients with SD as best response, progression-free survival (PFS) was considerably higher: 129 days with 8 patients censored at the date of last study visit (Table 5A). Among the 16 efficacy-evaluable patients enrolled in part 2, 5 patients presented to the study with a diagnosis of hepatocellular carcinoma and had progressed on or were intolerant of sorafenib. In these 5 patients, prolonged disease stabilisation with an average PFS of 166 days (6 months) was observed (Table 5B); in patients with HCC who received more than 2 cycles of treatment, the PFS was 209 days, approaching 7.5 months
Click to Show/Hide
|
||||
| Experiment 9 Reporting the Activity Data of This PDC | [95] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Progression-free survival (PFS) |
119 day
|
|||
| Patients Enrolled |
Patients with prostate cancer.
|
||||
| Administration Time | Day 1/2/3 | ||||
| Administration Dosage | Dose level ( mg/m2) 88 | ||||
| Description |
Among the 42 patients considered to be evaluable for efficacy, there were no observations of objective response. However, 12 (28.6%) of patients had disease stabilisation. Overall, PFS was a median of 52.0 days (n1/442; censored 8; Q1, Q3: 50.0, 112.0). However, among the 12 patients with SD as best response, progression-free survival (PFS) was considerably higher: 129 days with 8 patients censored at the date of last study visit (Table 5A). Among the 16 efficacy-evaluable patients enrolled in part 2, 5 patients presented to the study with a diagnosis of hepatocellular carcinoma and had progressed on or were intolerant of sorafenib. In these 5 patients, prolonged disease stabilisation with an average PFS of 166 days (6 months) was observed (Table 5B); in patients with HCC who received more than 2 cycles of treatment, the PFS was 209 days, approaching 7.5 months
Click to Show/Hide
|
||||
| Experiment 10 Reporting the Activity Data of This PDC | [95] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Progression-free survival (PFS) |
121 day
|
|||
| Patients Enrolled |
Patients with prostate cancer.
|
||||
| Administration Time | Day 1/2/3 | ||||
| Administration Dosage | Dose level ( mg/m2) 40/66.8/66.8 | ||||
| Description |
Among the 42 patients considered to be evaluable for efficacy, there were no observations of objective response. However, 12 (28.6%) of patients had disease stabilisation. Overall, PFS was a median of 52.0 days (n1/442; censored 8; Q1, Q3: 50.0, 112.0). However, among the 12 patients with SD as best response, progression-free survival (PFS) was considerably higher: 129 days with 8 patients censored at the date of last study visit (Table 5A). Among the 16 efficacy-evaluable patients enrolled in part 2, 5 patients presented to the study with a diagnosis of hepatocellular carcinoma and had progressed on or were intolerant of sorafenib. In these 5 patients, prolonged disease stabilisation with an average PFS of 166 days (6 months) was observed (Table 5B); in patients with HCC who received more than 2 cycles of treatment, the PFS was 209 days, approaching 7.5 months
Click to Show/Hide
|
||||
| Experiment 11 Reporting the Activity Data of This PDC | [95] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Progression-free survival (PFS) |
133 day
|
|||
| Patients Enrolled |
Patients with hepatocellular cancer.
|
||||
| Administration Time | Day 1/2/3 | ||||
| Administration Dosage | Dose level ( mg/m2) 40/66.8/66.8 | ||||
| Description |
Among the 42 patients considered to be evaluable for efficacy, there were no observations of objective response. However, 12 (28.6%) of patients had disease stabilisation. Overall, PFS was a median of 52.0 days (n1/442; censored 8; Q1, Q3: 50.0, 112.0). However, among the 12 patients with SD as best response, progression-free survival (PFS) was considerably higher: 129 days with 8 patients censored at the date of last study visit (Table 5A). Among the 16 efficacy-evaluable patients enrolled in part 2, 5 patients presented to the study with a diagnosis of hepatocellular carcinoma and had progressed on or were intolerant of sorafenib. In these 5 patients, prolonged disease stabilisation with an average PFS of 166 days (6 months) was observed (Table 5B); in patients with HCC who received more than 2 cycles of treatment, the PFS was 209 days, approaching 7.5 months
Click to Show/Hide
|
||||
| Experiment 12 Reporting the Activity Data of This PDC | [95] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Progression-free survival (PFS) |
277 day
|
|||
| Patients Enrolled |
Patients with hepatocellular cancer.
|
||||
| Administration Time | Day 1/2/3 | ||||
| Administration Dosage | Dose level ( mg/m2) 40/66.8/66.8 | ||||
| Description |
Among the 42 patients considered to be evaluable for efficacy, there were no observations of objective response. However, 12 (28.6%) of patients had disease stabilisation. Overall, PFS was a median of 52.0 days (n1/442; censored 8; Q1, Q3: 50.0, 112.0). However, among the 12 patients with SD as best response, progression-free survival (PFS) was considerably higher: 129 days with 8 patients censored at the date of last study visit (Table 5A). Among the 16 efficacy-evaluable patients enrolled in part 2, 5 patients presented to the study with a diagnosis of hepatocellular carcinoma and had progressed on or were intolerant of sorafenib. In these 5 patients, prolonged disease stabilisation with an average PFS of 166 days (6 months) was observed (Table 5B); in patients with HCC who received more than 2 cycles of treatment, the PFS was 209 days, approaching 7.5 months
Click to Show/Hide
|
||||
| Experiment 13 Reporting the Activity Data of This PDC | [95] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Progression-free survival (PFS) |
336 day
|
|||
| Patients Enrolled |
Patients with hepatocellular cancer.
|
||||
| Administration Time | Day 1/2/3 | ||||
| Administration Dosage | Dose level ( mg/m2) 40/66.8/66.8 | ||||
| Description |
Among the 42 patients considered to be evaluable for efficacy, there were no observations of objective response. However, 12 (28.6%) of patients had disease stabilisation. Overall, PFS was a median of 52.0 days (n1/442; censored 8; Q1, Q3: 50.0, 112.0). However, among the 12 patients with SD as best response, progression-free survival (PFS) was considerably higher: 129 days with 8 patients censored at the date of last study visit (Table 5A). Among the 16 efficacy-evaluable patients enrolled in part 2, 5 patients presented to the study with a diagnosis of hepatocellular carcinoma and had progressed on or were intolerant of sorafenib. In these 5 patients, prolonged disease stabilisation with an average PFS of 166 days (6 months) was observed (Table 5B); in patients with HCC who received more than 2 cycles of treatment, the PFS was 209 days, approaching 7.5 months
Click to Show/Hide
|
||||
| Experiment 14 Reporting the Activity Data of This PDC | [94] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Progression-free survival (PFS) |
129
|
|||
| Patients Enrolled |
Patients with hepatocellular carcinoma who progressed on or after treatment with sorafenib or intolerant of sorafenib.
|
||||
| Administration Time | Days 1, 2, and 3 of a 28-day cycle | ||||
| Administration Dosage | 40 mg/m2 | ||||
| Description |
The median TTP was 134.0 days, median PFS was 129.0 days, and median OS was 205.0 days.
|
||||
| Experiment 15 Reporting the Activity Data of This PDC | [94] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Median overall survival (mOS) |
205
|
|||
| Patients Enrolled |
Patients with hepatocellular carcinoma who progressed on or after treatment with sorafenib or intolerant of sorafenib.
|
||||
| Administration Time | Days 1, 2, and 3 of a 28-day cycle | ||||
| Administration Dosage | 40 mg/m2 | ||||
| Description |
The median TTP was 134.0 days, median PFS was 129.0 days, and median OS was 205.0 days.
|
||||
References