
Peptide-drug Conjugate Information
General Information of This Peptide-drug Conjugate (PDC)
| PDC ID |
PDC_02125
|
|||||
|---|---|---|---|---|---|---|
| PDC Name |
cyclo[DKP-isoDGR]-PEG4-GPLG-PABC-MMAE
|
|||||
| PDC Status |
Investigative
|
|||||
| Indication |
In total 4 Indication(s)
|
|||||
| Structure |
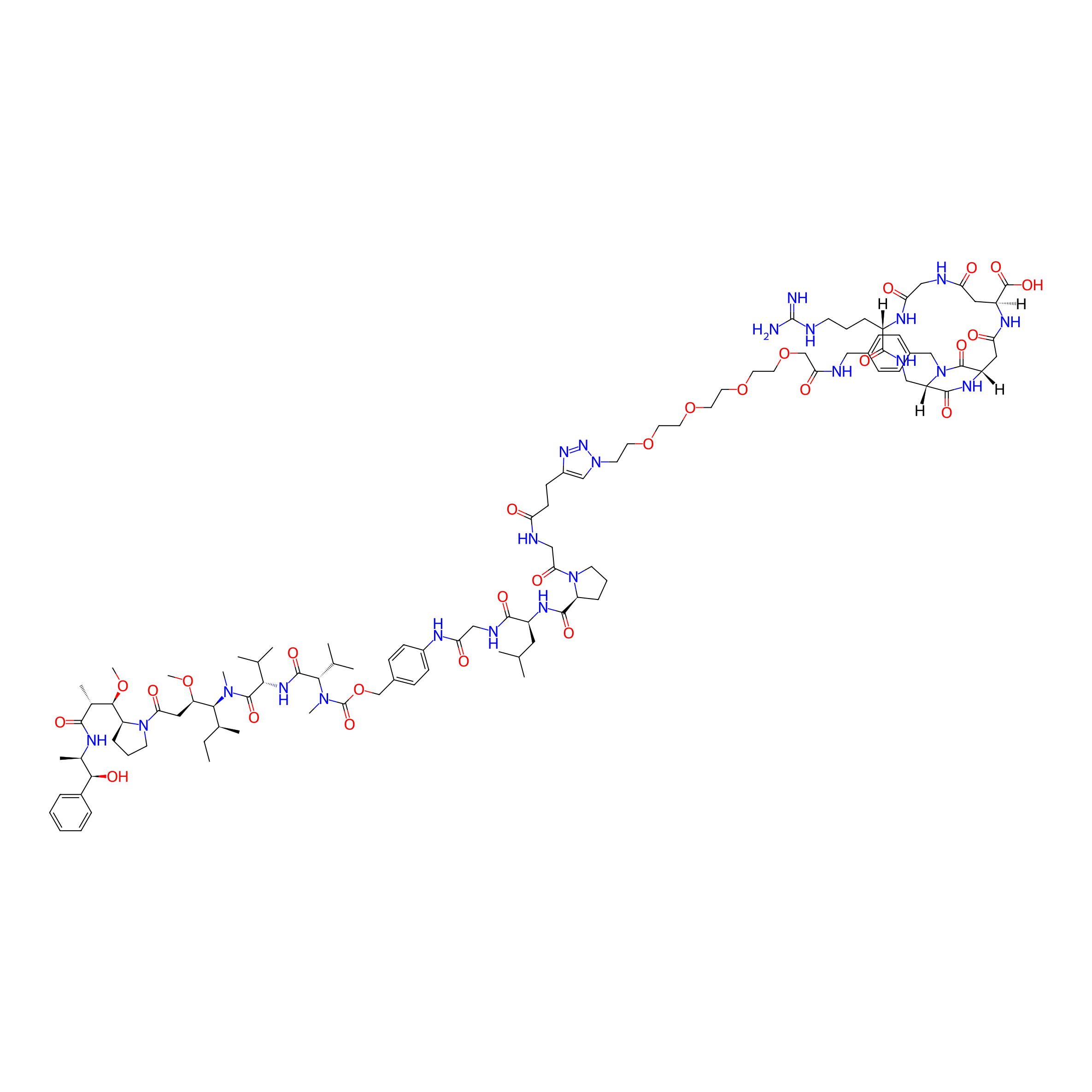
|
|||||
| Peptide Name |
cyclo[DKP-isoDGR]
|
Peptide Info | ||||
| Receptor Name |
Integrin alpha-V (ITGAV)
|
Receptor Info | ||||
| Drug Name |
Monomethyl auristatin E
|
Drug Info | ||||
| Therapeutic Target |
Microtubule (MT)
|
Target Info | ||||
| Linker Name |
PEG4-GPLG-PABC
|
Linker Info | ||||
| Formula |
C104H157N23O27
|
|||||
| #Ro5 Violations (Lipinski): 4 | Molecular Weight | 2161.534 | ||||
| Lipid-water partition coefficient (xlogp) | -0.58183 | |||||
| Hydrogen Bond Donor Count (hbonddonor) | 17 | |||||
| Hydrogen Bond Acceptor Count (hbondacc) | 30 | |||||
| Rotatable Bond Count (rotbonds) | 58 | |||||
Full List of Activity Data of This Peptide-drug Conjugate
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Ovarian serous cystadenocarcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
3.7 ± 0.3 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Here, we show that the replacement of the Val-Ala-p-aminobenzyloxycarbamate linker with the Gly-Pro-Leu-Gly-p-aminobenzyloxycarbamate (GPLG-PABC) sequence as enzymatically cleavable linker in the SMDC bearing the cyclo[DKP-isoDGR] αVβ3 integrin ligand as tumor homing moiety and the monomethyl auristatin E (MMAE) as cytotoxic payload led to a 4-fold more potent anti-tumoral effect of the final conjugate on different cancer cell lines. In addition, the synthesized conjugate resulted to be significantly more potent than the free MMAE when tested following the kiss-and-run protocol, and the relative potency were clearly consistent with the expression of the αVβ3 integrin receptor in the considered cancer cell lines. In vitro enzymatic cleavage tests showed that the GPLG-PABC linker is cleaved by lysosomal enzymes, and that the released drug is observable already after 15 min of incubation. Although additional data are needed to fully characterize the releasing capacity of GPLG-PABC linker, our findings are of therapeutic significance since we are introducing an alternative to other well-established enzymatically sensitive peptide sequences that might be used in the future for generating more efficient and less toxic drug delivery systems.
Click to Show/Hide
|
||||
| Description |
In a first instance we performed an in vitro antiproliferative assay in which the different cell lines were incubated with different concentrations of the conjugate 2 for 72 h. As reported in Figure 3, 2 showed a potent antitumor activity in a low nanomolar range, from 3.7 to 70.4 nM depending on the cancer cell line, whereas the treatment with the free drug MMAE led to an antitumor activity of 0.03 nM for SK-OV-3 and of 0.23 nM for U87MG, and in very low nanomolar range on SK-MEL-28 cell line. Interestingly, the relative potency (RP) of the conjugate, normalized with respect to MMAE (i.e., the ratio of the IC50 values of 2 vs. MMAE) in U87MG cancer cell line after 72 h was much lower than the one observed with the previously reported cyclo[DKP-isoDGR]-PEG4-VA-PABC-MMAE (33-fold loss of potency vs. 151-fold loss of potency, respectively) bearing the VA linker. This data could suggest a positive effect of the GPLG cleavable linker on the release of the MMAE. Furthermore, a drop of potency between the free drug and the conjugate can be noticed in A549 from the RP value, where the integrin receptor level is significantly lower than in the other cancer cell lines. To better explore the targeting ability of the conjugate 2 towards different level of integrin expression, we decided to further evaluate the antiproliferative activity of the GPLG-conjugate by changing the incubation time.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
7.5 ± 0.9 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Here, we show that the replacement of the Val-Ala-p-aminobenzyloxycarbamate linker with the Gly-Pro-Leu-Gly-p-aminobenzyloxycarbamate (GPLG-PABC) sequence as enzymatically cleavable linker in the SMDC bearing the cyclo[DKP-isoDGR] αVβ3 integrin ligand as tumor homing moiety and the monomethyl auristatin E (MMAE) as cytotoxic payload led to a 4-fold more potent anti-tumoral effect of the final conjugate on different cancer cell lines. In addition, the synthesized conjugate resulted to be significantly more potent than the free MMAE when tested following the kiss-and-run protocol, and the relative potency were clearly consistent with the expression of the αVβ3 integrin receptor in the considered cancer cell lines. In vitro enzymatic cleavage tests showed that the GPLG-PABC linker is cleaved by lysosomal enzymes, and that the released drug is observable already after 15 min of incubation. Although additional data are needed to fully characterize the releasing capacity of GPLG-PABC linker, our findings are of therapeutic significance since we are introducing an alternative to other well-established enzymatically sensitive peptide sequences that might be used in the future for generating more efficient and less toxic drug delivery systems.
Click to Show/Hide
|
||||
| Description |
In a first instance we performed an in vitro antiproliferative assay in which the different cell lines were incubated with different concentrations of the conjugate 2 for 72 h. As reported in Figure 3, 2 showed a potent antitumor activity in a low nanomolar range, from 3.7 to 70.4 nM depending on the cancer cell line, whereas the treatment with the free drug MMAE led to an antitumor activity of 0.03 nM for SK-OV-3 and of 0.23 nM for U87MG, and in very low nanomolar range on SK-MEL-28 cell line. Interestingly, the relative potency (RP) of the conjugate, normalized with respect to MMAE (i.e., the ratio of the IC50 values of 2 vs. MMAE) in U87MG cancer cell line after 72 h was much lower than the one observed with the previously reported cyclo[DKP-isoDGR]-PEG4-VA-PABC-MMAE (33-fold loss of potency vs. 151-fold loss of potency, respectively) bearing the VA linker. This data could suggest a positive effect of the GPLG cleavable linker on the release of the MMAE. Furthermore, a drop of potency between the free drug and the conjugate can be noticed in A549 from the RP value, where the integrin receptor level is significantly lower than in the other cancer cell lines. To better explore the targeting ability of the conjugate 2 towards different level of integrin expression, we decided to further evaluate the antiproliferative activity of the GPLG-conjugate by changing the incubation time.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Lung adenocarcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
20.8 ± 1.8 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Here, we show that the replacement of the Val-Ala-p-aminobenzyloxycarbamate linker with the Gly-Pro-Leu-Gly-p-aminobenzyloxycarbamate (GPLG-PABC) sequence as enzymatically cleavable linker in the SMDC bearing the cyclo[DKP-isoDGR] αVβ3 integrin ligand as tumor homing moiety and the monomethyl auristatin E (MMAE) as cytotoxic payload led to a 4-fold more potent anti-tumoral effect of the final conjugate on different cancer cell lines. In addition, the synthesized conjugate resulted to be significantly more potent than the free MMAE when tested following the kiss-and-run protocol, and the relative potency were clearly consistent with the expression of the αVβ3 integrin receptor in the considered cancer cell lines. In vitro enzymatic cleavage tests showed that the GPLG-PABC linker is cleaved by lysosomal enzymes, and that the released drug is observable already after 15 min of incubation. Although additional data are needed to fully characterize the releasing capacity of GPLG-PABC linker, our findings are of therapeutic significance since we are introducing an alternative to other well-established enzymatically sensitive peptide sequences that might be used in the future for generating more efficient and less toxic drug delivery systems.
Click to Show/Hide
|
||||
| Description |
In a first instance we performed an in vitro antiproliferative assay in which the different cell lines were incubated with different concentrations of the conjugate 2 for 72 h. As reported in Figure 3, 2 showed a potent antitumor activity in a low nanomolar range, from 3.7 to 70.4 nM depending on the cancer cell line, whereas the treatment with the free drug MMAE led to an antitumor activity of 0.03 nM for SK-OV-3 and of 0.23 nM for U87MG, and in very low nanomolar range on SK-MEL-28 cell line. Interestingly, the relative potency (RP) of the conjugate, normalized with respect to MMAE (i.e., the ratio of the IC50 values of 2 vs. MMAE) in U87MG cancer cell line after 72 h was much lower than the one observed with the previously reported cyclo[DKP-isoDGR]-PEG4-VA-PABC-MMAE (33-fold loss of potency vs. 151-fold loss of potency, respectively) bearing the VA linker. This data could suggest a positive effect of the GPLG cleavable linker on the release of the MMAE. Furthermore, a drop of potency between the free drug and the conjugate can be noticed in A549 from the RP value, where the integrin receptor level is significantly lower than in the other cancer cell lines. To better explore the targeting ability of the conjugate 2 towards different level of integrin expression, we decided to further evaluate the antiproliferative activity of the GPLG-conjugate by changing the incubation time.
Click to Show/Hide
|
||||
| In Vitro Model | Lung adenocarcinoma | A-549 cell | CVCL_0023 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
70.4 ± 7.1 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Here, we show that the replacement of the Val-Ala-p-aminobenzyloxycarbamate linker with the Gly-Pro-Leu-Gly-p-aminobenzyloxycarbamate (GPLG-PABC) sequence as enzymatically cleavable linker in the SMDC bearing the cyclo[DKP-isoDGR] αVβ3 integrin ligand as tumor homing moiety and the monomethyl auristatin E (MMAE) as cytotoxic payload led to a 4-fold more potent anti-tumoral effect of the final conjugate on different cancer cell lines. In addition, the synthesized conjugate resulted to be significantly more potent than the free MMAE when tested following the kiss-and-run protocol, and the relative potency were clearly consistent with the expression of the αVβ3 integrin receptor in the considered cancer cell lines. In vitro enzymatic cleavage tests showed that the GPLG-PABC linker is cleaved by lysosomal enzymes, and that the released drug is observable already after 15 min of incubation. Although additional data are needed to fully characterize the releasing capacity of GPLG-PABC linker, our findings are of therapeutic significance since we are introducing an alternative to other well-established enzymatically sensitive peptide sequences that might be used in the future for generating more efficient and less toxic drug delivery systems.
Click to Show/Hide
|
||||
| Description |
In a first instance we performed an in vitro antiproliferative assay in which the different cell lines were incubated with different concentrations of the conjugate 2 for 72 h. As reported in Figure 3, 2 showed a potent antitumor activity in a low nanomolar range, from 3.7 to 70.4 nM depending on the cancer cell line, whereas the treatment with the free drug MMAE led to an antitumor activity of 0.03 nM for SK-OV-3 and of 0.23 nM for U87MG, and in very low nanomolar range on SK-MEL-28 cell line. Interestingly, the relative potency (RP) of the conjugate, normalized with respect to MMAE (i.e., the ratio of the IC50 values of 2 vs. MMAE) in U87MG cancer cell line after 72 h was much lower than the one observed with the previously reported cyclo[DKP-isoDGR]-PEG4-VA-PABC-MMAE (33-fold loss of potency vs. 151-fold loss of potency, respectively) bearing the VA linker. This data could suggest a positive effect of the GPLG cleavable linker on the release of the MMAE. Furthermore, a drop of potency between the free drug and the conjugate can be noticed in A549 from the RP value, where the integrin receptor level is significantly lower than in the other cancer cell lines. To better explore the targeting ability of the conjugate 2 towards different level of integrin expression, we decided to further evaluate the antiproliferative activity of the GPLG-conjugate by changing the incubation time.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | SK-MEL-202 cell | CVCL_6106 | ||
References