
Peptide-drug Conjugate Information
General Information of This Peptide-drug Conjugate (PDC)
| PDC ID |
PDC_00322
|
|||||
|---|---|---|---|---|---|---|
| PDC Name |
RGD-GFLG-DOX
|
|||||
| PDC Status |
Investigative
|
|||||
| Indication |
In total 5 Indication(s)
|
|||||
| Structure |
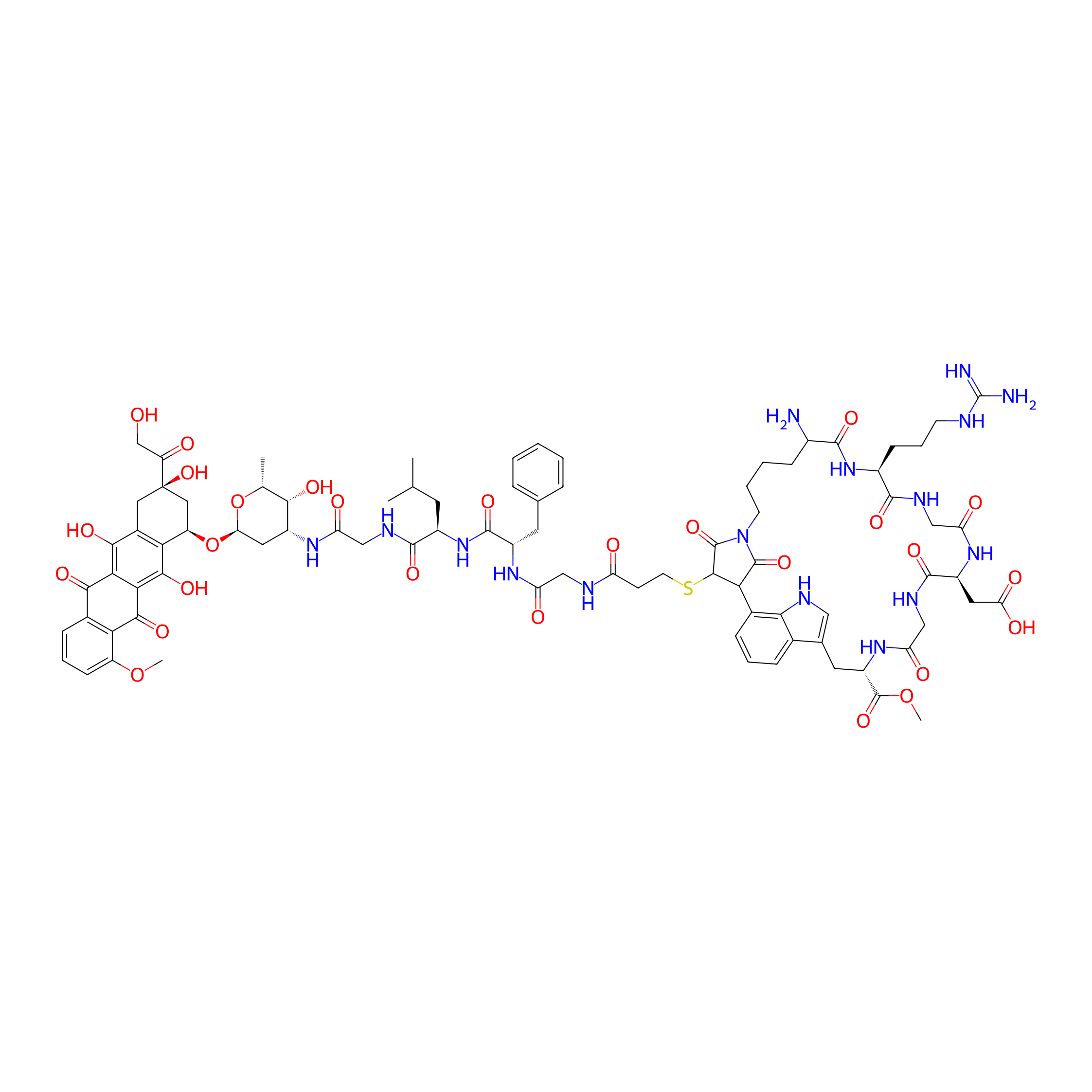
|
|||||
| Peptide Name |
GFLG
|
Peptide Info | ||||
| Receptor Name |
Integrin alpha-V; Integrin beta-3 (ITGAV; ITGB3)
|
Receptor Info | ||||
| Drug Name |
Doxorubicin
|
Drug Info | ||||
| Therapeutic Target |
DNA topoisomerase 2-alpha (TOP2A)
|
Target Info | ||||
| Linker Name |
GFLG
|
Linker Info | ||||
| Peptide Modified Type |
Cyclization modification
|
|||||
| Modified Segment |
Head-to-tail cyclization
|
|||||
| Formula |
C85H106N16O27S
|
|||||
| #Ro5 Violations (Lipinski): 5 | Molecular Weight | 1815.935 | ||||
| Lipid-water partition coefficient (xlogp) | -3.57313 | |||||
| Hydrogen Bond Donor Count (hbonddonor) | 21 | |||||
| Hydrogen Bond Acceptor Count (hbondacc) | 29 | |||||
| Rotatable Bond Count (rotbonds) | 29 | |||||
Full List of Activity Data of This Peptide-drug Conjugate
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Lung cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4 µM
|
|||
| Administration Time | 36 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In conclusion, we developed a robust and regioselective rhodium-catalyzed methodology for C(7)-H Trp maleimidation. This reaction served as an efficient tool for peptide/drug modification, ligation, and particularly peptide cyclization, confirming its promising potential in pharmaceutical chemistry and drug synthesis. Notably, this catalytical system is not limited by the Trp position in the peptides. We also demonstrated that tryptophan-substituted maleimide could be used as an effective click functional group to rapidly react with sulfhydryl groups. Moreover, the introduced N-pivaloyl directing group and protecting groups of the peptides could be removed in a single step, providing a more convenient approach compared to the previous methods, which require multi-step removal of the corresponding directing groups and peptide protection groups. Additionally, cyclic peptide 10a exhibited excellent binding affinity to integrin vβ3, indicating its good drug-like properties. With rational design, RGD- GFLG -DOX, which is a stapled PDC, displayed higher selectivity, stronger binding affinity and better cell penetrability than the more commonly used DOX. The proposed strategy for rapid preparation of stapled peptides is expected to further improve PDC formulation.
Click to Show/Hide
|
||||
| Description |
Doxorubicin (DOX) is one of the most effective anticancer drugs and has been successfully used in clinical practice. However, DOX cannot differentiate between cancer cells and normal cells, which may induce unwanted side effects and severe toxicity. Compared with traditional small-molecule anticancer drugs, the peptide-drug conjugates (PDCs) have enhanced targeting specificity and water solubility. Based on these advantages, to further explore the function of 10a, we designed and prepared a anticancer PDC drug compound RGD-GFLG-DOX containing the tetrapeptide linker Gly-Phe-Leu-Gly, which can be cleaved in presence of cathepsin B, a highly upregulated enzyme in malignant tumors, to release the drug. RGD-GFLG was synthesized as a control. The inhibitory effects of RGD-GFLG-DOX on cancer cell lines were assessed using cytotoxicity assay. Specifically, the effects of RGD-GFLG-DOX were evaluated on integrin v3-positive cancer cell lines, including A549 and U87MG cells, integrin v3-negative cancer cell lines such as HeLa and MCF-7 cells, as well as normal cell lines, namely LO2 and MIHA cells. RGD-GFLG-DOX exhibited a lower cytotoxicity on HeLa, MCF-7, LO2 and MIHA cells, but a stronger cytotoxicity than DOX on A549 and U87MG cells. For comparison, RGD-GFLG demonstrated minimal cytotoxicity. In addition, the cytotoxicity of RGD-GFLG-DOX with various concentrations (0-40 uM) on A549 and U87MG cells was studied. The results showed that the cytotoxicity of RGD-GFLG-DOX on A549 and U87MG cells was dose-dependent. These indicate that RGD-GFLG-DOX has a good specificity and inhibitory activity toward integrin v3-overexpressed A549 and U87MG cells.
Click to Show/Hide
|
||||
| In Vitro Model | Lung adenocarcinoma | A-549 cell | CVCL_0023 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
4 µM
|
|||
| Administration Time | 36 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In conclusion, we developed a robust and regioselective rhodium-catalyzed methodology for C(7)-H Trp maleimidation. This reaction served as an efficient tool for peptide/drug modification, ligation, and particularly peptide cyclization, confirming its promising potential in pharmaceutical chemistry and drug synthesis. Notably, this catalytical system is not limited by the Trp position in the peptides. We also demonstrated that tryptophan-substituted maleimide could be used as an effective click functional group to rapidly react with sulfhydryl groups. Moreover, the introduced N-pivaloyl directing group and protecting groups of the peptides could be removed in a single step, providing a more convenient approach compared to the previous methods, which require multi-step removal of the corresponding directing groups and peptide protection groups. Additionally, cyclic peptide 10a exhibited excellent binding affinity to integrin vβ3, indicating its good drug-like properties. With rational design, RGD- GFLG -DOX, which is a stapled PDC, displayed higher selectivity, stronger binding affinity and better cell penetrability than the more commonly used DOX. The proposed strategy for rapid preparation of stapled peptides is expected to further improve PDC formulation.
Click to Show/Hide
|
||||
| Description |
Doxorubicin (DOX) is one of the most effective anticancer drugs and has been successfully used in clinical practice. However, DOX cannot differentiate between cancer cells and normal cells, which may induce unwanted side effects and severe toxicity. Compared with traditional small-molecule anticancer drugs, the peptide-drug conjugates (PDCs) have enhanced targeting specificity and water solubility. Based on these advantages, to further explore the function of 10a, we designed and prepared a anticancer PDC drug compound RGD-GFLG-DOX containing the tetrapeptide linker Gly-Phe-Leu-Gly, which can be cleaved in presence of cathepsin B, a highly upregulated enzyme in malignant tumors, to release the drug. RGD-GFLG was synthesized as a control. The inhibitory effects of RGD-GFLG-DOX on cancer cell lines were assessed using cytotoxicity assay. Specifically, the effects of RGD-GFLG-DOX were evaluated on integrin v3-positive cancer cell lines, including A549 and U87MG cells, integrin v3-negative cancer cell lines such as HeLa and MCF-7 cells, as well as normal cell lines, namely LO2 and MIHA cells. RGD-GFLG-DOX exhibited a lower cytotoxicity on HeLa, MCF-7, LO2 and MIHA cells, but a stronger cytotoxicity than DOX on A549 and U87MG cells. For comparison, RGD-GFLG demonstrated minimal cytotoxicity. In addition, the cytotoxicity of RGD-GFLG-DOX with various concentrations (0-40 uM) on A549 and U87MG cells was studied. The results showed that the cytotoxicity of RGD-GFLG-DOX on A549 and U87MG cells was dose-dependent. These indicate that RGD-GFLG-DOX has a good specificity and inhibitory activity toward integrin v3-overexpressed A549 and U87MG cells.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Cervical carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 40 µM | |||
| Administration Time | 36 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In conclusion, we developed a robust and regioselective rhodium-catalyzed methodology for C(7)-H Trp maleimidation. This reaction served as an efficient tool for peptide/drug modification, ligation, and particularly peptide cyclization, confirming its promising potential in pharmaceutical chemistry and drug synthesis. Notably, this catalytical system is not limited by the Trp position in the peptides. We also demonstrated that tryptophan-substituted maleimide could be used as an effective click functional group to rapidly react with sulfhydryl groups. Moreover, the introduced N-pivaloyl directing group and protecting groups of the peptides could be removed in a single step, providing a more convenient approach compared to the previous methods, which require multi-step removal of the corresponding directing groups and peptide protection groups. Additionally, cyclic peptide 10a exhibited excellent binding affinity to integrin vβ3, indicating its good drug-like properties. With rational design, RGD- GFLG -DOX, which is a stapled PDC, displayed higher selectivity, stronger binding affinity and better cell penetrability than the more commonly used DOX. The proposed strategy for rapid preparation of stapled peptides is expected to further improve PDC formulation.
Click to Show/Hide
|
||||
| Description |
Doxorubicin (DOX) is one of the most effective anticancer drugs and has been successfully used in clinical practice. However, DOX cannot differentiate between cancer cells and normal cells, which may induce unwanted side effects and severe toxicity. Compared with traditional small-molecule anticancer drugs, the peptide-drug conjugates (PDCs) have enhanced targeting specificity and water solubility. Based on these advantages, to further explore the function of 10a, we designed and prepared a anticancer PDC drug compound RGD-GFLG-DOX containing the tetrapeptide linker Gly-Phe-Leu-Gly, which can be cleaved in presence of cathepsin B, a highly upregulated enzyme in malignant tumors, to release the drug. RGD-GFLG was synthesized as a control. The inhibitory effects of RGD-GFLG-DOX on cancer cell lines were assessed using cytotoxicity assay. Specifically, the effects of RGD-GFLG-DOX were evaluated on integrin v3-positive cancer cell lines, including A549 and U87MG cells, integrin v3-negative cancer cell lines such as HeLa and MCF-7 cells, as well as normal cell lines, namely LO2 and MIHA cells. RGD-GFLG-DOX exhibited a lower cytotoxicity on HeLa, MCF-7, LO2 and MIHA cells, but a stronger cytotoxicity than DOX on A549 and U87MG cells. For comparison, RGD-GFLG demonstrated minimal cytotoxicity. In addition, the cytotoxicity of RGD-GFLG-DOX with various concentrations (0-40 uM) on A549 and U87MG cells was studied. The results showed that the cytotoxicity of RGD-GFLG-DOX on A549 and U87MG cells was dose-dependent. These indicate that RGD-GFLG-DOX has a good specificity and inhibitory activity toward integrin v3-overexpressed A549 and U87MG cells.
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Liver cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 40 µM | |||
| Administration Time | 36 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In conclusion, we developed a robust and regioselective rhodium-catalyzed methodology for C(7)-H Trp maleimidation. This reaction served as an efficient tool for peptide/drug modification, ligation, and particularly peptide cyclization, confirming its promising potential in pharmaceutical chemistry and drug synthesis. Notably, this catalytical system is not limited by the Trp position in the peptides. We also demonstrated that tryptophan-substituted maleimide could be used as an effective click functional group to rapidly react with sulfhydryl groups. Moreover, the introduced N-pivaloyl directing group and protecting groups of the peptides could be removed in a single step, providing a more convenient approach compared to the previous methods, which require multi-step removal of the corresponding directing groups and peptide protection groups. Additionally, cyclic peptide 10a exhibited excellent binding affinity to integrin vβ3, indicating its good drug-like properties. With rational design, RGD- GFLG -DOX, which is a stapled PDC, displayed higher selectivity, stronger binding affinity and better cell penetrability than the more commonly used DOX. The proposed strategy for rapid preparation of stapled peptides is expected to further improve PDC formulation.
Click to Show/Hide
|
||||
| Description |
Doxorubicin (DOX) is one of the most effective anticancer drugs and has been successfully used in clinical practice. However, DOX cannot differentiate between cancer cells and normal cells, which may induce unwanted side effects and severe toxicity. Compared with traditional small-molecule anticancer drugs, the peptide-drug conjugates (PDCs) have enhanced targeting specificity and water solubility. Based on these advantages, to further explore the function of 10a, we designed and prepared a anticancer PDC drug compound RGD-GFLG-DOX containing the tetrapeptide linker Gly-Phe-Leu-Gly, which can be cleaved in presence of cathepsin B, a highly upregulated enzyme in malignant tumors, to release the drug. RGD-GFLG was synthesized as a control. The inhibitory effects of RGD-GFLG-DOX on cancer cell lines were assessed using cytotoxicity assay. Specifically, the effects of RGD-GFLG-DOX were evaluated on integrin v3-positive cancer cell lines, including A549 and U87MG cells, integrin v3-negative cancer cell lines such as HeLa and MCF-7 cells, as well as normal cell lines, namely LO2 and MIHA cells. RGD-GFLG-DOX exhibited a lower cytotoxicity on HeLa, MCF-7, LO2 and MIHA cells, but a stronger cytotoxicity than DOX on A549 and U87MG cells. For comparison, RGD-GFLG demonstrated minimal cytotoxicity. In addition, the cytotoxicity of RGD-GFLG-DOX with various concentrations (0-40 uM) on A549 and U87MG cells was studied. The results showed that the cytotoxicity of RGD-GFLG-DOX on A549 and U87MG cells was dose-dependent. These indicate that RGD-GFLG-DOX has a good specificity and inhibitory activity toward integrin v3-overexpressed A549 and U87MG cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MIHA cell | CVCL_SA11 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 40 µM | |||
| Administration Time | 36 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In conclusion, we developed a robust and regioselective rhodium-catalyzed methodology for C(7)-H Trp maleimidation. This reaction served as an efficient tool for peptide/drug modification, ligation, and particularly peptide cyclization, confirming its promising potential in pharmaceutical chemistry and drug synthesis. Notably, this catalytical system is not limited by the Trp position in the peptides. We also demonstrated that tryptophan-substituted maleimide could be used as an effective click functional group to rapidly react with sulfhydryl groups. Moreover, the introduced N-pivaloyl directing group and protecting groups of the peptides could be removed in a single step, providing a more convenient approach compared to the previous methods, which require multi-step removal of the corresponding directing groups and peptide protection groups. Additionally, cyclic peptide 10a exhibited excellent binding affinity to integrin vβ3, indicating its good drug-like properties. With rational design, RGD- GFLG -DOX, which is a stapled PDC, displayed higher selectivity, stronger binding affinity and better cell penetrability than the more commonly used DOX. The proposed strategy for rapid preparation of stapled peptides is expected to further improve PDC formulation.
Click to Show/Hide
|
||||
| Description |
Doxorubicin (DOX) is one of the most effective anticancer drugs and has been successfully used in clinical practice. However, DOX cannot differentiate between cancer cells and normal cells, which may induce unwanted side effects and severe toxicity. Compared with traditional small-molecule anticancer drugs, the peptide-drug conjugates (PDCs) have enhanced targeting specificity and water solubility. Based on these advantages, to further explore the function of 10a, we designed and prepared a anticancer PDC drug compound RGD-GFLG-DOX containing the tetrapeptide linker Gly-Phe-Leu-Gly, which can be cleaved in presence of cathepsin B, a highly upregulated enzyme in malignant tumors, to release the drug. RGD-GFLG was synthesized as a control. The inhibitory effects of RGD-GFLG-DOX on cancer cell lines were assessed using cytotoxicity assay. Specifically, the effects of RGD-GFLG-DOX were evaluated on integrin v3-positive cancer cell lines, including A549 and U87MG cells, integrin v3-negative cancer cell lines such as HeLa and MCF-7 cells, as well as normal cell lines, namely LO2 and MIHA cells. RGD-GFLG-DOX exhibited a lower cytotoxicity on HeLa, MCF-7, LO2 and MIHA cells, but a stronger cytotoxicity than DOX on A549 and U87MG cells. For comparison, RGD-GFLG demonstrated minimal cytotoxicity. In addition, the cytotoxicity of RGD-GFLG-DOX with various concentrations (0-40 uM) on A549 and U87MG cells was studied. The results showed that the cytotoxicity of RGD-GFLG-DOX on A549 and U87MG cells was dose-dependent. These indicate that RGD-GFLG-DOX has a good specificity and inhibitory activity toward integrin v3-overexpressed A549 and U87MG cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Liver cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 40 µM | |||
| Administration Time | 36 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
In conclusion, we developed a robust and regioselective rhodium-catalyzed methodology for C(7)-H Trp maleimidation. This reaction served as an efficient tool for peptide/drug modification, ligation, and particularly peptide cyclization, confirming its promising potential in pharmaceutical chemistry and drug synthesis. Notably, this catalytical system is not limited by the Trp position in the peptides. We also demonstrated that tryptophan-substituted maleimide could be used as an effective click functional group to rapidly react with sulfhydryl groups. Moreover, the introduced N-pivaloyl directing group and protecting groups of the peptides could be removed in a single step, providing a more convenient approach compared to the previous methods, which require multi-step removal of the corresponding directing groups and peptide protection groups. Additionally, cyclic peptide 10a exhibited excellent binding affinity to integrin vβ3, indicating its good drug-like properties. With rational design, RGD- GFLG -DOX, which is a stapled PDC, displayed higher selectivity, stronger binding affinity and better cell penetrability than the more commonly used DOX. The proposed strategy for rapid preparation of stapled peptides is expected to further improve PDC formulation.
Click to Show/Hide
|
||||
| Description |
Doxorubicin (DOX) is one of the most effective anticancer drugs and has been successfully used in clinical practice. However, DOX cannot differentiate between cancer cells and normal cells, which may induce unwanted side effects and severe toxicity. Compared with traditional small-molecule anticancer drugs, the peptide-drug conjugates (PDCs) have enhanced targeting specificity and water solubility. Based on these advantages, to further explore the function of 10a, we designed and prepared a anticancer PDC drug compound RGD-GFLG-DOX containing the tetrapeptide linker Gly-Phe-Leu-Gly, which can be cleaved in presence of cathepsin B, a highly upregulated enzyme in malignant tumors, to release the drug. RGD-GFLG was synthesized as a control. The inhibitory effects of RGD-GFLG-DOX on cancer cell lines were assessed using cytotoxicity assay. Specifically, the effects of RGD-GFLG-DOX were evaluated on integrin v3-positive cancer cell lines, including A549 and U87MG cells, integrin v3-negative cancer cell lines such as HeLa and MCF-7 cells, as well as normal cell lines, namely LO2 and MIHA cells. RGD-GFLG-DOX exhibited a lower cytotoxicity on HeLa, MCF-7, LO2 and MIHA cells, but a stronger cytotoxicity than DOX on A549 and U87MG cells. For comparison, RGD-GFLG demonstrated minimal cytotoxicity. In addition, the cytotoxicity of RGD-GFLG-DOX with various concentrations (0-40 uM) on A549 and U87MG cells was studied. The results showed that the cytotoxicity of RGD-GFLG-DOX on A549 and U87MG cells was dose-dependent. These indicate that RGD-GFLG-DOX has a good specificity and inhibitory activity toward integrin v3-overexpressed A549 and U87MG cells.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | LO #2 cell | CVCL_C7SD | ||
References