
Linker Information
General Information of This Linker
| Linker ID |
LIN00002
|
|||||
|---|---|---|---|---|---|---|
| Linker Name |
GFLG
|
|||||
| Linker Type |
Enzyme-sensitive linkers
|
|||||
| Structure |
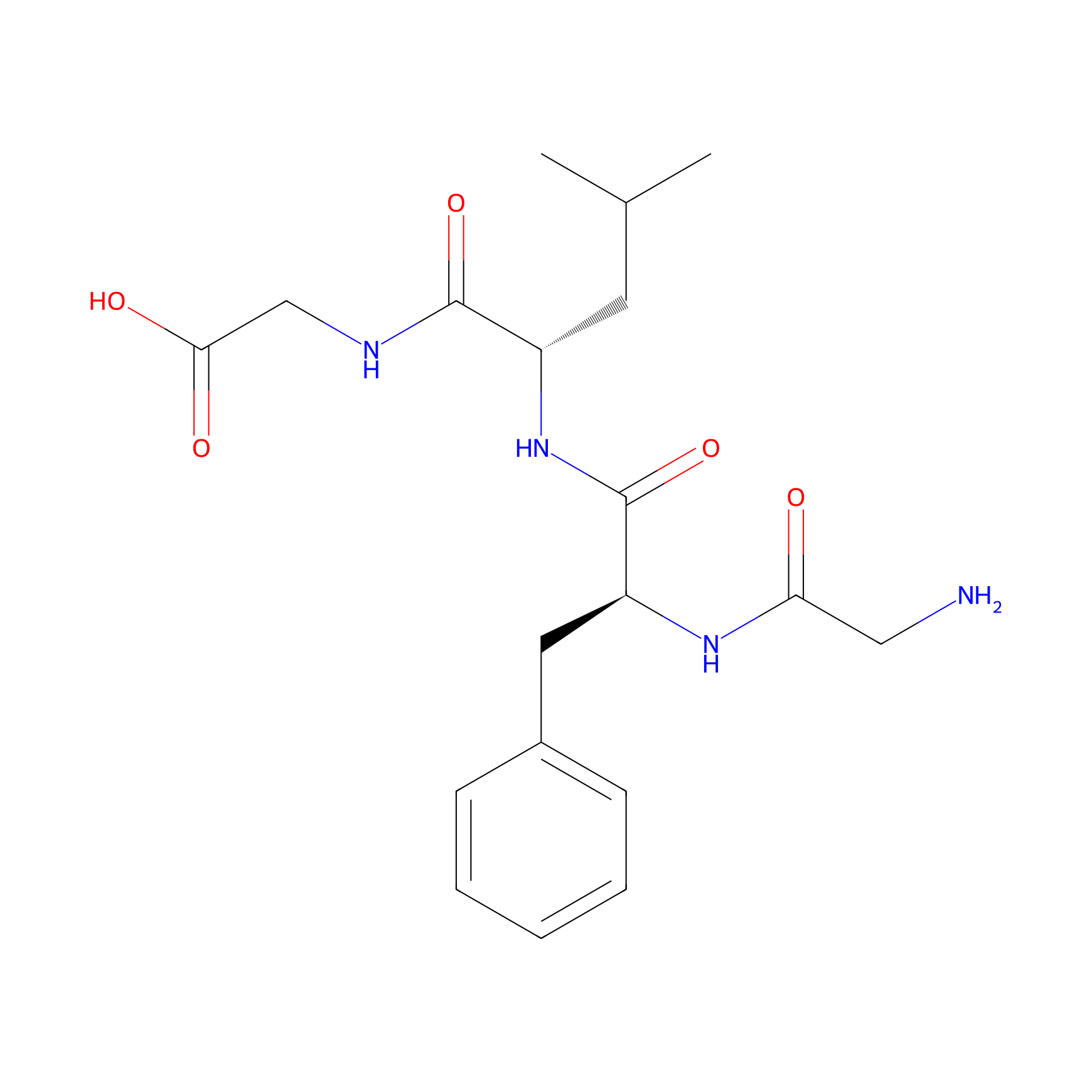
|
|||||
| Formula |
C19H28N4O5
|
|||||
| #Ro5 Violations (Lipinski): 1 | Molecular Weight (mw) | 392.4 | ||||
| Lipid-water partition coefficient (xlogp) | -1.9 | |||||
| Hydrogen Bond Donor Count (hbonddonor) | 5 | |||||
| Hydrogen Bond Acceptor Count (hbondacc) | 6 | |||||
| Rotatable Bond Count (rotbonds) | 11 | |||||
| PubChem CID | ||||||
| Canonical smiles |
CC(C)CC(C(=O)NCC(=O)O)NC(=O)C(CC1=CC=CC=C1)NC(=O)CN
|
|||||
| InChI |
InChI=1S/C19H28N4O5/c1-12(2)8-14(18(27)21-11-17(25)26)23-19(28)15(22-16(24)10-20)9-13-6-4-3-5-7-13/h3-7,12,14-15H,8-11,20H2,1-2H3,(H,21,27)(H,22,24)(H,23,28)(H,25,26)/t14-,15-/m0/s1
|
|||||
| InChIKey |
WEZDRVHTDXTVLT-GJZGRUSLSA-N
|
|||||
| IUPAC Name |
2-[[(2S)-2-[[(2S)-2-[(2-aminoacetyl)amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]acetic acid
|
|||||
Each Peptide-drug Conjugate Related to This Linker
Full Information of The Activity Data of The PDC(s) Related to This Linker
Tesaglitazar-[F7, P34]-NPY [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Type 2 diabetes | ||||
| Efficacy Data | Triglycerides level |
0.65
|
|||
| Administration Time | Every day; over 8 days | ||||
| Administration Dosage | 2.5 µM | ||||
| MOA of PDC |
In vitro studies revealed that the tesaglitazar-[F7, P34]-NPY conjugate selectively activates PPARγ in NPY1R-expressing cells and enhances adipocyte differentiation and adiponectin expression in adipocyte precursor cells.Additionally, tesa-NPY induces adipocyte differentiation in vivo.
|
||||
| Description |
The influence of tesa, tesa-NPY (3), and [F7, P34]-NPY (2) on the plasma lipids was also analyzed (Figure 9). The vehicle/untreated db/db mice showed elevated levels of triglycerides and free fatty acids (FFA) compared to the lean C57BL/6N mice. Treatment with tesa and tesa-NPY (3) led to a normalization of the triglycerides, FFA, whereas [F7, P34]-NPY (2) and vehicle/untreated had no influence on the lipid metabolism (Figure 9A/B). The cholesterol levels were unchanged by any treatment as these levels were also comparable in the untreated db/db mice compared to the lean mice (Figure 9C).
Click to Show/Hide
|
||||
| In Vivo Model | db/db mice model. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Type 2 diabetes | ||||
| Efficacy Data | Triglycerides level |
0.9
|
|||
| Administration Time | Every day; over 8 days | ||||
| Administration Dosage | 2.5 µM | ||||
| MOA of PDC |
In vitro studies revealed that the tesaglitazar-[F7, P34]-NPY conjugate selectively activates PPARγ in NPY1R-expressing cells and enhances adipocyte differentiation and adiponectin expression in adipocyte precursor cells.Additionally, tesa-NPY induces adipocyte differentiation in vivo.
|
||||
| Description |
The influence of tesa, tesa-NPY (3), and [F7, P34]-NPY (2) on the plasma lipids was also analyzed (Figure 9). The vehicle/untreated db/db mice showed elevated levels of triglycerides and free fatty acids (FFA) compared to the lean C57BL/6N mice. Treatment with tesa and tesa-NPY (3) led to a normalization of the triglycerides, FFA, whereas [F7, P34]-NPY (2) and vehicle/untreated had no influence on the lipid metabolism (Figure 9A/B). The cholesterol levels were unchanged by any treatment as these levels were also comparable in the untreated db/db mice compared to the lean mice (Figure 9C).
Click to Show/Hide
|
||||
| In Vivo Model | db/db mice model. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Type 2 diabetes | ||||
| Efficacy Data | Mcp-1 level |
305 pg/mL
|
|||
| Administration Time | Every day; over 8 days | ||||
| Administration Dosage | 2.5 µM | ||||
| MOA of PDC |
In vitro studies revealed that the tesaglitazar-[F7, P34]-NPY conjugate selectively activates PPARγ in NPY1R-expressing cells and enhances adipocyte differentiation and adiponectin expression in adipocyte precursor cells.Additionally, tesa-NPY induces adipocyte differentiation in vivo.
|
||||
| Description |
Whereas in the vehicle/untreated mice, the HbA1C values increased by approximately 2%, a graduated reduced increase was seen for [F7, P34]-NPY (2), peptide conjugate (3), and tesa (Figure 8A). Body temperature, which decreased to 35 C in the vehicle/untreated db/db controls, was normalized to 36 C in all of the treated mice including the mice treated with [F7, P34]-NPY (2) (Figure 8B). Treatment with tesa and tesa-NPY (3) led to normalization of the plasma concentration of ketone bodies and adiponectin, whereas [F7, P34]-NPY (2) and vehicle/untreated showed no effect (Figure 8C/E). Treatment had no major influence on the insulin and Mcp-1 levels (Figure 8D/G). The serum leptin concentration was reduced in the mice treated with tesa, whereas no reduction was detectable in all of the other treated mice (Figure 8F).
Click to Show/Hide
|
||||
| In Vivo Model | db/db mice model. | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Type 2 diabetes | ||||
| Efficacy Data | Leptin level |
82 ng/mL
|
|||
| Administration Time | Every day; over 8 days | ||||
| Administration Dosage | 2.5 µM | ||||
| MOA of PDC |
In vitro studies revealed that the tesaglitazar-[F7, P34]-NPY conjugate selectively activates PPARγ in NPY1R-expressing cells and enhances adipocyte differentiation and adiponectin expression in adipocyte precursor cells.Additionally, tesa-NPY induces adipocyte differentiation in vivo.
|
||||
| Description |
Whereas in the vehicle/untreated mice, the HbA1C values increased by approximately 2%, a graduated reduced increase was seen for [F7, P34]-NPY (2), peptide conjugate (3), and tesa (Figure 8A). Body temperature, which decreased to 35 C in the vehicle/untreated db/db controls, was normalized to 36 C in all of the treated mice including the mice treated with [F7, P34]-NPY (2) (Figure 8B). Treatment with tesa and tesa-NPY (3) led to normalization of the plasma concentration of ketone bodies and adiponectin, whereas [F7, P34]-NPY (2) and vehicle/untreated showed no effect (Figure 8C/E). Treatment had no major influence on the insulin and Mcp-1 levels (Figure 8D/G). The serum leptin concentration was reduced in the mice treated with tesa, whereas no reduction was detectable in all of the other treated mice (Figure 8F).
Click to Show/Hide
|
||||
| In Vivo Model | db/db mice model. | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Type 2 diabetes | ||||
| Efficacy Data | Ketone level |
0.5 mM/L
|
|||
| Administration Time | Every day; over 8 days | ||||
| Administration Dosage | 2.5 µM | ||||
| MOA of PDC |
In vitro studies revealed that the tesaglitazar-[F7, P34]-NPY conjugate selectively activates PPARγ in NPY1R-expressing cells and enhances adipocyte differentiation and adiponectin expression in adipocyte precursor cells.Additionally, tesa-NPY induces adipocyte differentiation in vivo.
|
||||
| Description |
Whereas in the vehicle/untreated mice, the HbA1C values increased by approximately 2%, a graduated reduced increase was seen for [F7, P34]-NPY (2), peptide conjugate (3), and tesa (Figure 8A). Body temperature, which decreased to 35 C in the vehicle/untreated db/db controls, was normalized to 36 C in all of the treated mice including the mice treated with [F7, P34]-NPY (2) (Figure 8B). Treatment with tesa and tesa-NPY (3) led to normalization of the plasma concentration of ketone bodies and adiponectin, whereas [F7, P34]-NPY (2) and vehicle/untreated showed no effect (Figure 8C/E). Treatment had no major influence on the insulin and Mcp-1 levels (Figure 8D/G). The serum leptin concentration was reduced in the mice treated with tesa, whereas no reduction was detectable in all of the other treated mice (Figure 8F).
Click to Show/Hide
|
||||
| In Vivo Model | db/db mice model. | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Type 2 diabetes | ||||
| Efficacy Data | Insulin level |
2 ng/mL
|
|||
| Administration Time | Every day; over 8 days | ||||
| Administration Dosage | 2.5 µM | ||||
| MOA of PDC |
In vitro studies revealed that the tesaglitazar-[F7, P34]-NPY conjugate selectively activates PPARγ in NPY1R-expressing cells and enhances adipocyte differentiation and adiponectin expression in adipocyte precursor cells.Additionally, tesa-NPY induces adipocyte differentiation in vivo.
|
||||
| Description |
Whereas in the vehicle/untreated mice, the HbA1C values increased by approximately 2%, a graduated reduced increase was seen for [F7, P34]-NPY (2), peptide conjugate (3), and tesa (Figure 8A). Body temperature, which decreased to 35 C in the vehicle/untreated db/db controls, was normalized to 36 C in all of the treated mice including the mice treated with [F7, P34]-NPY (2) (Figure 8B). Treatment with tesa and tesa-NPY (3) led to normalization of the plasma concentration of ketone bodies and adiponectin, whereas [F7, P34]-NPY (2) and vehicle/untreated showed no effect (Figure 8C/E). Treatment had no major influence on the insulin and Mcp-1 levels (Figure 8D/G). The serum leptin concentration was reduced in the mice treated with tesa, whereas no reduction was detectable in all of the other treated mice (Figure 8F).
Click to Show/Hide
|
||||
| In Vivo Model | db/db mice model. | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Type 2 diabetes | ||||
| Efficacy Data | HbA1C change |
1.05%
|
|||
| Administration Time | Every day; over 8 days | ||||
| Administration Dosage | 2.5 µM | ||||
| MOA of PDC |
In vitro studies revealed that the tesaglitazar-[F7, P34]-NPY conjugate selectively activates PPARγ in NPY1R-expressing cells and enhances adipocyte differentiation and adiponectin expression in adipocyte precursor cells.Additionally, tesa-NPY induces adipocyte differentiation in vivo.
|
||||
| Description |
Whereas in the vehicle/untreated mice, the HbA1C values increased by approximately 2%, a graduated reduced increase was seen for [F7, P34]-NPY (2), peptide conjugate (3), and tesa (Figure 8A). Body temperature, which decreased to 35 C in the vehicle/untreated db/db controls, was normalized to 36 C in all of the treated mice including the mice treated with [F7, P34]-NPY (2) (Figure 8B). Treatment with tesa and tesa-NPY (3) led to normalization of the plasma concentration of ketone bodies and adiponectin, whereas [F7, P34]-NPY (2) and vehicle/untreated showed no effect (Figure 8C/E). Treatment had no major influence on the insulin and Mcp-1 levels (Figure 8D/G). The serum leptin concentration was reduced in the mice treated with tesa, whereas no reduction was detectable in all of the other treated mice (Figure 8F).
Click to Show/Hide
|
||||
| In Vivo Model | db/db mice model. | ||||
| Experiment 8 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Type 2 diabetes | ||||
| Efficacy Data | Cholesterol level |
2.8
|
|||
| Administration Time | Every day; over 8 days | ||||
| Administration Dosage | 2.5 µM | ||||
| MOA of PDC |
In vitro studies revealed that the tesaglitazar-[F7, P34]-NPY conjugate selectively activates PPARγ in NPY1R-expressing cells and enhances adipocyte differentiation and adiponectin expression in adipocyte precursor cells.Additionally, tesa-NPY induces adipocyte differentiation in vivo.
|
||||
| Description |
The influence of tesa, tesa-NPY (3), and [F7, P34]-NPY (2) on the plasma lipids was also analyzed (Figure 9). The vehicle/untreated db/db mice showed elevated levels of triglycerides and free fatty acids (FFA) compared to the lean C57BL/6N mice. Treatment with tesa and tesa-NPY (3) led to a normalization of the triglycerides, FFA, whereas [F7, P34]-NPY (2) and vehicle/untreated had no influence on the lipid metabolism (Figure 9A/B). The cholesterol levels were unchanged by any treatment as these levels were also comparable in the untreated db/db mice compared to the lean mice (Figure 9C).
Click to Show/Hide
|
||||
| In Vivo Model | db/db mice model. | ||||
| Experiment 9 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Type 2 diabetes | ||||
| Efficacy Data | Body weigth change |
10%
|
|||
| Administration Time | Every day; over 8 days | ||||
| Administration Dosage | 2.5 µM | ||||
| Evaluation Method | Body weigth change assay | ||||
| MOA of PDC |
In vitro studies revealed that the tesaglitazar-[F7, P34]-NPY conjugate selectively activates PPARγ in NPY7R-expressing cells and enhances adipocyte differentiation and adiponectin expression in adipocyte precursor cells.Additionally, tesa-NPY induces adipocyte differentiation in vivo.
|
||||
| Description |
The mice treated with tesa and tesa-NPY (3) did not change significantly, whereas their littermates treated with [F7, P34]-NPY (2) or vehicle/untreated lost approximately 3% of their body weight (Figure 7A).
|
||||
| In Vivo Model | db/db mice model. | ||||
PDC-G2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Uptake Concentration (UC50) |
10.5 µM
|
|||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
We quantified the ability of the produced PDCs to promote the internalisation of daunorubicin after binding to GRP-R by flow cytometry. Each bioconjugate was incubated for 1.5 h in four concentrations (25 uM, 12.5 uM, 6.25 uM and 3.125 uM), with the three cell lines that we have also used for the evaluation of the cytostatic effect. For better comparison, the uptake was described as the necessary concentration to internalise 50% of the compound (UC50). The uptake reflects the GRP-R protein expression: the lower UC50 values, hence the highest uptakes, are noticed in the human breast cancer cell line MDA-MB-453, whereas they are comparable in the two other cell lines. As far as the internalisation of the individual conjugates is concerned, L5 and L6 are the most promising ones in these three cell lines. Notably, both hold the LRRY spacer and the Sta13. L1 and L2 have satisfactory UC50s, too. On the other hand, majority of the conjugates with the GFLG spacer are poorly internalised.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Uptake Concentration (UC50) |
17.04 µM
|
|||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
We quantified the ability of the produced PDCs to promote the internalisation of daunorubicin after binding to GRP-R by flow cytometry. Each bioconjugate was incubated for 1.5 h in four concentrations (25 uM, 12.5 uM, 6.25 uM and 3.125 uM), with the three cell lines that we have also used for the evaluation of the cytostatic effect. For better comparison, the uptake was described as the necessary concentration to internalise 50% of the compound (UC50). The uptake reflects the GRP-R protein expression: the lower UC50 values, hence the highest uptakes, are noticed in the human breast cancer cell line MDA-MB-453, whereas they are comparable in the two other cell lines. As far as the internalisation of the individual conjugates is concerned, L5 and L6 are the most promising ones in these three cell lines. Notably, both hold the LRRY spacer and the Sta13. L1 and L2 have satisfactory UC50s, too. On the other hand, majority of the conjugates with the GFLG spacer are poorly internalised.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-453 cell | CVCL_0418 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Uptake Concentration (UC50) | > 25 µM | |||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
We quantified the ability of the produced PDCs to promote the internalisation of daunorubicin after binding to GRP-R by flow cytometry. Each bioconjugate was incubated for 1.5 h in four concentrations (25 uM, 12.5 uM, 6.25 uM and 3.125 uM), with the three cell lines that we have also used for the evaluation of the cytostatic effect. For better comparison, the uptake was described as the necessary concentration to internalise 50% of the compound (UC50). The uptake reflects the GRP-R protein expression: the lower UC50 values, hence the highest uptakes, are noticed in the human breast cancer cell line MDA-MB-453, whereas they are comparable in the two other cell lines. As far as the internalisation of the individual conjugates is concerned, L5 and L6 are the most promising ones in these three cell lines. Notably, both hold the LRRY spacer and the Sta13. L1 and L2 have satisfactory UC50s, too. On the other hand, majority of the conjugates with the GFLG spacer are poorly internalised.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
5.98 ± 2.10 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
The cytostatic effect of the conjugates and the free peptides was evaluated on the three mentioned human cancer cell lines expressing GRP-R. The free Dau was used as a positive control for comparison purposes; it displays an IC50 in the high nanomolar range. Overall, the conjugates containing the LRRY spacer have a higher cytostatic effect than the ones with the GFLG spacer. L1, which has the original BBN (7-14) sequence, and L5, bearing a D-Phe in position 6 and the Sta13-Leu14 bond at the C-terminus, shows the best activity in all three cell lines, with IC50 values in the low micromolar range. The Gly11/-Ala11 and Leu13-Met14/Sta13-Nle14 substitutions, which led to a new BBN (6-14) peptide sequence, held by the conjugate L6, affect the activity only slightly. Contrarily, the two free peptides, bearing the sequences of L5 and L6, do not show any activity on any of the cell lines. The lack of toxicity of the conjugates on healthy cells was checked on MRC-5 human fibroblasts, proving that they are non-toxic on non-cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
7.29 ± 3.41 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
The cytostatic effect of the conjugates and the free peptides was evaluated on the three mentioned human cancer cell lines expressing GRP-R. The free Dau was used as a positive control for comparison purposes; it displays an IC50 in the high nanomolar range. Overall, the conjugates containing the LRRY spacer have a higher cytostatic effect than the ones with the GFLG spacer. L1, which has the original BBN (7-14) sequence, and L5, bearing a D-Phe in position 6 and the Sta13-Leu14 bond at the C-terminus, shows the best activity in all three cell lines, with IC50 values in the low micromolar range. The Gly11/-Ala11 and Leu13-Met14/Sta13-Nle14 substitutions, which led to a new BBN (6-14) peptide sequence, held by the conjugate L6, affect the activity only slightly. Contrarily, the two free peptides, bearing the sequences of L5 and L6, do not show any activity on any of the cell lines. The lack of toxicity of the conjugates on healthy cells was checked on MRC-5 human fibroblasts, proving that they are non-toxic on non-cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
12.72 ± 1.22 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
The cytostatic effect of the conjugates and the free peptides was evaluated on the three mentioned human cancer cell lines expressing GRP-R. The free Dau was used as a positive control for comparison purposes; it displays an IC50 in the high nanomolar range. Overall, the conjugates containing the LRRY spacer have a higher cytostatic effect than the ones with the GFLG spacer. L1, which has the original BBN (7-14) sequence, and L5, bearing a D-Phe in position 6 and the Sta13-Leu14 bond at the C-terminus, shows the best activity in all three cell lines, with IC50 values in the low micromolar range. The Gly11/-Ala11 and Leu13-Met14/Sta13-Nle14 substitutions, which led to a new BBN (6-14) peptide sequence, held by the conjugate L6, affect the activity only slightly. Contrarily, the two free peptides, bearing the sequences of L5 and L6, do not show any activity on any of the cell lines. The lack of toxicity of the conjugates on healthy cells was checked on MRC-5 human fibroblasts, proving that they are non-toxic on non-cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-453 cell | CVCL_0418 | ||
PDC-G4 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Uptake Concentration (UC50) |
20.22 µM
|
|||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
We quantified the ability of the produced PDCs to promote the internalisation of daunorubicin after binding to GRP-R by flow cytometry. Each bioconjugate was incubated for 1.5 h in four concentrations (25 uM, 12.5 uM, 6.25 uM and 3.125 uM), with the three cell lines that we have also used for the evaluation of the cytostatic effect. For better comparison, the uptake was described as the necessary concentration to internalise 50% of the compound (UC50). The uptake reflects the GRP-R protein expression: the lower UC50 values, hence the highest uptakes, are noticed in the human breast cancer cell line MDA-MB-453, whereas they are comparable in the two other cell lines. As far as the internalisation of the individual conjugates is concerned, L5 and L6 are the most promising ones in these three cell lines. Notably, both hold the LRRY spacer and the Sta13. L1 and L2 have satisfactory UC50s, too. On the other hand, majority of the conjugates with the GFLG spacer are poorly internalised.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-453 cell | CVCL_0418 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Uptake Concentration (UC50) |
23.33 µM
|
|||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
We quantified the ability of the produced PDCs to promote the internalisation of daunorubicin after binding to GRP-R by flow cytometry. Each bioconjugate was incubated for 1.5 h in four concentrations (25 uM, 12.5 uM, 6.25 uM and 3.125 uM), with the three cell lines that we have also used for the evaluation of the cytostatic effect. For better comparison, the uptake was described as the necessary concentration to internalise 50% of the compound (UC50). The uptake reflects the GRP-R protein expression: the lower UC50 values, hence the highest uptakes, are noticed in the human breast cancer cell line MDA-MB-453, whereas they are comparable in the two other cell lines. As far as the internalisation of the individual conjugates is concerned, L5 and L6 are the most promising ones in these three cell lines. Notably, both hold the LRRY spacer and the Sta13. L1 and L2 have satisfactory UC50s, too. On the other hand, majority of the conjugates with the GFLG spacer are poorly internalised.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Uptake Concentration (UC50) | > 25 µM | |||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
We quantified the ability of the produced PDCs to promote the internalisation of daunorubicin after binding to GRP-R by flow cytometry. Each bioconjugate was incubated for 1.5 h in four concentrations (25 uM, 12.5 uM, 6.25 uM and 3.125 uM), with the three cell lines that we have also used for the evaluation of the cytostatic effect. For better comparison, the uptake was described as the necessary concentration to internalise 50% of the compound (UC50). The uptake reflects the GRP-R protein expression: the lower UC50 values, hence the highest uptakes, are noticed in the human breast cancer cell line MDA-MB-453, whereas they are comparable in the two other cell lines. As far as the internalisation of the individual conjugates is concerned, L5 and L6 are the most promising ones in these three cell lines. Notably, both hold the LRRY spacer and the Sta13. L1 and L2 have satisfactory UC50s, too. On the other hand, majority of the conjugates with the GFLG spacer are poorly internalised.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
5.73 ± 0.24 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
The cytostatic effect of the conjugates and the free peptides was evaluated on the three mentioned human cancer cell lines expressing GRP-R. The free Dau was used as a positive control for comparison purposes; it displays an IC50 in the high nanomolar range. Overall, the conjugates containing the LRRY spacer have a higher cytostatic effect than the ones with the GFLG spacer. L1, which has the original BBN (7-14) sequence, and L5, bearing a D-Phe in position 6 and the Sta13-Leu14 bond at the C-terminus, shows the best activity in all three cell lines, with IC50 values in the low micromolar range. The Gly11/-Ala11 and Leu13-Met14/Sta13-Nle14 substitutions, which led to a new BBN (6-14) peptide sequence, held by the conjugate L6, affect the activity only slightly. Contrarily, the two free peptides, bearing the sequences of L5 and L6, do not show any activity on any of the cell lines. The lack of toxicity of the conjugates on healthy cells was checked on MRC-5 human fibroblasts, proving that they are non-toxic on non-cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
7.37 ± 1.28 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
The cytostatic effect of the conjugates and the free peptides was evaluated on the three mentioned human cancer cell lines expressing GRP-R. The free Dau was used as a positive control for comparison purposes; it displays an IC50 in the high nanomolar range. Overall, the conjugates containing the LRRY spacer have a higher cytostatic effect than the ones with the GFLG spacer. L1, which has the original BBN (7-14) sequence, and L5, bearing a D-Phe in position 6 and the Sta13-Leu14 bond at the C-terminus, shows the best activity in all three cell lines, with IC50 values in the low micromolar range. The Gly11/-Ala11 and Leu13-Met14/Sta13-Nle14 substitutions, which led to a new BBN (6-14) peptide sequence, held by the conjugate L6, affect the activity only slightly. Contrarily, the two free peptides, bearing the sequences of L5 and L6, do not show any activity on any of the cell lines. The lack of toxicity of the conjugates on healthy cells was checked on MRC-5 human fibroblasts, proving that they are non-toxic on non-cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-453 cell | CVCL_0418 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
20.98 ± 0.55 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
The cytostatic effect of the conjugates and the free peptides was evaluated on the three mentioned human cancer cell lines expressing GRP-R. The free Dau was used as a positive control for comparison purposes; it displays an IC50 in the high nanomolar range. Overall, the conjugates containing the LRRY spacer have a higher cytostatic effect than the ones with the GFLG spacer. L1, which has the original BBN (7-14) sequence, and L5, bearing a D-Phe in position 6 and the Sta13-Leu14 bond at the C-terminus, shows the best activity in all three cell lines, with IC50 values in the low micromolar range. The Gly11/-Ala11 and Leu13-Met14/Sta13-Nle14 substitutions, which led to a new BBN (6-14) peptide sequence, held by the conjugate L6, affect the activity only slightly. Contrarily, the two free peptides, bearing the sequences of L5 and L6, do not show any activity on any of the cell lines. The lack of toxicity of the conjugates on healthy cells was checked on MRC-5 human fibroblasts, proving that they are non-toxic on non-cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
PDC-G3 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Uptake Concentration (UC50) |
22.59 µM
|
|||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
We quantified the ability of the produced PDCs to promote the internalisation of daunorubicin after binding to GRP-R by flow cytometry. Each bioconjugate was incubated for 1.5 h in four concentrations (25 uM, 12.5 uM, 6.25 uM and 3.125 uM), with the three cell lines that we have also used for the evaluation of the cytostatic effect. For better comparison, the uptake was described as the necessary concentration to internalise 50% of the compound (UC50). The uptake reflects the GRP-R protein expression: the lower UC50 values, hence the highest uptakes, are noticed in the human breast cancer cell line MDA-MB-453, whereas they are comparable in the two other cell lines. As far as the internalisation of the individual conjugates is concerned, L5 and L6 are the most promising ones in these three cell lines. Notably, both hold the LRRY spacer and the Sta13. L1 and L2 have satisfactory UC50s, too. On the other hand, majority of the conjugates with the GFLG spacer are poorly internalised.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-453 cell | CVCL_0418 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Uptake Concentration (UC50) | > 25 µM | |||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
We quantified the ability of the produced PDCs to promote the internalisation of daunorubicin after binding to GRP-R by flow cytometry. Each bioconjugate was incubated for 1.5 h in four concentrations (25 uM, 12.5 uM, 6.25 uM and 3.125 uM), with the three cell lines that we have also used for the evaluation of the cytostatic effect. For better comparison, the uptake was described as the necessary concentration to internalise 50% of the compound (UC50). The uptake reflects the GRP-R protein expression: the lower UC50 values, hence the highest uptakes, are noticed in the human breast cancer cell line MDA-MB-453, whereas they are comparable in the two other cell lines. As far as the internalisation of the individual conjugates is concerned, L5 and L6 are the most promising ones in these three cell lines. Notably, both hold the LRRY spacer and the Sta13. L1 and L2 have satisfactory UC50s, too. On the other hand, majority of the conjugates with the GFLG spacer are poorly internalised.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Uptake Concentration (UC50) | > 25 µM | |||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
We quantified the ability of the produced PDCs to promote the internalisation of daunorubicin after binding to GRP-R by flow cytometry. Each bioconjugate was incubated for 1.5 h in four concentrations (25 uM, 12.5 uM, 6.25 uM and 3.125 uM), with the three cell lines that we have also used for the evaluation of the cytostatic effect. For better comparison, the uptake was described as the necessary concentration to internalise 50% of the compound (UC50). The uptake reflects the GRP-R protein expression: the lower UC50 values, hence the highest uptakes, are noticed in the human breast cancer cell line MDA-MB-453, whereas they are comparable in the two other cell lines. As far as the internalisation of the individual conjugates is concerned, L5 and L6 are the most promising ones in these three cell lines. Notably, both hold the LRRY spacer and the Sta13. L1 and L2 have satisfactory UC50s, too. On the other hand, majority of the conjugates with the GFLG spacer are poorly internalised.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4.55 ± 0.76 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
The cytostatic effect of the conjugates and the free peptides was evaluated on the three mentioned human cancer cell lines expressing GRP-R. The free Dau was used as a positive control for comparison purposes; it displays an IC50 in the high nanomolar range. Overall, the conjugates containing the LRRY spacer have a higher cytostatic effect than the ones with the GFLG spacer. L1, which has the original BBN (7-14) sequence, and L5, bearing a D-Phe in position 6 and the Sta13-Leu14 bond at the C-terminus, shows the best activity in all three cell lines, with IC50 values in the low micromolar range. The Gly11/-Ala11 and Leu13-Met14/Sta13-Nle14 substitutions, which led to a new BBN (6-14) peptide sequence, held by the conjugate L6, affect the activity only slightly. Contrarily, the two free peptides, bearing the sequences of L5 and L6, do not show any activity on any of the cell lines. The lack of toxicity of the conjugates on healthy cells was checked on MRC-5 human fibroblasts, proving that they are non-toxic on non-cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
8.78 ± 0.97 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
The cytostatic effect of the conjugates and the free peptides was evaluated on the three mentioned human cancer cell lines expressing GRP-R. The free Dau was used as a positive control for comparison purposes; it displays an IC50 in the high nanomolar range. Overall, the conjugates containing the LRRY spacer have a higher cytostatic effect than the ones with the GFLG spacer. L1, which has the original BBN (7-14) sequence, and L5, bearing a D-Phe in position 6 and the Sta13-Leu14 bond at the C-terminus, shows the best activity in all three cell lines, with IC50 values in the low micromolar range. The Gly11/-Ala11 and Leu13-Met14/Sta13-Nle14 substitutions, which led to a new BBN (6-14) peptide sequence, held by the conjugate L6, affect the activity only slightly. Contrarily, the two free peptides, bearing the sequences of L5 and L6, do not show any activity on any of the cell lines. The lack of toxicity of the conjugates on healthy cells was checked on MRC-5 human fibroblasts, proving that they are non-toxic on non-cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-453 cell | CVCL_0418 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
9.28 ± 0.13 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
The cytostatic effect of the conjugates and the free peptides was evaluated on the three mentioned human cancer cell lines expressing GRP-R. The free Dau was used as a positive control for comparison purposes; it displays an IC50 in the high nanomolar range. Overall, the conjugates containing the LRRY spacer have a higher cytostatic effect than the ones with the GFLG spacer. L1, which has the original BBN (7-14) sequence, and L5, bearing a D-Phe in position 6 and the Sta13-Leu14 bond at the C-terminus, shows the best activity in all three cell lines, with IC50 values in the low micromolar range. The Gly11/-Ala11 and Leu13-Met14/Sta13-Nle14 substitutions, which led to a new BBN (6-14) peptide sequence, held by the conjugate L6, affect the activity only slightly. Contrarily, the two free peptides, bearing the sequences of L5 and L6, do not show any activity on any of the cell lines. The lack of toxicity of the conjugates on healthy cells was checked on MRC-5 human fibroblasts, proving that they are non-toxic on non-cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
PDC-G1 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Uptake Concentration (UC50) | > 25 µM | |||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
We quantified the ability of the produced PDCs to promote the internalisation of daunorubicin after binding to GRP-R by flow cytometry. Each bioconjugate was incubated for 1.5 h in four concentrations (25 uM, 12.5 uM, 6.25 uM and 3.125 uM), with the three cell lines that we have also used for the evaluation of the cytostatic effect. For better comparison, the uptake was described as the necessary concentration to internalise 50% of the compound (UC50). The uptake reflects the GRP-R protein expression: the lower UC50 values, hence the highest uptakes, are noticed in the human breast cancer cell line MDA-MB-453, whereas they are comparable in the two other cell lines. As far as the internalisation of the individual conjugates is concerned, L5 and L6 are the most promising ones in these three cell lines. Notably, both hold the LRRY spacer and the Sta13. L1 and L2 have satisfactory UC50s, too. On the other hand, majority of the conjugates with the GFLG spacer are poorly internalised.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Uptake Concentration (UC50) | > 25 µM | |||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
We quantified the ability of the produced PDCs to promote the internalisation of daunorubicin after binding to GRP-R by flow cytometry. Each bioconjugate was incubated for 1.5 h in four concentrations (25 uM, 12.5 uM, 6.25 uM and 3.125 uM), with the three cell lines that we have also used for the evaluation of the cytostatic effect. For better comparison, the uptake was described as the necessary concentration to internalise 50% of the compound (UC50). The uptake reflects the GRP-R protein expression: the lower UC50 values, hence the highest uptakes, are noticed in the human breast cancer cell line MDA-MB-453, whereas they are comparable in the two other cell lines. As far as the internalisation of the individual conjugates is concerned, L5 and L6 are the most promising ones in these three cell lines. Notably, both hold the LRRY spacer and the Sta13. L1 and L2 have satisfactory UC50s, too. On the other hand, majority of the conjugates with the GFLG spacer are poorly internalised.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-453 cell | CVCL_0418 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Uptake Concentration (UC50) | > 25 µM | |||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
We quantified the ability of the produced PDCs to promote the internalisation of daunorubicin after binding to GRP-R by flow cytometry. Each bioconjugate was incubated for 1.5 h in four concentrations (25 uM, 12.5 uM, 6.25 uM and 3.125 uM), with the three cell lines that we have also used for the evaluation of the cytostatic effect. For better comparison, the uptake was described as the necessary concentration to internalise 50% of the compound (UC50). The uptake reflects the GRP-R protein expression: the lower UC50 values, hence the highest uptakes, are noticed in the human breast cancer cell line MDA-MB-453, whereas they are comparable in the two other cell lines. As far as the internalisation of the individual conjugates is concerned, L5 and L6 are the most promising ones in these three cell lines. Notably, both hold the LRRY spacer and the Sta13. L1 and L2 have satisfactory UC50s, too. On the other hand, majority of the conjugates with the GFLG spacer are poorly internalised.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
11.45 ± 1.53 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
The cytostatic effect of the conjugates and the free peptides was evaluated on the three mentioned human cancer cell lines expressing GRP-R. The free Dau was used as a positive control for comparison purposes; it displays an IC50 in the high nanomolar range. Overall, the conjugates containing the LRRY spacer have a higher cytostatic effect than the ones with the GFLG spacer. L1, which has the original BBN (7-14) sequence, and L5, bearing a D-Phe in position 6 and the Sta13-Leu14 bond at the C-terminus, shows the best activity in all three cell lines, with IC50 values in the low micromolar range. The Gly11/-Ala11 and Leu13-Met14/Sta13-Nle14 substitutions, which led to a new BBN (6-14) peptide sequence, held by the conjugate L6, affect the activity only slightly. Contrarily, the two free peptides, bearing the sequences of L5 and L6, do not show any activity on any of the cell lines. The lack of toxicity of the conjugates on healthy cells was checked on MRC-5 human fibroblasts, proving that they are non-toxic on non-cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-453 cell | CVCL_0418 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
11.83 ± 2.50 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
The cytostatic effect of the conjugates and the free peptides was evaluated on the three mentioned human cancer cell lines expressing GRP-R. The free Dau was used as a positive control for comparison purposes; it displays an IC50 in the high nanomolar range. Overall, the conjugates containing the LRRY spacer have a higher cytostatic effect than the ones with the GFLG spacer. L1, which has the original BBN (7-14) sequence, and L5, bearing a D-Phe in position 6 and the Sta13-Leu14 bond at the C-terminus, shows the best activity in all three cell lines, with IC50 values in the low micromolar range. The Gly11/-Ala11 and Leu13-Met14/Sta13-Nle14 substitutions, which led to a new BBN (6-14) peptide sequence, held by the conjugate L6, affect the activity only slightly. Contrarily, the two free peptides, bearing the sequences of L5 and L6, do not show any activity on any of the cell lines. The lack of toxicity of the conjugates on healthy cells was checked on MRC-5 human fibroblasts, proving that they are non-toxic on non-cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
22.80 ± 3.12 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
The cytostatic effect of the conjugates and the free peptides was evaluated on the three mentioned human cancer cell lines expressing GRP-R. The free Dau was used as a positive control for comparison purposes; it displays an IC50 in the high nanomolar range. Overall, the conjugates containing the LRRY spacer have a higher cytostatic effect than the ones with the GFLG spacer. L1, which has the original BBN (7-14) sequence, and L5, bearing a D-Phe in position 6 and the Sta13-Leu14 bond at the C-terminus, shows the best activity in all three cell lines, with IC50 values in the low micromolar range. The Gly11/-Ala11 and Leu13-Met14/Sta13-Nle14 substitutions, which led to a new BBN (6-14) peptide sequence, held by the conjugate L6, affect the activity only slightly. Contrarily, the two free peptides, bearing the sequences of L5 and L6, do not show any activity on any of the cell lines. The lack of toxicity of the conjugates on healthy cells was checked on MRC-5 human fibroblasts, proving that they are non-toxic on non-cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
PDC-G5 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Uptake Concentration (UC50) | > 25 µM | |||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
We quantified the ability of the produced PDCs to promote the internalisation of daunorubicin after binding to GRP-R by flow cytometry. Each bioconjugate was incubated for 1.5 h in four concentrations (25 uM, 12.5 uM, 6.25 uM and 3.125 uM), with the three cell lines that we have also used for the evaluation of the cytostatic effect. For better comparison, the uptake was described as the necessary concentration to internalise 50% of the compound (UC50). The uptake reflects the GRP-R protein expression: the lower UC50 values, hence the highest uptakes, are noticed in the human breast cancer cell line MDA-MB-453, whereas they are comparable in the two other cell lines. As far as the internalisation of the individual conjugates is concerned, L5 and L6 are the most promising ones in these three cell lines. Notably, both hold the LRRY spacer and the Sta13. L1 and L2 have satisfactory UC50s, too. On the other hand, majority of the conjugates with the GFLG spacer are poorly internalised.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Uptake Concentration (UC50) | > 25 µM | |||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
We quantified the ability of the produced PDCs to promote the internalisation of daunorubicin after binding to GRP-R by flow cytometry. Each bioconjugate was incubated for 1.5 h in four concentrations (25 uM, 12.5 uM, 6.25 uM and 3.125 uM), with the three cell lines that we have also used for the evaluation of the cytostatic effect. For better comparison, the uptake was described as the necessary concentration to internalise 50% of the compound (UC50). The uptake reflects the GRP-R protein expression: the lower UC50 values, hence the highest uptakes, are noticed in the human breast cancer cell line MDA-MB-453, whereas they are comparable in the two other cell lines. As far as the internalisation of the individual conjugates is concerned, L5 and L6 are the most promising ones in these three cell lines. Notably, both hold the LRRY spacer and the Sta13. L1 and L2 have satisfactory UC50s, too. On the other hand, majority of the conjugates with the GFLG spacer are poorly internalised.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-453 cell | CVCL_0418 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Uptake Concentration (UC50) | > 25 µM | |||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
We quantified the ability of the produced PDCs to promote the internalisation of daunorubicin after binding to GRP-R by flow cytometry. Each bioconjugate was incubated for 1.5 h in four concentrations (25 uM, 12.5 uM, 6.25 uM and 3.125 uM), with the three cell lines that we have also used for the evaluation of the cytostatic effect. For better comparison, the uptake was described as the necessary concentration to internalise 50% of the compound (UC50). The uptake reflects the GRP-R protein expression: the lower UC50 values, hence the highest uptakes, are noticed in the human breast cancer cell line MDA-MB-453, whereas they are comparable in the two other cell lines. As far as the internalisation of the individual conjugates is concerned, L5 and L6 are the most promising ones in these three cell lines. Notably, both hold the LRRY spacer and the Sta13. L1 and L2 have satisfactory UC50s, too. On the other hand, majority of the conjugates with the GFLG spacer are poorly internalised.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
9.69 ± 0.17 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
The cytostatic effect of the conjugates and the free peptides was evaluated on the three mentioned human cancer cell lines expressing GRP-R. The free Dau was used as a positive control for comparison purposes; it displays an IC50 in the high nanomolar range. Overall, the conjugates containing the LRRY spacer have a higher cytostatic effect than the ones with the GFLG spacer. L1, which has the original BBN (7-14) sequence, and L5, bearing a D-Phe in position 6 and the Sta13-Leu14 bond at the C-terminus, shows the best activity in all three cell lines, with IC50 values in the low micromolar range. The Gly11/-Ala11 and Leu13-Met14/Sta13-Nle14 substitutions, which led to a new BBN (6-14) peptide sequence, held by the conjugate L6, affect the activity only slightly. Contrarily, the two free peptides, bearing the sequences of L5 and L6, do not show any activity on any of the cell lines. The lack of toxicity of the conjugates on healthy cells was checked on MRC-5 human fibroblasts, proving that they are non-toxic on non-cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
11.62 ± 3.33 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
The cytostatic effect of the conjugates and the free peptides was evaluated on the three mentioned human cancer cell lines expressing GRP-R. The free Dau was used as a positive control for comparison purposes; it displays an IC50 in the high nanomolar range. Overall, the conjugates containing the LRRY spacer have a higher cytostatic effect than the ones with the GFLG spacer. L1, which has the original BBN (7-14) sequence, and L5, bearing a D-Phe in position 6 and the Sta13-Leu14 bond at the C-terminus, shows the best activity in all three cell lines, with IC50 values in the low micromolar range. The Gly11/-Ala11 and Leu13-Met14/Sta13-Nle14 substitutions, which led to a new BBN (6-14) peptide sequence, held by the conjugate L6, affect the activity only slightly. Contrarily, the two free peptides, bearing the sequences of L5 and L6, do not show any activity on any of the cell lines. The lack of toxicity of the conjugates on healthy cells was checked on MRC-5 human fibroblasts, proving that they are non-toxic on non-cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-453 cell | CVCL_0418 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
18.29 ± 1.46 µM
|
|||
| Administration Time | 24 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
We decided to select some of the described peptides bearing the mentioned substitutions and having affirmed affinity for GRP-R in the low nanomolar range, together with the original BBN, and use them as targeting moiety to deliver a cytotoxic payload, daunorubicin (Dau), to prostate and breast cancers. Compared to the BBN sequence, our targeting peptides are elongated by a D-Phe in position 6 and comprise substitutions in positions 11, 13 and 14. Starting from these variations, we have also generated a new sequence: [D-Phe6, β-Ala11, Sta13, Nle14]BBN. The bombesin analogues have been published in the works of different research groups throughout many years, however, a direct comparison between them in terms of drug delivery has never been done. Once the targeting peptides bind to receptors that are overexpressed on the surface of cancer cells, such as GRP-R, the PDC can be carried inside the cells via receptor-mediated endocytosis. Briefly, the receptor-ligand complex internalises via a special, coated vesicle that later fuses with an early endosome and matures into a late endosome. After the late endosome fuses with a lysosome, the PDC is also digested, the drug is released into the cytoplasm and binds to its target compartment inside the cell. To ensure this release, we have decided to insert cathepsin B cleavable tetrapeptide linkers, namely either GFLG or LRRY, between the homing peptide and Dau. Indeed, cathepsin B is highly expressed by lysosomes and can cleave between Gly-Phe and Leu-Arg, liberating the active metabolites Dau=Aoa-Gly-OH or Dau=Aoa-Leu-OH. Dau is part of the family of anthracyclines and elicits its cytostatic activity by intercalating between DNA base pairs. This prevents the topoisomerase II from resealing the DNA double helix, resulting in reduced cell proliferation. Its attachment to a peptide can be easily performed through an oxime linkage in a very simple and straightforward reaction that occurs between an aminooxy moiety and the C13 ketone on Dau. The high yields of the reaction are advantageous, especially when it comes to the production of an increased amount of conjugate for in vivo studies. Furthermore, thanks to its intrinsic fluorescence, it is possible to evaluate the internalisation of the Dau-conjugates by fluorescence-activated cell sorting (FACS) and confocal laser scanning microscopy (CLSM), without the need to produce alternative peptide conjugates. To the best of our knowledge, so far, the delivery of a payload by bombesin-related peptides has only been studied using fluorescent molecules such as TAMRA, radioligands or nanocarriers. Drugs were attached to the peptides less often, and mainly using the original or truncated bombesin sequence instead of more promising analogues, or ones that are non-selective for GRP-R. Oppositely, investigating features such as the cellular uptake directly, through a conjugate attached to a chemotherapeutic, gives the most realistic picture of the success of a homing peptide acting as a drug carrier. Thus, the generated structures provide valuable tools for the selection of targeting peptides, aiming to the further development of new tumour-selective conjugates containing different linker-payload systems.
Click to Show/Hide
|
||||
| Description |
The cytostatic effect of the conjugates and the free peptides was evaluated on the three mentioned human cancer cell lines expressing GRP-R. The free Dau was used as a positive control for comparison purposes; it displays an IC50 in the high nanomolar range. Overall, the conjugates containing the LRRY spacer have a higher cytostatic effect than the ones with the GFLG spacer. L1, which has the original BBN (7-14) sequence, and L5, bearing a D-Phe in position 6 and the Sta13-Leu14 bond at the C-terminus, shows the best activity in all three cell lines, with IC50 values in the low micromolar range. The Gly11/-Ala11 and Leu13-Met14/Sta13-Nle14 substitutions, which led to a new BBN (6-14) peptide sequence, held by the conjugate L6, affect the activity only slightly. Contrarily, the two free peptides, bearing the sequences of L5 and L6, do not show any activity on any of the cell lines. The lack of toxicity of the conjugates on healthy cells was checked on MRC-5 human fibroblasts, proving that they are non-toxic on non-cancerous cells.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
DauQ=AoaGFLGGE11PEG [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.2 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | xCELLigence SP system assay | ||||
| MOA of PDC |
As observed, the cathepsin B labile spacer (GFLG), which was incorporated between the targeting peptide and the daunomycin to ensure the efficient release of the active metabolite, highly increases the hydrophobicity of the conjugates. As demonstrated, the solubility problem of these conjugates can be solved by hydrophilic polymer coupling, not only by using the well-known PEG but also by using the amino-monofunctional HbPG. To the best of our knowledge, 1:1 covalent peptide-polymer conjugates with well-defined monofunctional HbPG have been reported here for the first time. The results of the in vitro cell viability and cellular uptake measurements on HT-29 human colon adenocarcinoma cells prove that the HbPG and the PEG highly influenced the biological activity of the drug-peptide-polymer conjugates. In both peptide conjugate series, one GE11 and one D4 targeting peptide-based conjugate were found with outstanding cellular uptake and cytotoxicity values, namely Dau[double bond, length as m-dash]Aoa-GFLG-GE11-PEG and Dau[double bond, length as m-dash]Aoa-GFLG-D4-G5-HbPG. According to our results, the PEG is suitable for longer targeting peptides (e.g. GE11), but the G5 spacer is not suitable irrespective of the length of the peptide because it may decrease the biological effect by increasing the flexibility of the polymer and shading of the targeting moiety. In contrast, the use of the hydrophilic hyperbranched polyglycerol (HbPG) is advantageous for short targeting peptides (e.g. D4) but only with a G5 spacer, which provides accessibility of the peptide for receptor binding and cellular uptake resulting in outstanding cytotoxicity.
Click to Show/Hide
|
||||
| Description |
The polymers influenced the cellular uptake of the conjugates in a different manner, but in both groups (GE11 and D4 containing conjugates), one of the compounds was outstanding. The HT-29 cells could uptake Dau[double bond, length as m-dash]Aoa-GFLG-GE11-PEG and Dau[double bond, length as m-dash]Aoa-GFLG-D4-G5-HbPG to the highest extent. The G5 spacer increased the uptake of the D4-HbPG derivative, presumably because the increased distance between the globular HbPG and the very short peptide sequence provides a better receptor binding. In sharp contrast, the G5 spacer decreases the uptake of the conjugates in all other cases. The most significant difference was observed in the case of GE11-PEG, where the G5 spacer completely demolished the internalization. Probably, here the G5 spacer provides more flexibility for the linear PEG chain resulting in decreased receptor binding. There was one outstanding conjugate from each group (Dau[double bond, length as m-dash]Aoa-GFLG-GE11-PEG and Dau[double bond, length as m-dash]Aoa-GFLG-D4-G5-HbPG) in the cytotoxicity measurements that correlated well with the results of the internalization studies. These conjugates were found to be the most potent ones in the viability measurement and were proved to be taken up by HT-29 cells the most effectively. Depending on the type of polymer, the incorporation of the G5 spacer had an opposite effect on the cytotoxic activity of the conjugates. In the presence of the G5 spacer, the antitumour activity of the PEGylated conjugates decreased, while the cytotoxicity of the HbPG-containing conjugates increased, especially in the case of those with the D4 targeting peptide. We observed that some of the conjugates (Dau[double bond, length as m-dash]Aoa-GFLG-D4-HbPG and Dau[double bond, length as m-dash]Aoa-GFLG-GE11-G5-PEG) could not cause complete cell death, i.e., ˜0% viability value - characteristic for cell-free culturing medium - was not achieved even at the highest concentration, since their dose-response curves reached a plateau in a lower concentration range. In our opinion, this can be explained by the different characteristics of the highly hydrophobic peptide chain and the highly hydrophilic polymer segment. Due to this amphiphilic character, self-aggregation of the conjugates may occur, which then may block the accessibility of the targeting peptide for receptor binding, thereby decreasing the efficiency of the conjugate as well. This assumption is also confirmed by the turbidity results, since the observed low turbidity values may be caused by the possible formation of nanosized aggregates.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
Cyclic NGR peptidedaunomycin conjugates 5 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.4 ± 0.1 µM
|
|||
| Administration Time | 72 h | ||||
| Description |
In contrast, conjugate 2 (containing a disulfide bridge) showed preferential toxicity to HT-1080 cells in the cytostatic experiments, resulting in the highest selectivity. The antitumor effect of 2 increased in time (72 h treatment), but its selectivity decreased significantly. In comparison to conjugates 3 (thioether linkage) and 5 (amide bond), both containing free Lys in the cycle, conjugate 5 had slightly better activity against HT-1080 cells. However, the selectivity of conjugate 5 to CD13 receptors (as judged by the relative toxicity against the two cell lines) was not significant after 6 h treatment, whereas it was significantly more toxic to HT-1080 cells in the 72 h experiment. This can also be explained by its higher chemostability that results in longer exposure the cells to the unmodified NGR peptide-drug conjugate. In contrast, their analogs in which the drug molecule is connected to the side chain of Lys in the cycle (conjugates 4 and 6) were more specific but less toxic, especially in the cytotoxicity experiments. Interestingly, there was no correlation between the specificity and the chemostability of the conjugates.
Click to Show/Hide
|
||||
| In Vitro Model | Fibrosarcoma | HT-1080 cell | CVCL_0317 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.0 ± 0.6 µM
|
|||
| Administration Time | 72 h | ||||
| Description |
In contrast, conjugate 2 (containing a disulfide bridge) showed preferential toxicity to HT-1080 cells in the cytostatic experiments, resulting in the highest selectivity. The antitumor effect of 2 increased in time (72 h treatment), but its selectivity decreased significantly. In comparison to conjugates 3 (thioether linkage) and 5 (amide bond), both containing free Lys in the cycle, conjugate 5 had slightly better activity against HT-1080 cells. However, the selectivity of conjugate 5 to CD13 receptors (as judged by the relative toxicity against the two cell lines) was not significant after 6 h treatment, whereas it was significantly more toxic to HT-1080 cells in the 72 h experiment. This can also be explained by its higher chemostability that results in longer exposure the cells to the unmodified NGR peptide-drug conjugate. In contrast, their analogs in which the drug molecule is connected to the side chain of Lys in the cycle (conjugates 4 and 6) were more specific but less toxic, especially in the cytotoxicity experiments. Interestingly, there was no correlation between the specificity and the chemostability of the conjugates.
Click to Show/Hide
|
||||
| In Vitro Model | Colon adenocarcinoma | CD13-negative HT29 cell | CVCL_0320 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
5.7 ± 0.5 µM
|
|||
| Administration Time | 6 h | ||||
| Description |
In contrast, conjugate 2 (containing a disulfide bridge) showed preferential toxicity to HT-1080 cells in the cytostatic experiments, resulting in the highest selectivity. The antitumor effect of 2 increased in time (72 h treatment), but its selectivity decreased significantly. In comparison to conjugates 3 (thioether linkage) and 5 (amide bond), both containing free Lys in the cycle, conjugate 5 had slightly better activity against HT-1080 cells. However, the selectivity of conjugate 5 to CD13 receptors (as judged by the relative toxicity against the two cell lines) was not significant after 6 h treatment, whereas it was significantly more toxic to HT-1080 cells in the 72 h experiment. This can also be explained by its higher chemostability that results in longer exposure the cells to the unmodified NGR peptide-drug conjugate. In contrast, their analogs in which the drug molecule is connected to the side chain of Lys in the cycle (conjugates 4 and 6) were more specific but less toxic, especially in the cytotoxicity experiments. Interestingly, there was no correlation between the specificity and the chemostability of the conjugates.
Click to Show/Hide
|
||||
| In Vitro Model | Fibrosarcoma | HT-1080 cell | CVCL_0317 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
8.7 ± 1.2 µM
|
|||
| Administration Time | 6 h | ||||
| Description |
In contrast, conjugate 2 (containing a disulfide bridge) showed preferential toxicity to HT-1080 cells in the cytostatic experiments, resulting in the highest selectivity. The antitumor effect of 2 increased in time (72 h treatment), but its selectivity decreased significantly. In comparison to conjugates 3 (thioether linkage) and 5 (amide bond), both containing free Lys in the cycle, conjugate 5 had slightly better activity against HT-1080 cells. However, the selectivity of conjugate 5 to CD13 receptors (as judged by the relative toxicity against the two cell lines) was not significant after 6 h treatment, whereas it was significantly more toxic to HT-1080 cells in the 72 h experiment. This can also be explained by its higher chemostability that results in longer exposure the cells to the unmodified NGR peptide-drug conjugate. In contrast, their analogs in which the drug molecule is connected to the side chain of Lys in the cycle (conjugates 4 and 6) were more specific but less toxic, especially in the cytotoxicity experiments. Interestingly, there was no correlation between the specificity and the chemostability of the conjugates.
Click to Show/Hide
|
||||
| In Vitro Model | Colon adenocarcinoma | CD13-negative HT29 cell | CVCL_0320 | ||
Cyclic NGR peptidedaunomycin conjugates 2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.0 ± 0.3 µM
|
|||
| Administration Time | 72 h | ||||
| Description |
In contrast, conjugate 2 (containing a disulfide bridge) showed preferential toxicity to HT-1080 cells in the cytostatic experiments, resulting in the highest selectivity. The antitumor effect of 2 increased in time (72 h treatment), but its selectivity decreased significantly. In comparison to conjugates 3 (thioether linkage) and 5 (amide bond), both containing free Lys in the cycle, conjugate 5 had slightly better activity against HT-1080 cells. However, the selectivity of conjugate 5 to CD13 receptors (as judged by the relative toxicity against the two cell lines) was not significant after 6 h treatment, whereas it was significantly more toxic to HT-1080 cells in the 72 h experiment. This can also be explained by its higher chemostability that results in longer exposure the cells to the unmodified NGR peptide-drug conjugate. In contrast, their analogs in which the drug molecule is connected to the side chain of Lys in the cycle (conjugates 4 and 6) were more specific but less toxic, especially in the cytotoxicity experiments. Interestingly, there was no correlation between the specificity and the chemostability of the conjugates.
Click to Show/Hide
|
||||
| In Vitro Model | Fibrosarcoma | HT-1080 cell | CVCL_0317 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.3 ± 0.9 µM
|
|||
| Administration Time | 72 h | ||||
| Description |
In contrast, conjugate 2 (containing a disulfide bridge) showed preferential toxicity to HT-1080 cells in the cytostatic experiments, resulting in the highest selectivity. The antitumor effect of 2 increased in time (72 h treatment), but its selectivity decreased significantly. In comparison to conjugates 3 (thioether linkage) and 5 (amide bond), both containing free Lys in the cycle, conjugate 5 had slightly better activity against HT-1080 cells. However, the selectivity of conjugate 5 to CD13 receptors (as judged by the relative toxicity against the two cell lines) was not significant after 6 h treatment, whereas it was significantly more toxic to HT-1080 cells in the 72 h experiment. This can also be explained by its higher chemostability that results in longer exposure the cells to the unmodified NGR peptide-drug conjugate. In contrast, their analogs in which the drug molecule is connected to the side chain of Lys in the cycle (conjugates 4 and 6) were more specific but less toxic, especially in the cytotoxicity experiments. Interestingly, there was no correlation between the specificity and the chemostability of the conjugates.
Click to Show/Hide
|
||||
| In Vitro Model | Colon adenocarcinoma | CD13-negative HT29 cell | CVCL_0320 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
6.8 ± 0.7 µM
|
|||
| Administration Time | 6 h | ||||
| Description |
In contrast, conjugate 2 (containing a disulfide bridge) showed preferential toxicity to HT-1080 cells in the cytostatic experiments, resulting in the highest selectivity. The antitumor effect of 2 increased in time (72 h treatment), but its selectivity decreased significantly. In comparison to conjugates 3 (thioether linkage) and 5 (amide bond), both containing free Lys in the cycle, conjugate 5 had slightly better activity against HT-1080 cells. However, the selectivity of conjugate 5 to CD13 receptors (as judged by the relative toxicity against the two cell lines) was not significant after 6 h treatment, whereas it was significantly more toxic to HT-1080 cells in the 72 h experiment. This can also be explained by its higher chemostability that results in longer exposure the cells to the unmodified NGR peptide-drug conjugate. In contrast, their analogs in which the drug molecule is connected to the side chain of Lys in the cycle (conjugates 4 and 6) were more specific but less toxic, especially in the cytotoxicity experiments. Interestingly, there was no correlation between the specificity and the chemostability of the conjugates.
Click to Show/Hide
|
||||
| In Vitro Model | Fibrosarcoma | HT-1080 cell | CVCL_0317 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
19.0 ± 1.4 µM
|
|||
| Administration Time | 6 h | ||||
| Description |
In contrast, conjugate 2 (containing a disulfide bridge) showed preferential toxicity to HT-1080 cells in the cytostatic experiments, resulting in the highest selectivity. The antitumor effect of 2 increased in time (72 h treatment), but its selectivity decreased significantly. In comparison to conjugates 3 (thioether linkage) and 5 (amide bond), both containing free Lys in the cycle, conjugate 5 had slightly better activity against HT-1080 cells. However, the selectivity of conjugate 5 to CD13 receptors (as judged by the relative toxicity against the two cell lines) was not significant after 6 h treatment, whereas it was significantly more toxic to HT-1080 cells in the 72 h experiment. This can also be explained by its higher chemostability that results in longer exposure the cells to the unmodified NGR peptide-drug conjugate. In contrast, their analogs in which the drug molecule is connected to the side chain of Lys in the cycle (conjugates 4 and 6) were more specific but less toxic, especially in the cytotoxicity experiments. Interestingly, there was no correlation between the specificity and the chemostability of the conjugates.
Click to Show/Hide
|
||||
| In Vitro Model | Colon adenocarcinoma | CD13-negative HT29 cell | CVCL_0320 | ||
PDC-5d [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.0 (5.7 ± 0.1) μM
|
|||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-468 cell | CVCL_0419 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4.2 (5.4 ± 0.1) μM
|
|||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
5.0 (5.3 ± 0.1) μM
|
|||
| In Vitro Model | Invasive breast carcinoma | T-47D cell | CVCL_0553 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
8.9 nM
|
|||
| In Vitro Model | Normal | COS-7 cell | CVCL_0224 | ||
Cyclic NGR peptidedaunomycin conjugates 3 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.3 ± 0.6 µM
|
|||
| Administration Time | 72 h | ||||
| Description |
In contrast, conjugate 2 (containing a disulfide bridge) showed preferential toxicity to HT-1080 cells in the cytostatic experiments, resulting in the highest selectivity. The antitumor effect of 2 increased in time (72 h treatment), but its selectivity decreased significantly. In comparison to conjugates 3 (thioether linkage) and 5 (amide bond), both containing free Lys in the cycle, conjugate 5 had slightly better activity against HT-1080 cells. However, the selectivity of conjugate 5 to CD13 receptors (as judged by the relative toxicity against the two cell lines) was not significant after 6 h treatment, whereas it was significantly more toxic to HT-1080 cells in the 72 h experiment. This can also be explained by its higher chemostability that results in longer exposure the cells to the unmodified NGR peptide-drug conjugate. In contrast, their analogs in which the drug molecule is connected to the side chain of Lys in the cycle (conjugates 4 and 6) were more specific but less toxic, especially in the cytotoxicity experiments. Interestingly, there was no correlation between the specificity and the chemostability of the conjugates.
Click to Show/Hide
|
||||
| In Vitro Model | Fibrosarcoma | HT-1080 cell | CVCL_0317 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
5.7 ± 0.9 µM
|
|||
| Administration Time | 72 h | ||||
| Description |
In contrast, conjugate 2 (containing a disulfide bridge) showed preferential toxicity to HT-1080 cells in the cytostatic experiments, resulting in the highest selectivity. The antitumor effect of 2 increased in time (72 h treatment), but its selectivity decreased significantly. In comparison to conjugates 3 (thioether linkage) and 5 (amide bond), both containing free Lys in the cycle, conjugate 5 had slightly better activity against HT-1080 cells. However, the selectivity of conjugate 5 to CD13 receptors (as judged by the relative toxicity against the two cell lines) was not significant after 6 h treatment, whereas it was significantly more toxic to HT-1080 cells in the 72 h experiment. This can also be explained by its higher chemostability that results in longer exposure the cells to the unmodified NGR peptide-drug conjugate. In contrast, their analogs in which the drug molecule is connected to the side chain of Lys in the cycle (conjugates 4 and 6) were more specific but less toxic, especially in the cytotoxicity experiments. Interestingly, there was no correlation between the specificity and the chemostability of the conjugates.
Click to Show/Hide
|
||||
| In Vitro Model | Colon adenocarcinoma | CD13-negative HT29 cell | CVCL_0320 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
8.0 ± 0.1 µM
|
|||
| Administration Time | 6 h | ||||
| Description |
In contrast, conjugate 2 (containing a disulfide bridge) showed preferential toxicity to HT-1080 cells in the cytostatic experiments, resulting in the highest selectivity. The antitumor effect of 2 increased in time (72 h treatment), but its selectivity decreased significantly. In comparison to conjugates 3 (thioether linkage) and 5 (amide bond), both containing free Lys in the cycle, conjugate 5 had slightly better activity against HT-1080 cells. However, the selectivity of conjugate 5 to CD13 receptors (as judged by the relative toxicity against the two cell lines) was not significant after 6 h treatment, whereas it was significantly more toxic to HT-1080 cells in the 72 h experiment. This can also be explained by its higher chemostability that results in longer exposure the cells to the unmodified NGR peptide-drug conjugate. In contrast, their analogs in which the drug molecule is connected to the side chain of Lys in the cycle (conjugates 4 and 6) were more specific but less toxic, especially in the cytotoxicity experiments. Interestingly, there was no correlation between the specificity and the chemostability of the conjugates.
Click to Show/Hide
|
||||
| In Vitro Model | Fibrosarcoma | HT-1080 cell | CVCL_0317 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
10.3 ± 2.2 µM
|
|||
| Administration Time | 6 h | ||||
| Description |
In contrast, conjugate 2 (containing a disulfide bridge) showed preferential toxicity to HT-1080 cells in the cytostatic experiments, resulting in the highest selectivity. The antitumor effect of 2 increased in time (72 h treatment), but its selectivity decreased significantly. In comparison to conjugates 3 (thioether linkage) and 5 (amide bond), both containing free Lys in the cycle, conjugate 5 had slightly better activity against HT-1080 cells. However, the selectivity of conjugate 5 to CD13 receptors (as judged by the relative toxicity against the two cell lines) was not significant after 6 h treatment, whereas it was significantly more toxic to HT-1080 cells in the 72 h experiment. This can also be explained by its higher chemostability that results in longer exposure the cells to the unmodified NGR peptide-drug conjugate. In contrast, their analogs in which the drug molecule is connected to the side chain of Lys in the cycle (conjugates 4 and 6) were more specific but less toxic, especially in the cytotoxicity experiments. Interestingly, there was no correlation between the specificity and the chemostability of the conjugates.
Click to Show/Hide
|
||||
| In Vitro Model | Colon adenocarcinoma | CD13-negative HT29 cell | CVCL_0320 | ||
PDC-Z3 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.47 ± 0.67 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
5.51 ± 1.74 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
30.12 ± 1.38 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
32.44 ± 5.49 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
PDC-5b [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.6 (5.4 ± 0.1) μM
|
|||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-468 cell | CVCL_0419 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
7.7 (5.1 ± 0.1) μM
|
|||
| In Vitro Model | Invasive breast carcinoma | T-47D cell | CVCL_0553 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
8.1 (5.1 ± 0.1) μM
|
|||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
3.0 (8.5 ± 0.1) nM
|
|||
| In Vitro Model | Normal | COS-7 cell | CVCL_0224 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
226 (6.6 ± 0.1) nM
|
|||
| In Vitro Model | Normal | COS-7 cell | CVCL_0224 | ||
DauQ=AoaGFLGD4G5HbPG [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.8 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | xCELLigence SP system assay | ||||
| MOA of PDC |
As observed, the cathepsin B labile spacer (GFLG), which was incorporated between the targeting peptide and the daunomycin to ensure the efficient release of the active metabolite, highly increases the hydrophobicity of the conjugates. As demonstrated, the solubility problem of these conjugates can be solved by hydrophilic polymer coupling, not only by using the well-known PEG but also by using the amino-monofunctional HbPG. To the best of our knowledge, 1:1 covalent peptide-polymer conjugates with well-defined monofunctional HbPG have been reported here for the first time. The results of the in vitro cell viability and cellular uptake measurements on HT-29 human colon adenocarcinoma cells prove that the HbPG and the PEG highly influenced the biological activity of the drug-peptide-polymer conjugates. In both peptide conjugate series, one GE11 and one D4 targeting peptide-based conjugate were found with outstanding cellular uptake and cytotoxicity values, namely Dau[double bond, length as m-dash]Aoa-GFLG-GE11-PEG and Dau[double bond, length as m-dash]Aoa-GFLG-D4-G5-HbPG. According to our results, the PEG is suitable for longer targeting peptides (e.g. GE11), but the G5 spacer is not suitable irrespective of the length of the peptide because it may decrease the biological effect by increasing the flexibility of the polymer and shading of the targeting moiety. In contrast, the use of the hydrophilic hyperbranched polyglycerol (HbPG) is advantageous for short targeting peptides (e.g. D4) but only with a G5 spacer, which provides accessibility of the peptide for receptor binding and cellular uptake resulting in outstanding cytotoxicity.
Click to Show/Hide
|
||||
| Description |
The polymers influenced the cellular uptake of the conjugates in a different manner, but in both groups (GE11 and D4 containing conjugates), one of the compounds was outstanding. The HT-29 cells could uptake Dau[double bond, length as m-dash]Aoa-GFLG-GE11-PEG and Dau[double bond, length as m-dash]Aoa-GFLG-D4-G5-HbPG to the highest extent. The G5 spacer increased the uptake of the D4-HbPG derivative, presumably because the increased distance between the globular HbPG and the very short peptide sequence provides a better receptor binding. In sharp contrast, the G5 spacer decreases the uptake of the conjugates in all other cases. The most significant difference was observed in the case of GE11-PEG, where the G5 spacer completely demolished the internalization. Probably, here the G5 spacer provides more flexibility for the linear PEG chain resulting in decreased receptor binding. There was one outstanding conjugate from each group (Dau[double bond, length as m-dash]Aoa-GFLG-GE11-PEG and Dau[double bond, length as m-dash]Aoa-GFLG-D4-G5-HbPG) in the cytotoxicity measurements that correlated well with the results of the internalization studies. These conjugates were found to be the most potent ones in the viability measurement and were proved to be taken up by HT-29 cells the most effectively. Depending on the type of polymer, the incorporation of the G5 spacer had an opposite effect on the cytotoxic activity of the conjugates. In the presence of the G5 spacer, the antitumour activity of the PEGylated conjugates decreased, while the cytotoxicity of the HbPG-containing conjugates increased, especially in the case of those with the D4 targeting peptide. We observed that some of the conjugates (Dau[double bond, length as m-dash]Aoa-GFLG-D4-HbPG and Dau[double bond, length as m-dash]Aoa-GFLG-GE11-G5-PEG) could not cause complete cell death, i.e., ˜0% viability value - characteristic for cell-free culturing medium - was not achieved even at the highest concentration, since their dose-response curves reached a plateau in a lower concentration range. In our opinion, this can be explained by the different characteristics of the highly hydrophobic peptide chain and the highly hydrophilic polymer segment. Due to this amphiphilic character, self-aggregation of the conjugates may occur, which then may block the accessibility of the targeting peptide for receptor binding, thereby decreasing the efficiency of the conjugate as well. This assumption is also confirmed by the turbidity results, since the observed low turbidity values may be caused by the possible formation of nanosized aggregates.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
RGD-GFLG-DOX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Lung cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4 µM
|
|||
| Administration Time | 36 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In conclusion, we developed a robust and regioselective rhodium-catalyzed methodology for C(7)-H Trp maleimidation. This reaction served as an efficient tool for peptide/drug modification, ligation, and particularly peptide cyclization, confirming its promising potential in pharmaceutical chemistry and drug synthesis. Notably, this catalytical system is not limited by the Trp position in the peptides. We also demonstrated that tryptophan-substituted maleimide could be used as an effective click functional group to rapidly react with sulfhydryl groups. Moreover, the introduced N-pivaloyl directing group and protecting groups of the peptides could be removed in a single step, providing a more convenient approach compared to the previous methods, which require multi-step removal of the corresponding directing groups and peptide protection groups. Additionally, cyclic peptide 10a exhibited excellent binding affinity to integrin vβ3, indicating its good drug-like properties. With rational design, RGD- GFLG -DOX, which is a stapled PDC, displayed higher selectivity, stronger binding affinity and better cell penetrability than the more commonly used DOX. The proposed strategy for rapid preparation of stapled peptides is expected to further improve PDC formulation.
Click to Show/Hide
|
||||
| Description |
Doxorubicin (DOX) is one of the most effective anticancer drugs and has been successfully used in clinical practice. However, DOX cannot differentiate between cancer cells and normal cells, which may induce unwanted side effects and severe toxicity. Compared with traditional small-molecule anticancer drugs, the peptide-drug conjugates (PDCs) have enhanced targeting specificity and water solubility. Based on these advantages, to further explore the function of 10a, we designed and prepared a anticancer PDC drug compound RGD-GFLG-DOX containing the tetrapeptide linker Gly-Phe-Leu-Gly, which can be cleaved in presence of cathepsin B, a highly upregulated enzyme in malignant tumors, to release the drug. RGD-GFLG was synthesized as a control. The inhibitory effects of RGD-GFLG-DOX on cancer cell lines were assessed using cytotoxicity assay. Specifically, the effects of RGD-GFLG-DOX were evaluated on integrin v3-positive cancer cell lines, including A549 and U87MG cells, integrin v3-negative cancer cell lines such as HeLa and MCF-7 cells, as well as normal cell lines, namely LO2 and MIHA cells. RGD-GFLG-DOX exhibited a lower cytotoxicity on HeLa, MCF-7, LO2 and MIHA cells, but a stronger cytotoxicity than DOX on A549 and U87MG cells. For comparison, RGD-GFLG demonstrated minimal cytotoxicity. In addition, the cytotoxicity of RGD-GFLG-DOX with various concentrations (0-40 uM) on A549 and U87MG cells was studied. The results showed that the cytotoxicity of RGD-GFLG-DOX on A549 and U87MG cells was dose-dependent. These indicate that RGD-GFLG-DOX has a good specificity and inhibitory activity toward integrin v3-overexpressed A549 and U87MG cells.
Click to Show/Hide
|
||||
| In Vitro Model | Lung adenocarcinoma | A-549 cell | CVCL_0023 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4 µM
|
|||
| Administration Time | 36 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In conclusion, we developed a robust and regioselective rhodium-catalyzed methodology for C(7)-H Trp maleimidation. This reaction served as an efficient tool for peptide/drug modification, ligation, and particularly peptide cyclization, confirming its promising potential in pharmaceutical chemistry and drug synthesis. Notably, this catalytical system is not limited by the Trp position in the peptides. We also demonstrated that tryptophan-substituted maleimide could be used as an effective click functional group to rapidly react with sulfhydryl groups. Moreover, the introduced N-pivaloyl directing group and protecting groups of the peptides could be removed in a single step, providing a more convenient approach compared to the previous methods, which require multi-step removal of the corresponding directing groups and peptide protection groups. Additionally, cyclic peptide 10a exhibited excellent binding affinity to integrin vβ3, indicating its good drug-like properties. With rational design, RGD- GFLG -DOX, which is a stapled PDC, displayed higher selectivity, stronger binding affinity and better cell penetrability than the more commonly used DOX. The proposed strategy for rapid preparation of stapled peptides is expected to further improve PDC formulation.
Click to Show/Hide
|
||||
| Description |
Doxorubicin (DOX) is one of the most effective anticancer drugs and has been successfully used in clinical practice. However, DOX cannot differentiate between cancer cells and normal cells, which may induce unwanted side effects and severe toxicity. Compared with traditional small-molecule anticancer drugs, the peptide-drug conjugates (PDCs) have enhanced targeting specificity and water solubility. Based on these advantages, to further explore the function of 10a, we designed and prepared a anticancer PDC drug compound RGD-GFLG-DOX containing the tetrapeptide linker Gly-Phe-Leu-Gly, which can be cleaved in presence of cathepsin B, a highly upregulated enzyme in malignant tumors, to release the drug. RGD-GFLG was synthesized as a control. The inhibitory effects of RGD-GFLG-DOX on cancer cell lines were assessed using cytotoxicity assay. Specifically, the effects of RGD-GFLG-DOX were evaluated on integrin v3-positive cancer cell lines, including A549 and U87MG cells, integrin v3-negative cancer cell lines such as HeLa and MCF-7 cells, as well as normal cell lines, namely LO2 and MIHA cells. RGD-GFLG-DOX exhibited a lower cytotoxicity on HeLa, MCF-7, LO2 and MIHA cells, but a stronger cytotoxicity than DOX on A549 and U87MG cells. For comparison, RGD-GFLG demonstrated minimal cytotoxicity. In addition, the cytotoxicity of RGD-GFLG-DOX with various concentrations (0-40 uM) on A549 and U87MG cells was studied. The results showed that the cytotoxicity of RGD-GFLG-DOX on A549 and U87MG cells was dose-dependent. These indicate that RGD-GFLG-DOX has a good specificity and inhibitory activity toward integrin v3-overexpressed A549 and U87MG cells.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Cervical carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 40 µM | |||
| Administration Time | 36 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In conclusion, we developed a robust and regioselective rhodium-catalyzed methodology for C(7)-H Trp maleimidation. This reaction served as an efficient tool for peptide/drug modification, ligation, and particularly peptide cyclization, confirming its promising potential in pharmaceutical chemistry and drug synthesis. Notably, this catalytical system is not limited by the Trp position in the peptides. We also demonstrated that tryptophan-substituted maleimide could be used as an effective click functional group to rapidly react with sulfhydryl groups. Moreover, the introduced N-pivaloyl directing group and protecting groups of the peptides could be removed in a single step, providing a more convenient approach compared to the previous methods, which require multi-step removal of the corresponding directing groups and peptide protection groups. Additionally, cyclic peptide 10a exhibited excellent binding affinity to integrin vβ3, indicating its good drug-like properties. With rational design, RGD- GFLG -DOX, which is a stapled PDC, displayed higher selectivity, stronger binding affinity and better cell penetrability than the more commonly used DOX. The proposed strategy for rapid preparation of stapled peptides is expected to further improve PDC formulation.
Click to Show/Hide
|
||||
| Description |
Doxorubicin (DOX) is one of the most effective anticancer drugs and has been successfully used in clinical practice. However, DOX cannot differentiate between cancer cells and normal cells, which may induce unwanted side effects and severe toxicity. Compared with traditional small-molecule anticancer drugs, the peptide-drug conjugates (PDCs) have enhanced targeting specificity and water solubility. Based on these advantages, to further explore the function of 10a, we designed and prepared a anticancer PDC drug compound RGD-GFLG-DOX containing the tetrapeptide linker Gly-Phe-Leu-Gly, which can be cleaved in presence of cathepsin B, a highly upregulated enzyme in malignant tumors, to release the drug. RGD-GFLG was synthesized as a control. The inhibitory effects of RGD-GFLG-DOX on cancer cell lines were assessed using cytotoxicity assay. Specifically, the effects of RGD-GFLG-DOX were evaluated on integrin v3-positive cancer cell lines, including A549 and U87MG cells, integrin v3-negative cancer cell lines such as HeLa and MCF-7 cells, as well as normal cell lines, namely LO2 and MIHA cells. RGD-GFLG-DOX exhibited a lower cytotoxicity on HeLa, MCF-7, LO2 and MIHA cells, but a stronger cytotoxicity than DOX on A549 and U87MG cells. For comparison, RGD-GFLG demonstrated minimal cytotoxicity. In addition, the cytotoxicity of RGD-GFLG-DOX with various concentrations (0-40 uM) on A549 and U87MG cells was studied. The results showed that the cytotoxicity of RGD-GFLG-DOX on A549 and U87MG cells was dose-dependent. These indicate that RGD-GFLG-DOX has a good specificity and inhibitory activity toward integrin v3-overexpressed A549 and U87MG cells.
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Liver cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 40 µM | |||
| Administration Time | 36 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In conclusion, we developed a robust and regioselective rhodium-catalyzed methodology for C(7)-H Trp maleimidation. This reaction served as an efficient tool for peptide/drug modification, ligation, and particularly peptide cyclization, confirming its promising potential in pharmaceutical chemistry and drug synthesis. Notably, this catalytical system is not limited by the Trp position in the peptides. We also demonstrated that tryptophan-substituted maleimide could be used as an effective click functional group to rapidly react with sulfhydryl groups. Moreover, the introduced N-pivaloyl directing group and protecting groups of the peptides could be removed in a single step, providing a more convenient approach compared to the previous methods, which require multi-step removal of the corresponding directing groups and peptide protection groups. Additionally, cyclic peptide 10a exhibited excellent binding affinity to integrin vβ3, indicating its good drug-like properties. With rational design, RGD- GFLG -DOX, which is a stapled PDC, displayed higher selectivity, stronger binding affinity and better cell penetrability than the more commonly used DOX. The proposed strategy for rapid preparation of stapled peptides is expected to further improve PDC formulation.
Click to Show/Hide
|
||||
| Description |
Doxorubicin (DOX) is one of the most effective anticancer drugs and has been successfully used in clinical practice. However, DOX cannot differentiate between cancer cells and normal cells, which may induce unwanted side effects and severe toxicity. Compared with traditional small-molecule anticancer drugs, the peptide-drug conjugates (PDCs) have enhanced targeting specificity and water solubility. Based on these advantages, to further explore the function of 10a, we designed and prepared a anticancer PDC drug compound RGD-GFLG-DOX containing the tetrapeptide linker Gly-Phe-Leu-Gly, which can be cleaved in presence of cathepsin B, a highly upregulated enzyme in malignant tumors, to release the drug. RGD-GFLG was synthesized as a control. The inhibitory effects of RGD-GFLG-DOX on cancer cell lines were assessed using cytotoxicity assay. Specifically, the effects of RGD-GFLG-DOX were evaluated on integrin v3-positive cancer cell lines, including A549 and U87MG cells, integrin v3-negative cancer cell lines such as HeLa and MCF-7 cells, as well as normal cell lines, namely LO2 and MIHA cells. RGD-GFLG-DOX exhibited a lower cytotoxicity on HeLa, MCF-7, LO2 and MIHA cells, but a stronger cytotoxicity than DOX on A549 and U87MG cells. For comparison, RGD-GFLG demonstrated minimal cytotoxicity. In addition, the cytotoxicity of RGD-GFLG-DOX with various concentrations (0-40 uM) on A549 and U87MG cells was studied. The results showed that the cytotoxicity of RGD-GFLG-DOX on A549 and U87MG cells was dose-dependent. These indicate that RGD-GFLG-DOX has a good specificity and inhibitory activity toward integrin v3-overexpressed A549 and U87MG cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MIHA cell | CVCL_SA11 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 40 µM | |||
| Administration Time | 36 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In conclusion, we developed a robust and regioselective rhodium-catalyzed methodology for C(7)-H Trp maleimidation. This reaction served as an efficient tool for peptide/drug modification, ligation, and particularly peptide cyclization, confirming its promising potential in pharmaceutical chemistry and drug synthesis. Notably, this catalytical system is not limited by the Trp position in the peptides. We also demonstrated that tryptophan-substituted maleimide could be used as an effective click functional group to rapidly react with sulfhydryl groups. Moreover, the introduced N-pivaloyl directing group and protecting groups of the peptides could be removed in a single step, providing a more convenient approach compared to the previous methods, which require multi-step removal of the corresponding directing groups and peptide protection groups. Additionally, cyclic peptide 10a exhibited excellent binding affinity to integrin vβ3, indicating its good drug-like properties. With rational design, RGD- GFLG -DOX, which is a stapled PDC, displayed higher selectivity, stronger binding affinity and better cell penetrability than the more commonly used DOX. The proposed strategy for rapid preparation of stapled peptides is expected to further improve PDC formulation.
Click to Show/Hide
|
||||
| Description |
Doxorubicin (DOX) is one of the most effective anticancer drugs and has been successfully used in clinical practice. However, DOX cannot differentiate between cancer cells and normal cells, which may induce unwanted side effects and severe toxicity. Compared with traditional small-molecule anticancer drugs, the peptide-drug conjugates (PDCs) have enhanced targeting specificity and water solubility. Based on these advantages, to further explore the function of 10a, we designed and prepared a anticancer PDC drug compound RGD-GFLG-DOX containing the tetrapeptide linker Gly-Phe-Leu-Gly, which can be cleaved in presence of cathepsin B, a highly upregulated enzyme in malignant tumors, to release the drug. RGD-GFLG was synthesized as a control. The inhibitory effects of RGD-GFLG-DOX on cancer cell lines were assessed using cytotoxicity assay. Specifically, the effects of RGD-GFLG-DOX were evaluated on integrin v3-positive cancer cell lines, including A549 and U87MG cells, integrin v3-negative cancer cell lines such as HeLa and MCF-7 cells, as well as normal cell lines, namely LO2 and MIHA cells. RGD-GFLG-DOX exhibited a lower cytotoxicity on HeLa, MCF-7, LO2 and MIHA cells, but a stronger cytotoxicity than DOX on A549 and U87MG cells. For comparison, RGD-GFLG demonstrated minimal cytotoxicity. In addition, the cytotoxicity of RGD-GFLG-DOX with various concentrations (0-40 uM) on A549 and U87MG cells was studied. The results showed that the cytotoxicity of RGD-GFLG-DOX on A549 and U87MG cells was dose-dependent. These indicate that RGD-GFLG-DOX has a good specificity and inhibitory activity toward integrin v3-overexpressed A549 and U87MG cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Liver cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 40 µM | |||
| Administration Time | 36 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In conclusion, we developed a robust and regioselective rhodium-catalyzed methodology for C(7)-H Trp maleimidation. This reaction served as an efficient tool for peptide/drug modification, ligation, and particularly peptide cyclization, confirming its promising potential in pharmaceutical chemistry and drug synthesis. Notably, this catalytical system is not limited by the Trp position in the peptides. We also demonstrated that tryptophan-substituted maleimide could be used as an effective click functional group to rapidly react with sulfhydryl groups. Moreover, the introduced N-pivaloyl directing group and protecting groups of the peptides could be removed in a single step, providing a more convenient approach compared to the previous methods, which require multi-step removal of the corresponding directing groups and peptide protection groups. Additionally, cyclic peptide 10a exhibited excellent binding affinity to integrin vβ3, indicating its good drug-like properties. With rational design, RGD- GFLG -DOX, which is a stapled PDC, displayed higher selectivity, stronger binding affinity and better cell penetrability than the more commonly used DOX. The proposed strategy for rapid preparation of stapled peptides is expected to further improve PDC formulation.
Click to Show/Hide
|
||||
| Description |
Doxorubicin (DOX) is one of the most effective anticancer drugs and has been successfully used in clinical practice. However, DOX cannot differentiate between cancer cells and normal cells, which may induce unwanted side effects and severe toxicity. Compared with traditional small-molecule anticancer drugs, the peptide-drug conjugates (PDCs) have enhanced targeting specificity and water solubility. Based on these advantages, to further explore the function of 10a, we designed and prepared a anticancer PDC drug compound RGD-GFLG-DOX containing the tetrapeptide linker Gly-Phe-Leu-Gly, which can be cleaved in presence of cathepsin B, a highly upregulated enzyme in malignant tumors, to release the drug. RGD-GFLG was synthesized as a control. The inhibitory effects of RGD-GFLG-DOX on cancer cell lines were assessed using cytotoxicity assay. Specifically, the effects of RGD-GFLG-DOX were evaluated on integrin v3-positive cancer cell lines, including A549 and U87MG cells, integrin v3-negative cancer cell lines such as HeLa and MCF-7 cells, as well as normal cell lines, namely LO2 and MIHA cells. RGD-GFLG-DOX exhibited a lower cytotoxicity on HeLa, MCF-7, LO2 and MIHA cells, but a stronger cytotoxicity than DOX on A549 and U87MG cells. For comparison, RGD-GFLG demonstrated minimal cytotoxicity. In addition, the cytotoxicity of RGD-GFLG-DOX with various concentrations (0-40 uM) on A549 and U87MG cells was studied. The results showed that the cytotoxicity of RGD-GFLG-DOX on A549 and U87MG cells was dose-dependent. These indicate that RGD-GFLG-DOX has a good specificity and inhibitory activity toward integrin v3-overexpressed A549 and U87MG cells.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | LO #2 cell | CVCL_C7SD | ||
DauQ=AoaGFLGD4PEG [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
5.1 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | xCELLigence SP system assay | ||||
| MOA of PDC |
As observed, the cathepsin B labile spacer (GFLG), which was incorporated between the targeting peptide and the daunomycin to ensure the efficient release of the active metabolite, highly increases the hydrophobicity of the conjugates. As demonstrated, the solubility problem of these conjugates can be solved by hydrophilic polymer coupling, not only by using the well-known PEG but also by using the amino-monofunctional HbPG. To the best of our knowledge, 1:1 covalent peptide-polymer conjugates with well-defined monofunctional HbPG have been reported here for the first time. The results of the in vitro cell viability and cellular uptake measurements on HT-29 human colon adenocarcinoma cells prove that the HbPG and the PEG highly influenced the biological activity of the drug-peptide-polymer conjugates. In both peptide conjugate series, one GE11 and one D4 targeting peptide-based conjugate were found with outstanding cellular uptake and cytotoxicity values, namely Dau[double bond, length as m-dash]Aoa-GFLG-GE11-PEG and Dau[double bond, length as m-dash]Aoa-GFLG-D4-G5-HbPG. According to our results, the PEG is suitable for longer targeting peptides (e.g. GE11), but the G5 spacer is not suitable irrespective of the length of the peptide because it may decrease the biological effect by increasing the flexibility of the polymer and shading of the targeting moiety. In contrast, the use of the hydrophilic hyperbranched polyglycerol (HbPG) is advantageous for short targeting peptides (e.g. D4) but only with a G5 spacer, which provides accessibility of the peptide for receptor binding and cellular uptake resulting in outstanding cytotoxicity.
Click to Show/Hide
|
||||
| Description |
The polymers influenced the cellular uptake of the conjugates in a different manner, but in both groups (GE11 and D4 containing conjugates), one of the compounds was outstanding. The HT-29 cells could uptake Dau[double bond, length as m-dash]Aoa-GFLG-GE11-PEG and Dau[double bond, length as m-dash]Aoa-GFLG-D4-G5-HbPG to the highest extent. The G5 spacer increased the uptake of the D4-HbPG derivative, presumably because the increased distance between the globular HbPG and the very short peptide sequence provides a better receptor binding. In sharp contrast, the G5 spacer decreases the uptake of the conjugates in all other cases. The most significant difference was observed in the case of GE11-PEG, where the G5 spacer completely demolished the internalization. Probably, here the G5 spacer provides more flexibility for the linear PEG chain resulting in decreased receptor binding. There was one outstanding conjugate from each group (Dau[double bond, length as m-dash]Aoa-GFLG-GE11-PEG and Dau[double bond, length as m-dash]Aoa-GFLG-D4-G5-HbPG) in the cytotoxicity measurements that correlated well with the results of the internalization studies. These conjugates were found to be the most potent ones in the viability measurement and were proved to be taken up by HT-29 cells the most effectively. Depending on the type of polymer, the incorporation of the G5 spacer had an opposite effect on the cytotoxic activity of the conjugates. In the presence of the G5 spacer, the antitumour activity of the PEGylated conjugates decreased, while the cytotoxicity of the HbPG-containing conjugates increased, especially in the case of those with the D4 targeting peptide. We observed that some of the conjugates (Dau[double bond, length as m-dash]Aoa-GFLG-D4-HbPG and Dau[double bond, length as m-dash]Aoa-GFLG-GE11-G5-PEG) could not cause complete cell death, i.e., ˜0% viability value - characteristic for cell-free culturing medium - was not achieved even at the highest concentration, since their dose-response curves reached a plateau in a lower concentration range. In our opinion, this can be explained by the different characteristics of the highly hydrophobic peptide chain and the highly hydrophilic polymer segment. Due to this amphiphilic character, self-aggregation of the conjugates may occur, which then may block the accessibility of the targeting peptide for receptor binding, thereby decreasing the efficiency of the conjugate as well. This assumption is also confirmed by the turbidity results, since the observed low turbidity values may be caused by the possible formation of nanosized aggregates.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
DauQ=AoaGFLGGE11G5HbPG [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
6 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | xCELLigence SP system assay | ||||
| MOA of PDC |
As observed, the cathepsin B labile spacer (GFLG), which was incorporated between the targeting peptide and the daunomycin to ensure the efficient release of the active metabolite, highly increases the hydrophobicity of the conjugates. As demonstrated, the solubility problem of these conjugates can be solved by hydrophilic polymer coupling, not only by using the well-known PEG but also by using the amino-monofunctional HbPG. To the best of our knowledge, 1:1 covalent peptide-polymer conjugates with well-defined monofunctional HbPG have been reported here for the first time. The results of the in vitro cell viability and cellular uptake measurements on HT-29 human colon adenocarcinoma cells prove that the HbPG and the PEG highly influenced the biological activity of the drug-peptide-polymer conjugates. In both peptide conjugate series, one GE11 and one D4 targeting peptide-based conjugate were found with outstanding cellular uptake and cytotoxicity values, namely Dau[double bond, length as m-dash]Aoa-GFLG-GE11-PEG and Dau[double bond, length as m-dash]Aoa-GFLG-D4-G5-HbPG. According to our results, the PEG is suitable for longer targeting peptides (e.g. GE11), but the G5 spacer is not suitable irrespective of the length of the peptide because it may decrease the biological effect by increasing the flexibility of the polymer and shading of the targeting moiety. In contrast, the use of the hydrophilic hyperbranched polyglycerol (HbPG) is advantageous for short targeting peptides (e.g. D4) but only with a G5 spacer, which provides accessibility of the peptide for receptor binding and cellular uptake resulting in outstanding cytotoxicity.
Click to Show/Hide
|
||||
| Description |
The polymers influenced the cellular uptake of the conjugates in a different manner, but in both groups (GE11 and D4 containing conjugates), one of the compounds was outstanding. The HT-29 cells could uptake Dau[double bond, length as m-dash]Aoa-GFLG-GE11-PEG and Dau[double bond, length as m-dash]Aoa-GFLG-D4-G5-HbPG to the highest extent. The G5 spacer increased the uptake of the D4-HbPG derivative, presumably because the increased distance between the globular HbPG and the very short peptide sequence provides a better receptor binding. In sharp contrast, the G5 spacer decreases the uptake of the conjugates in all other cases. The most significant difference was observed in the case of GE11-PEG, where the G5 spacer completely demolished the internalization. Probably, here the G5 spacer provides more flexibility for the linear PEG chain resulting in decreased receptor binding. There was one outstanding conjugate from each group (Dau[double bond, length as m-dash]Aoa-GFLG-GE11-PEG and Dau[double bond, length as m-dash]Aoa-GFLG-D4-G5-HbPG) in the cytotoxicity measurements that correlated well with the results of the internalization studies. These conjugates were found to be the most potent ones in the viability measurement and were proved to be taken up by HT-29 cells the most effectively. Depending on the type of polymer, the incorporation of the G5 spacer had an opposite effect on the cytotoxic activity of the conjugates. In the presence of the G5 spacer, the antitumour activity of the PEGylated conjugates decreased, while the cytotoxicity of the HbPG-containing conjugates increased, especially in the case of those with the D4 targeting peptide. We observed that some of the conjugates (Dau[double bond, length as m-dash]Aoa-GFLG-D4-HbPG and Dau[double bond, length as m-dash]Aoa-GFLG-GE11-G5-PEG) could not cause complete cell death, i.e., ˜0% viability value - characteristic for cell-free culturing medium - was not achieved even at the highest concentration, since their dose-response curves reached a plateau in a lower concentration range. In our opinion, this can be explained by the different characteristics of the highly hydrophobic peptide chain and the highly hydrophilic polymer segment. Due to this amphiphilic character, self-aggregation of the conjugates may occur, which then may block the accessibility of the targeting peptide for receptor binding, thereby decreasing the efficiency of the conjugate as well. This assumption is also confirmed by the turbidity results, since the observed low turbidity values may be caused by the possible formation of nanosized aggregates.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
DauQ=AoaGFLGGE11G5PEG [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
9.1 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | xCELLigence SP system assay | ||||
| MOA of PDC |
As observed, the cathepsin B labile spacer (GFLG), which was incorporated between the targeting peptide and the daunomycin to ensure the efficient release of the active metabolite, highly increases the hydrophobicity of the conjugates. As demonstrated, the solubility problem of these conjugates can be solved by hydrophilic polymer coupling, not only by using the well-known PEG but also by using the amino-monofunctional HbPG. To the best of our knowledge, 1:1 covalent peptide-polymer conjugates with well-defined monofunctional HbPG have been reported here for the first time. The results of the in vitro cell viability and cellular uptake measurements on HT-29 human colon adenocarcinoma cells prove that the HbPG and the PEG highly influenced the biological activity of the drug-peptide-polymer conjugates. In both peptide conjugate series, one GE11 and one D4 targeting peptide-based conjugate were found with outstanding cellular uptake and cytotoxicity values, namely Dau[double bond, length as m-dash]Aoa-GFLG-GE11-PEG and Dau[double bond, length as m-dash]Aoa-GFLG-D4-G5-HbPG. According to our results, the PEG is suitable for longer targeting peptides (e.g. GE11), but the G5 spacer is not suitable irrespective of the length of the peptide because it may decrease the biological effect by increasing the flexibility of the polymer and shading of the targeting moiety. In contrast, the use of the hydrophilic hyperbranched polyglycerol (HbPG) is advantageous for short targeting peptides (e.g. D4) but only with a G5 spacer, which provides accessibility of the peptide for receptor binding and cellular uptake resulting in outstanding cytotoxicity.
Click to Show/Hide
|
||||
| Description |
The polymers influenced the cellular uptake of the conjugates in a different manner, but in both groups (GE11 and D4 containing conjugates), one of the compounds was outstanding. The HT-29 cells could uptake Dau[double bond, length as m-dash]Aoa-GFLG-GE11-PEG and Dau[double bond, length as m-dash]Aoa-GFLG-D4-G5-HbPG to the highest extent. The G5 spacer increased the uptake of the D4-HbPG derivative, presumably because the increased distance between the globular HbPG and the very short peptide sequence provides a better receptor binding. In sharp contrast, the G5 spacer decreases the uptake of the conjugates in all other cases. The most significant difference was observed in the case of GE11-PEG, where the G5 spacer completely demolished the internalization. Probably, here the G5 spacer provides more flexibility for the linear PEG chain resulting in decreased receptor binding. There was one outstanding conjugate from each group (Dau[double bond, length as m-dash]Aoa-GFLG-GE11-PEG and Dau[double bond, length as m-dash]Aoa-GFLG-D4-G5-HbPG) in the cytotoxicity measurements that correlated well with the results of the internalization studies. These conjugates were found to be the most potent ones in the viability measurement and were proved to be taken up by HT-29 cells the most effectively. Depending on the type of polymer, the incorporation of the G5 spacer had an opposite effect on the cytotoxic activity of the conjugates. In the presence of the G5 spacer, the antitumour activity of the PEGylated conjugates decreased, while the cytotoxicity of the HbPG-containing conjugates increased, especially in the case of those with the D4 targeting peptide. We observed that some of the conjugates (Dau[double bond, length as m-dash]Aoa-GFLG-D4-HbPG and Dau[double bond, length as m-dash]Aoa-GFLG-GE11-G5-PEG) could not cause complete cell death, i.e., ˜0% viability value - characteristic for cell-free culturing medium - was not achieved even at the highest concentration, since their dose-response curves reached a plateau in a lower concentration range. In our opinion, this can be explained by the different characteristics of the highly hydrophobic peptide chain and the highly hydrophilic polymer segment. Due to this amphiphilic character, self-aggregation of the conjugates may occur, which then may block the accessibility of the targeting peptide for receptor binding, thereby decreasing the efficiency of the conjugate as well. This assumption is also confirmed by the turbidity results, since the observed low turbidity values may be caused by the possible formation of nanosized aggregates.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
DauQ=AoaGFLGGE11HbPG [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
9.3 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | xCELLigence SP system assay | ||||
| MOA of PDC |
As observed, the cathepsin B labile spacer (GFLG), which was incorporated between the targeting peptide and the daunomycin to ensure the efficient release of the active metabolite, highly increases the hydrophobicity of the conjugates. As demonstrated, the solubility problem of these conjugates can be solved by hydrophilic polymer coupling, not only by using the well-known PEG but also by using the amino-monofunctional HbPG. To the best of our knowledge, 1:1 covalent peptide-polymer conjugates with well-defined monofunctional HbPG have been reported here for the first time. The results of the in vitro cell viability and cellular uptake measurements on HT-29 human colon adenocarcinoma cells prove that the HbPG and the PEG highly influenced the biological activity of the drug-peptide-polymer conjugates. In both peptide conjugate series, one GE11 and one D4 targeting peptide-based conjugate were found with outstanding cellular uptake and cytotoxicity values, namely Dau[double bond, length as m-dash]Aoa-GFLG-GE11-PEG and Dau[double bond, length as m-dash]Aoa-GFLG-D4-G5-HbPG. According to our results, the PEG is suitable for longer targeting peptides (e.g. GE11), but the G5 spacer is not suitable irrespective of the length of the peptide because it may decrease the biological effect by increasing the flexibility of the polymer and shading of the targeting moiety. In contrast, the use of the hydrophilic hyperbranched polyglycerol (HbPG) is advantageous for short targeting peptides (e.g. D4) but only with a G5 spacer, which provides accessibility of the peptide for receptor binding and cellular uptake resulting in outstanding cytotoxicity.
Click to Show/Hide
|
||||
| Description |
The polymers influenced the cellular uptake of the conjugates in a different manner, but in both groups (GE11 and D4 containing conjugates), one of the compounds was outstanding. The HT-29 cells could uptake Dau[double bond, length as m-dash]Aoa-GFLG-GE11-PEG and Dau[double bond, length as m-dash]Aoa-GFLG-D4-G5-HbPG to the highest extent. The G5 spacer increased the uptake of the D4-HbPG derivative, presumably because the increased distance between the globular HbPG and the very short peptide sequence provides a better receptor binding. In sharp contrast, the G5 spacer decreases the uptake of the conjugates in all other cases. The most significant difference was observed in the case of GE11-PEG, where the G5 spacer completely demolished the internalization. Probably, here the G5 spacer provides more flexibility for the linear PEG chain resulting in decreased receptor binding. There was one outstanding conjugate from each group (Dau[double bond, length as m-dash]Aoa-GFLG-GE11-PEG and Dau[double bond, length as m-dash]Aoa-GFLG-D4-G5-HbPG) in the cytotoxicity measurements that correlated well with the results of the internalization studies. These conjugates were found to be the most potent ones in the viability measurement and were proved to be taken up by HT-29 cells the most effectively. Depending on the type of polymer, the incorporation of the G5 spacer had an opposite effect on the cytotoxic activity of the conjugates. In the presence of the G5 spacer, the antitumour activity of the PEGylated conjugates decreased, while the cytotoxicity of the HbPG-containing conjugates increased, especially in the case of those with the D4 targeting peptide. We observed that some of the conjugates (Dau[double bond, length as m-dash]Aoa-GFLG-D4-HbPG and Dau[double bond, length as m-dash]Aoa-GFLG-GE11-G5-PEG) could not cause complete cell death, i.e., ˜0% viability value - characteristic for cell-free culturing medium - was not achieved even at the highest concentration, since their dose-response curves reached a plateau in a lower concentration range. In our opinion, this can be explained by the different characteristics of the highly hydrophobic peptide chain and the highly hydrophilic polymer segment. Due to this amphiphilic character, self-aggregation of the conjugates may occur, which then may block the accessibility of the targeting peptide for receptor binding, thereby decreasing the efficiency of the conjugate as well. This assumption is also confirmed by the turbidity results, since the observed low turbidity values may be caused by the possible formation of nanosized aggregates.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
DauQ=AoaGFLGD4G5PEG [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
13.5 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | xCELLigence SP system assay | ||||
| MOA of PDC |
As observed, the cathepsin B labile spacer (GFLG), which was incorporated between the targeting peptide and the daunomycin to ensure the efficient release of the active metabolite, highly increases the hydrophobicity of the conjugates. As demonstrated, the solubility problem of these conjugates can be solved by hydrophilic polymer coupling, not only by using the well-known PEG but also by using the amino-monofunctional HbPG. To the best of our knowledge, 1:1 covalent peptide-polymer conjugates with well-defined monofunctional HbPG have been reported here for the first time. The results of the in vitro cell viability and cellular uptake measurements on HT-29 human colon adenocarcinoma cells prove that the HbPG and the PEG highly influenced the biological activity of the drug-peptide-polymer conjugates. In both peptide conjugate series, one GE11 and one D4 targeting peptide-based conjugate were found with outstanding cellular uptake and cytotoxicity values, namely Dau[double bond, length as m-dash]Aoa-GFLG-GE11-PEG and Dau[double bond, length as m-dash]Aoa-GFLG-D4-G5-HbPG. According to our results, the PEG is suitable for longer targeting peptides (e.g. GE11), but the G5 spacer is not suitable irrespective of the length of the peptide because it may decrease the biological effect by increasing the flexibility of the polymer and shading of the targeting moiety. In contrast, the use of the hydrophilic hyperbranched polyglycerol (HbPG) is advantageous for short targeting peptides (e.g. D4) but only with a G5 spacer, which provides accessibility of the peptide for receptor binding and cellular uptake resulting in outstanding cytotoxicity.
Click to Show/Hide
|
||||
| Description |
The polymers influenced the cellular uptake of the conjugates in a different manner, but in both groups (GE11 and D4 containing conjugates), one of the compounds was outstanding. The HT-29 cells could uptake Dau[double bond, length as m-dash]Aoa-GFLG-GE11-PEG and Dau[double bond, length as m-dash]Aoa-GFLG-D4-G5-HbPG to the highest extent. The G5 spacer increased the uptake of the D4-HbPG derivative, presumably because the increased distance between the globular HbPG and the very short peptide sequence provides a better receptor binding. In sharp contrast, the G5 spacer decreases the uptake of the conjugates in all other cases. The most significant difference was observed in the case of GE11-PEG, where the G5 spacer completely demolished the internalization. Probably, here the G5 spacer provides more flexibility for the linear PEG chain resulting in decreased receptor binding. There was one outstanding conjugate from each group (Dau[double bond, length as m-dash]Aoa-GFLG-GE11-PEG and Dau[double bond, length as m-dash]Aoa-GFLG-D4-G5-HbPG) in the cytotoxicity measurements that correlated well with the results of the internalization studies. These conjugates were found to be the most potent ones in the viability measurement and were proved to be taken up by HT-29 cells the most effectively. Depending on the type of polymer, the incorporation of the G5 spacer had an opposite effect on the cytotoxic activity of the conjugates. In the presence of the G5 spacer, the antitumour activity of the PEGylated conjugates decreased, while the cytotoxicity of the HbPG-containing conjugates increased, especially in the case of those with the D4 targeting peptide. We observed that some of the conjugates (Dau[double bond, length as m-dash]Aoa-GFLG-D4-HbPG and Dau[double bond, length as m-dash]Aoa-GFLG-GE11-G5-PEG) could not cause complete cell death, i.e., ˜0% viability value - characteristic for cell-free culturing medium - was not achieved even at the highest concentration, since their dose-response curves reached a plateau in a lower concentration range. In our opinion, this can be explained by the different characteristics of the highly hydrophobic peptide chain and the highly hydrophilic polymer segment. Due to this amphiphilic character, self-aggregation of the conjugates may occur, which then may block the accessibility of the targeting peptide for receptor binding, thereby decreasing the efficiency of the conjugate as well. This assumption is also confirmed by the turbidity results, since the observed low turbidity values may be caused by the possible formation of nanosized aggregates.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
Cyclic NGR peptidedaunomycin conjugates 4 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
14.5 ± 2.0 µM
|
|||
| Administration Time | 72 h | ||||
| Description |
In contrast, conjugate 2 (containing a disulfide bridge) showed preferential toxicity to HT-1080 cells in the cytostatic experiments, resulting in the highest selectivity. The antitumor effect of 2 increased in time (72 h treatment), but its selectivity decreased significantly. In comparison to conjugates 3 (thioether linkage) and 5 (amide bond), both containing free Lys in the cycle, conjugate 5 had slightly better activity against HT-1080 cells. However, the selectivity of conjugate 5 to CD13 receptors (as judged by the relative toxicity against the two cell lines) was not significant after 6 h treatment, whereas it was significantly more toxic to HT-1080 cells in the 72 h experiment. This can also be explained by its higher chemostability that results in longer exposure the cells to the unmodified NGR peptide-drug conjugate. In contrast, their analogs in which the drug molecule is connected to the side chain of Lys in the cycle (conjugates 4 and 6) were more specific but less toxic, especially in the cytotoxicity experiments. Interestingly, there was no correlation between the specificity and the chemostability of the conjugates.
Click to Show/Hide
|
||||
| In Vitro Model | Fibrosarcoma | HT-1080 cell | CVCL_0317 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
45.2 ± 13.2 µM
|
|||
| Administration Time | 6 h | ||||
| Description |
In contrast, conjugate 2 (containing a disulfide bridge) showed preferential toxicity to HT-1080 cells in the cytostatic experiments, resulting in the highest selectivity. The antitumor effect of 2 increased in time (72 h treatment), but its selectivity decreased significantly. In comparison to conjugates 3 (thioether linkage) and 5 (amide bond), both containing free Lys in the cycle, conjugate 5 had slightly better activity against HT-1080 cells. However, the selectivity of conjugate 5 to CD13 receptors (as judged by the relative toxicity against the two cell lines) was not significant after 6 h treatment, whereas it was significantly more toxic to HT-1080 cells in the 72 h experiment. This can also be explained by its higher chemostability that results in longer exposure the cells to the unmodified NGR peptide-drug conjugate. In contrast, their analogs in which the drug molecule is connected to the side chain of Lys in the cycle (conjugates 4 and 6) were more specific but less toxic, especially in the cytotoxicity experiments. Interestingly, there was no correlation between the specificity and the chemostability of the conjugates.
Click to Show/Hide
|
||||
| In Vitro Model | Fibrosarcoma | HT-1080 cell | CVCL_0317 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | » 50 µM | |||
| Administration Time | 6 h | ||||
| Description |
In contrast, conjugate 2 (containing a disulfide bridge) showed preferential toxicity to HT-1080 cells in the cytostatic experiments, resulting in the highest selectivity. The antitumor effect of 2 increased in time (72 h treatment), but its selectivity decreased significantly. In comparison to conjugates 3 (thioether linkage) and 5 (amide bond), both containing free Lys in the cycle, conjugate 5 had slightly better activity against HT-1080 cells. However, the selectivity of conjugate 5 to CD13 receptors (as judged by the relative toxicity against the two cell lines) was not significant after 6 h treatment, whereas it was significantly more toxic to HT-1080 cells in the 72 h experiment. This can also be explained by its higher chemostability that results in longer exposure the cells to the unmodified NGR peptide-drug conjugate. In contrast, their analogs in which the drug molecule is connected to the side chain of Lys in the cycle (conjugates 4 and 6) were more specific but less toxic, especially in the cytotoxicity experiments. Interestingly, there was no correlation between the specificity and the chemostability of the conjugates.
Click to Show/Hide
|
||||
| In Vitro Model | Colon adenocarcinoma | CD13-negative HT29 cell | CVCL_0320 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | » 50 µM | |||
| Administration Time | 72 h | ||||
| Description |
In contrast, conjugate 2 (containing a disulfide bridge) showed preferential toxicity to HT-1080 cells in the cytostatic experiments, resulting in the highest selectivity. The antitumor effect of 2 increased in time (72 h treatment), but its selectivity decreased significantly. In comparison to conjugates 3 (thioether linkage) and 5 (amide bond), both containing free Lys in the cycle, conjugate 5 had slightly better activity against HT-1080 cells. However, the selectivity of conjugate 5 to CD13 receptors (as judged by the relative toxicity against the two cell lines) was not significant after 6 h treatment, whereas it was significantly more toxic to HT-1080 cells in the 72 h experiment. This can also be explained by its higher chemostability that results in longer exposure the cells to the unmodified NGR peptide-drug conjugate. In contrast, their analogs in which the drug molecule is connected to the side chain of Lys in the cycle (conjugates 4 and 6) were more specific but less toxic, especially in the cytotoxicity experiments. Interestingly, there was no correlation between the specificity and the chemostability of the conjugates.
Click to Show/Hide
|
||||
| In Vitro Model | Colon adenocarcinoma | CD13-negative HT29 cell | CVCL_0320 | ||
Cyclic NGR peptidedaunomycin conjugates 6 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
15.8 ± 4.4 µM
|
|||
| Administration Time | 72 h | ||||
| Description |
In contrast, conjugate 2 (containing a disulfide bridge) showed preferential toxicity to HT-1080 cells in the cytostatic experiments, resulting in the highest selectivity. The antitumor effect of 2 increased in time (72 h treatment), but its selectivity decreased significantly. In comparison to conjugates 3 (thioether linkage) and 5 (amide bond), both containing free Lys in the cycle, conjugate 5 had slightly better activity against HT-1080 cells. However, the selectivity of conjugate 5 to CD13 receptors (as judged by the relative toxicity against the two cell lines) was not significant after 6 h treatment, whereas it was significantly more toxic to HT-1080 cells in the 72 h experiment. This can also be explained by its higher chemostability that results in longer exposure the cells to the unmodified NGR peptide-drug conjugate. In contrast, their analogs in which the drug molecule is connected to the side chain of Lys in the cycle (conjugates 4 and 6) were more specific but less toxic, especially in the cytotoxicity experiments. Interestingly, there was no correlation between the specificity and the chemostability of the conjugates.
Click to Show/Hide
|
||||
| In Vitro Model | Fibrosarcoma | HT-1080 cell | CVCL_0317 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
43.7 ± 4.1 µM
|
|||
| Administration Time | 6 h | ||||
| Description |
In contrast, conjugate 2 (containing a disulfide bridge) showed preferential toxicity to HT-1080 cells in the cytostatic experiments, resulting in the highest selectivity. The antitumor effect of 2 increased in time (72 h treatment), but its selectivity decreased significantly. In comparison to conjugates 3 (thioether linkage) and 5 (amide bond), both containing free Lys in the cycle, conjugate 5 had slightly better activity against HT-1080 cells. However, the selectivity of conjugate 5 to CD13 receptors (as judged by the relative toxicity against the two cell lines) was not significant after 6 h treatment, whereas it was significantly more toxic to HT-1080 cells in the 72 h experiment. This can also be explained by its higher chemostability that results in longer exposure the cells to the unmodified NGR peptide-drug conjugate. In contrast, their analogs in which the drug molecule is connected to the side chain of Lys in the cycle (conjugates 4 and 6) were more specific but less toxic, especially in the cytotoxicity experiments. Interestingly, there was no correlation between the specificity and the chemostability of the conjugates.
Click to Show/Hide
|
||||
| In Vitro Model | Fibrosarcoma | HT-1080 cell | CVCL_0317 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | » 50 µM | |||
| Administration Time | 6 h | ||||
| Description |
In contrast, conjugate 2 (containing a disulfide bridge) showed preferential toxicity to HT-1080 cells in the cytostatic experiments, resulting in the highest selectivity. The antitumor effect of 2 increased in time (72 h treatment), but its selectivity decreased significantly. In comparison to conjugates 3 (thioether linkage) and 5 (amide bond), both containing free Lys in the cycle, conjugate 5 had slightly better activity against HT-1080 cells. However, the selectivity of conjugate 5 to CD13 receptors (as judged by the relative toxicity against the two cell lines) was not significant after 6 h treatment, whereas it was significantly more toxic to HT-1080 cells in the 72 h experiment. This can also be explained by its higher chemostability that results in longer exposure the cells to the unmodified NGR peptide-drug conjugate. In contrast, their analogs in which the drug molecule is connected to the side chain of Lys in the cycle (conjugates 4 and 6) were more specific but less toxic, especially in the cytotoxicity experiments. Interestingly, there was no correlation between the specificity and the chemostability of the conjugates.
Click to Show/Hide
|
||||
| In Vitro Model | Colon adenocarcinoma | CD13-negative HT29 cell | CVCL_0320 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | » 50 µM | |||
| Administration Time | 72 h | ||||
| Description |
In contrast, conjugate 2 (containing a disulfide bridge) showed preferential toxicity to HT-1080 cells in the cytostatic experiments, resulting in the highest selectivity. The antitumor effect of 2 increased in time (72 h treatment), but its selectivity decreased significantly. In comparison to conjugates 3 (thioether linkage) and 5 (amide bond), both containing free Lys in the cycle, conjugate 5 had slightly better activity against HT-1080 cells. However, the selectivity of conjugate 5 to CD13 receptors (as judged by the relative toxicity against the two cell lines) was not significant after 6 h treatment, whereas it was significantly more toxic to HT-1080 cells in the 72 h experiment. This can also be explained by its higher chemostability that results in longer exposure the cells to the unmodified NGR peptide-drug conjugate. In contrast, their analogs in which the drug molecule is connected to the side chain of Lys in the cycle (conjugates 4 and 6) were more specific but less toxic, especially in the cytotoxicity experiments. Interestingly, there was no correlation between the specificity and the chemostability of the conjugates.
Click to Show/Hide
|
||||
| In Vitro Model | Colon adenocarcinoma | CD13-negative HT29 cell | CVCL_0320 | ||
H-TFFYGGSRGK(Dau=Aoa-GFLG)RNNFKTEEY-OH [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
16.9 ± 5.6 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The blood-brain barrier (BBB) is a semipermeable system, and, therefore, most of the active substances are poorly transported through this barrier, resulting in decreased therapeutic effects. Angiopep-2 (TFFYGGSRGKRNNFKTEEY) is a peptide ligand of low-density lipoprotein receptor-related protein-1 (LRP1), which can cross the BBB via receptor-mediated transcytosis and simultaneously target glioblastomas. Angiopep-2 contains three amino groups that have previously been used to produce drug-peptide conjugates, although the role and importance of each position have not yet been investigated. Thus, we studied the number and position of drug molecules in Angiopep-2 based conjugates. Conjugates containing one, two, and three daunomycin molecules conjugated via oxime linkage in all possible variations were prepared. The in vitro cytostatic effect and cellular uptake of the conjugates were investigated on U87 human glioblastoma cells. Degradation studies in the presence of rat liver lysosomal homogenates were also performed in order for us to better understand the structure-activity relationship and to determine the smallest metabolites. Conjugates with the best cytostatic effects had a drug molecule at the N-terminus. We demonstrated that the increasing number of drug molecules does not necessarily increase the efficacy of the conjugates, and proved that modification of the different conjugation sites results in differing biological effectiveness.
Click to Show/Hide
|
||||
| Description |
The in vitro cytostatic effect and the in vitro cellular uptake of the spacer-containing daunomycin-peptide conjugates (9-11) were studied on U87 human glioblastoma cells as previously described. The measured IC50 values are shown in Table 2, while the measured cellular uptake at 10 μM and 50 μM concentration are given in Figure 4. The fluorescence intensity values showed good correlation with the percentage of daunomycin-positive cells in the case of these conjugates as well. Conjugate 9 showed slight toxicity during the measurements at a concentration of 50 μM.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
Cyclic NGR peptidedaunomycin conjugates 1 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
20.6 ± 5.6 µM
|
|||
| Administration Time | 72 h | ||||
| Description |
In contrast, conjugate 2 (containing a disulfide bridge) showed preferential toxicity to HT-1080 cells in the cytostatic experiments, resulting in the highest selectivity. The antitumor effect of 2 increased in time (72 h treatment), but its selectivity decreased significantly. In comparison to conjugates 3 (thioether linkage) and 5 (amide bond), both containing free Lys in the cycle, conjugate 5 had slightly better activity against HT-1080 cells. However, the selectivity of conjugate 5 to CD13 receptors (as judged by the relative toxicity against the two cell lines) was not significant after 6 h treatment, whereas it was significantly more toxic to HT-1080 cells in the 72 h experiment. This can also be explained by its higher chemostability that results in longer exposure the cells to the unmodified NGR peptide-drug conjugate. In contrast, their analogs in which the drug molecule is connected to the side chain of Lys in the cycle (conjugates 4 and 6) were more specific but less toxic, especially in the cytotoxicity experiments. Interestingly, there was no correlation between the specificity and the chemostability of the conjugates.
Click to Show/Hide
|
||||
| In Vitro Model | Colon adenocarcinoma | CD13-negative HT29 cell | CVCL_0320 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
23.8 ± 0.7 µM
|
|||
| Administration Time | 72 h | ||||
| Description |
In contrast, conjugate 2 (containing a disulfide bridge) showed preferential toxicity to HT-1080 cells in the cytostatic experiments, resulting in the highest selectivity. The antitumor effect of 2 increased in time (72 h treatment), but its selectivity decreased significantly. In comparison to conjugates 3 (thioether linkage) and 5 (amide bond), both containing free Lys in the cycle, conjugate 5 had slightly better activity against HT-1080 cells. However, the selectivity of conjugate 5 to CD13 receptors (as judged by the relative toxicity against the two cell lines) was not significant after 6 h treatment, whereas it was significantly more toxic to HT-1080 cells in the 72 h experiment. This can also be explained by its higher chemostability that results in longer exposure the cells to the unmodified NGR peptide-drug conjugate. In contrast, their analogs in which the drug molecule is connected to the side chain of Lys in the cycle (conjugates 4 and 6) were more specific but less toxic, especially in the cytotoxicity experiments. Interestingly, there was no correlation between the specificity and the chemostability of the conjugates.
Click to Show/Hide
|
||||
| In Vitro Model | Fibrosarcoma | HT-1080 cell | CVCL_0317 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | » 50 µM | |||
| Administration Time | 6 h | ||||
| Description |
In contrast, conjugate 2 (containing a disulfide bridge) showed preferential toxicity to HT-1080 cells in the cytostatic experiments, resulting in the highest selectivity. The antitumor effect of 2 increased in time (72 h treatment), but its selectivity decreased significantly. In comparison to conjugates 3 (thioether linkage) and 5 (amide bond), both containing free Lys in the cycle, conjugate 5 had slightly better activity against HT-1080 cells. However, the selectivity of conjugate 5 to CD13 receptors (as judged by the relative toxicity against the two cell lines) was not significant after 6 h treatment, whereas it was significantly more toxic to HT-1080 cells in the 72 h experiment. This can also be explained by its higher chemostability that results in longer exposure the cells to the unmodified NGR peptide-drug conjugate. In contrast, their analogs in which the drug molecule is connected to the side chain of Lys in the cycle (conjugates 4 and 6) were more specific but less toxic, especially in the cytotoxicity experiments. Interestingly, there was no correlation between the specificity and the chemostability of the conjugates.
Click to Show/Hide
|
||||
| In Vitro Model | Fibrosarcoma | HT-1080 cell | CVCL_0317 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | » 50 µM | |||
| Administration Time | 6 h | ||||
| Description |
In contrast, conjugate 2 (containing a disulfide bridge) showed preferential toxicity to HT-1080 cells in the cytostatic experiments, resulting in the highest selectivity. The antitumor effect of 2 increased in time (72 h treatment), but its selectivity decreased significantly. In comparison to conjugates 3 (thioether linkage) and 5 (amide bond), both containing free Lys in the cycle, conjugate 5 had slightly better activity against HT-1080 cells. However, the selectivity of conjugate 5 to CD13 receptors (as judged by the relative toxicity against the two cell lines) was not significant after 6 h treatment, whereas it was significantly more toxic to HT-1080 cells in the 72 h experiment. This can also be explained by its higher chemostability that results in longer exposure the cells to the unmodified NGR peptide-drug conjugate. In contrast, their analogs in which the drug molecule is connected to the side chain of Lys in the cycle (conjugates 4 and 6) were more specific but less toxic, especially in the cytotoxicity experiments. Interestingly, there was no correlation between the specificity and the chemostability of the conjugates.
Click to Show/Hide
|
||||
| In Vitro Model | Colon adenocarcinoma | CD13-negative HT29 cell | CVCL_0320 | ||
DauQ=AoaGFLGD4HbPG [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Colon adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
450 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | xCELLigence SP system assay | ||||
| MOA of PDC |
As observed, the cathepsin B labile spacer (GFLG), which was incorporated between the targeting peptide and the daunomycin to ensure the efficient release of the active metabolite, highly increases the hydrophobicity of the conjugates. As demonstrated, the solubility problem of these conjugates can be solved by hydrophilic polymer coupling, not only by using the well-known PEG but also by using the amino-monofunctional HbPG. To the best of our knowledge, 1:1 covalent peptide-polymer conjugates with well-defined monofunctional HbPG have been reported here for the first time. The results of the in vitro cell viability and cellular uptake measurements on HT-29 human colon adenocarcinoma cells prove that the HbPG and the PEG highly influenced the biological activity of the drug-peptide-polymer conjugates. In both peptide conjugate series, one GE11 and one D4 targeting peptide-based conjugate were found with outstanding cellular uptake and cytotoxicity values, namely Dau[double bond, length as m-dash]Aoa-GFLG-GE11-PEG and Dau[double bond, length as m-dash]Aoa-GFLG-D4-G5-HbPG. According to our results, the PEG is suitable for longer targeting peptides (e.g. GE11), but the G5 spacer is not suitable irrespective of the length of the peptide because it may decrease the biological effect by increasing the flexibility of the polymer and shading of the targeting moiety. In contrast, the use of the hydrophilic hyperbranched polyglycerol (HbPG) is advantageous for short targeting peptides (e.g. D4) but only with a G5 spacer, which provides accessibility of the peptide for receptor binding and cellular uptake resulting in outstanding cytotoxicity.
Click to Show/Hide
|
||||
| Description |
The polymers influenced the cellular uptake of the conjugates in a different manner, but in both groups (GE11 and D4 containing conjugates), one of the compounds was outstanding. The HT-29 cells could uptake Dau[double bond, length as m-dash]Aoa-GFLG-GE11-PEG and Dau[double bond, length as m-dash]Aoa-GFLG-D4-G5-HbPG to the highest extent. The G5 spacer increased the uptake of the D4-HbPG derivative, presumably because the increased distance between the globular HbPG and the very short peptide sequence provides a better receptor binding. In sharp contrast, the G5 spacer decreases the uptake of the conjugates in all other cases. The most significant difference was observed in the case of GE11-PEG, where the G5 spacer completely demolished the internalization. Probably, here the G5 spacer provides more flexibility for the linear PEG chain resulting in decreased receptor binding. There was one outstanding conjugate from each group (Dau[double bond, length as m-dash]Aoa-GFLG-GE11-PEG and Dau[double bond, length as m-dash]Aoa-GFLG-D4-G5-HbPG) in the cytotoxicity measurements that correlated well with the results of the internalization studies. These conjugates were found to be the most potent ones in the viability measurement and were proved to be taken up by HT-29 cells the most effectively. Depending on the type of polymer, the incorporation of the G5 spacer had an opposite effect on the cytotoxic activity of the conjugates. In the presence of the G5 spacer, the antitumour activity of the PEGylated conjugates decreased, while the cytotoxicity of the HbPG-containing conjugates increased, especially in the case of those with the D4 targeting peptide. We observed that some of the conjugates (Dau[double bond, length as m-dash]Aoa-GFLG-D4-HbPG and Dau[double bond, length as m-dash]Aoa-GFLG-GE11-G5-PEG) could not cause complete cell death, i.e., ˜0% viability value - characteristic for cell-free culturing medium - was not achieved even at the highest concentration, since their dose-response curves reached a plateau in a lower concentration range. In our opinion, this can be explained by the different characteristics of the highly hydrophobic peptide chain and the highly hydrophilic polymer segment. Due to this amphiphilic character, self-aggregation of the conjugates may occur, which then may block the accessibility of the targeting peptide for receptor binding, thereby decreasing the efficiency of the conjugate as well. This assumption is also confirmed by the turbidity results, since the observed low turbidity values may be caused by the possible formation of nanosized aggregates.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
c[DKP-RGD]-PEG4-sC18(dau=Aoa-GFLG-Lys8) [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
2.7 μM
|
|||
| Administration Time | 72 h | ||||
| Description |
All dau-loaded conjugates showed high toxicity in all cell lines tested with EC50 values in the lower micromolar range.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
3.9 μM
|
|||
| Administration Time | 72 h | ||||
| Description |
All dau-loaded conjugates showed high toxicity in all cell lines tested with EC50 values in the lower micromolar range.
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
7.8 μM
|
|||
| Administration Time | 15 min | ||||
| Description |
More specifically, compounds1band3bdemonstrated significantly higher activity against U87 cells compared to MCF-7 and HT-29 cells (Figure6). Importantly,1bwas more active than3bdemonstrating that the attached CPP is indeed necessary to increase the overall cellular uptake and thus cytotoxic activity of the conjugates. Of note is also that2bwas less efficient than1bbut marginally more active than3b, although with no selectivity. For the two conjugates bearing the GFLG motif, it was seen that3cis still significantly more active in U87 cells expressing integrin receptors. By contrast,1ckept its selectivity against HT-29 cells, but was pronouncedly more active in MCF-7 cells compared to1b.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
11 μM
|
|||
| Administration Time | 72 h | ||||
| Description |
All dau-loaded conjugates showed high toxicity in all cell lines tested with EC50 values in the lower micromolar range.
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
| Experiment 5 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
23.8 μM
|
|||
| Administration Time | 15 min | ||||
| Description |
More specifically, compounds1band3bdemonstrated significantly higher activity against U87 cells compared to MCF-7 and HT-29 cells (Figure6). Importantly,1bwas more active than3bdemonstrating that the attached CPP is indeed necessary to increase the overall cellular uptake and thus cytotoxic activity of the conjugates. Of note is also that2bwas less efficient than1bbut marginally more active than3b, although with no selectivity. For the two conjugates bearing the GFLG motif, it was seen that3cis still significantly more active in U87 cells expressing integrin receptors. By contrast,1ckept its selectivity against HT-29 cells, but was pronouncedly more active in MCF-7 cells compared to1b.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) |
89.1 μM
|
|||
| Administration Time | 15 min | ||||
| Description |
More specifically, compounds1band3bdemonstrated significantly higher activity against U87 cells compared to MCF-7 and HT-29 cells (Figure6). Importantly,1bwas more active than3bdemonstrating that the attached CPP is indeed necessary to increase the overall cellular uptake and thus cytotoxic activity of the conjugates. Of note is also that2bwas less efficient than1bbut marginally more active than3b, although with no selectivity. For the two conjugates bearing the GFLG motif, it was seen that3cis still significantly more active in U87 cells expressing integrin receptors. By contrast,1ckept its selectivity against HT-29 cells, but was pronouncedly more active in MCF-7 cells compared to1b.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
References