
Peptide-drug Conjugate Information
General Information of This Peptide-drug Conjugate (PDC)
| PDC ID |
PDC_01083
|
|||||
|---|---|---|---|---|---|---|
| PDC Name |
Dipeptide 23 - 4-Amino-7-chloroquinoline conjugate 12
|
|||||
| PDC Status |
Investigative
|
|||||
| Indication |
In total 1 Indication(s)
|
|||||
| Structure |
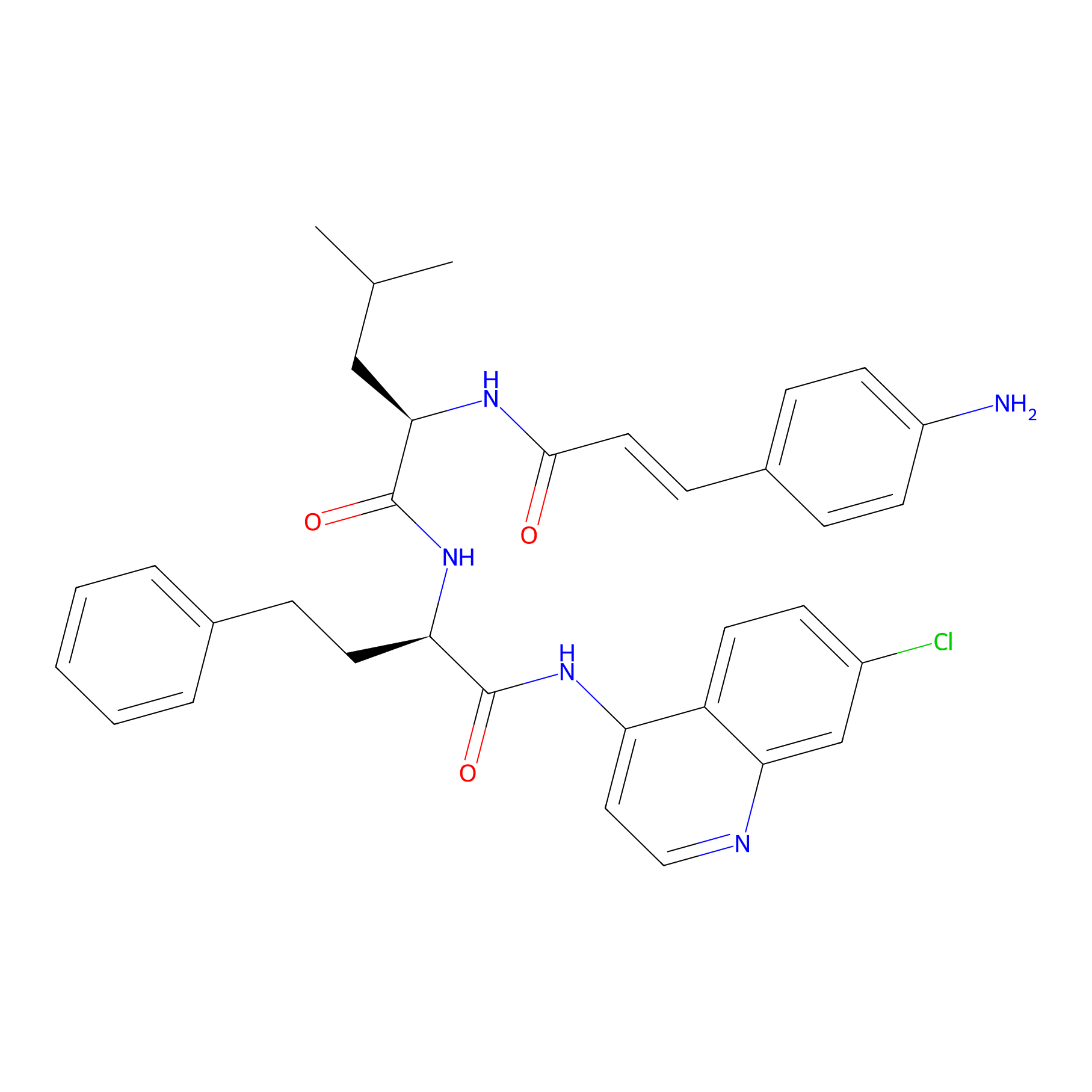
|
|||||
| Peptide Name |
LFX 8e
|
Peptide Info | ||||
| Receptor Name |
Falcipain-1 (FP1)
|
Receptor Info | ||||
| Drug Name |
4-Amino-7-chloroquinoline
|
Drug Info | ||||
| Linker Name |
Amide bond
|
Linker Info | ||||
| Formula |
C34H36ClN5O3
|
|||||
| #Ro5 Violations (Lipinski): 3 | Molecular Weight | 598.147 | ||||
| Lipid-water partition coefficient (xlogp) | 5.7708 | |||||
| Hydrogen Bond Donor Count (hbonddonor) | 4 | |||||
| Hydrogen Bond Acceptor Count (hbondacc) | 5 | |||||
| Rotatable Bond Count (rotbonds) | 12 | |||||
Full List of Activity Data of This Peptide-drug Conjugate
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Haemozoin inhibition | ≥ 75% | |||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
The capacity of test compounds 8 and 9 to inhibit heme polymerization in vitro was assessed by previously reported methods, given in detail under Experimental. The assays were run in 96-well microplates, where negative controls (water, DMSO) and positive controls (1 mM CQ) were included. Test compounds were assayed at 1 mM and data are given in Table 1. Interestingly, the dipeptide spacer was required to block heme polymerization, i.e., while HECINs 9 were not active, HEDICINs 8 displayed variable inhibitory efficiencies, with four out of the twelve compounds (8b, 8e, 8j and 8l) highly active (i.e., comparable to the reference drug, CQ). Though no clear trend could be established regarding stereoelectronic properties of the aryl substituent in compounds 8, it was clear that: (i) hydrogen (i.e., absence of a substituent) or halogens in the para position were detrimental for activity, whereas (ii) nitrogenated groups at either the ortho (8j, o-NO2) or the para (8e, p-NH2; 8l, p-NO2) position of the aryl ring was beneficial, but (iii) detrimental if placed in the meta position (8k, m-NO2). Furthermore, while small alkyl groups in para (8b, p-Me) were advantageous, bulkier groups as in 8c (p-iPr) led to complete loss of activity. Thus, replacement of CQ's aliphatic chain by an adequate dipeptidyl-cinnamoyl moiety as in 8b, 8e, 8j and 8l appears to preserve the parent drug's ability to inhibit hemozoin formation, suggesting that these novel compounds could be promising leads for new CQ surrogates.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum strain W2 | 5833 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
3.28 μM
|
|||
| Administration Time | 48 h | ||||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
Activity against blood-stage CQ-resistant P. falciparum strain W2 was assessed as previously reported and given in detail in Experimental. Results demonstrated a complete lack of activity displayed by HECINs 9, correlating with their inability to inhibit heme polymerization. In turn, eleven out of the twelve HEDICINs 8 had IC50 values under 10 μM. Interestingly, three of the four most active HEDICIN blockers of heme polymerization (8b, 8j and 8l) were also among the four most active antiplasmodials, with IC50 below 2 μM. These results suggest that inhibition of heme polymerization is, at least in part, responsible for the anti-plasmodial activity of HEDICINs. An obvious exception to correlation between inhibition of heme polymerization and anti-plasmodial activity in HEDICINs is compound 8c; this bears a bulky electron-donating p-isopropyl group and did not inhibit heme polymerization in vitro, but displayed the highest anti-plasmodial activity. The inability of 8c to inhibit heme polymerization could be related to the bulkiness of the isopropyl group, but due to the higher hydrophobicity of this substituent, 8c was the most lipophilic HEDICIN assayed. Though we could not establish a full correlation between HEDICIN anti-plasmodial activity and estimated clogP values (not shown), the markedly higher lipophilicity of 8c, as compared to the other analogs, could promote a higher permeabilization of this compound into the infected RBC. Kirk and co-workers have demonstrated that P. falciparum parasites create new permeability pathways in host RBC, leading to increased permeability to small organic cations. In summary, though clean correlations could not be drawn between the anti-plasmodial activities displayed by the different HEDICINs (8) in vitro and molecular descriptors such as stereoelectronic factors (aryl substituents) or lipophilicity, it is demonstrated that these compounds displayed anti-plasmodial activity, whereas their HECIN counterparts (9), lacking the dipeptide spacer, did not. HEDICINs (8) inhibited heme polymerization in vitro, suggesting that this inhibitory activity is at least in part responsible for their anti-plasmodial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum | 5833 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
20.3 μM
|
|||
| Administration Time | 30 min | ||||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
The very different activities of compounds 8 and 9 suggest that the retro-enantio dipeptide spacer has a relevant role in determining anti-plasmodial activity. In view of this, we hypothesized that the anti-plasmodial activity of HEDICINs could also be partly due to falcipain inhibition. Therefore, both HEDICINs 8 and HECINs 9 were evaluated in vitro for inhibition of falcipains, using previously described methods, given in detail under Experimental. Only those compounds with IC50 < 50 μM against FP2 were assayed against FP3, as it has been established that FP2 has a larger catalytic cavity that accommodates a wider range of inhibitors than FP3. Consistent with this assumption, none of the compounds that inhibited FP2 in vitro displayed IC50 < 50 μM against FP3. Falcipain inhibition results contrasted with those for inhibition of heme polymerization or parasite development. HECINs 9 generally displayed more potent inhibition of falcipain than did HEDICINs 8. The ability of the test compounds to inhibit FP2 did not correlate with their anti-plasmodial activity. Although none of the HECINs 9 displayed anti-plasmodial activity, many inhibited FP2 and one of them, 9j, was actually the best FP2 inhibitor amongst the test compounds. Concerning HEDICINs, 8a, 8f and 8k, derived from cinnamic acid, m-fluorocinnamic acid, and m-nitrocinnamic acid, respectively, were the most active HEDICINs against FP2, with IC50 values of 19.7, 23.1 and 28.1 μM. Thus, stereoelectronic effects from aryl substituents did not correlate with inhibitory activities, as in HEDICINs 8 the most active compound was unsubstituted (8a), followed by the meta-fluorinated compound (8f); in turn, in HECINs 9, the meta-fluorinated derivative was inactive, whereas the most active of the set was 9j, which bears a meta-nitro substituent. The effect of amino acid configuration on HEDICIN activity was also assessed through synthesis and evaluation of 10a, the L-amino acid analog of 8a; interestingly, replacement of the D-amino acids by their natural L counterparts led to a clear decrease in both anti-plasmodial and falcipain-inhibitory activity. Therefore, amino acid configuration does influence compound behavior as either anti-plasmodial agent or falcipain inhibitor and, in the particular case of HEDICINs, data suggests that D-amino acids are preferable. Of note, the compound with highest anti-plasmodial activity, 8c, was completely devoid of inhibitory activity against FP2. This compound likely exerts its anti-plasmodial action by mechanisms other than inhibition of hemozoin formation or falcipain activity. Data on compound 8e reinforce the idea that falcipain or hemozoin inhibition are not the main mechanisms of action responsible for HEDICINs anti-plasmodial activity: 8e, bearing a p-amino substituent in the aryl ring, was the test compound which best reached our original goal of a dual-action inhibitor, by joining high hemozoin inhibitory activity with an IC50 20 μM against FP2; however, such was not translated into the highest anti-plasmodial activity being observed for 8e. Taken together, data from in vitro falcipain inhibition and parasite development assays suggest that the dipeptide spacer in HEDICINs 8 promotes uptake into infected RBCs. This hypothesis could explain why HEDICINs perform better than HECINs as antiplasmodials, despite the observation that HECINs were better falcipain-inhibitors than the HEDICINs. In addition, lipophilicity could have a role in compound uptake and anti-plasmodial action, as HEDICINs 8 are more lipophilic than HECINs 9 (due to the D-Leu and the D-hPhe hydrophobic side chains), and the most lipophilic compound, 8c, had the greatest anti-plasmodial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum strain W2 | 5833 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Malaria | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 50 μM | |||
| Administration Time | 30 min | ||||
| MOA of PDC |
HEterocyclic-DIpeptide-CINnamic acid conjugates (HEDICINs) were designed to link, through a suitable spacer, (i) the CQ heterocyclic core, known as relevant to inhibit hemozoin formation, to a (ii) trans-cinnamic acid motif, as cinnamic acids have been described to exhibit both antimalarial activity and inhibiting enzyme catalytic Cys residues. Cinnamic acid derivatives, due to their a,β-unsaturated carbonyl moiety, can act as Michael acceptors and inhibit cysteine proteases through S-alkylation. Irreversible S-alkylation of the falcipain catalytic Cys has been considered the major mechanism behind the inhibitory and in vitro anti-plasmodial activity of peptidyl inhibitors including leupeptin and vinyl sulfones developed by Rosenthal and co-workers. The most active vinyl sulfones contained a dipeptide Leu-hPhe spacer between a bulky moiety and the S-alkylating motif. Unfortunately, peptide-based inhibitors are prone to proteolytic degradation, a problem that can be overcome by use of suitable peptide delivery systems. Still, such systems are likely to impair efficient inhibition of the target enzyme. One way to circumvent the limitations of peptidyl inhibitors is the use of retro-enantio peptides, i.e., analogs where all amino acids have a D configuration and are assembled in reversed order. As these molecules have native side chain topology but reversed amide bonds, they theoretically allow enzyme-ligand contacts identical to those displayed by native peptides, while eluding recognition by other proteases. The retro-enantio analog of Leu-hPhe seems the best mimic of falcipain P2-P1 sites, linking the putative P3 motif (heterocyclic core of CQ) to the Michael acceptor in P1 (cinnamic acid). However, preliminary computational studies by our group on HEDICINs 1 and 8f (structures given below, Fig. 2A and B, respectively), differing only in the order of the two D-amino acids, suggested that the inverse sequence, i.e., D-hPhe-D-Leu, would allow a closer approximation of the electrophilic moiety to the catalytic center. These preliminary observations in silico were later supported in vitro by anti-plasmodial activity tests against the CQ-resistant Plasmodium falciparum strain W2, which revealed that 1 did not display any antimalarial activity up to 10 μM, while 8f inhibited parasite development with an IC50 of 5.43 μM. Therefore, we engaged in the synthesis of HEDICINs with general structure 8 as potential dual-action drugs against blood-stage parasites. Analogs lacking the dipeptide spacer, HECINs 9, were also prepared to assess the relevance of that spacer. Compounds 8 and 9 were evaluated in vitro concerning their ability to inhibit (i) heme polymerization to hemozoin; (ii) falcipain activity; and (iii) development of blood-stage P. falciparum. In order to support observed SAR and to rationalize the activity profile of the novel compounds, we also performed molecular modeling calculations on computational models derived from X-ray structures of FP2 (PDB code: 3BPF) and FP3 (PDB code: 3BWK) co-crystalized with E64 and K11017, respectively.
Click to Show/Hide
|
||||
| Description |
The very different activities of compounds 8 and 9 suggest that the retro-enantio dipeptide spacer has a relevant role in determining anti-plasmodial activity. In view of this, we hypothesized that the anti-plasmodial activity of HEDICINs could also be partly due to falcipain inhibition. Therefore, both HEDICINs 8 and HECINs 9 were evaluated in vitro for inhibition of falcipains, using previously described methods, given in detail under Experimental. Only those compounds with IC50 < 50 μM against FP2 were assayed against FP3, as it has been established that FP2 has a larger catalytic cavity that accommodates a wider range of inhibitors than FP3. Consistent with this assumption, none of the compounds that inhibited FP2 in vitro displayed IC50 < 50 μM against FP3. Falcipain inhibition results contrasted with those for inhibition of heme polymerization or parasite development. HECINs 9 generally displayed more potent inhibition of falcipain than did HEDICINs 8. The ability of the test compounds to inhibit FP2 did not correlate with their anti-plasmodial activity. Although none of the HECINs 9 displayed anti-plasmodial activity, many inhibited FP2 and one of them, 9j, was actually the best FP2 inhibitor amongst the test compounds. Concerning HEDICINs, 8a, 8f and 8k, derived from cinnamic acid, m-fluorocinnamic acid, and m-nitrocinnamic acid, respectively, were the most active HEDICINs against FP2, with IC50 values of 19.7, 23.1 and 28.1 μM. Thus, stereoelectronic effects from aryl substituents did not correlate with inhibitory activities, as in HEDICINs 8 the most active compound was unsubstituted (8a), followed by the meta-fluorinated compound (8f); in turn, in HECINs 9, the meta-fluorinated derivative was inactive, whereas the most active of the set was 9j, which bears a meta-nitro substituent. The effect of amino acid configuration on HEDICIN activity was also assessed through synthesis and evaluation of 10a, the L-amino acid analog of 8a; interestingly, replacement of the D-amino acids by their natural L counterparts led to a clear decrease in both anti-plasmodial and falcipain-inhibitory activity. Therefore, amino acid configuration does influence compound behavior as either anti-plasmodial agent or falcipain inhibitor and, in the particular case of HEDICINs, data suggests that D-amino acids are preferable. Of note, the compound with highest anti-plasmodial activity, 8c, was completely devoid of inhibitory activity against FP2. This compound likely exerts its anti-plasmodial action by mechanisms other than inhibition of hemozoin formation or falcipain activity. Data on compound 8e reinforce the idea that falcipain or hemozoin inhibition are not the main mechanisms of action responsible for HEDICINs anti-plasmodial activity: 8e, bearing a p-amino substituent in the aryl ring, was the test compound which best reached our original goal of a dual-action inhibitor, by joining high hemozoin inhibitory activity with an IC50 20 μM against FP2; however, such was not translated into the highest anti-plasmodial activity being observed for 8e. Taken together, data from in vitro falcipain inhibition and parasite development assays suggest that the dipeptide spacer in HEDICINs 8 promotes uptake into infected RBCs. This hypothesis could explain why HEDICINs perform better than HECINs as antiplasmodials, despite the observation that HECINs were better falcipain-inhibitors than the HEDICINs. In addition, lipophilicity could have a role in compound uptake and anti-plasmodial action, as HEDICINs 8 are more lipophilic than HECINs 9 (due to the D-Leu and the D-hPhe hydrophobic side chains), and the most lipophilic compound, 8c, had the greatest anti-plasmodial activity.
Click to Show/Hide
|
||||
| In Vitro Model | Plasmodium falciparum infection | Plasmodium falciparum strain W2 | 5833 | ||
References