
Peptide Information
General Information of This Peptide
| Peptide ID |
PEP00123
|
|||||
|---|---|---|---|---|---|---|
| Peptide Name |
RGDKLAK
|
|||||
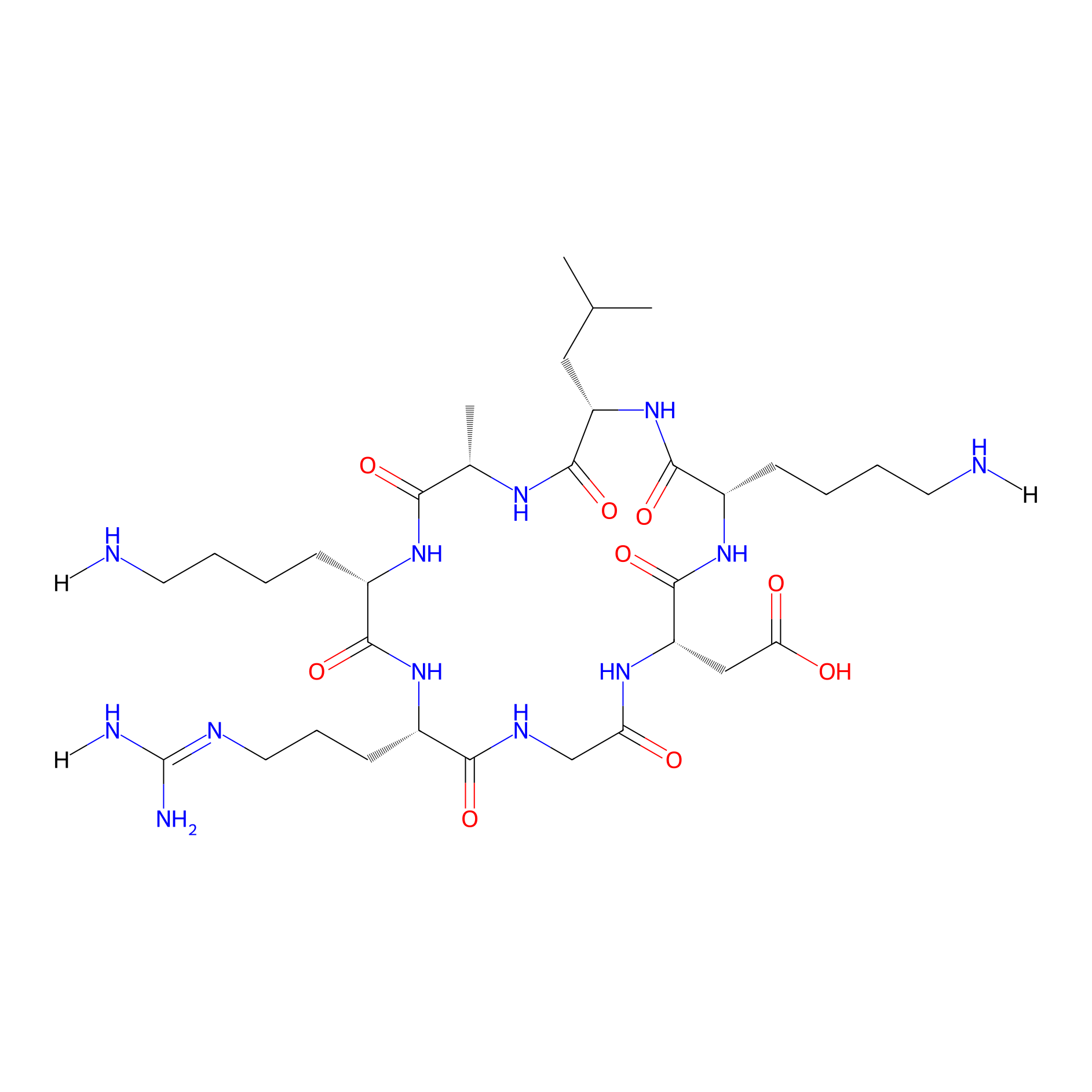
| Structure |
|
|||||
| Sequence |
RGDKLAK
|
|||||
| Peptide Type |
Cyclic
|
|||||
| Receptor Name |
Integrin alpha-V; Integrin beta-3 (ITGAV; ITGB3)
|
Receptor Info | ||||
| PDC Transmembrane Types | Cell-penetrating peptides (CPPs) | |||||
| Formula |
C33H60N12O9
|
|||||
| Isosmiles |
[H]NCCCC[C@@H]1NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCCN[H])NC(=O)[C@H](CC(=O)O)NC(=O)CNC(=O)[C@H](CCC/N=C(\N)N[H])NC1=O
|
|||||
| InChI |
InChI=1S/C33H60N12O9/c1-18(2)15-23-31(53)40-19(3)27(49)42-21(9-4-6-12-34)29(51)43-20(11-8-14-38-33(36)37)28(50)39-17-25(46)41-24(16-26(47)48)32(54)44-22(30(52)45-23)10-5-7-13-35/h18-24H,4-17,34-35H2,1-3H3,(H,39,50)(H,40,53)(H,41,46)(H,42,49)(H,43,51)(H,44,54)(H,45,52)(H,47,48)(H4,36,37,38)/t19-,20-,21-,22-,23-,24-/m0/s1
|
|||||
| InChIKey |
SIEBFGSFCKANAJ-BTNSXGMBSA-N
|
|||||
| Pharmaceutical Properties |
Molecule Weight
|
768.918
|
Polar area
|
357.44
|
||
|
Complexity
|
768.4606215
|
xlogp Value
|
-4.1227
|
|||
|
Heavy Count
|
54
|
Rot Bonds
|
18
|
|||
|
Hbond acc
|
11
|
Hbond Donor
|
12
|
|||
Each Peptide-drug Conjugate Related to This Peptide
Full Information of The Activity Data of The PDC(s) Related to This Peptide
CPP-SA-PTX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Malignant glioblastoma | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
20.55 ± 1.02 nM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
A highly sensitive, nontoxic, hydrophilic cell-penetrating peptide (CPP = c[RGDKLAK]) was selected for the construction of an effective peptide-drug conjugate (PDC). A hydrophobic drug paclitaxel (PTX) was successfully conjugated with CPP via ester linkage with succinic acid (SA) as a pH-cleavable linker moiety. The characterization techniques employed in this study indicate the >95% purity of the resulting PDC (CPP-SA-PTX). The in vitro studies show that our proposed PDC exhibits enhanced stability (˜90%) and cytotoxicity (EC50 = 8.32 ± 0.09 nM). Besides the excellent solubility of PDC in water, the PTX effect on positive β-tubulin-III indicates that the drug releases retained pharmacological properties. Additionally, in vivo, therapeutic-dose treatment reveals the prominent tumor-growth inhibitory effects (2.82-3.24-fold) of PDC in tumor mice models. Subsequently, these observations confirmed that our novel-designed PDC (CPP-SA-PTX) adduct may serve as a promising therapeutic agent to treat glioblastoma.
Click to Show/Hide
|
||||
| Description |
Furthermore, it was also observed that PDC shows time-dependent cytotoxicity against U87MG cells, as at 12 h drug treatment. PDC shows a slightly lower inhibitory effect on cell survival (EC50 = 25.82 ± 0.31 nM) than PTX alone (EC50 = 12.25 ± 0.13 nM), and CPP-SA alone shows higher EC50 = 54.37 ± 0.24 in U87MG cells. Besides this, it was previously observed that CPP-treated healthy VERO cells showed 88.05 ± 0.86% viable cells even using 10 uM concentration, indicating the specificity of CPP for glioblastoma cells. However, at 24 h incubation period, our PDC shows a significantly enhanced cell survival inhibitory effect with EC50 = 8.32 ± 0.09 nM, suggesting the delayed effect of PDC because of the time required for the intracellular pH to cause maximal cleavage of PTX from the PDC. Moreover, it also shows that CPP-SA has a lower cytotoxicity effect on glioblastoma cells compared with PTX alone and PDC, indicating its specificity as a carrier and selectivity for integrin receptors overexpressed on the cell surface. Additionally, we have also studied the potential of our novel-designed PDC to internalize into PTX-resistant glioblastoma (U87MG-PR) cells to show cytotoxic activity upon intracellular cleavage of PTX from CPP. The data presented in Figure 3b reveal approximately 15% viability increases in U87MG-PR cells compared with parent U87MG cells. It can be seen in the inset in Figure 3b that PTX alone (EC50 = 41.3 ± 1.5 nM) showed significantly 2-fold reduced cytotoxic activity in U87MG-PR cells in comparison with our novel-designed PDC having EC50 = 20.55 ± 1.02 nM. It was also observed in Figure 3b that at lower concentrations (0-10 nM) the viable cell count was ˜75%; however, it significantly decreased to ˜20% viability with an increase in concentration (40 nM) of the test samples, highlighting the dose-dependent cytotoxicity behavior of PDC in a 24 h incubation period.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U87MG-PR cell | CVCL_0022 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Malignant glioblastoma | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
25.82 ± 0.31 nM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
A highly sensitive, nontoxic, hydrophilic cell-penetrating peptide (CPP = c[RGDKLAK]) was selected for the construction of an effective peptide-drug conjugate (PDC). A hydrophobic drug paclitaxel (PTX) was successfully conjugated with CPP via ester linkage with succinic acid (SA) as a pH-cleavable linker moiety. The characterization techniques employed in this study indicate the >95% purity of the resulting PDC (CPP-SA-PTX). The in vitro studies show that our proposed PDC exhibits enhanced stability (˜90%) and cytotoxicity (EC50 = 8.32 ± 0.09 nM). Besides the excellent solubility of PDC in water, the PTX effect on positive β-tubulin-III indicates that the drug releases retained pharmacological properties. Additionally, in vivo, therapeutic-dose treatment reveals the prominent tumor-growth inhibitory effects (2.82-3.24-fold) of PDC in tumor mice models. Subsequently, these observations confirmed that our novel-designed PDC (CPP-SA-PTX) adduct may serve as a promising therapeutic agent to treat glioblastoma.
Click to Show/Hide
|
||||
| Description |
Furthermore, it was also observed that PDC shows time-dependent cytotoxicity against U87MG cells, as at 12 h drug treatment. PDC shows a slightly lower inhibitory effect on cell survival (EC50 = 25.82 ± 0.31 nM) than PTX alone (EC50 = 12.25 ± 0.13 nM), and CPP-SA alone shows higher EC50 = 54.37 ± 0.24 in U87MG cells. Besides this, it was previously observed that CPP-treated healthy VERO cells showed 88.05 ± 0.86% viable cells even using 10 uM concentration, indicating the specificity of CPP for glioblastoma cells. However, at 24 h incubation period, our PDC shows a significantly enhanced cell survival inhibitory effect with EC50 = 8.32 ± 0.09 nM, suggesting the delayed effect of PDC because of the time required for the intracellular pH to cause maximal cleavage of PTX from the PDC. Moreover, it also shows that CPP-SA has a lower cytotoxicity effect on glioblastoma cells compared with PTX alone and PDC, indicating its specificity as a carrier and selectivity for integrin receptors overexpressed on the cell surface. Additionally, we have also studied the potential of our novel-designed PDC to internalize into PTX-resistant glioblastoma (U87MG-PR) cells to show cytotoxic activity upon intracellular cleavage of PTX from CPP. The data presented in Figure 3b reveal approximately 15% viability increases in U87MG-PR cells compared with parent U87MG cells. It can be seen in the inset in Figure 3b that PTX alone (EC50 = 41.3 ± 1.5 nM) showed significantly 2-fold reduced cytotoxic activity in U87MG-PR cells in comparison with our novel-designed PDC having EC50 = 20.55 ± 1.02 nM. It was also observed in Figure 3b that at lower concentrations (0-10 nM) the viable cell count was ˜75%; however, it significantly decreased to ˜20% viability with an increase in concentration (40 nM) of the test samples, highlighting the dose-dependent cytotoxicity behavior of PDC in a 24 h incubation period.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
References