
Peptide Information
General Information of This Peptide
| Peptide ID |
PEP00063
|
|||||
|---|---|---|---|---|---|---|
| Peptide Name |
DEVD
|
|||||
| Structure |
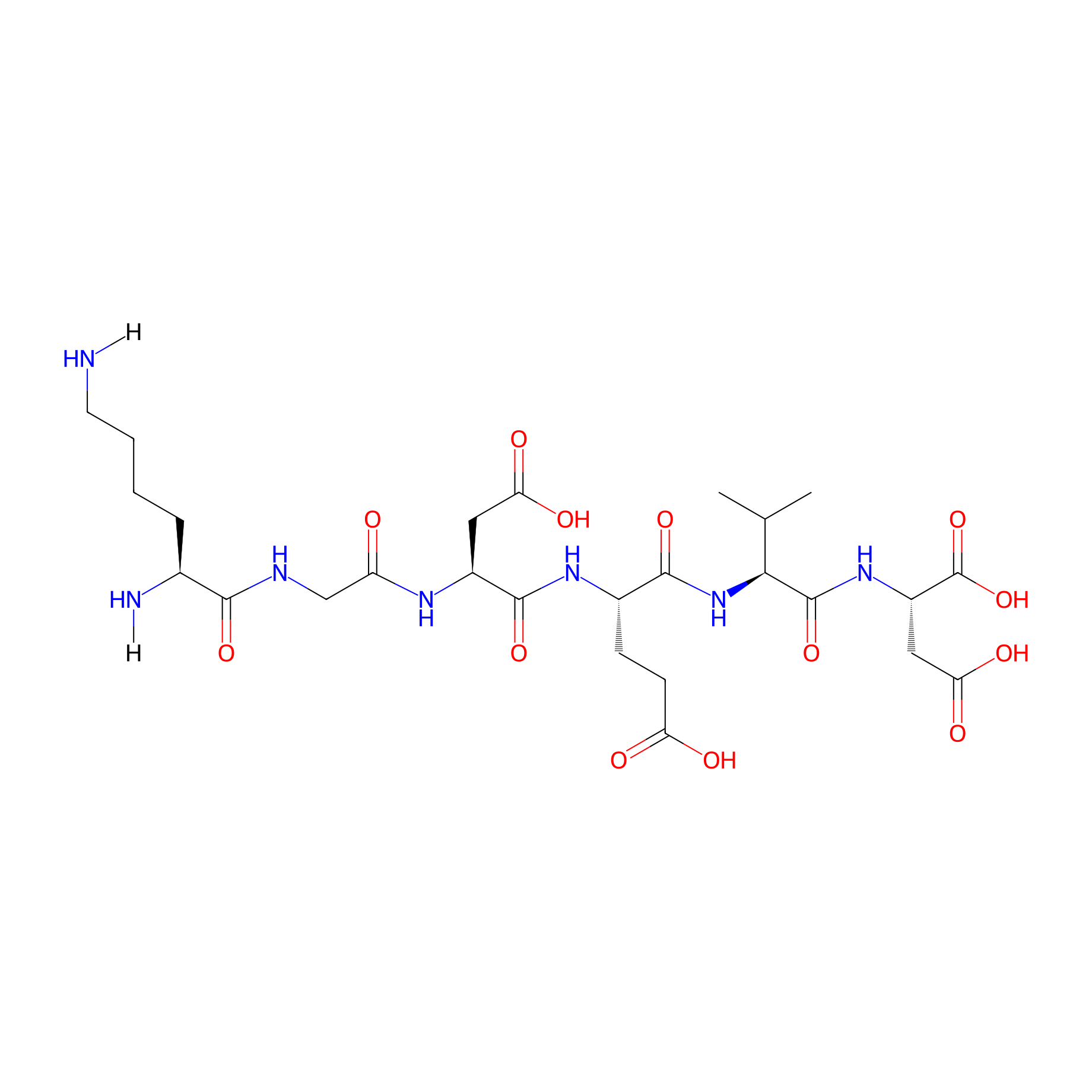
|
|||||
| Sequence |
KGDEVD
|
|||||
| Peptide Type |
Linear
|
|||||
| Receptor Name |
Caspase-3 (CASP3)
|
Receptor Info | ||||
| PDC Transmembrane Types | Cell targeting peptides (CTPs) | |||||
| Formula |
C26H43N7O13
|
|||||
| Isosmiles |
[H]NCCCC[C@H](N[H])C(=O)NCC(=O)N[C@@H](CC(=O)O)C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@H](C(=O)N[C@@H](CC(=O)O)C(=O)O)C(C)C
|
|||||
| InChI |
InChI=1S/C26H43N7O13/c1-12(2)21(25(44)32-16(26(45)46)10-20(39)40)33-23(42)14(6-7-18(35)36)31-24(43)15(9-19(37)38)30-17(34)11-29-22(41)13(28)5-3-4-8-27/h12-16,21H,3-11,27-28H2,1-2H3,(H,29,41)(H,30,34)(H,31,43)(H,32,44)(H,33,42)(H,35,36)(H,37,38)(H,39,40)(H,45,46)/t13-,14-,15-,16-,21-/m0/s1
|
|||||
| InChIKey |
WXMASBPOWHWEMR-GVZUZGFXSA-N
|
|||||
| Pharmaceutical Properties |
Molecule Weight
|
661.666
|
Polar area
|
346.74
|
||
|
Complexity
|
661.2918844
|
xlogp Value
|
-3.9471
|
|||
|
Heavy Count
|
46
|
Rot Bonds
|
25
|
|||
|
Hbond acc
|
11
|
Hbond Donor
|
11
|
|||
Each Peptide-drug Conjugate Related to This Peptide
Full Information of The Activity Data of The PDC(s) Related to This Peptide
MPD1 [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
12.50%
|
|||
| Administration Time | 30 days | ||||
| Administration Dosage | 10 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BxPC-3 cells (KRAS wild type) xenografted mice. | ||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | BxPC-3 cell | CVCL_0186 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
69.10%
|
|||
| Administration Time | 30 days | ||||
| Administration Dosage | 5 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | AsPC-1 cells (G12D KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | AsPC-1 (KRAS G12D) cell | L-929 cell line | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Lung cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
80.00%
|
|||
| Administration Time | 30 days | ||||
| Administration Dosage | 5 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | A549 cells (G12S KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Lung adenocarcinoma | A-549 (KRAS G12S) cell | CVCL_0023 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
86.80%
|
|||
| Administration Time | 30 days | ||||
| Administration Dosage | 5 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | HCT116 cells (G13D KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Colon carcinoma | HCT 116 (KRAS G13D) cell | CVCL_0291 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
95.10%
|
|||
| Administration Time | 30 days | ||||
| Administration Dosage | 10 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | AsPC-1 cells (G12D KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | AsPC-1 (KRAS G12D) cell | L-929 cell line | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Lung cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
95.10%
|
|||
| Administration Time | 30 days | ||||
| Administration Dosage | 10 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | A549 cells (G12S KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Lung adenocarcinoma | A-549 (KRAS G12S) cell | CVCL_0023 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
96.50%
|
|||
| Administration Time | 30 days | ||||
| Administration Dosage | 5 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | MDA-MB-231 cells (G13D KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (KRAS G13D) cell | CVCL_0062 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 8 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
97.80%
|
|||
| Administration Time | 30 days | ||||
| Administration Dosage | 10 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | HCT116 cells (G13D KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Colon carcinoma | HCT 116 (KRAS G13D) cell | CVCL_0291 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 9 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
99.90%
|
|||
| Administration Time | 30 days | ||||
| Administration Dosage | 5 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | MIA PaCa-2 cells (G12C KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | MIA PaCa-2 (KRAS G12C) cell | CVCL_0428 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 10 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
100.00%
|
|||
| Administration Time | 30 days | ||||
| Administration Dosage | 10 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | MIA PaCa-2 cells (G12C KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | MIA PaCa-2 (KRAS G12C) cell | CVCL_0428 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 11 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
100.00%
|
|||
| Administration Time | 30 days | ||||
| Administration Dosage | 10 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | MDA-MB-231 cells (G13D KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (KRAS G13D) cell | CVCL_0062 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 12 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Albumin uptake rate |
6.67%
|
|||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | AsPC-1 cells (G12D KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | AsPC-1 (KRAS G12D) cell | L-929 cell line | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 13 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Lung cancer | ||||
| Efficacy Data | Albumin uptake rate |
12.80%
|
|||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | A549 cells (G12S KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Lung adenocarcinoma | A-549 (KRAS G12S) cell | CVCL_0023 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 14 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Albumin uptake rate |
51.57%
|
|||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | MDA-MB-231 cells (G13D KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (KRAS G13D) cell | CVCL_0062 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 15 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Albumin uptake rate |
52.30%
|
|||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | HCT116 cells (G13D KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Colon carcinoma | HCT 116 (KRAS G13D) cell | CVCL_0291 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 16 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Albumin uptake rate |
68.43%
|
|||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | MIA PaCa-2 cells (G12C KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | MIA PaCa-2 (KRAS G12C) cell | CVCL_0428 | ||
| Half life period | 8.51 ± 0.50 h | ||||
MPD3 [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Metastatic triple-negative breast cancer | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
48.40%
|
|||
| Administration Time | 3 weeks | ||||
| Administration Dosage | 3 mg/kg | ||||
| MOA of PDC |
To demonstrate our strategy, we developed a peptide-drug conjugate (PDC) composed of the caspase-3 cleavablepeptide sequence(DEVD), an albumin-binding functional moiety, and docetaxel linked to the peptide via self-immolated linker. Functionalization of albumin-binding domain enables intravenously injected PDC to spontaneously bind to circulatingserum albumin, which is employed as a drug carrier capable of inducing macropinocytic uptake of the conjugate. Furthermore, in order to combat tumoral heterogeneity, a unique peptide sequence of DEVD is deployed, which is a core component of the PDC that propagates bystander killing of cancer cells. More specifically, caspase-3 is triggered from albumin-metabolism induced apoptosis in PTEN-loss cancer cells, which in turn recognizes and activates extracellular PDC to release the payload. The released hydrophobic payload subsequently penetrates neighboring cancer cells, thereby inducing bystander killing in a non-selective manner, leading to the continual amplification ofin-situapoptosis. Additionally, to boost the apoptosis of initial cancer cells and caspase-3 expression, we used olaparib as a combination therapeutic agent. Given that olaparib is the first targeted therapy approved against metastatic TNBC with BRCA mutation, we reasoned that combination therapy with olaparib has the potential to induce apoptosis in BRCA mutant cancer cells and intensify tumoral apoptosis. In this study, we demonstrate the therapeutic potential of exploiting PTEN-loss drivenbiomimeticdrug delivery and the significant therapeutic benefit of combination therapy in the treatment of metastatic TNBC with altered PTEN and BRCA using our novel PDC.
Click to Show/Hide
|
||||
| Description |
To examine whether BRCA and PTEN alteration status affect the anti-tumor efficacy of olaparib/MPD3 combination, we performedin vivoexperiments on Hs578t-tumor and BT549-tumor bearing balb/cnude mice. As expected, olaparib monotherapy in Hs578t or BT549 tumors did not show detectable tumor suppressing efficacy, which could be ascribed to the poor sensitivity of olaparib in BRCA wild-type tumors. Interestingly, Hs578t tumors showed delayed onset of tumor growth inhibition effect by MPD3 monotherapy, producing 48.4% TGI rate on the last day of observation. Following an unexpected response to MPD3 in PTEN wild-type tumors, different control tumor specimens at day 1, 16, and 28 were examined by immunohistochemical staining to investigate tumor microenvironmental changes during the drug treatment course. We found evidence of increasing pattern of active caspase-3 expression over the course of time, which is thought to be associated with activation of localized extracellular prodrug and late tumor inhibiting efficacy in Hs578t tumors. Owing to the poor response to olaparib therapy, both MPD3 and olaparib/MPD3 displayed similar tumor inhibiting efficacies in Hs578t tumor bearing mice, suggesting that olaparib triggered apoptosis is needed to generate synergistic efficacy with MPD3. Lastly,in vivoexperiment results with BT549 tumor xenografted model confirmed the contribution of PTEN-loss alteration to thein vivoactivity of MPD3. MPD3 monotherapy was able to induce substantial tumor growth inhibition with 63.4% TGI. We ascribe these results mainly to the PTEN-loss induced upregulation of the macropinocytosis level, witnessed by significantly increased albumin uptake level in BT549cells. However, as with Hs578t xenograft model, the addition of olaparib did not cause significant tumor growth inhibition. Note that despite the comparable anti-cancer activity of olaparib/docetaxel to that of olaparib/MPD3, docetaxel combination treatment caused severebody weight loss, with 60% and 20% lethality in Hs578t and in BT549 tumor bearing mice, respectively. The results ofin vivoanti-cancer efficacies of MPD3 and olaparib in TNBC tumor models expressing different biomarkers are summarized inTable 1. Coefficients of drug interaction (CDI) values of the combination of olaparib/MPD3 were 0.04, 1.22 and 0.42 in MDA-MB-436, Hs578t, BT549 xenograft models, respectively, where CDI of <1, =1, or >1 indicates that the combination therapy is synergistic, additive, or antagonistic. These results provide compelling evidence that the BRCA and PTEN alteration status influence the efficacy of our combination strategy, showing that BRCA mutation and PTEN-loss contribute to the favorable anti-cancer efficacy of the strategy.
Click to Show/Hide
|
||||
| In Vivo Model | BRCA/PTEN wt Hs578t cells female BALB/cnude mice xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma of no special type | BRCA/PTEN WT Hs578t cell | CVCL_0332 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Metastatic triple-negative breast cancer | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
63.40%
|
|||
| Administration Time | 3 weeks | ||||
| Administration Dosage | 3 mg/kg | ||||
| MOA of PDC |
To demonstrate our strategy, we developed a peptide-drug conjugate (PDC) composed of the caspase-3 cleavablepeptide sequence(DEVD), an albumin-binding functional moiety, and docetaxel linked to the peptide via self-immolated linker. Functionalization of albumin-binding domain enables intravenously injected PDC to spontaneously bind to circulatingserum albumin, which is employed as a drug carrier capable of inducing macropinocytic uptake of the conjugate. Furthermore, in order to combat tumoral heterogeneity, a unique peptide sequence of DEVD is deployed, which is a core component of the PDC that propagates bystander killing of cancer cells. More specifically, caspase-3 is triggered from albumin-metabolism induced apoptosis in PTEN-loss cancer cells, which in turn recognizes and activates extracellular PDC to release the payload. The released hydrophobic payload subsequently penetrates neighboring cancer cells, thereby inducing bystander killing in a non-selective manner, leading to the continual amplification ofin-situapoptosis. Additionally, to boost the apoptosis of initial cancer cells and caspase-3 expression, we used olaparib as a combination therapeutic agent. Given that olaparib is the first targeted therapy approved against metastatic TNBC with BRCA mutation, we reasoned that combination therapy with olaparib has the potential to induce apoptosis in BRCA mutant cancer cells and intensify tumoral apoptosis. In this study, we demonstrate the therapeutic potential of exploiting PTEN-loss drivenbiomimeticdrug delivery and the significant therapeutic benefit of combination therapy in the treatment of metastatic TNBC with altered PTEN and BRCA using our novel PDC.
Click to Show/Hide
|
||||
| Description |
To examine whether BRCA and PTEN alteration status affect the anti-tumor efficacy of olaparib/MPD3 combination, we performedin vivoexperiments on Hs578t-tumor and BT549-tumor bearing balb/cnude mice. As expected, olaparib monotherapy in Hs578t or BT549 tumors did not show detectable tumor suppressing efficacy, which could be ascribed to the poor sensitivity of olaparib in BRCA wild-type tumors. Interestingly, Hs578t tumors showed delayed onset of tumor growth inhibition effect by MPD3 monotherapy, producing 48.4% TGI rate on the last day of observation. Following an unexpected response to MPD3 in PTEN wild-type tumors, different control tumor specimens at day 1, 16, and 28 were examined by immunohistochemical staining to investigate tumor microenvironmental changes during the drug treatment course. We found evidence of increasing pattern of active caspase-3 expression over the course of time, which is thought to be associated with activation of localized extracellular prodrug and late tumor inhibiting efficacy in Hs578t tumors. Owing to the poor response to olaparib therapy, both MPD3 and olaparib/MPD3 displayed similar tumor inhibiting efficacies in Hs578t tumor bearing mice, suggesting that olaparib triggered apoptosis is needed to generate synergistic efficacy with MPD3. Lastly,in vivoexperiment results with BT549 tumor xenografted model confirmed the contribution of PTEN-loss alteration to thein vivoactivity of MPD3. MPD3 monotherapy was able to induce substantial tumor growth inhibition with 63.4% TGI. We ascribe these results mainly to the PTEN-loss induced upregulation of the macropinocytosis level, witnessed by significantly increased albumin uptake level in BT549cells. However, as with Hs578t xenograft model, the addition of olaparib did not cause significant tumor growth inhibition. Note that despite the comparable anti-cancer activity of olaparib/docetaxel to that of olaparib/MPD3, docetaxel combination treatment caused severebody weight loss, with 60% and 20% lethality in Hs578t and in BT549 tumor bearing mice, respectively. The results ofin vivoanti-cancer efficacies of MPD3 and olaparib in TNBC tumor models expressing different biomarkers are summarized inTable 1. Coefficients of drug interaction (CDI) values of the combination of olaparib/MPD3 were 0.04, 1.22 and 0.42 in MDA-MB-436, Hs578t, BT549 xenograft models, respectively, where CDI of <1, =1, or >1 indicates that the combination therapy is synergistic, additive, or antagonistic. These results provide compelling evidence that the BRCA and PTEN alteration status influence the efficacy of our combination strategy, showing that BRCA mutation and PTEN-loss contribute to the favorable anti-cancer efficacy of the strategy.
Click to Show/Hide
|
||||
| In Vivo Model | BRCA wt BT549 cells female BALB/cnude mice xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma of no special type | BRCA WT BT-549 cell | CVCL_1092 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Metastatic triple-negative breast cancer | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
64.60%
|
|||
| Administration Time | 3 weeks | ||||
| Administration Dosage | 3 mg/kg | ||||
| MOA of PDC |
To demonstrate our strategy, we developed a peptide-drug conjugate (PDC) composed of the caspase-3 cleavablepeptide sequence(DEVD), an albumin-binding functional moiety, and docetaxel linked to the peptide via self-immolated linker. Functionalization of albumin-binding domain enables intravenously injected PDC to spontaneously bind to circulatingserum albumin, which is employed as a drug carrier capable of inducing macropinocytic uptake of the conjugate. Furthermore, in order to combat tumoral heterogeneity, a unique peptide sequence of DEVD is deployed, which is a core component of the PDC that propagates bystander killing of cancer cells. More specifically, caspase-3 is triggered from albumin-metabolism induced apoptosis in PTEN-loss cancer cells, which in turn recognizes and activates extracellular PDC to release the payload. The released hydrophobic payload subsequently penetrates neighboring cancer cells, thereby inducing bystander killing in a non-selective manner, leading to the continual amplification ofin-situapoptosis. Additionally, to boost the apoptosis of initial cancer cells and caspase-3 expression, we used olaparib as a combination therapeutic agent. Given that olaparib is the first targeted therapy approved against metastatic TNBC with BRCA mutation, we reasoned that combination therapy with olaparib has the potential to induce apoptosis in BRCA mutant cancer cells and intensify tumoral apoptosis. In this study, we demonstrate the therapeutic potential of exploiting PTEN-loss drivenbiomimeticdrug delivery and the significant therapeutic benefit of combination therapy in the treatment of metastatic TNBC with altered PTEN and BRCA using our novel PDC.
Click to Show/Hide
|
||||
| Description |
To examine whether BRCA and PTEN alteration status affect the anti-tumor efficacy of olaparib/MPD3 combination, we performedin vivoexperiments on Hs578t-tumor and BT549-tumor bearing balb/cnude mice. As expected, olaparib monotherapy in Hs578t or BT549 tumors did not show detectable tumor suppressing efficacy, which could be ascribed to the poor sensitivity of olaparib in BRCA wild-type tumors. Interestingly, Hs578t tumors showed delayed onset of tumor growth inhibition effect by MPD3 monotherapy, producing 48.4% TGI rate on the last day of observation. Following an unexpected response to MPD3 in PTEN wild-type tumors, different control tumor specimens at day 1, 16, and 28 were examined by immunohistochemical staining to investigate tumor microenvironmental changes during the drug treatment course. We found evidence of increasing pattern of active caspase-3 expression over the course of time, which is thought to be associated with activation of localized extracellular prodrug and late tumor inhibiting efficacy in Hs578t tumors. Owing to the poor response to olaparib therapy, both MPD3 and olaparib/MPD3 displayed similar tumor inhibiting efficacies in Hs578t tumor bearing mice, suggesting that olaparib triggered apoptosis is needed to generate synergistic efficacy with MPD3. Lastly,in vivoexperiment results with BT549 tumor xenografted model confirmed the contribution of PTEN-loss alteration to thein vivoactivity of MPD3. MPD3 monotherapy was able to induce substantial tumor growth inhibition with 63.4% TGI. We ascribe these results mainly to the PTEN-loss induced upregulation of the macropinocytosis level, witnessed by significantly increased albumin uptake level in BT549cells. However, as with Hs578t xenograft model, the addition of olaparib did not cause significant tumor growth inhibition. Note that despite the comparable anti-cancer activity of olaparib/docetaxel to that of olaparib/MPD3, docetaxel combination treatment caused severebody weight loss, with 60% and 20% lethality in Hs578t and in BT549 tumor bearing mice, respectively. The results ofin vivoanti-cancer efficacies of MPD3 and olaparib in TNBC tumor models expressing different biomarkers are summarized inTable 1. Coefficients of drug interaction (CDI) values of the combination of olaparib/MPD3 were 0.04, 1.22 and 0.42 in MDA-MB-436, Hs578t, BT549 xenograft models, respectively, where CDI of <1, =1, or >1 indicates that the combination therapy is synergistic, additive, or antagonistic. These results provide compelling evidence that the BRCA and PTEN alteration status influence the efficacy of our combination strategy, showing that BRCA mutation and PTEN-loss contribute to the favorable anti-cancer efficacy of the strategy.
Click to Show/Hide
|
||||
| In Vivo Model | BRCA1 mut MDA-MB-436 cells female BALB/cnude mice xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma of no special type | BRCA1 mut MDA-MB-436 cell | CVCL_0623 | ||
References