
Drug Information
General Information of This Drug
| Drug ID | DRG00011 | |||||
|---|---|---|---|---|---|---|
| Drug Name | Doxorubicin | |||||
| Synonyms |
doxorubicin; Adriamycin; 23214-92-8; Adriablastin; Doxil; Doxorubicine; Adriblastina; Doxorubicinum; 14-Hydroxydaunomycin; Doxorubicina; 14-Hydroxydaunorubicine; Adriamycin semiquinone; Doxorubicine [INN-French]; Doxorubicinum [INN-Latin]; Doxorubicina [INN-Spanish]; Hydroxydaunorubicin; CCRIS 739; HSDB 3070; NCI-C01514; NDC 38242-874; EINECS 245-495-6; FI 106; NSC 123127; CHEBI:28748; UNII-80168379AG; NSC-759155; CHEMBL53463; Caelyx (liposomal doxorubicin); (7S,9S)-7-[(2R,4S,5S,6S)-4-amino-5-hydroxy-6-methyloxan-2-yl]oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-8,10-dihydro-7H-tetracene-5,12-dione; 5,12-Naphthacenedione, 10-((3-amino-2,3,6-trideoxy-alpha-L-lyxo-hexopyranosyl)oxy)-7,8,9,10-tetrahydro-6,8,11-trihydroxy-8-(hydroxyacetyl)-1-methoxy-, (8S-cis)-; 80168379AG; DTXSID8021480; (1S,3S)-3-Glycoloyl-1,2,3,4,6,11-hexahydro-3,5,12-trihydroxy-10-methoxy-6,11-dioxo-1-naphthacenyl-(3-amino-2,3,6-tridesoxy-alpha-L-lyxo-hexopyranosid); (1S,3S)-3-glycoloyl-3,5,12-trihydroxy-10-methoxy-6,11-dioxo-1,2,3,4,6,11-hexahydrotetracen-1-yl 3-amino-2,3,6-trideoxy-alpha-L-lyxo-hexopyranoside; (8S,10S)-10-((3-Amino-2,3,6-trideoxy-alpha-L-lyxo-hexopyranosyl)oxy)-8-glycoloyl-7,8,9,10-tetrahydro-6,8,11-trihydroxy-1-methoxy-5,12-naphthacenedione; 1,2,3,4,6,11-Hexahydro-4beta,5,12-trihydroxy-4-(hydroxyacetyl)-10-methoxy-6,11-dioxonaphthacen-1beta-yl-3-amino-2,3,6-trideoxy-alpha-L-lyxohexopyranoside; ADR; ADM; Doxorubicine (INN-French); Doxorubicinum (INN-Latin); NSC-123127; Doxorubicina (INN-Spanish); DOXORUBICIN (MART.); DOXORUBICIN [MART.]; (1S,3S)-3,5,12-trihydroxy-3-(hydroxyacetyl)-10-methoxy-6,11-dioxo-1,2,3,4,6,11-hexahydrotetracen-1-yl 3-amino-2,3,6-trideoxy-alpha-L-lyxo-hexopyranoside; (8S,10S)-10-(((2R,4S,5S,6S)-4-amino-5-hydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)-6,8,11-trihydroxy-8-(2-hydroxyacetyl)-1-methoxy-7,8,9,10-tetrahydrotetracene-5,12-dione; (8S,10S)-10-{[(2R,4S,5S,6S)-4-amino-5-hydroxy-6-methyloxan-2-yl]oxy}-6,8,11-trihydroxy-8-(2-hydroxyacetyl)-1-methoxy-5,7,8,9,10,12-hexahydrotetracene-5,12-dione; (8S-cis)-10-((3-amino-2,3,6-trideoxy-alpha-L-lyxo-hexopyranosyl)oxy)-7,8,9,10-tetrahydro-6,8,11-trihydroxy-8-(hydroxyacetyl)-1-methoxy-5,12-naphthacenedione; Doxorubicin [USAN:INN:BAN]; ThermoDox; hydroxydaunomycin; MLS000028393; DM2; Doxorubicin-hLL1; (1S,3S)-3,5,12-trihydroxy-3-(hydroxyacetyl)-10-(methyloxy)-6,11-dioxo-1,2,3,4,6,11-hexahydrotetracen-1-yl 3-amino-2,3,6-trideoxy-alpha-L-lyxo-hexopyranoside; (7S,9S)-7-[(2R,4S,5S,6S)-4-amino-5-hydroxy-6-methyl-tetrahydropyran-2-yl]oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-8,10-dihydro-7H-tetracene-5,12-dione; 5,12-Naphthacenedione, 10-((3-amino-2,3,6-trideoxy-alpha-L-lyxo-hexopyranosyl)oxy)-7,8,9,10-tetrahydro-6,8,11-trihydroxy-8-(hydroxyacetyl)-1-methoxy-, (8S,10S)-; 5,12-Naphthacenedione, 10-[(3-amino-2,3,6-trideoxy-alpha-L-lyxo-hexopyranosyl)oxy]-7,8,9,10-tetrahydro-6,8,11-trihydroxy-8-(2-hydroxyacetyl)-1-methoxy-, (8S,10S)-; Adriblastina (TN); Doxorubicin-P4/D10; Doxorubicin (USAN/INN); VALRUBICIN IMPURITY, DOXORUBICIN (USP IMPURITY); VALRUBICIN IMPURITY, DOXORUBICIN [USP IMPURITY]; Doxorubicin-hLL1 conjugate; doxorrubicina; Doxorubicin-P4/D10 conjugate; Hydroxyldaunorubicin; Hydroxyl Daunorubicin; NSC123127; DOXORUBICIN [MI]; (7S,9S)-7-[(2R,4S,5S,6S)-4-amino-5-hydroxy-6-methyloxan-2-yl]oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-8,10-dihydro-7H-tetracene-5,12-dione;hydrochloride; Prestwick0_000438; Prestwick1_000438; Prestwick2_000438; Prestwick3_000438; DOXORUBICIN [INN]; DOXORUBICIN [HSDB]; DOXORUBICIN [USAN]; Probes1_000151; Probes2_000129; DOXORUBICIN [VANDF]; SCHEMBL3243; BSPBio_000456; BSPBio_001031; DOXORUBICIN [WHO-DD]; 10-((3-Amino-2,3,6-trideoxy-D-lyxohexopyranosyl)oxy)-8-glycolcyl-7,8,9,10-tetrahydro-6,8,11-trihydroxy-1-methoxy-5,12-naphthacenedione; SPBio_002395; (8S-cis)-10-; BPBio1_000502; cid_443939; DTXCID301480; GTPL7069; Valrubicin impurity, doxorubicin; BDBM22984; BDBM32022; L01DB01; HMS2089H06; (8S,10S)-10-((3-AMINO-2,3,6-TRIDEOXY-.ALPHA.-L-LYXO-HEXOPYRANOSYL)OXY)-8-GLYCOLOYL-7,8,9,10-TETRAHYDRO-6,8,11-TRIHYDROXY-1-METHOXY-5,12-NAPHTHACENEDIONE; 5,12-NAPHTHACENEDIONE, 10-((3-AMINO-2,3,6-TRIDEOXY-.ALPHA.-L-LYXO-HEXOPYRANOSYL)OXY)-7,8,9,10-TETRAHYDRO-6,8,11-TRIHYDROXY-8-(HYDROXYACETYL)-1-METHOXY-, (8S-CIS)-; GR-319; HY-15142A; LMPK13050001; AKOS015951330; Conjugate of doxorubicin with humanized monoclonal antibody LL1 against CD74; Conjugate of doxorubicin with monoclonal antibody P4/D10 against GP120; DB00997; SMP1_000106; NCGC00024415-35; NCGC00024415-37; NCGC00024415-38; NCGC00024415-40; NCGC00024415-41; NCGC00024415-42; NCGC00024415-61; BP-23114; NS00002473; (8S,10S)-10; (8S,10S)-10-; C01661; D03899; EN300-120698; Epirubicin hydrochloride impurity, doxorubicin-; H11954; Q18936; A816625; BRD-K92093830-003-04-3; BRD-K92093830-003-25-8; EPIRUBICIN HYDROCHLORIDE IMPURITY C [EP IMPURITY]; DAUNORUBICIN HYDROCHLORIDE IMPURITY D [EP IMPURITY]; EPIRUBICIN HYDROCHLORIDE IMPURITY, DOXORUBICIN- [USP IMPURITY]; (7S,9R)-7-[(2S,4S,5S,6S)-4-Amino-5-hydroxy-6-methyl-oxan-2-yl]oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-8,10-dihydro-7H-tetracene-5,12-dione; (7S,9S)-7-[(2R,4S,5S,6S)-4-amino-5-hydroxy-6-methyl-tetrahydropyran-2-yl]oxy-9-glycoloyl-6,9,11-trihydroxy-4-methoxy-8,10-dihydro-7H-tetracene-5,12-quinone;hydrochloride; (7S,9S)-7-[(2R,4S,5S,6S)-4-azanyl-6-methyl-5-oxidanyl-oxan-2-yl]oxy-4-methoxy-6,9,11-tris(oxidanyl)-9-(2-oxidanylethanoyl)-8,10-dihydro-7H-tetracene-5,12-dione;hydrochloride; (7S,9S)-7-[(4S,5S,6S)-4-amino-5-hydroxy-6-methyloxan-2-yl]oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-8,10-dihydro-7H-tetracene-5,12-dione; (7S,9S)-7-[[(2R,4S,5S,6S)-4-amino-5-hydroxy-6-methyl-2-oxanyl]oxy]-6,9,11-trihydroxy-9-(2-hydroxy-1-oxoethyl)-4-methoxy-8,10-dihydro-7H-tetracene-5,12-dione;hydrochloride; (8S,10S)-10-(((2R,4S,5S,6S)-4-amino-5-hydroxy-6-methyltetrahydro-2H-pyran-2-yl)oxy)-6,8,11-trihydroxy-8-(2-hydroxyacetyl)-1-methoxy-7,8,9,10-tetrahydrotetracene-5,12-dione;(7S,9S)-7-[(4S,5S,6S)-4-amino-5-hydroxy-6-methyl-tetrahydropyran-2-yl]oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-8,10-dihydro-7H-tetracene-5,12-dione; (8S,10S)-10-((2R,4S,5S,6S)-4-amino-5-hydroxy-6-methyltetrahydro-2H-pyran-2-yloxy)-6,8,11-trihydroxy-8-(2-hydroxyacetyl)-1-methoxy-7,8,9,10-tetrahydrotetracene-5,12-dione; (8S-cis)-10-((3-Amino-2,3,6-trideoxy-alpha-L-lyxo-hexopyranosyl)oxy)-7,8,9,10-tetrahydro-6,8,11-trihydroxy-8-(hydroacetyl)-1-methoxy-5,12-naphthacenedione; (8S-cis)-10-[(3-Amino-2,3,6-trideoxy-.alpha.-L-lyxo-hexopyranosyl]-7,8,9,10-tetrahydro-6,8,11-trihydroxy-8-(hydroxyacetyl)-1-methoxy-5,12-naphthacenedione; 1,2,3,4,6,11-hexahydro-4beta,5,12-trihydroxy-4-(hydroxyacetyl)-10-methoxy-6, 11-Dioxonaphthacen-1beta-yl-3-amino-2,3,6-trideoxy-alpha-l-lyxohexopyranoside; 1392315-46-6; 5,12-naphthacenedione, 10-((3-Amino-2,3,6-trideoxy-alpha-l-lyxo-hexopyranosyl)oxy)-7,8,9,10-tetrahydro- 6,8,11-trihydroxy-1-methoxy-5,12-naphthacenedione
Click to Show/Hide
|
|||||
| Target(s) | DNA topoisomerase 2-alpha (TOP2A) | Target Info | ||||
| Structure |
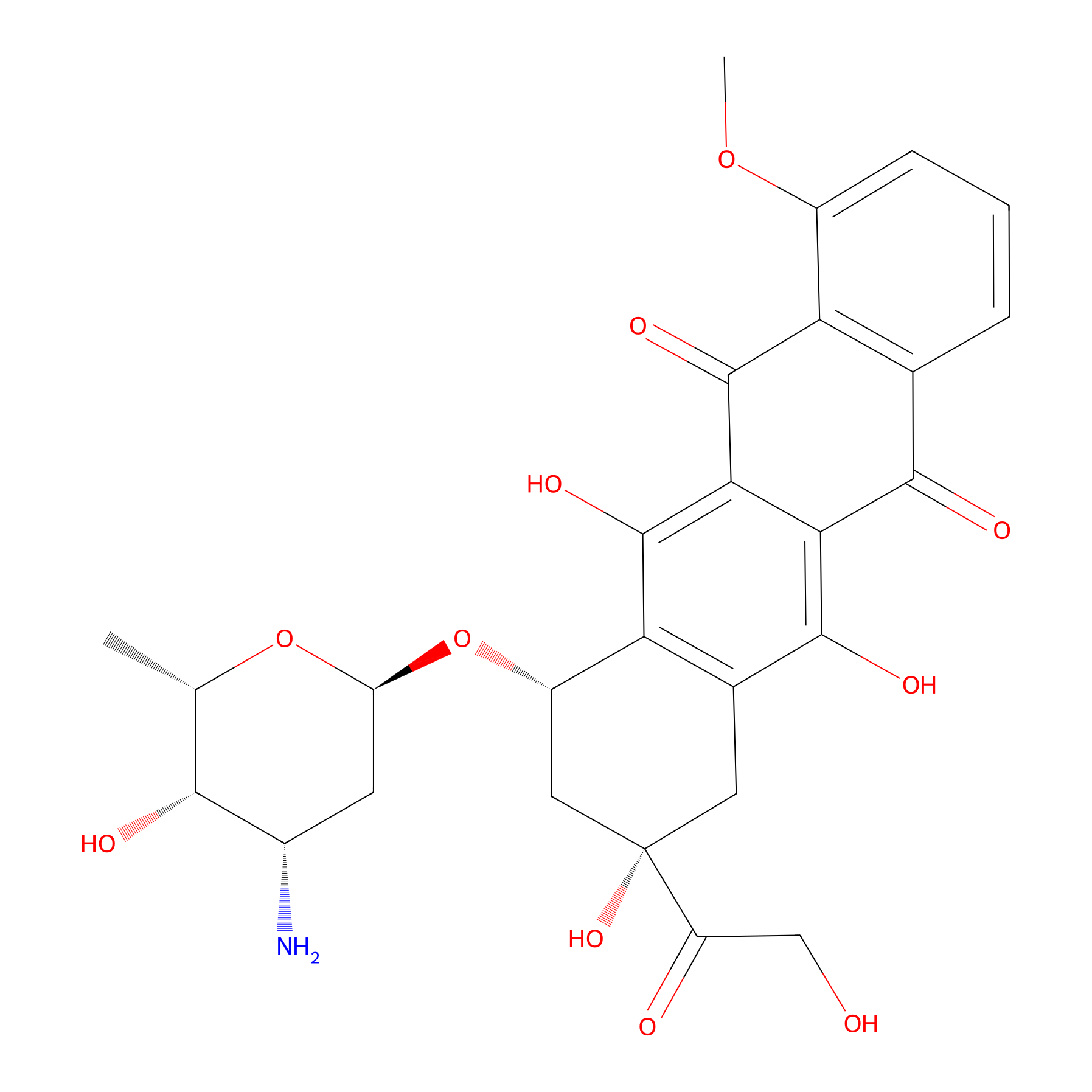
|
|||||
| Formula |
C27H29NO11
|
|||||
| #Ro5 Violations (Lipinski): 3 | Molecular Weight (mw) | 543.5 | ||||
| Lipid-water partition coefficient (xlogp) | 1.3 | |||||
| Hydrogen Bond Donor Count (hbonddonor) | 6 | |||||
| Hydrogen Bond Acceptor Count (hbondacc) | 12 | |||||
| Rotatable Bond Count (rotbonds) | 5 | |||||
| PubChem CID | ||||||
| Canonical smiles |
CC1C(C(CC(O1)OC2CC(CC3=C2C(=C4C(=C3O)C(=O)C5=C(C4=O)C(=CC=C5)OC)O)(C(=O)CO)O)N)O
|
|||||
| InChI |
InChI=1S/C27H29NO11/c1-10-22(31)13(28)6-17(38-10)39-15-8-27(36,16(30)9-29)7-12-19(15)26(35)21-20(24(12)33)23(32)11-4-3-5-14(37-2)18(11)25(21)34/h3-5,10,13,15,17,22,29,31,33,35-36H,6-9,28H2,1-2H3/t10-,13-,15-,17-,22+,27-/m0/s1
|
|||||
| InChIKey |
AOJJSUZBOXZQNB-TZSSRYMLSA-N
|
|||||
| IUPAC Name |
(7S,9S)-7-[(2R,4S,5S,6S)-4-amino-5-hydroxy-6-methyloxan-2-yl]oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-8,10-dihydro-7H-tetracene-5,12-dione
|
|||||
The activity data of This Drug
| Standard Type | Value | Administration times | Administration dosage | Cell line | Cell line ID | Ref. |
|---|---|---|---|---|---|---|
| Cell survival rate | 20% | 24 h | 20 μg/mL | MCF-7 cell | CVCL_0031 | [1] |
| Cell survival rate | 37% | 24 h | 15 μg/mL | MCF-7 cell | CVCL_0031 | [1] |
| Cell survival rate | 55% | 24 h | 10 μg/mL | MCF-7 cell | CVCL_0031 | [1] |
| Cell survival rate | 76% | 24 h | 5 μg/mL | MCF-7 cell | CVCL_0031 | [1] |
| Cell viability | 1% | 48 h | 15 μM | MDA-MB-231 cell | CVCL_0062 | [2] |
| Cell viability | 1% | 48 h | 1.87 μM | SK-OV-3 cell | CVCL_0532 | [2] |
| Cell viability | 1% | 48 h | 15 μM | SK-OV-3 cell | CVCL_0532 | [2] |
| Cell viability | 1% | 48 h | 3.75 μM | SK-OV-3 cell | CVCL_0532 | [2] |
| Cell viability | 1% | 48 h | 7.5 μM | SK-OV-3 cell | CVCL_0532 | [2] |
| Cell viability | 1% | 48 h | 1.87 μM | SK-OV-3 cell | CVCL_0532 | [2] |
| Cell viability | 1% | 48 h | 15 μM | SK-OV-3 cell | CVCL_0532 | [2] |
| Cell viability | 1% | 48 h | 3.75 μM | SK-OV-3 cell | CVCL_0532 | [2] |
| Cell viability | 1% | 48 h | 7.5 μM | SK-OV-3 cell | CVCL_0532 | [2] |
| Cell viability | 2% | 48 h | 7.5 μM | MDA-MB-231 cell | CVCL_0062 | [2] |
| Cell viability | 3% | 48 h | 3.75 μM | MDA-MB-231 cell | CVCL_0062 | [2] |
| Cell viability | 6% | 48 h | 0.94 μM | SK-OV-3 cell | CVCL_0532 | [2] |
| Cell viability | 6% | 48 h | 0.94 μM | SK-OV-3 cell | CVCL_0532 | [2] |
| Cell viability | 9% | 48 h | 1.87 μM | MDA-MB-231 cell | CVCL_0062 | [2] |
| Cell viability | 21% | 48 h | 0.94 μM | MDA-MB-231 cell | CVCL_0062 | [2] |
| Cell viability | 32% | 48 h | 0.47 μM | SK-OV-3 cell | CVCL_0532 | [2] |
| Cell viability | 32% | 48 h | 0.47 μM | SK-OV-3 cell | CVCL_0532 | [2] |
| Cell viability | 37% | 48 h | 0.47 μM | MDA-MB-231 cell | CVCL_0062 | [2] |
| Tumor Growth Inhibition value (TGI) | 23.13±2.4% | 14 days | 5 mg/kg | SK-BR-3 cell | CVCL_0033 | [3] |
| Tumor volume | 1100 mm3 | 24 days | 6 mg/kg | HCCLM3-Luc cell | CVCL_6832 | [4] |
| Cell viability | 62.90% | 24 h | N.A. | OCM-3 cell | CVCL_6937 | [5] |
| Cell viability | 89.70% | 48 h | N.A. | OCM-4 cell | N.A. | [5] |
| Half Maximal Effective Concentration (EC50) | 6.78 µM | 24 h | N.A. | HeLa cell | CVCL_0030 | [6] |
| Half Maximal Inhibitory Concentration (IC50) | 0.03 µM | 48 h | N.A. | Human umbilical vein endothelial cell | N.A. | [7] |
| Half Maximal Inhibitory Concentration (IC50) | 0.06 µM | 48 h | N.A. | U87 cell | CVCL_3429 | [7] |
| Half Maximal Inhibitory Concentration (IC50) | 0.18±0.08 µM | 72 h | N.A. | Kelly-WT cell | CVCL_2092 | [8] |
| Half Maximal Inhibitory Concentration (IC50) | 0.65±0.20 µM | 48 h | N.A. | U87 cell | CVCL_3429 | [9] |
| Half Maximal Inhibitory Concentration (IC50) | 0.90±0.12 µM | 72 h | N.A. | MCF-7 cell | CVCL_0031 | [8] |
| Half Maximal Inhibitory Concentration (IC50) | 1.29±0.18 µM | 48 h | N.A. | LO #2 cell | CVCL_C7SD | [9] |
| Half Maximal Inhibitory Concentration (IC50) | 1.29±0.18 µM | 48 h | N.A. | LO #2 cell | CVCL_C7SD | [9] |
| Half Maximal Inhibitory Concentration (IC50) | 1.58±0.19 µM | 48 h | N.A. | Hep-G2 cell | CVCL_0027 | [9] |
| Half Maximal Inhibitory Concentration (IC50) | 3 µM | 36 h | N.A. | LO #2 cell | CVCL_C7SD | [10] |
| Half Maximal Inhibitory Concentration (IC50) | 3 µM | 36 h | N.A. | LO #2 cell | CVCL_C7SD | [10] |
| Half Maximal Inhibitory Concentration (IC50) | 3.55±0.21 µM | 48 h | N.A. | A-549 cell | CVCL_0023 | [9] |
| Half Maximal Inhibitory Concentration (IC50) | 6 µM | 36 h | N.A. | U-87MG cell | CVCL_0022 | [10] |
| Half Maximal Inhibitory Concentration (IC50) | 6 µM | 36 h | N.A. | A-549 cell | CVCL_0023 | [10] |
| Half Maximal Inhibitory Concentration (IC50) | 6 µM | 36 h | N.A. | MIHA cell | CVCL_SA11 | [10] |
| Half Maximal Inhibitory Concentration (IC50) | 6.57±0.61 µM | 72 h | N.A. | Kelly-ADR cell | CVCL_2092 | [8] |
| Half Maximal Inhibitory Concentration (IC50) | 10 µM | 36 h | N.A. | MCF-7 cell | CVCL_0031 | [10] |
| Half Maximal Inhibitory Concentration (IC50) | 11 µM | 36 h | N.A. | HeLa cell | CVCL_0030 | [10] |
| Half Maximal Inhibitory Concentration (IC50) | 410 nM | 24 h | N.A. | SK-BR-3 cell | CVCL_0033 | [3] |
| 30% Inhibitory Concentration (IC30) | 2.13 ug/mL | N.A. | N.A. | Jurkat cell | CVCL_0065 | [11] |
| 90% Lethal Concentration (IC50) | 6.82 ug/mL | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [12] |
| 90% Lethal Concentration (IC50) | 7.32 ug/mL | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [12] |
| Half Maximal Cytotoxicity Concentration (CC50) | 2 ug/mL | N.A. | N.A. | HeLa cell | CVCL_0030 | [13] |
| Half Maximal Cytotoxicity Concentration (CC50) | 20 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [14] |
| Half Maximal Cytotoxicity Concentration (CC50) | 200 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [15] |
| Half Maximal Cytotoxicity Concentration (CC50) | <500 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [16] |
| Half Maximal Cytotoxicity Concentration (CC50) | 700 nM | N.A. | N.A. | THP-1 cell | CVCL_0006 | [15] |
| Half Maximal Cytotoxicity Concentration (CC50) | 2 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [17] |
| Half Maximal Cytotoxicity Concentration (CC50) | 13.9 uM | N.A. | N.A. | MCF-10A cell | CVCL_0598 | [18] |
| Half Maximal Effective Concentration (EC50) | 10 ng/mL | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [19] |
| Half Maximal Effective Concentration (EC50) | 10 ng/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [19] |
| Half Maximal Effective Concentration (EC50) | 17.62 µM | N.A. | N.A. | SMMC-7721 cell | CVCL_0534 | [4] |
| Half Maximal Effective Concentration (EC50) | 17.62 µM | N.A. | N.A. | SMMC-7721 cell | CVCL_0534 | [4] |
| Half Maximal Effective Concentration (EC50) | 30 ng/mL | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [19] |
| Half Maximal Effective Concentration (EC50) | 9 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [20] |
| Half Maximal Effective Concentration (EC50) | 9 nM | N.A. | N.A. | A-375 cell | CVCL_0132 | [21] |
| Half Maximal Effective Concentration (EC50) | 100 nM | N.A. | N.A. | A2780-1A9 cell | CVCL_H619 | [22] |
| Half Maximal Effective Concentration (EC50) | 400 nM | N.A. | N.A. | KB cell | CVCL_0372 | [22] |
| Half Maximal Effective Concentration (EC50) | 700 nM | N.A. | N.A. | LNCaP cell | CVCL_0395 | [23] |
| Half Maximal Effective Concentration (EC50) | 900 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [22] |
| Half Maximal Effective Concentration (EC50) | 1.508 uM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [24] |
| Half Maximal Effective Concentration (EC50) | 1.8 uM | N.A. | N.A. | DuPro cell | CVCL_4738 | [23] |
| Half Maximal Effective Dosage (ED50) | 0.1 ug/mL | N.A. | N.A. | NCI-H23 cell | CVCL_1547 | [25] |
| Half Maximal Effective Dosage (ED50) | 0.1 ug/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [26] |
| Half Maximal Effective Dosage (ED50) | 0.1 ug/mL | N.A. | N.A. | XF498 cell | CVCL_8928 | [27] |
| Half Maximal Effective Dosage (ED50) | 0.1 ug/mL | N.A. | N.A. | SW620 cell | CVCL_0547 | [25] |
| Half Maximal Effective Dosage (ED50) | 0.1 ug/mL | N.A. | N.A. | DLD-1 cell | CVCL_0248 | [26] |
| Half Maximal Effective Dosage (ED50) | 0.104 ng/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [28] |
| Half Maximal Effective Dosage (ED50) | 0.105 ng/ml | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [29] |
| Half Maximal Effective Dosage (ED50) | 0.11 ug/mL | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [27] |
| Half Maximal Effective Dosage (ED50) | 0.11 ug/mL | N.A. | N.A. | SK-MEL-2 cell | CVCL_0069 | [30] |
| Half Maximal Effective Dosage (ED50) | 0.112 ng/ml | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [29] |
| Half Maximal Effective Dosage (ED50) | 0.113 ug/ml | N.A. | N.A. | A498 cell | CVCL_1056 | [31] |
| Half Maximal Effective Dosage (ED50) | 0.12 ug/mL | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [32] |
| Half Maximal Effective Dosage (ED50) | 0.12 ug/mL | N.A. | N.A. | XF498 cell | CVCL_8928 | [33] |
| Half Maximal Effective Dosage (ED50) | 0.12 ug/mL | N.A. | N.A. | WiDr cell | CVCL_2760 | [34] |
| Half Maximal Effective Dosage (ED50) | 0.12 ug/mL | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [35] |
| Half Maximal Effective Dosage (ED50) | 0.124 ug/ml | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [36] |
| Half Maximal Effective Dosage (ED50) | 0.126 ug/ml | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [37] |
| Half Maximal Effective Dosage (ED50) | 0.129 ug/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [38] |
| Half Maximal Effective Dosage (ED50) | 0.13 ug/mL | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [39] |
| Half Maximal Effective Dosage (ED50) | 0.13 ug/mL | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [40] |
| Half Maximal Effective Dosage (ED50) | 0.142 ug/ml | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [41] |
| Half Maximal Effective Dosage (ED50) | 0.15 ug/mL | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [35] |
| Half Maximal Effective Dosage (ED50) | 0.152 ug/ml | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [42] |
| Half Maximal Effective Dosage (ED50) | 0.153 ng/mL | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [28] |
| Half Maximal Effective Dosage (ED50) | 0.16 ug/mL | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [43] |
| Half Maximal Effective Dosage (ED50) | 0.17 ug/ml | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [44] |
| Half Maximal Effective Dosage (ED50) | 0.17 ug/mL | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [45] |
| Half Maximal Effective Dosage (ED50) | 0.17 ug/mL | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [33] |
| Half Maximal Effective Dosage (ED50) | 0.179 ug/ml | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [46] |
| Half Maximal Effective Dosage (ED50) | 0.18 ng/mL | N.A. | N.A. | MIA PaCa-2 cell | CVCL_0428 | [47] |
| Half Maximal Effective Dosage (ED50) | 0.18 ug/mL | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [48] |
| Half Maximal Effective Dosage (ED50) | 0.189 ug/ml | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [49] |
| Half Maximal Effective Dosage (ED50) | 0.19 ug/mL | N.A. | N.A. | XF498 cell | CVCL_8928 | [50] |
| Half Maximal Effective Dosage (ED50) | 0.198 ng/ml | N.A. | N.A. | A-549 cell | CVCL_0023 | [51] |
| Half Maximal Effective Dosage (ED50) | 0.2 ug/mL | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [52] |
| Half Maximal Effective Dosage (ED50) | 0.2 ug/mL | N.A. | N.A. | SK-MEL-2 cell | CVCL_0069 | [52] |
| Half Maximal Effective Dosage (ED50) | 0.2 ug/mL | N.A. | N.A. | SF539 cell | CVCL_1691 | [25] |
| Half Maximal Effective Dosage (ED50) | 0.2 ug/mL | N.A. | N.A. | XF498 cell | CVCL_8928 | [52] |
| Half Maximal Effective Dosage (ED50) | 0.208 ng/ml | N.A. | N.A. | A-549 cell | CVCL_0023 | [53] |
| Half Maximal Effective Dosage (ED50) | 0.209 ug/ml | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [54] |
| Half Maximal Effective Dosage (ED50) | 0.23 ug/ml | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [55] |
| Half Maximal Effective Dosage (ED50) | 0.24 ug/mL | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [33] |
| Half Maximal Effective Dosage (ED50) | 0.25 ug/mL | N.A. | N.A. | KB cell | CVCL_0372 | [56] |
| Half Maximal Effective Dosage (ED50) | 0.28 ng/ml | N.A. | N.A. | SK-MEL-5 cell | CVCL_0527 | [57] |
| Half Maximal Effective Dosage (ED50) | 0.287 ug/mL | N.A. | N.A. | PC-3 cell | CVCL_0035 | [58] |
| Half Maximal Effective Dosage (ED50) | 0.29 ug/mL | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [59] |
| Half Maximal Effective Dosage (ED50) | 0.3 ug/mL | N.A. | N.A. | UO-31 cell | CVCL_1911 | [60] |
| Half Maximal Effective Dosage (ED50) | 0.3 ug/mL | N.A. | N.A. | KB cell | CVCL_0372 | [61] |
| Half Maximal Effective Dosage (ED50) | 0.329 ug/ml | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [62] |
| Half Maximal Effective Dosage (ED50) | 0.35 ug/mL | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [32] |
| Half Maximal Effective Dosage (ED50) | 0.366 ng/ml | N.A. | N.A. | A498 cell | CVCL_1056 | [63] |
| Half Maximal Effective Dosage (ED50) | 0.56 ng/ml | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [57] |
| Half Maximal Effective Dosage (ED50) | 0.57 ug/mL | N.A. | N.A. | DLD-1 cell | CVCL_0248 | [56] |
| Half Maximal Effective Dosage (ED50) | 0.79 ug/mL | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [64] |
| Half Maximal Effective Dosage (ED50) | 0.953 ug/ml | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [65] |
| Half Maximal Effective Dosage (ED50) | 1 ng/mL | N.A. | N.A. | LOX IMVI cell | CVCL_1381 | [66] |
| Half Maximal Effective Dosage (ED50) | 1 ng/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [66] |
| Half Maximal Effective Dosage (ED50) | 1 ng/mL | N.A. | N.A. | U-251MG cell | CVCL_0021 | [66] |
| Half Maximal Effective Dosage (ED50) | 1.01 ng/ml | N.A. | N.A. | A-549 cell | CVCL_0023 | [63] |
| Half Maximal Effective Dosage (ED50) | 1.1 ng/ml | N.A. | N.A. | A498 cell | CVCL_1056 | [67] |
| Half Maximal Effective Dosage (ED50) | 1.1 ng/ml | N.A. | N.A. | MIA PaCa-2 cell | CVCL_0428 | [68] |
| Half Maximal Effective Dosage (ED50) | 1.1 ng/ml | N.A. | N.A. | A-549 cell | CVCL_0023 | [69] |
| Half Maximal Effective Dosage (ED50) | 1.32 ng/ml | N.A. | N.A. | MIA PaCa-2 cell | CVCL_0428 | [63] |
| Half Maximal Effective Dosage (ED50) | 1.36 ng/mL | N.A. | N.A. | MIA PaCa-2 cell | CVCL_0428 | [70] |
| Half Maximal Effective Dosage (ED50) | 1.57 ng/ml | N.A. | N.A. | A-549 cell | CVCL_0023 | [71] |
| Half Maximal Effective Dosage (ED50) | 1.78 ng/ml | N.A. | N.A. | A498 cell | CVCL_1056 | [44] |
| Half Maximal Effective Dosage (ED50) | 1.9 ng/ml | N.A. | N.A. | A498 cell | CVCL_1056 | [72] |
| Half Maximal Effective Dosage (ED50) | 1.95 ng/ml | N.A. | N.A. | MIA PaCa-2 cell | CVCL_0428 | [62] |
| Half Maximal Effective Dosage (ED50) | 1.97 ng/ml | N.A. | N.A. | A498 cell | CVCL_1056 | [62] |
| Half Maximal Effective Dosage (ED50) | 2 ng/mL | N.A. | N.A. | SK-MEL-2 cell | CVCL_0069 | [66] |
| Half Maximal Effective Dosage (ED50) | 2 ng/ml | N.A. | N.A. | A-549 cell | CVCL_0023 | [73] |
| Half Maximal Effective Dosage (ED50) | 2.02 ng/ml | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [51] |
| Half Maximal Effective Dosage (ED50) | 2.1 ng/ml | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [74] |
| Half Maximal Effective Dosage (ED50) | 2.26 ng/ml | N.A. | N.A. | A498 cell | CVCL_1056 | [75] |
| Half Maximal Effective Dosage (ED50) | 2.4 ug/mL | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [30] |
| Half Maximal Effective Dosage (ED50) | 2.43 ng/ml | N.A. | N.A. | MIA PaCa-2 cell | CVCL_0428 | [44] |
| Half Maximal Effective Dosage (ED50) | 2.7 ng/ml | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [76] |
| Half Maximal Effective Dosage (ED50) | 2.86 ng/mL | N.A. | N.A. | A498 cell | CVCL_1056 | [77] |
| Half Maximal Effective Dosage (ED50) | 2.88 ng/ml | N.A. | N.A. | MIA PaCa-2 cell | CVCL_0428 | [67] |
| Half Maximal Effective Dosage (ED50) | 2.94 ng/ml | N.A. | N.A. | A-549 cell | CVCL_0023 | [36] |
| Half Maximal Effective Dosage (ED50) | 3 ng/mL | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [78] |
| Half Maximal Effective Dosage (ED50) | 3.41 ng/ml | N.A. | N.A. | A-549 cell | CVCL_0023 | [62] |
| Half Maximal Effective Dosage (ED50) | 3.56 ng/ml | N.A. | N.A. | A-549 cell | CVCL_0023 | [79] |
| Half Maximal Effective Dosage (ED50) | 3.67 ng/ml | N.A. | N.A. | A-549 cell | CVCL_0023 | [80] |
| Half Maximal Effective Dosage (ED50) | 3.9 ug/mL | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [81] |
| Half Maximal Effective Dosage (ED50) | 3.93 ng/ml | N.A. | N.A. | A-549 cell | CVCL_0023 | [31] |
| Half Maximal Effective Dosage (ED50) | 4.24 ng/ml | N.A. | N.A. | A498 cell | CVCL_1056 | [82] |
| Half Maximal Effective Dosage (ED50) | 4.28 ng/ml | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [44] |
| Half Maximal Effective Dosage (ED50) | 4.3 ng/ml | N.A. | N.A. | MIA PaCa-2 cell | CVCL_0428 | [72] |
| Half Maximal Effective Dosage (ED50) | 4.45 ng/ml | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [83] |
| Half Maximal Effective Dosage (ED50) | 4.56 ng/ml | N.A. | N.A. | MIA PaCa-2 cell | CVCL_0428 | [36] |
| Half Maximal Effective Dosage (ED50) | 4.83 ng/ml | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [84] |
| Half Maximal Effective Dosage (ED50) | 4.99 ng/ml | N.A. | N.A. | A-549 cell | CVCL_0023 | [82] |
| Half Maximal Effective Dosage (ED50) | 5.6 ng/ml | N.A. | N.A. | PC-3 cell | CVCL_0035 | [68] |
| Half Maximal Effective Dosage (ED50) | 6 ng/mL | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [66] |
| Half Maximal Effective Dosage (ED50) | 6.1 ng/ml | N.A. | N.A. | MIA PaCa-2 cell | CVCL_0428 | [65] |
| Half Maximal Effective Dosage (ED50) | 6.22 ng/ml | N.A. | N.A. | A-549 cell | CVCL_0023 | [65] |
| Half Maximal Effective Dosage (ED50) | 6.37 ug/mL | N.A. | N.A. | PC-3 cell | CVCL_0035 | [81] |
| Half Maximal Effective Dosage (ED50) | 6.49 ng/ml | N.A. | N.A. | A498 cell | CVCL_1056 | [54] |
| Half Maximal Effective Dosage (ED50) | 6.69 ng/ml | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [85] |
| Half Maximal Effective Dosage (ED50) | 6.84 ng/ml | N.A. | N.A. | A-549 cell | CVCL_0023 | [86] |
| Half Maximal Effective Dosage (ED50) | 7 ng/ml | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [87] |
| Half Maximal Effective Dosage (ED50) | 7.9 ng/ml | N.A. | N.A. | MIA PaCa-2 cell | CVCL_0428 | [54] |
| Half Maximal Effective Dosage (ED50) | 9.2 ng/ml | N.A. | N.A. | A-549 cell | CVCL_0023 | [55] |
| Half Maximal Effective Dosage (ED50) | 9.66 ng/ml | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [80] |
| Half Maximal Effective Dosage (ED50) | 10 ng/ml | N.A. | N.A. | A498 cell | CVCL_1056 | [42] |
| Half Maximal Effective Dosage (ED50) | 10 ng/ml | N.A. | N.A. | PC-3 cell | CVCL_0035 | [57] |
| Half Maximal Effective Dosage (ED50) | 10 ng/mL | N.A. | N.A. | SK-MEL-2 cell | CVCL_0069 | [88] |
| Half Maximal Effective Dosage (ED50) | 10 ng/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [89] |
| Half Maximal Effective Dosage (ED50) | 10 ng/mL | N.A. | N.A. | XF498 cell | CVCL_8928 | [88] |
| Half Maximal Effective Dosage (ED50) | 10 ng/ml | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [72] |
| Half Maximal Effective Dosage (ED50) | 10.1 ng/ml | N.A. | N.A. | A-549 cell | CVCL_0023 | [37] |
| Half Maximal Effective Dosage (ED50) | 10.3 ng/ml | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [63] |
| Half Maximal Effective Dosage (ED50) | 10.8 ng/ml | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [84] |
| Half Maximal Effective Dosage (ED50) | 11.6 ng/ml | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [62] |
| Half Maximal Effective Dosage (ED50) | 12 ng/ml | N.A. | N.A. | A431 cell | CVCL_0037 | [57] |
| Half Maximal Effective Dosage (ED50) | 12.2 ng/ml | N.A. | N.A. | A498 cell | CVCL_1056 | [90] |
| Half Maximal Effective Dosage (ED50) | 13.3 ng/ml | N.A. | N.A. | MIA PaCa-2 cell | CVCL_0428 | [82] |
| Half Maximal Effective Dosage (ED50) | 13.8 ng/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [91] |
| Half Maximal Effective Dosage (ED50) | 17.7 ng/ml | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [86] |
| Half Maximal Effective Dosage (ED50) | 18 ng/ml | N.A. | N.A. | MIA PaCa-2 cell | CVCL_0428 | [92] |
| Half Maximal Effective Dosage (ED50) | 18.2 ng/ml | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [75] |
| Half Maximal Effective Dosage (ED50) | 18.9 ng/ml | N.A. | N.A. | PC-3 cell | CVCL_0035 | [44] |
| Half Maximal Effective Dosage (ED50) | 19.6 ng/ml | N.A. | N.A. | PC-3 cell | CVCL_0035 | [63] |
| Half Maximal Effective Dosage (ED50) | 20 ng/ml | N.A. | N.A. | PC-3 cell | CVCL_0035 | [72] |
| Half Maximal Effective Dosage (ED50) | 20 ng/mL | N.A. | N.A. | SK-MEL3 cell | CVCL_0550 | [93] |
| Half Maximal Effective Dosage (ED50) | 20 ng/mL | N.A. | N.A. | SK-MEL-2 cell | CVCL_0069 | [43] |
| Half Maximal Effective Dosage (ED50) | 20 ng/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [94] |
| Half Maximal Effective Dosage (ED50) | 20 ng/mL | N.A. | N.A. | MOLT-4F cell | CVCL_2792 | [60] |
| Half Maximal Effective Dosage (ED50) | 20 ng/ml | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [57] |
| Half Maximal Effective Dosage (ED50) | 20 ng/mL | N.A. | N.A. | XF498 cell | CVCL_8928 | [64] |
| Half Maximal Effective Dosage (ED50) | 20 ng/ml | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [92] |
| Half Maximal Effective Dosage (ED50) | 20 ng/mL | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [88] |
| Half Maximal Effective Dosage (ED50) | 22.7 ng/ml | N.A. | N.A. | PC-3 cell | CVCL_0035 | [80] |
| Half Maximal Effective Dosage (ED50) | 22.8 ng/ml | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [69] |
| Half Maximal Effective Dosage (ED50) | 23 ng/ml | N.A. | N.A. | MIA PaCa-2 cell | CVCL_0428 | [95] |
| Half Maximal Effective Dosage (ED50) | 24.3 ng/ml | N.A. | N.A. | A-549 cell | CVCL_0023 | [54] |
| Half Maximal Effective Dosage (ED50) | 24.3 ng/mL | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [70] |
| Half Maximal Effective Dosage (ED50) | 26.2 ng/ml | N.A. | N.A. | PC-3 cell | CVCL_0035 | [54] |
| Half Maximal Effective Dosage (ED50) | 26.2 ng/ml | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [63] |
| Half Maximal Effective Dosage (ED50) | 27.5 ng/ml | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [37] |
| Half Maximal Effective Dosage (ED50) | 27.6 ng/ml | N.A. | N.A. | PC-3 cell | CVCL_0035 | [82] |
| Half Maximal Effective Dosage (ED50) | 28.4 ng/ml | N.A. | N.A. | A-549 cell | CVCL_0023 | [96] |
| Half Maximal Effective Dosage (ED50) | 28.7 ng/ml | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [65] |
| Half Maximal Effective Dosage (ED50) | 29 ng/ml | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [97] |
| Half Maximal Effective Dosage (ED50) | 29 ng/mL | N.A. | N.A. | XF498 cell | CVCL_8928 | [78] |
| Half Maximal Effective Dosage (ED50) | 29.2 ng/ml | N.A. | N.A. | A-549 cell | CVCL_0023 | [98] |
| Half Maximal Effective Dosage (ED50) | 30 ng/mL | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [99] |
| Half Maximal Effective Dosage (ED50) | 30 ng/mL | N.A. | N.A. | SK-MEL-2 cell | CVCL_0069 | [100] |
| Half Maximal Effective Dosage (ED50) | 30 ng/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [101] |
| Half Maximal Effective Dosage (ED50) | 30.1 ng/ml | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [79] |
| Half Maximal Effective Dosage (ED50) | 34.9 ng/mL | N.A. | N.A. | PC-3 cell | CVCL_0035 | [70] |
| Half Maximal Effective Dosage (ED50) | 35.1 ng/ml | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [82] |
| Half Maximal Effective Dosage (ED50) | 35.7 ng/ml | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [49] |
| Half Maximal Effective Dosage (ED50) | 39.2 ng/ml | N.A. | N.A. | MIA PaCa-2 cell | CVCL_0428 | [90] |
| Half Maximal Effective Dosage (ED50) | 40 ng/mL | N.A. | N.A. | SK-MEL-2 cell | CVCL_0069 | [39] |
| Half Maximal Effective Dosage (ED50) | 40 ng/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [48] |
| Half Maximal Effective Dosage (ED50) | 40 ng/ml | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [73] |
| Half Maximal Effective Dosage (ED50) | 40 ng/mL | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [102] |
| Half Maximal Effective Dosage (ED50) | 41.6 ng/ml | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [96] |
| Half Maximal Effective Dosage (ED50) | 49.4 ng/mL | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [58] |
| Half Maximal Effective Dosage (ED50) | 49.8 ng/ml | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [103] |
| Half Maximal Effective Dosage (ED50) | 50 ng/mL | N.A. | N.A. | SK-MEL-2 cell | CVCL_0069 | [35] |
| Half Maximal Effective Dosage (ED50) | 50 ng/mL | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [89] |
| Half Maximal Effective Dosage (ED50) | 51 ng/ml | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [104] |
| Half Maximal Effective Dosage (ED50) | 57.7 ng/ml | N.A. | N.A. | PC-3 cell | CVCL_0035 | [65] |
| Half Maximal Effective Dosage (ED50) | 60 ng/mL | N.A. | N.A. | SK-MEL-2 cell | CVCL_0069 | [50] |
| Half Maximal Effective Dosage (ED50) | 60 ng/mL | N.A. | N.A. | XF498 cell | CVCL_8928 | [39] |
| Half Maximal Effective Dosage (ED50) | 60 ng/mL | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [39] |
| Half Maximal Effective Dosage (ED50) | 66.3 ng/mL | N.A. | N.A. | A498 cell | CVCL_1056 | [58] |
| Half Maximal Effective Dosage (ED50) | 70 ng/mL | N.A. | N.A. | XF498 cell | CVCL_8928 | [40] |
| Half Maximal Effective Dosage (ED50) | 71 ng/mL | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [78] |
| Half Maximal Effective Dosage (ED50) | 80 ng/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [40] |
| Half Maximal Effective Dosage (ED50) | 80 ng/mL | N.A. | N.A. | XF498 cell | CVCL_8928 | [102] |
| Half Maximal Effective Dosage (ED50) | 89.7 ng/ml | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [67] |
| Half Maximal Effective Dosage (ED50) | 90 ng/mL | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [64] |
| Half Maximal Effective Dosage (ED50) | 90 ng/mL | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [100] |
| Half Maximal Effective Dosage (ED50) | <0.1 nM | N.A. | N.A. | EMT6 cell | CVCL_1923 | [105] |
| Half Maximal Effective Dosage (ED50) | 6.3 nM | N.A. | N.A. | MIA PaCa-2 cell | CVCL_0428 | [106] |
| Half Maximal Effective Dosage (ED50) | 29 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [107] |
| Half Maximal Effective Dosage (ED50) | 37 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [106] |
| Half Maximal Effective Dosage (ED50) | 40 nM | N.A. | N.A. | LNCaP cell | CVCL_0395 | [108] |
| Half Maximal Effective Dosage (ED50) | 40 nM | N.A. | N.A. | ZR-75-1 cell | CVCL_0588 | [108] |
| Half Maximal Effective Dosage (ED50) | 55 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [107] |
| Half Maximal Effective Dosage (ED50) | 56 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [109] |
| Half Maximal Effective Dosage (ED50) | 57 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [106] |
| Half Maximal Effective Dosage (ED50) | 59.5 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [110] |
| Half Maximal Effective Dosage (ED50) | 0.15 uM | N.A. | N.A. | DU145 cell | CVCL_0105 | [108] |
| Half Maximal Effective Dosage (ED50) | 0.18 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [108] |
| Half Maximal Effective Dosage (ED50) | 0.58 uM | N.A. | N.A. | L1210 cell | CVCL_0382 | [111] |
| Half Maximal Effective Dosage (ED50) | 1.2 uM | N.A. | N.A. | HCT-8 cell | CVCL_2478 | [108] |
| Half Maximal Effective Dosage (ED50) | 1.5 uM | N.A. | N.A. | L1210 cell | CVCL_0382 | [112] |
| Half Maximal Effective Dosage (ED50) | 1.6 uM | N.A. | N.A. | L1210 cell | CVCL_0382 | [113] |
| Half Maximal Effective Dosage (ED50) | 2.9 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [114] |
| Half Maximal Effective Dosage (ED50) | 3.1 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [114] |
| Half Maximal Effective Dosage (ED50) | 3.2 uM | N.A. | N.A. | SK-MEL-5 cell | CVCL_0527 | [114] |
| Half Maximal Effective Dosage (ED50) | 5.5 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [114] |
| Half Maximal Effective Dosage (ED50) | 8 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [115] |
| Half Maximal Growth Inhibition (GI50) | 0.11 ug/mL | N.A. | N.A. | NUGC-3 cell | CVCL_1612 | [116] |
| Half Maximal Growth Inhibition (GI50) | 0.25 ug/mL | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [117] |
| Half Maximal Growth Inhibition (GI50) | 0.27 ug/mL | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [118] |
| Half Maximal Growth Inhibition (GI50) | 0.28 ug/mL | N.A. | N.A. | PC-3 cell | CVCL_0035 | [119] |
| Half Maximal Growth Inhibition (GI50) | 0.33 ug/mL | N.A. | N.A. | NCI-ADR-RES cell | CVCL_1452 | [119] |
| Half Maximal Growth Inhibition (GI50) | 0.36 pg/mL | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [120] |
| Half Maximal Growth Inhibition (GI50) | 0.39 ug/mL | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [116] |
| Half Maximal Growth Inhibition (GI50) | 0.53 pg/mL | N.A. | N.A. | SF268 cell | CVCL_1689 | [120] |
| Half Maximal Growth Inhibition (GI50) | 0.62 pg/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [120] |
| Half Maximal Growth Inhibition (GI50) | 0.65 pg/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [121] |
| Half Maximal Growth Inhibition (GI50) | 0.85 ug/mL | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [117] |
| Half Maximal Growth Inhibition (GI50) | 0.86 pg/mL | N.A. | N.A. | SF268 cell | CVCL_1689 | [122] |
| Half Maximal Growth Inhibition (GI50) | 1 ug/mL | N.A. | N.A. | K562 cell | CVCL_0004 | [123] |
| Half Maximal Growth Inhibition (GI50) | 1.45 mM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [124] |
| Half Maximal Growth Inhibition (GI50) | 2.36 mM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [124] |
| Half Maximal Growth Inhibition (GI50) | 4.6 ng/mL | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [125] |
| Half Maximal Growth Inhibition (GI50) | 5.48 mM | N.A. | N.A. | LoVo cell | CVCL_0399 | [124] |
| Half Maximal Growth Inhibition (GI50) | 6.4 ng/mL | N.A. | N.A. | A498 cell | CVCL_1056 | [125] |
| Half Maximal Growth Inhibition (GI50) | <10 ug/mL | N.A. | N.A. | NCI-H226 cell | CVCL_1544 | [126] |
| Half Maximal Growth Inhibition (GI50) | <25 ng/mL | N.A. | N.A. | HaCaT cell | CVCL_0038 | [127] |
| Half Maximal Growth Inhibition (GI50) | <25 ng/mL | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [128] |
| Half Maximal Growth Inhibition (GI50) | <25 ng/mL | N.A. | N.A. | U-251MG cell | CVCL_0021 | [127] |
| Half Maximal Growth Inhibition (GI50) | 29 ng/mL | N.A. | N.A. | PC-3 cell | CVCL_0035 | [125] |
| Half Maximal Growth Inhibition (GI50) | 30 ng/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [119] |
| Half Maximal Growth Inhibition (GI50) | 38 ng/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [128] |
| Half Maximal Growth Inhibition (GI50) | 40 ng/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [129] |
| Half Maximal Growth Inhibition (GI50) | 60 ng/mL | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [129] |
| Half Maximal Growth Inhibition (GI50) | 70 ng/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [117] |
| Half Maximal Growth Inhibition (GI50) | 70 ng/mL | N.A. | N.A. | SK-HEP1 cell | CVCL_0525 | [116] |
| Half Maximal Growth Inhibition (GI50) | 70 ng/mL | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [119] |
| Half Maximal Growth Inhibition (GI50) | 71 ng/mL | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [125] |
| Half Maximal Growth Inhibition (GI50) | 73 ng/mL | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [125] |
| Half Maximal Growth Inhibition (GI50) | 90 ng/mL | N.A. | N.A. | UACC-62 cell | CVCL_1780 | [119] |
| Half Maximal Growth Inhibition (GI50) | 0.8 pM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [130] |
| Half Maximal Growth Inhibition (GI50) | 0.85 pM | N.A. | N.A. | SF268 cell | CVCL_1689 | [130] |
| Half Maximal Growth Inhibition (GI50) | 1.2 nM | N.A. | N.A. | A431 cell | CVCL_0037 | [131] |
| Half Maximal Growth Inhibition (GI50) | 3 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [131] |
| Half Maximal Growth Inhibition (GI50) | 5 nM | N.A. | N.A. | NCI-H23 cell | CVCL_1547 | [132] |
| Half Maximal Growth Inhibition (GI50) | 7 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [133] |
| Half Maximal Growth Inhibition (GI50) | 8.7 nM | N.A. | N.A. | OVCAR-3 cell | CVCL_0465 | [134] |
| Half Maximal Growth Inhibition (GI50) | <10 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [135] |
| Half Maximal Growth Inhibition (GI50) | 10 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [136] |
| Half Maximal Growth Inhibition (GI50) | 10 nM | N.A. | N.A. | LoVo cell | CVCL_0399 | [136] |
| Half Maximal Growth Inhibition (GI50) | <10 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [135] |
| Half Maximal Growth Inhibition (GI50) | 10 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [137] |
| Half Maximal Growth Inhibition (GI50) | 10.86 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [138] |
| Half Maximal Growth Inhibition (GI50) | 15.8 nM | N.A. | N.A. | TRAMP-C2H cell | CVCL_H591 | [139] |
| Half Maximal Growth Inhibition (GI50) | 18 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [140] |
| Half Maximal Growth Inhibition (GI50) | 20 nM | N.A. | N.A. | LNCaP cell | CVCL_0395 | [136] |
| Half Maximal Growth Inhibition (GI50) | 20 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [141] |
| Half Maximal Growth Inhibition (GI50) | 20 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [142] |
| Half Maximal Growth Inhibition (GI50) | 20.4 nM | N.A. | N.A. | TRAMP-C1A cell | CVCL_H593 | [143] |
| Half Maximal Growth Inhibition (GI50) | 23 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [144] |
| Half Maximal Growth Inhibition (GI50) | 28 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [145] |
| Half Maximal Growth Inhibition (GI50) | 30 nM | N.A. | N.A. | MOLT-4 cell | CVCL_0013 | [146] |
| Half Maximal Growth Inhibition (GI50) | 30 nM | N.A. | N.A. | NCI-H522 cell | CVCL_1567 | [146] |
| Half Maximal Growth Inhibition (GI50) | 30 nM | N.A. | N.A. | SR cell | CVCL_1711 | [146] |
| Half Maximal Growth Inhibition (GI50) | 30 nM | N.A. | N.A. | SW620 cell | CVCL_0547 | [147] |
| Half Maximal Growth Inhibition (GI50) | 30 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [148] |
| Half Maximal Growth Inhibition (GI50) | 35.6 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [149] |
| Half Maximal Growth Inhibition (GI50) | 38 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [150] |
| Half Maximal Growth Inhibition (GI50) | 40 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [151] |
| Half Maximal Growth Inhibition (GI50) | 40 nM | N.A. | N.A. | SNB-19 cell | CVCL_0535 | [152] |
| Half Maximal Growth Inhibition (GI50) | 42.8 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [138] |
| Half Maximal Growth Inhibition (GI50) | 45.3 nM | N.A. | N.A. | LNCaP cell | CVCL_0395 | [139] |
| Half Maximal Growth Inhibition (GI50) | <46 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [153] |
| Half Maximal Growth Inhibition (GI50) | <46 nM | N.A. | N.A. | U-251MG cell | CVCL_0021 | [154] |
| Half Maximal Growth Inhibition (GI50) | 50 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [155] |
| Half Maximal Growth Inhibition (GI50) | 50 nM | N.A. | N.A. | MDA-MB-468 cell | CVCL_0419 | [141] |
| Half Maximal Growth Inhibition (GI50) | 55 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [156] |
| Half Maximal Growth Inhibition (GI50) | 56 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [144] |
| Half Maximal Growth Inhibition (GI50) | 60 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [157] |
| Half Maximal Growth Inhibition (GI50) | 60 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [146] |
| Half Maximal Growth Inhibition (GI50) | 60 nM | N.A. | N.A. | T-47D cell | CVCL_0553 | [141] |
| Half Maximal Growth Inhibition (GI50) | 60 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [133] |
| Half Maximal Growth Inhibition (GI50) | 68.8 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [158] |
| Half Maximal Growth Inhibition (GI50) | 70 nM | N.A. | N.A. | SN12C cell | CVCL_1705 | [146] |
| Half Maximal Growth Inhibition (GI50) | 70 nM | N.A. | N.A. | OVCAR-3 cell | CVCL_0465 | [148] |
| Half Maximal Growth Inhibition (GI50) | 70 nM | N.A. | N.A. | LOX IMVI cell | CVCL_1381 | [146] |
| Half Maximal Growth Inhibition (GI50) | 70 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [159] |
| Half Maximal Growth Inhibition (GI50) | 70 nM | N.A. | N.A. | SNB-75 cell | CVCL_1706 | [146] |
| Half Maximal Growth Inhibition (GI50) | 70 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [160] |
| Half Maximal Growth Inhibition (GI50) | 73 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [161] |
| Half Maximal Growth Inhibition (GI50) | 78.9 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [162] |
| Half Maximal Growth Inhibition (GI50) | 80 nM | N.A. | N.A. | ACHN cell | CVCL_1067 | [163] |
| Half Maximal Growth Inhibition (GI50) | 80 nM | N.A. | N.A. | SK-MEL-28 cell | CVCL_0526 | [146] |
| Half Maximal Growth Inhibition (GI50) | 80 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [141] |
| Half Maximal Growth Inhibition (GI50) | 85 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [161] |
| Half Maximal Growth Inhibition (GI50) | 87.5 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [164] |
| Half Maximal Growth Inhibition (GI50) | 90 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [165] |
| Half Maximal Growth Inhibition (GI50) | 90 nM | N.A. | N.A. | NCI-H23 cell | CVCL_1547 | [166] |
| Half Maximal Growth Inhibition (GI50) | 90 nM | N.A. | N.A. | SW620 cell | CVCL_0547 | [141] |
| Half Maximal Growth Inhibition (GI50) | 90 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [167] |
| Half Maximal Growth Inhibition (GI50) | 90 nM | N.A. | N.A. | SF268 cell | CVCL_1689 | [151] |
| Half Maximal Growth Inhibition (GI50) | 93 nM | N.A. | N.A. | SF268 cell | CVCL_1689 | [168] |
| Half Maximal Growth Inhibition (GI50) | 94 nM | N.A. | N.A. | UACC-62 cell | CVCL_1780 | [169] |
| Half Maximal Growth Inhibition (GI50) | 94 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [170] |
| Half Maximal Growth Inhibition (GI50) | 94 nM | N.A. | N.A. | SF268 cell | CVCL_1689 | [138] |
| Half Maximal Growth Inhibition (GI50) | 100 nM | N.A. | N.A. | RXF 393 cell | CVCL_1673 | [146] |
| Half Maximal Growth Inhibition (GI50) | <100 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [171] |
| Half Maximal Growth Inhibition (GI50) | <100 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [172] |
| Half Maximal Growth Inhibition (GI50) | <100 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [173] |
| Half Maximal Growth Inhibition (GI50) | <100 nM | N.A. | N.A. | COLO205 cell | CVCL_F402 | [174] |
| Half Maximal Growth Inhibition (GI50) | 100 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [175] |
| Half Maximal Growth Inhibition (GI50) | <100 nM | N.A. | N.A. | ZR-75-1 cell | CVCL_0588 | [173] |
| Half Maximal Growth Inhibition (GI50) | <100 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [172] |
| Half Maximal Growth Inhibition (GI50) | >100 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [176] |
| Half Maximal Growth Inhibition (GI50) | 100 nM | N.A. | N.A. | SF-295 cell | CVCL_1690 | [146] |
| Half Maximal Growth Inhibition (GI50) | <100 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [177] |
| Half Maximal Growth Inhibition (GI50) | 100 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [175] |
| Half Maximal Growth Inhibition (GI50) | 100 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [178] |
| Half Maximal Growth Inhibition (GI50) | 100 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [175] |
| Half Maximal Growth Inhibition (GI50) | 100 nM | N.A. | N.A. | SF268 cell | CVCL_1689 | [141] |
| Half Maximal Growth Inhibition (GI50) | 107.15 nM | N.A. | N.A. | HOP-62 cell | CVCL_1285 | [176] |
| Half Maximal Growth Inhibition (GI50) | 110 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [146] |
| Half Maximal Growth Inhibition (GI50) | 110 nM | N.A. | N.A. | SK-MEL-2 cell | CVCL_0069 | [157] |
| Half Maximal Growth Inhibition (GI50) | 110 nM | N.A. | N.A. | ZR-75-1 cell | CVCL_0588 | [174] |
| Half Maximal Growth Inhibition (GI50) | 110 nM | N.A. | N.A. | KB cell | CVCL_0372 | [179] |
| Half Maximal Growth Inhibition (GI50) | 120 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [157] |
| Half Maximal Growth Inhibition (GI50) | 120 nM | N.A. | N.A. | UACC-257 cell | CVCL_1779 | [146] |
| Half Maximal Growth Inhibition (GI50) | 120 nM | N.A. | N.A. | SF539 cell | CVCL_1691 | [146] |
| Half Maximal Growth Inhibition (GI50) | 120 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [141] |
| Half Maximal Growth Inhibition (GI50) | 120 nM | N.A. | N.A. | LoVo cell | CVCL_0399 | [180] |
| Half Maximal Growth Inhibition (GI50) | 123 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [162] |
| Half Maximal Growth Inhibition (GI50) | 123.03 nM | N.A. | N.A. | A498 cell | CVCL_1056 | [181] |
| Half Maximal Growth Inhibition (GI50) | 130 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [182] |
| Half Maximal Growth Inhibition (GI50) | 131.83 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [176] |
| Half Maximal Growth Inhibition (GI50) | 134 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [162] |
| Half Maximal Growth Inhibition (GI50) | 140 nM | N.A. | N.A. | COLO205 cell | CVCL_F402 | [183] |
| Half Maximal Growth Inhibition (GI50) | 140 nM | N.A. | N.A. | HOP-62 cell | CVCL_1285 | [183] |
| Half Maximal Growth Inhibition (GI50) | 140 nM | N.A. | N.A. | UACC-62 cell | CVCL_1780 | [141] |
| Half Maximal Growth Inhibition (GI50) | 140 nM | N.A. | N.A. | SK-MEL-5 cell | CVCL_0527 | [146] |
| Half Maximal Growth Inhibition (GI50) | 140 nM | N.A. | N.A. | SiHa cell | CVCL_0032 | [184] |
| Half Maximal Growth Inhibition (GI50) | 141.25 nM | N.A. | N.A. | KB cell | CVCL_0372 | [176] |
| Half Maximal Growth Inhibition (GI50) | 150 nM | N.A. | N.A. | COLO205 cell | CVCL_F402 | [185] |
| Half Maximal Growth Inhibition (GI50) | 150 nM | N.A. | N.A. | NCI-H23 cell | CVCL_1547 | [146] |
| Half Maximal Growth Inhibition (GI50) | 150 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [179] |
| Half Maximal Growth Inhibition (GI50) | 160 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [183] |
| Half Maximal Growth Inhibition (GI50) | 160 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [157] |
| Half Maximal Growth Inhibition (GI50) | 170 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [179] |
| Half Maximal Growth Inhibition (GI50) | 170 nM | N.A. | N.A. | IGROV-1 cell | CVCL_1304 | [146] |
| Half Maximal Growth Inhibition (GI50) | 170 nM | N.A. | N.A. | SK-MEL-2 cell | CVCL_0069 | [152] |
| Half Maximal Growth Inhibition (GI50) | 170 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [183] |
| Half Maximal Growth Inhibition (GI50) | 170 nM | N.A. | N.A. | KB cell | CVCL_0372 | [183] |
| Half Maximal Growth Inhibition (GI50) | 170 nM | N.A. | N.A. | SiHa cell | CVCL_0032 | [186] |
| Half Maximal Growth Inhibition (GI50) | 178 nM | N.A. | N.A. | NCI-H23 cell | CVCL_1547 | [187] |
| Half Maximal Growth Inhibition (GI50) | 180 nM | N.A. | N.A. | A431 cell | CVCL_0037 | [182] |
| Half Maximal Growth Inhibition (GI50) | 180 nM | N.A. | N.A. | TK-10 cell | CVCL_1773 | [152] |
| Half Maximal Growth Inhibition (GI50) | 180 nM | N.A. | N.A. | COLO205 cell | CVCL_F402 | [152] |
| Half Maximal Growth Inhibition (GI50) | 180 nM | N.A. | N.A. | M14 cell | CVCL_1395 | [146] |
| Half Maximal Growth Inhibition (GI50) | 182 nM | N.A. | N.A. | ACHN cell | CVCL_1067 | [188] |
| Half Maximal Growth Inhibition (GI50) | 190 nM | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [131] |
| Half Maximal Growth Inhibition (GI50) | 194 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [188] |
| Half Maximal Growth Inhibition (GI50) | 200 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [189] |
| Half Maximal Growth Inhibition (GI50) | 200 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [190] |
| Half Maximal Growth Inhibition (GI50) | 210 nM | N.A. | N.A. | SK-MEL-2 cell | CVCL_0069 | [146] |
| Half Maximal Growth Inhibition (GI50) | 210 nM | N.A. | N.A. | SK-MEL-28 cell | CVCL_0526 | [141] |
| Half Maximal Growth Inhibition (GI50) | 220 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [146] |
| Half Maximal Growth Inhibition (GI50) | 230 nM | N.A. | N.A. | BT-549 cell | CVCL_1092 | [146] |
| Half Maximal Growth Inhibition (GI50) | 250 nM | N.A. | N.A. | NUGC-3 cell | CVCL_1612 | [191] |
| Half Maximal Growth Inhibition (GI50) | 250 nM | N.A. | N.A. | MDA-MB-435 cell | CVCL_0417 | [152] |
| Half Maximal Growth Inhibition (GI50) | 260 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [153] |
| Half Maximal Growth Inhibition (GI50) | 260 nM | N.A. | N.A. | HCC 2998 cell | CVCL_1266 | [141] |
| Half Maximal Growth Inhibition (GI50) | 270 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [192] |
| Half Maximal Growth Inhibition (GI50) | 280 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [193] |
| Half Maximal Growth Inhibition (GI50) | 320 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [146] |
| Half Maximal Growth Inhibition (GI50) | 330 nM | N.A. | N.A. | Hs 578T cell | CVCL_0332 | [146] |
| Half Maximal Growth Inhibition (GI50) | 370 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [134] |
| Half Maximal Growth Inhibition (GI50) | 380 nM | N.A. | N.A. | TK-10 cell | CVCL_1773 | [141] |
| Half Maximal Growth Inhibition (GI50) | 400 nM | N.A. | N.A. | NCI-H23 cell | CVCL_1547 | [194] |
| Half Maximal Growth Inhibition (GI50) | 410 nM | N.A. | N.A. | ACHN cell | CVCL_1067 | [191] |
| Half Maximal Growth Inhibition (GI50) | 410 nM | N.A. | N.A. | EKVX cell | CVCL_1195 | [146] |
| Half Maximal Growth Inhibition (GI50) | 490 nM | N.A. | N.A. | UO-31 cell | CVCL_1911 | [146] |
| Half Maximal Growth Inhibition (GI50) | 510 nM | N.A. | N.A. | NCI-H23 cell | CVCL_1547 | [191] |
| Half Maximal Growth Inhibition (GI50) | 510 nM | N.A. | N.A. | NUGC-3 cell | CVCL_1612 | [195] |
| Half Maximal Growth Inhibition (GI50) | 510 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [146] |
| Half Maximal Growth Inhibition (GI50) | 540 nM | N.A. | N.A. | NCI-H322M cell | CVCL_1557 | [141] |
| Half Maximal Growth Inhibition (GI50) | 570 nM | N.A. | N.A. | TK-10 cell | CVCL_1773 | [156] |
| Half Maximal Growth Inhibition (GI50) | 660 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [196] |
| Half Maximal Growth Inhibition (GI50) | 730 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [191] |
| Half Maximal Growth Inhibition (GI50) | 930 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [159] |
| Half Maximal Growth Inhibition (GI50) | 950 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [197] |
| Half Maximal Growth Inhibition (GI50) | 950 nM | N.A. | N.A. | Caki-1 cell | CVCL_0234 | [146] |
| Half Maximal Growth Inhibition (GI50) | <1000 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [198] |
| Half Maximal Growth Inhibition (GI50) | 1000 nM | N.A. | N.A. | NUGC-3 cell | CVCL_1612 | [199] |
| Half Maximal Growth Inhibition (GI50) | 1000 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [17] |
| Half Maximal Growth Inhibition (GI50) | 1.1 uM | N.A. | N.A. | SK-BR-3 cell | CVCL_0033 | [136] |
| Half Maximal Growth Inhibition (GI50) | 1.2 uM | N.A. | N.A. | 184B5 cell | CVCL_4688 | [200] |
| Half Maximal Growth Inhibition (GI50) | 1.2 uM | N.A. | N.A. | L929 cell | CVCL_0462 | [17] |
| Half Maximal Growth Inhibition (GI50) | 1.41 uM | N.A. | N.A. | PANC-1 cell | CVCL_0480 | [201] |
| Half Maximal Growth Inhibition (GI50) | 1.65 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [199] |
| Half Maximal Growth Inhibition (GI50) | 1.72 uM | N.A. | N.A. | KB cell | CVCL_0372 | [202] |
| Half Maximal Growth Inhibition (GI50) | 1.79 uM | N.A. | N.A. | ZR-75-1 cell | CVCL_0588 | [186] |
| Half Maximal Growth Inhibition (GI50) | 1.79 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [203] |
| Half Maximal Growth Inhibition (GI50) | 1.81 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [184] |
| Half Maximal Growth Inhibition (GI50) | 1.9 uM | N.A. | N.A. | NCI-H23 cell | CVCL_1547 | [195] |
| Half Maximal Growth Inhibition (GI50) | 1.9 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [197] |
| Half Maximal Growth Inhibition (GI50) | 1.9 uM | N.A. | N.A. | SiHa cell | CVCL_0032 | [173] |
| Half Maximal Growth Inhibition (GI50) | 2.0893 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [176] |
| Half Maximal Growth Inhibition (GI50) | 2.1 uM | N.A. | N.A. | SiHa cell | CVCL_0032 | [179] |
| Half Maximal Growth Inhibition (GI50) | 2.177 uM | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [188] |
| Half Maximal Growth Inhibition (GI50) | 2.4 uM | N.A. | N.A. | IGROV-1 cell | CVCL_1304 | [136] |
| Half Maximal Growth Inhibition (GI50) | 4.1 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [204] |
| Half Maximal Growth Inhibition (GI50) | 4.4 uM | N.A. | N.A. | IGROV-1 cell | CVCL_1304 | [136] |
| Half Maximal Growth Inhibition (GI50) | >5 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [205] |
| Half Maximal Growth Inhibition (GI50) | 5.7 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [204] |
| Half Maximal Growth Inhibition (GI50) | 7.16 uM | N.A. | N.A. | NCI-ADR-RES cell | CVCL_1452 | [146] |
| Half Maximal Growth Inhibition (GI50) | 7.25 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [206] |
| Half Maximal Growth Inhibition (GI50) | 7.51 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [207] |
| Half Maximal Growth Inhibition (GI50) | 8.14 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [207] |
| Half Maximal Growth Inhibition (GI50) | 8.4 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [208] |
| Half Maximal Growth Inhibition (GI50) | 10.7 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [209] |
| Half Maximal Growth Inhibition (GI50) | 10.7 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [210] |
| Half Maximal Growth Inhibition (GI50) | 13.01 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [173] |
| Half Maximal Growth Inhibition (GI50) | 14.7 uM | N.A. | N.A. | COLO205 cell | CVCL_F402 | [173] |
| Half Maximal Growth Inhibition (GI50) | 16.7 uM | N.A. | N.A. | NCI-ADR-RES cell | CVCL_1452 | [144] |
| Half Maximal Growth Inhibition (GI50) | 35.48134 uM | N.A. | N.A. | HOP-62 cell | CVCL_1285 | [176] |
| Half Maximal Growth Inhibition (GI50) | 42.8 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [211] |
| Half Maximal Infective Dose (ID50) | 10 nM | N.A. | N.A. | Raji cell | CVCL_0511 | [212] |
| Half Maximal Infective Dose (ID50) | 14.2 uM | N.A. | N.A. | MCF7-VP cell | CVCL_5I65 | [213] |
| Half Maximal Inhibitory Concentration (IC50) | 0.1 ug/mL | N.A. | N.A. | SK-MEL-2 cell | CVCL_0069 | [214] |
| Half Maximal Inhibitory Concentration (IC50) | <0.1 ug/mL | N.A. | N.A. | L1210 cell | CVCL_0382 | [215] |
| Half Maximal Inhibitory Concentration (IC50) | 0.1 ug/mL | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [216] |
| Half Maximal Inhibitory Concentration (IC50) | 0.11 ug/mL | N.A. | N.A. | CHO cell | CVCL_0213 | [217] |
| Half Maximal Inhibitory Concentration (IC50) | 0.11 ug/mL | N.A. | N.A. | B16 cell | CVCL_F936 | [218] |
| Half Maximal Inhibitory Concentration (IC50) | 0.11 ug/mL | N.A. | N.A. | LOX IMVI cell | CVCL_1381 | [218] |
| Half Maximal Inhibitory Concentration (IC50) | 0.11 ug/mL | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [219] |
| Half Maximal Inhibitory Concentration (IC50) | 0.11 ug/mL | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [220] |
| Half Maximal Inhibitory Concentration (IC50) | 0.12 ug/mL | N.A. | N.A. | KB cell | CVCL_0372 | [221] |
| Half Maximal Inhibitory Concentration (IC50) | 0.13 ug/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [218] |
| Half Maximal Inhibitory Concentration (IC50) | 0.13 ug/mL | N.A. | N.A. | KB cell | CVCL_0372 | [222] |
| Half Maximal Inhibitory Concentration (IC50) | 0.136 ug/mL | N.A. | N.A. | DLD-1 cell | CVCL_0248 | [223] |
| Half Maximal Inhibitory Concentration (IC50) | 0.14 ug/mL | N.A. | N.A. | K562 cell | CVCL_0004 | [224] |
| Half Maximal Inhibitory Concentration (IC50) | 0.14 ug/mL | N.A. | N.A. | SF268 cell | CVCL_1689 | [225] |
| Half Maximal Inhibitory Concentration (IC50) | 0.144 ug/mL | N.A. | N.A. | KB cell | CVCL_0372 | [226] |
| Half Maximal Inhibitory Concentration (IC50) | 0.15 ug/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [227] |
| Half Maximal Inhibitory Concentration (IC50) | 0.16 ug/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [228] |
| Half Maximal Inhibitory Concentration (IC50) | 0.16 ug/mL | N.A. | N.A. | SNU-16 cell | CVCL_0076 | [229] |
| Half Maximal Inhibitory Concentration (IC50) | 0.163 ug/mL | N.A. | N.A. | HeLa cell | CVCL_0030 | [230] |
| Half Maximal Inhibitory Concentration (IC50) | 0.17 ug/mL | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [231] |
| Half Maximal Inhibitory Concentration (IC50) | 0.17 ug/mL | N.A. | N.A. | HCC 2998 cell | CVCL_1266 | [218] |
| Half Maximal Inhibitory Concentration (IC50) | 0.17 ug/mL | N.A. | N.A. | SW620 cell | CVCL_0547 | [218] |
| Half Maximal Inhibitory Concentration (IC50) | 0.18 ug/mL | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [225] |
| Half Maximal Inhibitory Concentration (IC50) | 0.18 ug/mL | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [232] |
| Half Maximal Inhibitory Concentration (IC50) | 0.19 ug/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [233] |
| Half Maximal Inhibitory Concentration (IC50) | 0.19 ug/mL | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [233] |
| Half Maximal Inhibitory Concentration (IC50) | 0.19 ug/mL | N.A. | N.A. | XF498 cell | CVCL_8928 | [234] |
| Half Maximal Inhibitory Concentration (IC50) | 0.19 ug/mL | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [235] |
| Half Maximal Inhibitory Concentration (IC50) | 0.2 ug/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [236] |
| Half Maximal Inhibitory Concentration (IC50) | 0.2 ug/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [237] |
| Half Maximal Inhibitory Concentration (IC50) | 0.2 ug/mL | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [238] |
| Half Maximal Inhibitory Concentration (IC50) | 0.21 ug/mL | N.A. | N.A. | A431 cell | CVCL_0037 | [218] |
| Half Maximal Inhibitory Concentration (IC50) | 0.21 ug/mL | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [218] |
| Half Maximal Inhibitory Concentration (IC50) | 0.22 ug/mL | N.A. | N.A. | KB cell | CVCL_0372 | [239] |
| Half Maximal Inhibitory Concentration (IC50) | 0.22 ug/mL | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [240] |
| Half Maximal Inhibitory Concentration (IC50) | 0.23 ug/mL | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [241] |
| Half Maximal Inhibitory Concentration (IC50) | 0.23 ug/mL | N.A. | N.A. | SF-295 cell | CVCL_1690 | [242] |
| Half Maximal Inhibitory Concentration (IC50) | 0.24 ug/mL | N.A. | N.A. | PC-3 cell | CVCL_0035 | [243] |
| Half Maximal Inhibitory Concentration (IC50) | 0.24 ug/mL | N.A. | N.A. | HeLa cell | CVCL_0030 | [228] |
| Half Maximal Inhibitory Concentration (IC50) | 0.24 ug/mL | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [218] |
| Half Maximal Inhibitory Concentration (IC50) | 0.241 ug/mL | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [244] |
| Half Maximal Inhibitory Concentration (IC50) | 0.25 ug/mL | N.A. | N.A. | SF-295 cell | CVCL_1690 | [245] |
| Half Maximal Inhibitory Concentration (IC50) | 0.26 ug/mL | N.A. | N.A. | K562 cell | CVCL_0004 | [246] |
| Half Maximal Inhibitory Concentration (IC50) | 0.28 ug/mL | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [247] |
| Half Maximal Inhibitory Concentration (IC50) | 0.28 ug/mL | N.A. | N.A. | C-33-A cell | CVCL_1094 | [248] |
| Half Maximal Inhibitory Concentration (IC50) | 0.286 ug/mL | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [249] |
| Half Maximal Inhibitory Concentration (IC50) | 0.29 ug/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [250] |
| Half Maximal Inhibitory Concentration (IC50) | 0.31 ug/mL | N.A. | N.A. | Ca9-22 cell | CVCL_1102 | [232] |
| Half Maximal Inhibitory Concentration (IC50) | 0.32 ug/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [251] |
| Half Maximal Inhibitory Concentration (IC50) | 0.32 ug/mL | N.A. | N.A. | DLD-1 cell | CVCL_0248 | [218] |
| Half Maximal Inhibitory Concentration (IC50) | 0.33 ug/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [248] |
| Half Maximal Inhibitory Concentration (IC50) | 0.339 ug/mL | N.A. | N.A. | HeLa cell | CVCL_0030 | [252] |
| Half Maximal Inhibitory Concentration (IC50) | 0.34 ug/mL | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [251] |
| Half Maximal Inhibitory Concentration (IC50) | 0.35 ug/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [253] |
| Half Maximal Inhibitory Concentration (IC50) | 0.38 ug/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [254] |
| Half Maximal Inhibitory Concentration (IC50) | 0.39 ug/mL | N.A. | N.A. | P388 cell | CVCL_7222 | [216] |
| Half Maximal Inhibitory Concentration (IC50) | 0.39 ug/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [255] |
| Half Maximal Inhibitory Concentration (IC50) | 0.4 ug/mL | N.A. | N.A. | DU145 cell | CVCL_0105 | [256] |
| Half Maximal Inhibitory Concentration (IC50) | 0.4 ug/mL | N.A. | N.A. | LLC-PK1 cell | CVCL_0391 | [257] |
| Half Maximal Inhibitory Concentration (IC50) | 0.4 ug/mL | N.A. | N.A. | HEp-2 cell | CVCL_1906 | [258] |
| Half Maximal Inhibitory Concentration (IC50) | 0.4 ug/mL | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [259] |
| Half Maximal Inhibitory Concentration (IC50) | 0.41 ug/mL | N.A. | N.A. | SF-295 cell | CVCL_1690 | [260] |
| Half Maximal Inhibitory Concentration (IC50) | 0.42 ug/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [235] |
| Half Maximal Inhibitory Concentration (IC50) | 0.43 ug/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [261] |
| Half Maximal Inhibitory Concentration (IC50) | 0.44±0.11 µM | N.A. | N.A. | L-02 cell | CVCL_6926 | [262] |
| Half Maximal Inhibitory Concentration (IC50) | 0.45 ug/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [248] |
| Half Maximal Inhibitory Concentration (IC50) | 0.45 ug/mL | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [263] |
| Half Maximal Inhibitory Concentration (IC50) | 0.45 ug/mL | N.A. | N.A. | K562 cell | CVCL_0004 | [239] |
| Half Maximal Inhibitory Concentration (IC50) | 0.457 ug/mL | N.A. | N.A. | HL-60 cell | CVCL_0002 | [249] |
| Half Maximal Inhibitory Concentration (IC50) | 0.46 ug/mL | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [235] |
| Half Maximal Inhibitory Concentration (IC50) | 0.47 ug/mL | N.A. | N.A. | MDA-MB-435 cell | CVCL_0417 | [243] |
| Half Maximal Inhibitory Concentration (IC50) | 0.48 ug/mL | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [264] |
| Half Maximal Inhibitory Concentration (IC50) | <0.5 ug/mL | N.A. | N.A. | SNU-1 cell | CVCL_0099 | [265] |
| Half Maximal Inhibitory Concentration (IC50) | 0.5 ug/mL | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [214] |
| Half Maximal Inhibitory Concentration (IC50) | 0.51 ng/mL | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [266] |
| Half Maximal Inhibitory Concentration (IC50) | 0.52 ug/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [231] |
| Half Maximal Inhibitory Concentration (IC50) | 0.52 ug/mL | N.A. | N.A. | LoVo cell | CVCL_0399 | [267] |
| Half Maximal Inhibitory Concentration (IC50) | 0.53 ug/mL | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [218] |
| Half Maximal Inhibitory Concentration (IC50) | 0.54 ug/mL | N.A. | N.A. | B16 cell | CVCL_F936 | [229] |
| Half Maximal Inhibitory Concentration (IC50) | 0.54 ug/mL | N.A. | N.A. | K562 cell | CVCL_0004 | [268] |
| Half Maximal Inhibitory Concentration (IC50) | <0.55 ug/mL | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [269] |
| Half Maximal Inhibitory Concentration (IC50) | 0.55 ug/mL | N.A. | N.A. | P388 cell | CVCL_7222 | [217] |
| Half Maximal Inhibitory Concentration (IC50) | <0.55 ug/mL | N.A. | N.A. | KB cell | CVCL_0372 | [269] |
| Half Maximal Inhibitory Concentration (IC50) | 0.55 ug/mL | N.A. | N.A. | LoVo cell | CVCL_0399 | [270] |
| Half Maximal Inhibitory Concentration (IC50) | 0.57 ug/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [271] |
| Half Maximal Inhibitory Concentration (IC50) | 0.58 ug/mL | N.A. | N.A. | LoVo cell | CVCL_0399 | [272] |
| Half Maximal Inhibitory Concentration (IC50) | 0.59 ug/mL | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [12] |
| Half Maximal Inhibitory Concentration (IC50) | 0.63 ug/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [253] |
| Half Maximal Inhibitory Concentration (IC50) | 0.65 ug/mL | N.A. | N.A. | LLC-PK1 cell | CVCL_0391 | [273] |
| Half Maximal Inhibitory Concentration (IC50) | 0.65 ug/mL | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [12] |
| Half Maximal Inhibitory Concentration (IC50) | 0.67 ug/mL | N.A. | N.A. | EL4 cell | CVCL_0255 | [216] |
| Half Maximal Inhibitory Concentration (IC50) | 0.69 ug/mL | N.A. | N.A. | MDA-MB-435 cell | CVCL_0417 | [260] |
| Half Maximal Inhibitory Concentration (IC50) | 0.7 ug/mL | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [274] |
| Half Maximal Inhibitory Concentration (IC50) | 0.71 ug/mL | N.A. | N.A. | WI-38 cell | CVCL_0579 | [275] |
| Half Maximal Inhibitory Concentration (IC50) | 0.72 ug/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [12] |
| Half Maximal Inhibitory Concentration (IC50) | 0.73 ug/mL | N.A. | N.A. | MG-22A cell | CVCL_U248 | [276] |
| Half Maximal Inhibitory Concentration (IC50) | 0.73 ug/mL | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [233] |
| Half Maximal Inhibitory Concentration (IC50) | 0.75 ug/mL | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [277] |
| Half Maximal Inhibitory Concentration (IC50) | 0.76 ug/mL | N.A. | N.A. | WI-38 cell | CVCL_0579 | [264] |
| Half Maximal Inhibitory Concentration (IC50) | 0.77 ug/mL | N.A. | N.A. | MRC5 cell | CVCL_0440 | [235] |
| Half Maximal Inhibitory Concentration (IC50) | 0.8 ug/mL | N.A. | N.A. | BT-549 cell | CVCL_1092 | [278] |
| Half Maximal Inhibitory Concentration (IC50) | 0.87 ug/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [279] |
| Half Maximal Inhibitory Concentration (IC50) | 0.899 ug/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [226] |
| Half Maximal Inhibitory Concentration (IC50) | 0.9 ug/mL | N.A. | N.A. | L1210 cell | CVCL_0382 | [280] |
| Half Maximal Inhibitory Concentration (IC50) | 0.988 ug/mL | N.A. | N.A. | HeLa cell | CVCL_0030 | [252] |
| Half Maximal Inhibitory Concentration (IC50) | <1 ng/mL | N.A. | N.A. | MG-22A cell | CVCL_U248 | [276] |
| Half Maximal Inhibitory Concentration (IC50) | 1 ng/mL | N.A. | N.A. | B16 cell | CVCL_F936 | [276] |
| Half Maximal Inhibitory Concentration (IC50) | 1 ug/mL | N.A. | N.A. | Caco-2 cell | CVCL_0025 | [258] |
| Half Maximal Inhibitory Concentration (IC50) | 1 ug/mL | N.A. | N.A. | BT-549 cell | CVCL_1092 | [281] |
| Half Maximal Inhibitory Concentration (IC50) | 1.03±0.13 µM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [262] |
| Half Maximal Inhibitory Concentration (IC50) | 1.05 ug/mL | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [251] |
| Half Maximal Inhibitory Concentration (IC50) | <1.1 ug/mL | N.A. | N.A. | KB cell | CVCL_0372 | [282] |
| Half Maximal Inhibitory Concentration (IC50) | 1.2 ug/mL | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [278] |
| Half Maximal Inhibitory Concentration (IC50) | 1.21 ug/mL | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [283] |
| Half Maximal Inhibitory Concentration (IC50) | 1.28 ug/mL | N.A. | N.A. | HeLa cell | CVCL_0030 | [284] |
| Half Maximal Inhibitory Concentration (IC50) | 1.3 mM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [285] |
| Half Maximal Inhibitory Concentration (IC50) | 1.3 ug/mL | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [286] |
| Half Maximal Inhibitory Concentration (IC50) | 1.3 ug/mL | N.A. | N.A. | BT-549 cell | CVCL_1092 | [287] |
| Half Maximal Inhibitory Concentration (IC50) | 1.32 ug/mL | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [253] |
| Half Maximal Inhibitory Concentration (IC50) | 1.35 ug/mL | N.A. | N.A. | KB cell | CVCL_0372 | [278] |
| Half Maximal Inhibitory Concentration (IC50) | 1.41 ug/mL | N.A. | N.A. | K562 cell | CVCL_0004 | [288] |
| Half Maximal Inhibitory Concentration (IC50) | 1.5 ug/mL | N.A. | N.A. | Vero cell | CVCL_0059 | [289] |
| Half Maximal Inhibitory Concentration (IC50) | 1.53 ug/mL | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [281] |
| Half Maximal Inhibitory Concentration (IC50) | 1.6 ug/mL | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [228] |
| Half Maximal Inhibitory Concentration (IC50) | 1.7 ug/mL | N.A. | N.A. | KB cell | CVCL_0372 | [281] |
| Half Maximal Inhibitory Concentration (IC50) | 1.8 ug/mL | N.A. | N.A. | A-375 cell | CVCL_0132 | [290] |
| Half Maximal Inhibitory Concentration (IC50) | 1.88 ug/mL | N.A. | N.A. | HL-60 cell | CVCL_0002 | [252] |
| Half Maximal Inhibitory Concentration (IC50) | 2 ng/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [291] |
| Half Maximal Inhibitory Concentration (IC50) | 2.2 ug/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [222] |
| Half Maximal Inhibitory Concentration (IC50) | 2.3 ug/mL | N.A. | N.A. | PC-3 cell | CVCL_0035 | [228] |
| Half Maximal Inhibitory Concentration (IC50) | 2.4 ng/mL | N.A. | N.A. | MIA PaCa-2 cell | CVCL_0428 | [292] |
| Half Maximal Inhibitory Concentration (IC50) | 2.5 ug/mL | N.A. | N.A. | HSC-2 cell | CVCL_1287 | [290] |
| Half Maximal Inhibitory Concentration (IC50) | 2.5 mM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [293] |
| Half Maximal Inhibitory Concentration (IC50) | 2.97 ug/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [294] |
| Half Maximal Inhibitory Concentration (IC50) | 2.97 ug/mL | N.A. | N.A. | HL-60 cell | CVCL_0002 | [252] |
| Half Maximal Inhibitory Concentration (IC50) | 3.1 ng/mL | N.A. | N.A. | B16-F10 cell | CVCL_0159 | [295] |
| Half Maximal Inhibitory Concentration (IC50) | 3.1 ng/mL | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [295] |
| Half Maximal Inhibitory Concentration (IC50) | 3.2 ug/mL | N.A. | N.A. | Vero cell | CVCL_0059 | [269] |
| Half Maximal Inhibitory Concentration (IC50) | 3.2 ug/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [284] |
| Half Maximal Inhibitory Concentration (IC50) | 3.3 ug/mL | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [296] |
| Half Maximal Inhibitory Concentration (IC50) | 3.73 ug/mL | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [297] |
| Half Maximal Inhibitory Concentration (IC50) | 3.73 ug/mL | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [298] |
| Half Maximal Inhibitory Concentration (IC50) | 4 ng/mL | N.A. | N.A. | N2a cell | CVCL_0470 | [276] |
| Half Maximal Inhibitory Concentration (IC50) | 4 ng/mL | N.A. | N.A. | P388 cell | CVCL_7222 | [299] |
| Half Maximal Inhibitory Concentration (IC50) | 4 ug/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [300] |
| Half Maximal Inhibitory Concentration (IC50) | 4 ug/mL | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [300] |
| Half Maximal Inhibitory Concentration (IC50) | 4.12 ug/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [301] |
| Half Maximal Inhibitory Concentration (IC50) | 4.24 ug/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [302] |
| Half Maximal Inhibitory Concentration (IC50) | 4.5 ug/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [303] |
| Half Maximal Inhibitory Concentration (IC50) | 4.57 ug/mL | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [304] |
| Half Maximal Inhibitory Concentration (IC50) | 4.7 ug/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [305] |
| Half Maximal Inhibitory Concentration (IC50) | 4.9 ng/mL | N.A. | N.A. | A498 cell | CVCL_1056 | [292] |
| Half Maximal Inhibitory Concentration (IC50) | 5 ug | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [306] |
| Half Maximal Inhibitory Concentration (IC50) | 5 mM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [293] |
| Half Maximal Inhibitory Concentration (IC50) | 5.4 ug/mL | N.A. | N.A. | Vero cell | CVCL_0059 | [307] |
| Half Maximal Inhibitory Concentration (IC50) | 5.49 mM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [308] |
| Half Maximal Inhibitory Concentration (IC50) | 5.5 ug/mL | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [298] |
| Half Maximal Inhibitory Concentration (IC50) | 6.24 ug/mL | N.A. | N.A. | BeWo cell | CVCL_0044 | [216] |
| Half Maximal Inhibitory Concentration (IC50) | 6.3 ug/mL | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [305] |
| Half Maximal Inhibitory Concentration (IC50) | 6.39 ng/mL | N.A. | N.A. | ZR-75-1 cell | CVCL_0588 | [223] |
| Half Maximal Inhibitory Concentration (IC50) | 6.71 ug/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [309] |
| Half Maximal Inhibitory Concentration (IC50) | 7.5 ug/mL | N.A. | N.A. | Vero cell | CVCL_0059 | [310] |
| Half Maximal Inhibitory Concentration (IC50) | 8 ng/mL | N.A. | N.A. | BT-549 cell | CVCL_1092 | [289] |
| Half Maximal Inhibitory Concentration (IC50) | 8.72 ug/mL | N.A. | N.A. | HeLa cell | CVCL_0030 | [309] |
| Half Maximal Inhibitory Concentration (IC50) | 8.92 ug/mL | N.A. | N.A. | BT-474 cell | CVCL_0179 | [311] |
| Half Maximal Inhibitory Concentration (IC50) | 9.6 ng/mL | N.A. | N.A. | P388 cell | CVCL_7222 | [229] |
| Half Maximal Inhibitory Concentration (IC50) | 10 ng/mL | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [312] |
| Half Maximal Inhibitory Concentration (IC50) | 10 ng/mL | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [291] |
| Half Maximal Inhibitory Concentration (IC50) | 10 ng/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [313] |
| Half Maximal Inhibitory Concentration (IC50) | 10 ng/mL | N.A. | N.A. | Bel-7402 cell | CVCL_5492 | [314] |
| Half Maximal Inhibitory Concentration (IC50) | 10 ng/mL | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [315] |
| Half Maximal Inhibitory Concentration (IC50) | 11 ng/mL | N.A. | N.A. | PC-3 cell | CVCL_0035 | [292] |
| Half Maximal Inhibitory Concentration (IC50) | 11 ng/mL | N.A. | N.A. | K562 cell | CVCL_0004 | [314] |
| Half Maximal Inhibitory Concentration (IC50) | 12.1 ng/mL | N.A. | N.A. | A2780 cell | CVCL_0134 | [223] |
| Half Maximal Inhibitory Concentration (IC50) | 17 ng/mL | N.A. | N.A. | P388 cell | CVCL_7222 | [220] |
| Half Maximal Inhibitory Concentration (IC50) | 20 ng/mL | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [237] |
| Half Maximal Inhibitory Concentration (IC50) | 20 ng/mL | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [280] |
| Half Maximal Inhibitory Concentration (IC50) | 20 ng/mL | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [234] |
| Half Maximal Inhibitory Concentration (IC50) | 20 ng/mL | N.A. | N.A. | HL-60 cell | CVCL_0002 | [316] |
| Half Maximal Inhibitory Concentration (IC50) | 24 ng/mL | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [234] |
| Half Maximal Inhibitory Concentration (IC50) | 24.8 ng/mL | N.A. | N.A. | NCI-H446 cell | CVCL_1562 | [249] |
| Half Maximal Inhibitory Concentration (IC50) | 25 ng/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [314] |
| Half Maximal Inhibitory Concentration (IC50) | 26.1 ug/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [317] |
| Half Maximal Inhibitory Concentration (IC50) | 28 ng/mL | N.A. | N.A. | HL-60 cell | CVCL_0002 | [252] |
| Half Maximal Inhibitory Concentration (IC50) | 30 ng/mL | N.A. | N.A. | HT-1197 cell | CVCL_1291 | [274] |
| Half Maximal Inhibitory Concentration (IC50) | 30 ng/mL | N.A. | N.A. | MDA-MB-435 cell | CVCL_0417 | [316] |
| Half Maximal Inhibitory Concentration (IC50) | 30 ng/mL | N.A. | N.A. | HL-60 cell | CVCL_0002 | [260] |
| Half Maximal Inhibitory Concentration (IC50) | 35 ng/mL | N.A. | N.A. | K562 cell | CVCL_0004 | [289] |
| Half Maximal Inhibitory Concentration (IC50) | 37.4 ug/mL | N.A. | N.A. | Ehrlich cell | CVCL_3873 | [305] |
| Half Maximal Inhibitory Concentration (IC50) | 37.57 ug/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [227] |
| Half Maximal Inhibitory Concentration (IC50) | 40 ng/mL | N.A. | N.A. | NCI-H187 cell | CVCL_1501 | [222] |
| Half Maximal Inhibitory Concentration (IC50) | 40 ng/mL | N.A. | N.A. | HCT-8 cell | CVCL_2478 | [243] |
| Half Maximal Inhibitory Concentration (IC50) | 40 ng/mL | N.A. | N.A. | SF-295 cell | CVCL_1690 | [316] |
| Half Maximal Inhibitory Concentration (IC50) | 42 ng/mL | N.A. | N.A. | NCI-H187 cell | CVCL_1501 | [221] |
| Half Maximal Inhibitory Concentration (IC50) | 50 ng/mL | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [318] |
| Half Maximal Inhibitory Concentration (IC50) | 50 ng/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [247] |
| Half Maximal Inhibitory Concentration (IC50) | 50 ng/mL | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [313] |
| Half Maximal Inhibitory Concentration (IC50) | 53 ng/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [220] |
| Half Maximal Inhibitory Concentration (IC50) | 53 ug/mL | N.A. | N.A. | LoVo cell | CVCL_0399 | [272] |
| Half Maximal Inhibitory Concentration (IC50) | 58 ng/mL | N.A. | N.A. | NCI-H187 cell | CVCL_1501 | [226] |
| Half Maximal Inhibitory Concentration (IC50) | 60 ng/mL | N.A. | N.A. | UACC-62 cell | CVCL_1780 | [218] |
| Half Maximal Inhibitory Concentration (IC50) | 60 ng/mL | N.A. | N.A. | HCT-8 cell | CVCL_2478 | [260] |
| Half Maximal Inhibitory Concentration (IC50) | 60 ng/mL | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [280] |
| Half Maximal Inhibitory Concentration (IC50) | 90 ng/mL | N.A. | N.A. | SK-MEL-2 cell | CVCL_0069 | [313] |
| Half Maximal Inhibitory Concentration (IC50) | 90 ng/mL | N.A. | N.A. | P388 cell | CVCL_7222 | [319] |
| Half Maximal Inhibitory Concentration (IC50) | 90 ng/mL | N.A. | N.A. | L1210 cell | CVCL_0382 | [217] |
| Half Maximal Inhibitory Concentration (IC50) | 90 ng/mL | N.A. | N.A. | XF498 cell | CVCL_8928 | [247] |
| Half Maximal Inhibitory Concentration (IC50) | 90 ng/mL | N.A. | N.A. | Ca9-22 cell | CVCL_1102 | [231] |
| Half Maximal Inhibitory Concentration (IC50) | 0.19 nM | N.A. | N.A. | MKN45 cell | CVCL_0434 | [320] |
| Half Maximal Inhibitory Concentration (IC50) | 0.38 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [320] |
| Half Maximal Inhibitory Concentration (IC50) | 0.414 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [321] |
| Half Maximal Inhibitory Concentration (IC50) | 0.7 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [322] |
| Half Maximal Inhibitory Concentration (IC50) | 1 nM | N.A. | N.A. | ACHN cell | CVCL_1067 | [323] |
| Half Maximal Inhibitory Concentration (IC50) | <1 nM | N.A. | N.A. | NCI-H647 cell | CVCL_1574 | [324] |
| Half Maximal Inhibitory Concentration (IC50) | 1 nM | N.A. | N.A. | KB cell | CVCL_0372 | [325] |
| Half Maximal Inhibitory Concentration (IC50) | 1.13 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [326] |
| Half Maximal Inhibitory Concentration (IC50) | 1.2 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [327] |
| Half Maximal Inhibitory Concentration (IC50) | 1.35 nM | N.A. | N.A. | A431 cell | CVCL_0037 | [328] |
| Half Maximal Inhibitory Concentration (IC50) | 2 nM | N.A. | N.A. | MOLT-4 cell | CVCL_0013 | [329] |
| Half Maximal Inhibitory Concentration (IC50) | 2 nM | N.A. | N.A. | SK-MEL-2 cell | CVCL_0069 | [330] |
| Half Maximal Inhibitory Concentration (IC50) | 2 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [331] |
| Half Maximal Inhibitory Concentration (IC50) | 2 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [332] |
| Half Maximal Inhibitory Concentration (IC50) | 2.7 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [327] |
| Half Maximal Inhibitory Concentration (IC50) | 3 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [333] |
| Half Maximal Inhibitory Concentration (IC50) | 3 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [334] |
| Half Maximal Inhibitory Concentration (IC50) | 3 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [335] |
| Half Maximal Inhibitory Concentration (IC50) | 3 nM | N.A. | N.A. | MT4 cell | CVCL_2632 | [332] |
| Half Maximal Inhibitory Concentration (IC50) | 3.9 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [336] |
| Half Maximal Inhibitory Concentration (IC50) | 4 nM | N.A. | N.A. | HOP-62 cell | CVCL_1285 | [337] |
| Half Maximal Inhibitory Concentration (IC50) | 4 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [324] |
| Half Maximal Inhibitory Concentration (IC50) | 4.16 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [338] |
| Half Maximal Inhibitory Concentration (IC50) | 4.45 nM | N.A. | N.A. | U-937 cell | CVCL_0007 | [326] |
| Half Maximal Inhibitory Concentration (IC50) | 4.73 nM | N.A. | N.A. | GOTO cell | CVCL_1234 | [338] |
| Half Maximal Inhibitory Concentration (IC50) | 5 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [339] |
| Half Maximal Inhibitory Concentration (IC50) | 5 nM | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [340] |
| Half Maximal Inhibitory Concentration (IC50) | 5 nM | N.A. | N.A. | MDA-MB-468 cell | CVCL_0419 | [324] |
| Half Maximal Inhibitory Concentration (IC50) | 5.1 nM | N.A. | N.A. | IMR-32 cell | CVCL_0346 | [341] |
| Half Maximal Inhibitory Concentration (IC50) | 5.15 nM | N.A. | N.A. | NB-1 cell | CVCL_GZ01 | [326] |
| Half Maximal Inhibitory Concentration (IC50) | 6 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [325] |
| Half Maximal Inhibitory Concentration (IC50) | 6 nM | N.A. | N.A. | A2780 cisR cell | CVCL_H745 | [342] |
| Half Maximal Inhibitory Concentration (IC50) | 6 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [324] |
| Half Maximal Inhibitory Concentration (IC50) | 6.2 nM | N.A. | N.A. | SJSA-1 cell | CVCL_1697 | [343] |
| Half Maximal Inhibitory Concentration (IC50) | 6.3 nM | N.A. | N.A. | CH1 cell | CVCL_D177 | [344] |
| Half Maximal Inhibitory Concentration (IC50) | 6.9 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [345] |
| Half Maximal Inhibitory Concentration (IC50) | 7 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [342] |
| Half Maximal Inhibitory Concentration (IC50) | 7 nM | N.A. | N.A. | T-47D cell | CVCL_0553 | [346] |
| Half Maximal Inhibitory Concentration (IC50) | 7 nM | N.A. | N.A. | KB cell | CVCL_0372 | [347] |
| Half Maximal Inhibitory Concentration (IC50) | 7 nM | N.A. | N.A. | MDA-MB-468 cell | CVCL_0419 | [346] |
| Half Maximal Inhibitory Concentration (IC50) | 7.1 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [348] |
| Half Maximal Inhibitory Concentration (IC50) | 7.2 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [312] |
| Half Maximal Inhibitory Concentration (IC50) | 7.6 nM | N.A. | N.A. | B16 cell | CVCL_F936 | [349] |
| Half Maximal Inhibitory Concentration (IC50) | 8 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [350] |
| Half Maximal Inhibitory Concentration (IC50) | 8 nM | N.A. | N.A. | LoVo cell | CVCL_0399 | [351] |
| Half Maximal Inhibitory Concentration (IC50) | 8 nM | N.A. | N.A. | THP-1 cell | CVCL_0006 | [352] |
| Half Maximal Inhibitory Concentration (IC50) | 8.4 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [256] |
| Half Maximal Inhibitory Concentration (IC50) | 8.8 nM | N.A. | N.A. | Daoy cell | CVCL_1167 | [353] |
| Half Maximal Inhibitory Concentration (IC50) | 9 nM | N.A. | N.A. | ZR-75-1 cell | CVCL_0588 | [324] |
| Half Maximal Inhibitory Concentration (IC50) | 9.6 nM | N.A. | N.A. | Jurkat cell | CVCL_0065 | [354] |
| Half Maximal Inhibitory Concentration (IC50) | 9.6 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [344] |
| Half Maximal Inhibitory Concentration (IC50) | 9.7 nM | N.A. | N.A. | MES-SA cell | CVCL_1404 | [355] |
| Half Maximal Inhibitory Concentration (IC50) | 10 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [356] |
| Half Maximal Inhibitory Concentration (IC50) | 10 nM | N.A. | N.A. | MIA PaCa-2 cell | CVCL_0428 | [357] |
| Half Maximal Inhibitory Concentration (IC50) | 10 nM | N.A. | N.A. | MT4 cell | CVCL_2632 | [323] |
| Half Maximal Inhibitory Concentration (IC50) | 10 nM | N.A. | N.A. | IMR-32 cell | CVCL_0346 | [358] |
| Half Maximal Inhibitory Concentration (IC50) | 10 nM | N.A. | N.A. | MC-38 cell | CVCL_B288 | [359] |
| Half Maximal Inhibitory Concentration (IC50) | 10 nM | N.A. | N.A. | SK-MEL-2 cell | CVCL_0069 | [360] |
| Half Maximal Inhibitory Concentration (IC50) | 10 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [361] |
| Half Maximal Inhibitory Concentration (IC50) | 10 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [362] |
| Half Maximal Inhibitory Concentration (IC50) | 10 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [324] |
| Half Maximal Inhibitory Concentration (IC50) | 10 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [356] |
| Half Maximal Inhibitory Concentration (IC50) | 10 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [363] |
| Half Maximal Inhibitory Concentration (IC50) | 10 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [364] |
| Half Maximal Inhibitory Concentration (IC50) | 10 nM | N.A. | N.A. | U-373MG ATCC cell | CVCL_2219 | [351] |
| Half Maximal Inhibitory Concentration (IC50) | 10 nM | N.A. | N.A. | A-375 cell | CVCL_0132 | [365] |
| Half Maximal Inhibitory Concentration (IC50) | 12 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [325] |
| Half Maximal Inhibitory Concentration (IC50) | 12 nM | N.A. | N.A. | MDA-MB-361 cell | CVCL_0620 | [366] |
| Half Maximal Inhibitory Concentration (IC50) | 14 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [340] |
| Half Maximal Inhibitory Concentration (IC50) | 15 nM | N.A. | N.A. | P388 cell | CVCL_7222 | [367] |
| Half Maximal Inhibitory Concentration (IC50) | 15 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [135] |
| Half Maximal Inhibitory Concentration (IC50) | 15 nM | N.A. | N.A. | KB cell | CVCL_0372 | [368] |
| Half Maximal Inhibitory Concentration (IC50) | 15.6 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [369] |
| Half Maximal Inhibitory Concentration (IC50) | 16 nM | N.A. | N.A. | MES-SA cell | CVCL_1404 | [370] |
| Half Maximal Inhibitory Concentration (IC50) | 17 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [371] |
| Half Maximal Inhibitory Concentration (IC50) | 17.9 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [372] |
| Half Maximal Inhibitory Concentration (IC50) | 18 nM | N.A. | N.A. | HEK293 cell | CVCL_0045 | [373] |
| Half Maximal Inhibitory Concentration (IC50) | 18 nM | N.A. | N.A. | MDA-MB-361 cell | CVCL_0620 | [366] |
| Half Maximal Inhibitory Concentration (IC50) | 18 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [374] |
| Half Maximal Inhibitory Concentration (IC50) | 19 nM | N.A. | N.A. | P388/S cell | CVCL_D640 | [375] |
| Half Maximal Inhibitory Concentration (IC50) | 19 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [372] |
| Half Maximal Inhibitory Concentration (IC50) | 19 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [376] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [377] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | Jurkat cell | CVCL_0065 | [378] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | MOLT-4 cell | CVCL_0013 | [379] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [380] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [337] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | OVCAR-3 cell | CVCL_0465 | [381] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [382] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | HEK293 cell | CVCL_0045 | [383] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | P388 cell | CVCL_7222 | [384] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | CCRF-SB cell | CVCL_1860 | [323] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [385] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | C8166 cell | CVCL_1099 | [323] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | HCT-8 cell | CVCL_2478 | [386] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | WIL2-NS cell | CVCL_2761 | [323] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | SK-MEL-28 cell | CVCL_0526 | [387] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [388] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [387] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | SW620 cell | CVCL_0547 | [357] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [389] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | SK-BR-3 cell | CVCL_0033 | [324] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | T24 cell | CVCL_0554 | [351] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | 5637 cell | CVCL_0126 | [358] |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [390] |
| Half Maximal Inhibitory Concentration (IC50) | 20.1 nM | N.A. | N.A. | COR-L23 cell | CVCL_1139 | [391] |
| Half Maximal Inhibitory Concentration (IC50) | 21 nM | N.A. | N.A. | KB 3-1 cell | CVCL_2088 | [392] |
| Half Maximal Inhibitory Concentration (IC50) | 22 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [393] |
| Half Maximal Inhibitory Concentration (IC50) | 22 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [394] |
| Half Maximal Inhibitory Concentration (IC50) | 23 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [395] |
| Half Maximal Inhibitory Concentration (IC50) | 23.4 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [396] |
| Half Maximal Inhibitory Concentration (IC50) | 24 nM | N.A. | N.A. | LoVo cell | CVCL_0399 | [397] |
| Half Maximal Inhibitory Concentration (IC50) | 25 nM | N.A. | N.A. | L1210 cell | CVCL_0382 | [392] |
| Half Maximal Inhibitory Concentration (IC50) | 25 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [398] |
| Half Maximal Inhibitory Concentration (IC50) | 26 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [397] |
| Half Maximal Inhibitory Concentration (IC50) | 27 nM | N.A. | N.A. | NCI-H69 cell | CVCL_1579 | [399] |
| Half Maximal Inhibitory Concentration (IC50) | 27 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [400] |
| Half Maximal Inhibitory Concentration (IC50) | 28 nM | N.A. | N.A. | SK-MEL-2 cell | CVCL_0069 | [401] |
| Half Maximal Inhibitory Concentration (IC50) | 28 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [402] |
| Half Maximal Inhibitory Concentration (IC50) | 28 nM | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [362] |
| Half Maximal Inhibitory Concentration (IC50) | 29 nM | N.A. | N.A. | L1210 cell | CVCL_0382 | [403] |
| Half Maximal Inhibitory Concentration (IC50) | 30 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [323] |
| Half Maximal Inhibitory Concentration (IC50) | 30 nM | N.A. | N.A. | Jurkat cell | CVCL_0065 | [404] |
| Half Maximal Inhibitory Concentration (IC50) | 30 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [358] |
| Half Maximal Inhibitory Concentration (IC50) | 30 nM | N.A. | N.A. | MIA PaCa-2 cell | CVCL_0428 | [405] |
| Half Maximal Inhibitory Concentration (IC50) | 30 nM | N.A. | N.A. | LXFL 529 cell | CVCL_D085 | [364] |
| Half Maximal Inhibitory Concentration (IC50) | 30 nM | N.A. | N.A. | CCRF-SB cell | CVCL_1860 | [332] |
| Half Maximal Inhibitory Concentration (IC50) | 30 nM | N.A. | N.A. | SK-MES-1 cell | CVCL_0630 | [358] |
| Half Maximal Inhibitory Concentration (IC50) | 30 nM | N.A. | N.A. | NCI-H187 cell | CVCL_1501 | [406] |
| Half Maximal Inhibitory Concentration (IC50) | 30 nM | N.A. | N.A. | KB cell | CVCL_0372 | [407] |
| Half Maximal Inhibitory Concentration (IC50) | 30 nM | N.A. | N.A. | Raji cell | CVCL_0511 | [408] |
| Half Maximal Inhibitory Concentration (IC50) | 30 nM | N.A. | N.A. | Caco-2 cell | CVCL_0025 | [409] |
| Half Maximal Inhibitory Concentration (IC50) | 30 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [410] |
| Half Maximal Inhibitory Concentration (IC50) | 30 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [411] |
| Half Maximal Inhibitory Concentration (IC50) | 30.3 nM | N.A. | N.A. | TRAMP-C2H cell | CVCL_H591 | [412] |
| Half Maximal Inhibitory Concentration (IC50) | 31 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [413] |
| Half Maximal Inhibitory Concentration (IC50) | 31 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [414] |
| Half Maximal Inhibitory Concentration (IC50) | 31.62 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [415] |
| Half Maximal Inhibitory Concentration (IC50) | 31.62 nM | N.A. | N.A. | H9c2 cell | CVCL_0286 | [415] |
| Half Maximal Inhibitory Concentration (IC50) | 31.62 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [415] |
| Half Maximal Inhibitory Concentration (IC50) | 33 nM | N.A. | N.A. | RPMI-8226 cell | CVCL_7353 | [416] |
| Half Maximal Inhibitory Concentration (IC50) | 33 nM | N.A. | N.A. | LoVo cell | CVCL_0399 | [342] |
| Half Maximal Inhibitory Concentration (IC50) | 33 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [417] |
| Half Maximal Inhibitory Concentration (IC50) | 33 nM | N.A. | N.A. | MDA-MB-361 cell | CVCL_0620 | [366] |
| Half Maximal Inhibitory Concentration (IC50) | 35 nM | N.A. | N.A. | A498 cell | CVCL_1056 | [418] |
| Half Maximal Inhibitory Concentration (IC50) | 35 nM | N.A. | N.A. | OVCAR-3 cell | CVCL_0465 | [402] |
| Half Maximal Inhibitory Concentration (IC50) | 35 nM | N.A. | N.A. | L1210 cell | CVCL_0382 | [419] |
| Half Maximal Inhibitory Concentration (IC50) | 35 nM | N.A. | N.A. | SF-295 cell | CVCL_1690 | [337] |
| Half Maximal Inhibitory Concentration (IC50) | 36 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [420] |
| Half Maximal Inhibitory Concentration (IC50) | 37 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [421] |
| Half Maximal Inhibitory Concentration (IC50) | 37 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [418] |
| Half Maximal Inhibitory Concentration (IC50) | 37 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [422] |
| Half Maximal Inhibitory Concentration (IC50) | 40 nM | N.A. | N.A. | MES-SA cell | CVCL_1404 | [423] |
| Half Maximal Inhibitory Concentration (IC50) | 40 nM | N.A. | N.A. | ACHN cell | CVCL_1067 | [358] |
| Half Maximal Inhibitory Concentration (IC50) | 40 nM | N.A. | N.A. | MIA PaCa-2 cell | CVCL_0428 | [424] |
| Half Maximal Inhibitory Concentration (IC50) | 40 nM | N.A. | N.A. | MRC5 cell | CVCL_0440 | [425] |
| Half Maximal Inhibitory Concentration (IC50) | 40 nM | N.A. | N.A. | LXFL 529 cell | CVCL_D085 | [426] |
| Half Maximal Inhibitory Concentration (IC50) | 40 nM | N.A. | N.A. | B16-F10-luc2 cell | CVCL_5J17 | [427] |
| Half Maximal Inhibitory Concentration (IC50) | 40 nM | N.A. | N.A. | L1210 cell | CVCL_0382 | [428] |
| Half Maximal Inhibitory Concentration (IC50) | 40 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [429] |
| Half Maximal Inhibitory Concentration (IC50) | 40 nM | N.A. | N.A. | HEp-2 cell | CVCL_1906 | [408] |
| Half Maximal Inhibitory Concentration (IC50) | 40 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [430] |
| Half Maximal Inhibitory Concentration (IC50) | 40 nM | N.A. | N.A. | KB cell | CVCL_0372 | [411] |
| Half Maximal Inhibitory Concentration (IC50) | 40 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [430] |
| Half Maximal Inhibitory Concentration (IC50) | 40 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [431] |
| Half Maximal Inhibitory Concentration (IC50) | 40 nM | N.A. | N.A. | PC-3M cell | CVCL_9555 | [380] |
| Half Maximal Inhibitory Concentration (IC50) | 40 nM | N.A. | N.A. | SF268 cell | CVCL_1689 | [361] |
| Half Maximal Inhibitory Concentration (IC50) | 40 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [432] |
| Half Maximal Inhibitory Concentration (IC50) | 41 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [393] |
| Half Maximal Inhibitory Concentration (IC50) | 43 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [433] |
| Half Maximal Inhibitory Concentration (IC50) | 44 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [434] |
| Half Maximal Inhibitory Concentration (IC50) | 46 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [401] |
| Half Maximal Inhibitory Concentration (IC50) | 46 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [435] |
| Half Maximal Inhibitory Concentration (IC50) | 49 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [436] |
| Half Maximal Inhibitory Concentration (IC50) | 49.8 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [327] |
| Half Maximal Inhibitory Concentration (IC50) | 50 nM | N.A. | N.A. | MOLT-4 cell | CVCL_0013 | [437] |
| Half Maximal Inhibitory Concentration (IC50) | 50 nM | N.A. | N.A. | MIA PaCa-2 cell | CVCL_0428 | [438] |
| Half Maximal Inhibitory Concentration (IC50) | 50 nM | N.A. | N.A. | MT4 cell | CVCL_2632 | [437] |
| Half Maximal Inhibitory Concentration (IC50) | 50 nM | N.A. | N.A. | NIH3T3 cell | CVCL_0594 | [439] |
| Half Maximal Inhibitory Concentration (IC50) | 50 nM | N.A. | N.A. | CCRF-SB cell | CVCL_1860 | [437] |
| Half Maximal Inhibitory Concentration (IC50) | 50 nM | N.A. | N.A. | L1210 cell | CVCL_0382 | [384] |
| Half Maximal Inhibitory Concentration (IC50) | 50 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [440] |
| Half Maximal Inhibitory Concentration (IC50) | 50 nM | N.A. | N.A. | C8166 cell | CVCL_1099 | [437] |
| Half Maximal Inhibitory Concentration (IC50) | 50 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [408] |
| Half Maximal Inhibitory Concentration (IC50) | 50 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [424] |
| Half Maximal Inhibitory Concentration (IC50) | 50 nM | N.A. | N.A. | Caco-2 cell | CVCL_0025 | [441] |
| Half Maximal Inhibitory Concentration (IC50) | 50 nM | N.A. | N.A. | U-251MG cell | CVCL_0021 | [442] |
| Half Maximal Inhibitory Concentration (IC50) | 50 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [443] |
| Half Maximal Inhibitory Concentration (IC50) | 50 nM | N.A. | N.A. | MV4-11 cell | CVCL_0064 | [444] |
| Half Maximal Inhibitory Concentration (IC50) | 50 nM | N.A. | N.A. | THP-1 cell | CVCL_0006 | [445] |
| Half Maximal Inhibitory Concentration (IC50) | 51 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [446] |
| Half Maximal Inhibitory Concentration (IC50) | 52 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [447] |
| Half Maximal Inhibitory Concentration (IC50) | 52 nM | N.A. | N.A. | U-251MG cell | CVCL_0021 | [376] |
| Half Maximal Inhibitory Concentration (IC50) | 52.8 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [369] |
| Half Maximal Inhibitory Concentration (IC50) | 53 nM | N.A. | N.A. | WiDr cell | CVCL_2760 | [402] |
| Half Maximal Inhibitory Concentration (IC50) | 53.3 nM | N.A. | N.A. | L02 cell | CVCL_6926 | [448] |
| Half Maximal Inhibitory Concentration (IC50) | 56 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [449] |
| Half Maximal Inhibitory Concentration (IC50) | 56.4 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [369] |
| Half Maximal Inhibitory Concentration (IC50) | 57 nM | N.A. | N.A. | LoVo cell | CVCL_0399 | [450] |
| Half Maximal Inhibitory Concentration (IC50) | 59 nM | N.A. | N.A. | HEp-2 cell | CVCL_1906 | [342] |
| Half Maximal Inhibitory Concentration (IC50) | 60 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [451] |
| Half Maximal Inhibitory Concentration (IC50) | 60 nM | N.A. | N.A. | RXF 944 cell | CVCL_D127 | [364] |
| Half Maximal Inhibitory Concentration (IC50) | 60 nM | N.A. | N.A. | CHO cell | CVCL_0213 | [452] |
| Half Maximal Inhibitory Concentration (IC50) | 60 nM | N.A. | N.A. | NCI-H187 cell | CVCL_1501 | [453] |
| Half Maximal Inhibitory Concentration (IC50) | 60 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [454] |
| Half Maximal Inhibitory Concentration (IC50) | 60 nM | N.A. | N.A. | HCT-8 cell | CVCL_2478 | [455] |
| Half Maximal Inhibitory Concentration (IC50) | 60 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [456] |
| Half Maximal Inhibitory Concentration (IC50) | 60 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [405] |
| Half Maximal Inhibitory Concentration (IC50) | 60 nM | N.A. | N.A. | SK-MEL-28 cell | CVCL_0526 | [358] |
| Half Maximal Inhibitory Concentration (IC50) | 60 nM | N.A. | N.A. | SW480 cell | CVCL_0546 | [457] |
| Half Maximal Inhibitory Concentration (IC50) | 60 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [458] |
| Half Maximal Inhibitory Concentration (IC50) | 60 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [459] |
| Half Maximal Inhibitory Concentration (IC50) | 61 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [335] |
| Half Maximal Inhibitory Concentration (IC50) | 61.8 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [327] |
| Half Maximal Inhibitory Concentration (IC50) | 64 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [342] |
| Half Maximal Inhibitory Concentration (IC50) | 68 nM | N.A. | N.A. | P388/S cell | CVCL_D640 | [460] |
| Half Maximal Inhibitory Concentration (IC50) | 69 nM | N.A. | N.A. | L1210 cell | CVCL_0382 | [461] |
| Half Maximal Inhibitory Concentration (IC50) | 70 nM | N.A. | N.A. | SK-MEL-2 cell | CVCL_0069 | [462] |
| Half Maximal Inhibitory Concentration (IC50) | 70 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [438] |
| Half Maximal Inhibitory Concentration (IC50) | 70 nM | N.A. | N.A. | SNU-638 cell | CVCL_0102 | [463] |
| Half Maximal Inhibitory Concentration (IC50) | 70 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [332] |
| Half Maximal Inhibitory Concentration (IC50) | 70 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [464] |
| Half Maximal Inhibitory Concentration (IC50) | 70 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [465] |
| Half Maximal Inhibitory Concentration (IC50) | 70 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [424] |
| Half Maximal Inhibitory Concentration (IC50) | 71 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [466] |
| Half Maximal Inhibitory Concentration (IC50) | 75.9 nM | N.A. | N.A. | TRAMP-C1A cell | CVCL_H593 | [412] |
| Half Maximal Inhibitory Concentration (IC50) | 76 nM | N.A. | N.A. | SK-UT-1 cell | CVCL_0533 | [467] |
| Half Maximal Inhibitory Concentration (IC50) | 77.6 nM | N.A. | N.A. | LNCaP cell | CVCL_0395 | [468] |
| Half Maximal Inhibitory Concentration (IC50) | 78 nM | N.A. | N.A. | Jurkat cell | CVCL_0065 | [469] |
| Half Maximal Inhibitory Concentration (IC50) | 78 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [344] |
| Half Maximal Inhibitory Concentration (IC50) | 79.4 nM | N.A. | N.A. | J774 cell | CVCL_4692 | [470] |
| Half Maximal Inhibitory Concentration (IC50) | 80 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [471] |
| Half Maximal Inhibitory Concentration (IC50) | 80 nM | N.A. | N.A. | CHO cell | CVCL_0213 | [439] |
| Half Maximal Inhibitory Concentration (IC50) | 80 nM | N.A. | N.A. | L1210 cell | CVCL_0382 | [370] |
| Half Maximal Inhibitory Concentration (IC50) | 80 nM | N.A. | N.A. | NCI-H187 cell | CVCL_1501 | [472] |
| Half Maximal Inhibitory Concentration (IC50) | 80 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [473] |
| Half Maximal Inhibitory Concentration (IC50) | 80 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [409] |
| Half Maximal Inhibitory Concentration (IC50) | 80 nM | N.A. | N.A. | LoVo cell | CVCL_0399 | [351] |
| Half Maximal Inhibitory Concentration (IC50) | 81 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [474] |
| Half Maximal Inhibitory Concentration (IC50) | 82 nM | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [475] |
| Half Maximal Inhibitory Concentration (IC50) | 82 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [476] |
| Half Maximal Inhibitory Concentration (IC50) | 90 nM | N.A. | N.A. | Jurkat cell | CVCL_0065 | [477] |
| Half Maximal Inhibitory Concentration (IC50) | 90 nM | N.A. | N.A. | G-361 cell | CVCL_1220 | [323] |
| Half Maximal Inhibitory Concentration (IC50) | 90 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [478] |
| Half Maximal Inhibitory Concentration (IC50) | 90 nM | N.A. | N.A. | OVCAR-8 cell | CVCL_1629 | [388] |
| Half Maximal Inhibitory Concentration (IC50) | 90 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [479] |
| Half Maximal Inhibitory Concentration (IC50) | 90 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [480] |
| Half Maximal Inhibitory Concentration (IC50) | 90 nM | N.A. | N.A. | U-251MG cell | CVCL_0021 | [481] |
| Half Maximal Inhibitory Concentration (IC50) | 91.2 nM | N.A. | N.A. | SF268 cell | CVCL_1689 | [482] |
| Half Maximal Inhibitory Concentration (IC50) | 92 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [483] |
| Half Maximal Inhibitory Concentration (IC50) | 92 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [484] |
| Half Maximal Inhibitory Concentration (IC50) | 93.33 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [485] |
| Half Maximal Inhibitory Concentration (IC50) | 95.6 nM | N.A. | N.A. | LoVo cell | CVCL_0399 | [486] |
| Half Maximal Inhibitory Concentration (IC50) | 97 nM | N.A. | N.A. | SK-MEL-2 cell | CVCL_0069 | [487] |
| Half Maximal Inhibitory Concentration (IC50) | 100 nM | N.A. | N.A. | UO-31 cell | CVCL_1911 | [418] |
| Half Maximal Inhibitory Concentration (IC50) | 100 nM | N.A. | N.A. | MRC5 cell | CVCL_0440 | [404] |
| Half Maximal Inhibitory Concentration (IC50) | 100 nM | N.A. | N.A. | HMEC-1 cell | CVCL_0307 | [488] |
| Half Maximal Inhibitory Concentration (IC50) | <100 nM | N.A. | N.A. | B16 cell | CVCL_F936 | [489] |
| Half Maximal Inhibitory Concentration (IC50) | 100 nM | N.A. | N.A. | B16 cell | CVCL_F936 | [490] |
| Half Maximal Inhibitory Concentration (IC50) | 100 nM | N.A. | N.A. | NCI-H187 cell | CVCL_1501 | [491] |
| Half Maximal Inhibitory Concentration (IC50) | 100 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [492] |
| Half Maximal Inhibitory Concentration (IC50) | 100 nM | N.A. | N.A. | T-47D cell | CVCL_0553 | [467] |
| Half Maximal Inhibitory Concentration (IC50) | 100 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [493] |
| Half Maximal Inhibitory Concentration (IC50) | 100 nM | N.A. | N.A. | KB cell | CVCL_0372 | [493] |
| Half Maximal Inhibitory Concentration (IC50) | 100 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [492] |
| Half Maximal Inhibitory Concentration (IC50) | 100 nM | N.A. | N.A. | Ishikawa cell | CVCL_2529 | [494] |
| Half Maximal Inhibitory Concentration (IC50) | 100 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [492] |
| Half Maximal Inhibitory Concentration (IC50) | 100 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [495] |
| Half Maximal Inhibitory Concentration (IC50) | 100 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [496] |
| Half Maximal Inhibitory Concentration (IC50) | 100 nM | N.A. | N.A. | DLD-1 cell | CVCL_0248 | [418] |
| Half Maximal Inhibitory Concentration (IC50) | 100 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [497] |
| Half Maximal Inhibitory Concentration (IC50) | 100 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [418] |
| Half Maximal Inhibitory Concentration (IC50) | 100 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [498] |
| Half Maximal Inhibitory Concentration (IC50) | 109 nM | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [499] |
| Half Maximal Inhibitory Concentration (IC50) | 109.65 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [482] |
| Half Maximal Inhibitory Concentration (IC50) | 110 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [500] |
| Half Maximal Inhibitory Concentration (IC50) | 110 nM | N.A. | N.A. | Jurkat cell | CVCL_0065 | [443] |
| Half Maximal Inhibitory Concentration (IC50) | 110 nM | N.A. | N.A. | SW620 cell | CVCL_0547 | [501] |
| Half Maximal Inhibitory Concentration (IC50) | 110 nM | N.A. | N.A. | BT-549 cell | CVCL_1092 | [467] |
| Half Maximal Inhibitory Concentration (IC50) | 112 nM | N.A. | N.A. | UACC-375 cell | CVCL_M457 | [419] |
| Half Maximal Inhibitory Concentration (IC50) | 114 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [502] |
| Half Maximal Inhibitory Concentration (IC50) | 117 nM | N.A. | N.A. | SK-MEL-2 cell | CVCL_0069 | [449] |
| Half Maximal Inhibitory Concentration (IC50) | 120 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [503] |
| Half Maximal Inhibitory Concentration (IC50) | 120 nM | N.A. | N.A. | MRC5 cell | CVCL_0440 | [504] |
| Half Maximal Inhibitory Concentration (IC50) | 120 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [457] |
| Half Maximal Inhibitory Concentration (IC50) | 120 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [505] |
| Half Maximal Inhibitory Concentration (IC50) | 120 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [358] |
| Half Maximal Inhibitory Concentration (IC50) | 120 nM | N.A. | N.A. | HeLa S3 cell | CVCL_0058 | [506] |
| Half Maximal Inhibitory Concentration (IC50) | 120 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [428] |
| Half Maximal Inhibitory Concentration (IC50) | 120 nM | N.A. | N.A. | SW620 cell | CVCL_0547 | [507] |
| Half Maximal Inhibitory Concentration (IC50) | 120 nM | N.A. | N.A. | LoVo cell | CVCL_0399 | [508] |
| Half Maximal Inhibitory Concentration (IC50) | 120 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [384] |
| Half Maximal Inhibitory Concentration (IC50) | 123 nM | N.A. | N.A. | P388/ADR cell | CVCL_IZ75 | [509] |
| Half Maximal Inhibitory Concentration (IC50) | 124 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [414] |
| Half Maximal Inhibitory Concentration (IC50) | 125.89 nM | N.A. | N.A. | SHP-77 cell | CVCL_1693 | [415] |
| Half Maximal Inhibitory Concentration (IC50) | 130 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [510] |
| Half Maximal Inhibitory Concentration (IC50) | 130 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [473] |
| Half Maximal Inhibitory Concentration (IC50) | 130 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [511] |
| Half Maximal Inhibitory Concentration (IC50) | 130 nM | N.A. | N.A. | KB cell | CVCL_0372 | [512] |
| Half Maximal Inhibitory Concentration (IC50) | 130 nM | N.A. | N.A. | SGC-7901 cell | CVCL_0520 | [513] |
| Half Maximal Inhibitory Concentration (IC50) | 130 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [514] |
| Half Maximal Inhibitory Concentration (IC50) | 130 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [515] |
| Half Maximal Inhibitory Concentration (IC50) | 137 nM | N.A. | N.A. | NCI-H69 cell | CVCL_1579 | [399] |
| Half Maximal Inhibitory Concentration (IC50) | 140 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [383] |
| Half Maximal Inhibitory Concentration (IC50) | 140 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [443] |
| Half Maximal Inhibitory Concentration (IC50) | 142 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [516] |
| Half Maximal Inhibitory Concentration (IC50) | 150 nM | N.A. | N.A. | MIA PaCa-2 cell | CVCL_0428 | [517] |
| Half Maximal Inhibitory Concentration (IC50) | 150 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [518] |
| Half Maximal Inhibitory Concentration (IC50) | 150 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [462] |
| Half Maximal Inhibitory Concentration (IC50) | 150 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [519] |
| Half Maximal Inhibitory Concentration (IC50) | 150 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [520] |
| Half Maximal Inhibitory Concentration (IC50) | 150 nM | N.A. | N.A. | LoVo cell | CVCL_0399 | [521] |
| Half Maximal Inhibitory Concentration (IC50) | 150 nM | N.A. | N.A. | MDA-MB-435 cell | CVCL_0417 | [522] |
| Half Maximal Inhibitory Concentration (IC50) | 153.95 nM | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [523] |
| Half Maximal Inhibitory Concentration (IC50) | 160 nM | N.A. | N.A. | MES-SA cell | CVCL_1404 | [524] |
| Half Maximal Inhibitory Concentration (IC50) | 160 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [525] |
| Half Maximal Inhibitory Concentration (IC50) | 160 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [526] |
| Half Maximal Inhibitory Concentration (IC50) | 160 nM | N.A. | N.A. | U-937/GTB cell | CVCL_U631 | [527] |
| Half Maximal Inhibitory Concentration (IC50) | 160 nM | N.A. | N.A. | XF498 cell | CVCL_8928 | [332] |
| Half Maximal Inhibitory Concentration (IC50) | 160 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [528] |
| Half Maximal Inhibitory Concentration (IC50) | 160 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [529] |
| Half Maximal Inhibitory Concentration (IC50) | 161 nM | N.A. | N.A. | L929 cell | CVCL_0462 | [530] |
| Half Maximal Inhibitory Concentration (IC50) | 164 nM | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [449] |
| Half Maximal Inhibitory Concentration (IC50) | 166 nM | N.A. | N.A. | BALB/3T3 cell | CVCL_0184 | [531] |
| Half Maximal Inhibitory Concentration (IC50) | 168 nM | N.A. | N.A. | MKN45 cell | CVCL_0434 | [532] |
| Half Maximal Inhibitory Concentration (IC50) | 170 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [533] |
| Half Maximal Inhibitory Concentration (IC50) | 170 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [484] |
| Half Maximal Inhibitory Concentration (IC50) | 170 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [534] |
| Half Maximal Inhibitory Concentration (IC50) | 170 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [535] |
| Half Maximal Inhibitory Concentration (IC50) | 170 nM | N.A. | N.A. | LoVo cell | CVCL_0399 | [536] |
| Half Maximal Inhibitory Concentration (IC50) | 170 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [537] |
| Half Maximal Inhibitory Concentration (IC50) | 170 nM | N.A. | N.A. | U-251MG cell | CVCL_0021 | [446] |
| Half Maximal Inhibitory Concentration (IC50) | 178 nM | N.A. | N.A. | SW620 cell | CVCL_0547 | [538] |
| Half Maximal Inhibitory Concentration (IC50) | 179 nM | N.A. | N.A. | L1210 cell | CVCL_0382 | [539] |
| Half Maximal Inhibitory Concentration (IC50) | 180 nM | N.A. | N.A. | B16 cell | CVCL_F936 | [540] |
| Half Maximal Inhibitory Concentration (IC50) | 180 nM | N.A. | N.A. | JeKo-1 cell | CVCL_1865 | [541] |
| Half Maximal Inhibitory Concentration (IC50) | 180 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [542] |
| Half Maximal Inhibitory Concentration (IC50) | 180 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [433] |
| Half Maximal Inhibitory Concentration (IC50) | 182 nM | N.A. | N.A. | L1210 cell | CVCL_0382 | [543] |
| Half Maximal Inhibitory Concentration (IC50) | 190 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [544] |
| Half Maximal Inhibitory Concentration (IC50) | 190 nM | N.A. | N.A. | NCI-H292 cell | CVCL_0455 | [545] |
| Half Maximal Inhibitory Concentration (IC50) | 190 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [546] |
| Half Maximal Inhibitory Concentration (IC50) | 190 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [547] |
| Half Maximal Inhibitory Concentration (IC50) | 190 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [548] |
| Half Maximal Inhibitory Concentration (IC50) | 190 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [400] |
| Half Maximal Inhibitory Concentration (IC50) | 190 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [545] |
| Half Maximal Inhibitory Concentration (IC50) | 199.53 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [415] |
| Half Maximal Inhibitory Concentration (IC50) | 200 nM | N.A. | N.A. | MOLT-4 cell | CVCL_0013 | [549] |
| Half Maximal Inhibitory Concentration (IC50) | 200 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [550] |
| Half Maximal Inhibitory Concentration (IC50) | 200 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [550] |
| Half Maximal Inhibitory Concentration (IC50) | 200 nM | N.A. | N.A. | B16 cell | CVCL_F936 | [551] |
| Half Maximal Inhibitory Concentration (IC50) | 200 nM | N.A. | N.A. | L1210 cell | CVCL_0382 | [423] |
| Half Maximal Inhibitory Concentration (IC50) | 200 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [552] |
| Half Maximal Inhibitory Concentration (IC50) | 200 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [520] |
| Half Maximal Inhibitory Concentration (IC50) | 200 nM | N.A. | N.A. | KB cell | CVCL_0372 | [553] |
| Half Maximal Inhibitory Concentration (IC50) | 200 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [554] |
| Half Maximal Inhibitory Concentration (IC50) | 200 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [555] |
| Half Maximal Inhibitory Concentration (IC50) | 200 nM | N.A. | N.A. | HuTu 80 cell | CVCL_1301 | [556] |
| Half Maximal Inhibitory Concentration (IC50) | 200 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [552] |
| Half Maximal Inhibitory Concentration (IC50) | 200 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [469] |
| Half Maximal Inhibitory Concentration (IC50) | 200 nM | N.A. | N.A. | PC-3M cell | CVCL_9555 | [557] |
| Half Maximal Inhibitory Concentration (IC50) | 200 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [497] |
| Half Maximal Inhibitory Concentration (IC50) | 202 nM | N.A. | N.A. | SK-BR-3 cell | CVCL_0033 | [371] |
| Half Maximal Inhibitory Concentration (IC50) | 211 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [558] |
| Half Maximal Inhibitory Concentration (IC50) | 220 nM | N.A. | N.A. | L929 cell | CVCL_0462 | [559] |
| Half Maximal Inhibitory Concentration (IC50) | 220 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [560] |
| Half Maximal Inhibitory Concentration (IC50) | 220 nM | N.A. | N.A. | KB cell | CVCL_0372 | [561] |
| Half Maximal Inhibitory Concentration (IC50) | 220 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [562] |
| Half Maximal Inhibitory Concentration (IC50) | 220 nM | N.A. | N.A. | SF268 cell | CVCL_1689 | [563] |
| Half Maximal Inhibitory Concentration (IC50) | 223 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [564] |
| Half Maximal Inhibitory Concentration (IC50) | 225 nM | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [487] |
| Half Maximal Inhibitory Concentration (IC50) | 230 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [565] |
| Half Maximal Inhibitory Concentration (IC50) | 230 nM | N.A. | N.A. | B16-F1 cell | CVCL_0158 | [371] |
| Half Maximal Inhibitory Concentration (IC50) | 230 nM | N.A. | N.A. | NCI-H187 cell | CVCL_1501 | [566] |
| Half Maximal Inhibitory Concentration (IC50) | 230 nM | N.A. | N.A. | NCI-H417 cell | CVCL_1602 | [567] |
| Half Maximal Inhibitory Concentration (IC50) | 230 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [568] |
| Half Maximal Inhibitory Concentration (IC50) | 230 nM | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [569] |
| Half Maximal Inhibitory Concentration (IC50) | 235 nM | N.A. | N.A. | HaCaT cell | CVCL_0038 | [570] |
| Half Maximal Inhibitory Concentration (IC50) | 240 nM | N.A. | N.A. | HBL-100 cell | CVCL_4362 | [517] |
| Half Maximal Inhibitory Concentration (IC50) | 240 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [546] |
| Half Maximal Inhibitory Concentration (IC50) | 240 nM | N.A. | N.A. | KB cell | CVCL_0372 | [472] |
| Half Maximal Inhibitory Concentration (IC50) | 240 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [325] |
| Half Maximal Inhibitory Concentration (IC50) | 240 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [435] |
| Half Maximal Inhibitory Concentration (IC50) | 240 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [571] |
| Half Maximal Inhibitory Concentration (IC50) | 240 nM | N.A. | N.A. | SW620 cell | CVCL_0547 | [439] |
| Half Maximal Inhibitory Concentration (IC50) | 240 nM | N.A. | N.A. | LoVo cell | CVCL_0399 | [572] |
| Half Maximal Inhibitory Concentration (IC50) | 240 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [548] |
| Half Maximal Inhibitory Concentration (IC50) | 250 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [573] |
| Half Maximal Inhibitory Concentration (IC50) | 250 nM | N.A. | N.A. | NCI-H187 cell | CVCL_1501 | [574] |
| Half Maximal Inhibitory Concentration (IC50) | 250 nM | N.A. | N.A. | T-47D cell | CVCL_0553 | [575] |
| Half Maximal Inhibitory Concentration (IC50) | 250 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [576] |
| Half Maximal Inhibitory Concentration (IC50) | 250 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [577] |
| Half Maximal Inhibitory Concentration (IC50) | 260 nM | N.A. | N.A. | COLO205 cell | CVCL_F402 | [379] |
| Half Maximal Inhibitory Concentration (IC50) | 260 nM | N.A. | N.A. | Ca9-22 cell | CVCL_1102 | [578] |
| Half Maximal Inhibitory Concentration (IC50) | 270 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [579] |
| Half Maximal Inhibitory Concentration (IC50) | 270 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [580] |
| Half Maximal Inhibitory Concentration (IC50) | 280 nM | N.A. | N.A. | V79 cell | CVCL_2234 | [380] |
| Half Maximal Inhibitory Concentration (IC50) | 280 nM | N.A. | N.A. | KB cell | CVCL_0372 | [574] |
| Half Maximal Inhibitory Concentration (IC50) | 280 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [368] |
| Half Maximal Inhibitory Concentration (IC50) | 280 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [581] |
| Half Maximal Inhibitory Concentration (IC50) | 280 nM | N.A. | N.A. | SK-BR-3 cell | CVCL_0033 | [582] |
| Half Maximal Inhibitory Concentration (IC50) | 290 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [342] |
| Half Maximal Inhibitory Concentration (IC50) | 290 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [583] |
| Half Maximal Inhibitory Concentration (IC50) | 300 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [584] |
| Half Maximal Inhibitory Concentration (IC50) | 300 nM | N.A. | N.A. | MIA PaCa-2 cell | CVCL_0428 | [584] |
| Half Maximal Inhibitory Concentration (IC50) | 300 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [584] |
| Half Maximal Inhibitory Concentration (IC50) | 300 nM | N.A. | N.A. | L2987 cell | CVCL_H586 | [585] |
| Half Maximal Inhibitory Concentration (IC50) | 300 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [586] |
| Half Maximal Inhibitory Concentration (IC50) | 300 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [587] |
| Half Maximal Inhibitory Concentration (IC50) | 300 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [588] |
| Half Maximal Inhibitory Concentration (IC50) | 300 nM | N.A. | N.A. | LoVo cell | CVCL_0399 | [589] |
| Half Maximal Inhibitory Concentration (IC50) | 300 nM | N.A. | N.A. | Daudi cell | CVCL_0008 | [590] |
| Half Maximal Inhibitory Concentration (IC50) | 300 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [522] |
| Half Maximal Inhibitory Concentration (IC50) | 300 nM | N.A. | N.A. | MDA-MB-435 cell | CVCL_0417 | [509] |
| Half Maximal Inhibitory Concentration (IC50) | 310 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [528] |
| Half Maximal Inhibitory Concentration (IC50) | 310 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [591] |
| Half Maximal Inhibitory Concentration (IC50) | 310 nM | N.A. | N.A. | LoVo cell | CVCL_0399 | [592] |
| Half Maximal Inhibitory Concentration (IC50) | 311 nM | N.A. | N.A. | SiHa cell | CVCL_0032 | [593] |
| Half Maximal Inhibitory Concentration (IC50) | 316.23 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [482] |
| Half Maximal Inhibitory Concentration (IC50) | 316.23 nM | N.A. | N.A. | BxPC-3 cell | CVCL_0186 | [482] |
| Half Maximal Inhibitory Concentration (IC50) | 316.23 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [415] |
| Half Maximal Inhibitory Concentration (IC50) | 319.3 nM | N.A. | N.A. | COR-L23 cell | CVCL_1139 | [391] |
| Half Maximal Inhibitory Concentration (IC50) | 320 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [569] |
| Half Maximal Inhibitory Concentration (IC50) | 320 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [440] |
| Half Maximal Inhibitory Concentration (IC50) | 320 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [594] |
| Half Maximal Inhibitory Concentration (IC50) | 320 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [595] |
| Half Maximal Inhibitory Concentration (IC50) | 320 nM | N.A. | N.A. | U-251MG cell | CVCL_0021 | [596] |
| Half Maximal Inhibitory Concentration (IC50) | 324 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [563] |
| Half Maximal Inhibitory Concentration (IC50) | 330 nM | N.A. | N.A. | KB cell | CVCL_0372 | [597] |
| Half Maximal Inhibitory Concentration (IC50) | 330 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [407] |
| Half Maximal Inhibitory Concentration (IC50) | 330 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [598] |
| Half Maximal Inhibitory Concentration (IC50) | 340 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [591] |
| Half Maximal Inhibitory Concentration (IC50) | 340 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [506] |
| Half Maximal Inhibitory Concentration (IC50) | 340 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [599] |
| Half Maximal Inhibitory Concentration (IC50) | 340 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [600] |
| Half Maximal Inhibitory Concentration (IC50) | 350 nM | N.A. | N.A. | NCI-H661 cell | CVCL_1577 | [601] |
| Half Maximal Inhibitory Concentration (IC50) | 350 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [473] |
| Half Maximal Inhibitory Concentration (IC50) | 350 nM | N.A. | N.A. | XF498 cell | CVCL_8928 | [401] |
| Half Maximal Inhibitory Concentration (IC50) | 350 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [602] |
| Half Maximal Inhibitory Concentration (IC50) | 357 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [339] |
| Half Maximal Inhibitory Concentration (IC50) | 360 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [603] |
| Half Maximal Inhibitory Concentration (IC50) | 360 nM | N.A. | N.A. | Huh-7 cell | CVCL_0336 | [604] |
| Half Maximal Inhibitory Concentration (IC50) | 360 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [605] |
| Half Maximal Inhibitory Concentration (IC50) | 360 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [606] |
| Half Maximal Inhibitory Concentration (IC50) | 370 nM | N.A. | N.A. | A431 cell | CVCL_0037 | [464] |
| Half Maximal Inhibitory Concentration (IC50) | 370 nM | N.A. | N.A. | L1210 cell | CVCL_0382 | [607] |
| Half Maximal Inhibitory Concentration (IC50) | 370 nM | N.A. | N.A. | SMMC-7721 cell | CVCL_0534 | [608] |
| Half Maximal Inhibitory Concentration (IC50) | 370 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [609] |
| Half Maximal Inhibitory Concentration (IC50) | 370 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [610] |
| Half Maximal Inhibitory Concentration (IC50) | 370 nM | N.A. | N.A. | KB 3-1 cell | CVCL_2088 | [611] |
| Half Maximal Inhibitory Concentration (IC50) | 370 nM | N.A. | N.A. | SK-BR-3 cell | CVCL_0033 | [572] |
| Half Maximal Inhibitory Concentration (IC50) | 380 nM | N.A. | N.A. | WI-38 VA13 cell | CVCL_2759 | [612] |
| Half Maximal Inhibitory Concentration (IC50) | 380 nM | N.A. | N.A. | NIH3T3 cell | CVCL_0594 | [439] |
| Half Maximal Inhibitory Concentration (IC50) | 380 nM | N.A. | N.A. | Karpas-299 cell | CVCL_1324 | [446] |
| Half Maximal Inhibitory Concentration (IC50) | 400 nM | N.A. | N.A. | ACHN cell | CVCL_1067 | [613] |
| Half Maximal Inhibitory Concentration (IC50) | 400 nM | N.A. | N.A. | COLO205 cell | CVCL_F402 | [614] |
| Half Maximal Inhibitory Concentration (IC50) | 400 nM | N.A. | N.A. | Farage cell | CVCL_0214 | [613] |
| Half Maximal Inhibitory Concentration (IC50) | 400 nM | N.A. | N.A. | L1210 cell | CVCL_0382 | [615] |
| Half Maximal Inhibitory Concentration (IC50) | 400 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [616] |
| Half Maximal Inhibitory Concentration (IC50) | 400 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [617] |
| Half Maximal Inhibitory Concentration (IC50) | 400 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [557] |
| Half Maximal Inhibitory Concentration (IC50) | 400 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [407] |
| Half Maximal Inhibitory Concentration (IC50) | 400 nM | N.A. | N.A. | MGC-803 cell | CVCL_5334 | [618] |
| Half Maximal Inhibitory Concentration (IC50) | 400 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [619] |
| Half Maximal Inhibitory Concentration (IC50) | 410 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [620] |
| Half Maximal Inhibitory Concentration (IC50) | 410 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [621] |
| Half Maximal Inhibitory Concentration (IC50) | 411 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [622] |
| Half Maximal Inhibitory Concentration (IC50) | 419.8 nM | N.A. | N.A. | NCI-N87 cell | CVCL_1603 | [372] |
| Half Maximal Inhibitory Concentration (IC50) | 420 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [623] |
| Half Maximal Inhibitory Concentration (IC50) | 420 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [624] |
| Half Maximal Inhibitory Concentration (IC50) | 420 nM | N.A. | N.A. | HEp-2 cell | CVCL_1906 | [365] |
| Half Maximal Inhibitory Concentration (IC50) | 420 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [624] |
| Half Maximal Inhibitory Concentration (IC50) | 420 nM | N.A. | N.A. | HCT-8 cell | CVCL_2478 | [625] |
| Half Maximal Inhibitory Concentration (IC50) | 420 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [626] |
| Half Maximal Inhibitory Concentration (IC50) | 424 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [496] |
| Half Maximal Inhibitory Concentration (IC50) | 430 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [627] |
| Half Maximal Inhibitory Concentration (IC50) | 430 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [628] |
| Half Maximal Inhibitory Concentration (IC50) | 450 nM | N.A. | N.A. | CH1 cell | CVCL_D177 | [629] |
| Half Maximal Inhibitory Concentration (IC50) | 450 nM | N.A. | N.A. | OVCAR-8 cell | CVCL_1629 | [378] |
| Half Maximal Inhibitory Concentration (IC50) | 450 nM | N.A. | N.A. | SF-295 cell | CVCL_1690 | [559] |
| Half Maximal Inhibitory Concentration (IC50) | 450 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [630] |
| Half Maximal Inhibitory Concentration (IC50) | 450 nM | N.A. | N.A. | SiHa cell | CVCL_0032 | [631] |
| Half Maximal Inhibitory Concentration (IC50) | 450 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [632] |
| Half Maximal Inhibitory Concentration (IC50) | 451 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [633] |
| Half Maximal Inhibitory Concentration (IC50) | 456 nM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [619] |
| Half Maximal Inhibitory Concentration (IC50) | 459 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [634] |
| Half Maximal Inhibitory Concentration (IC50) | 460 nM | N.A. | N.A. | HEK293 cell | CVCL_0045 | [635] |
| Half Maximal Inhibitory Concentration (IC50) | 460 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [562] |
| Half Maximal Inhibitory Concentration (IC50) | 467 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [636] |
| Half Maximal Inhibitory Concentration (IC50) | 470 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [637] |
| Half Maximal Inhibitory Concentration (IC50) | 477 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [638] |
| Half Maximal Inhibitory Concentration (IC50) | 480 nM | N.A. | N.A. | SF-295 cell | CVCL_1690 | [639] |
| Half Maximal Inhibitory Concentration (IC50) | 490 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [640] |
| Half Maximal Inhibitory Concentration (IC50) | 500 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [560] |
| Half Maximal Inhibitory Concentration (IC50) | 500 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [641] |
| Half Maximal Inhibitory Concentration (IC50) | 500 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [642] |
| Half Maximal Inhibitory Concentration (IC50) | 500 nM | N.A. | N.A. | KB cell | CVCL_0372 | [613] |
| Half Maximal Inhibitory Concentration (IC50) | 500 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [631] |
| Half Maximal Inhibitory Concentration (IC50) | 500 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [643] |
| Half Maximal Inhibitory Concentration (IC50) | 500 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [644] |
| Half Maximal Inhibitory Concentration (IC50) | 500 nM | N.A. | N.A. | ME-180 cell | CVCL_1401 | [645] |
| Half Maximal Inhibitory Concentration (IC50) | 500 nM | N.A. | N.A. | SH-SY5Y cell | CVCL_0019 | [646] |
| Half Maximal Inhibitory Concentration (IC50) | 501 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [633] |
| Half Maximal Inhibitory Concentration (IC50) | 510 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [647] |
| Half Maximal Inhibitory Concentration (IC50) | 510.6 nM | N.A. | N.A. | DLD-1 cell | CVCL_0248 | [648] |
| Half Maximal Inhibitory Concentration (IC50) | 520 nM | N.A. | N.A. | PC-12 cell | CVCL_S979 | [627] |
| Half Maximal Inhibitory Concentration (IC50) | 520 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [649] |
| Half Maximal Inhibitory Concentration (IC50) | 520 nM | N.A. | N.A. | Hs 578T cell | CVCL_0332 | [650] |
| Half Maximal Inhibitory Concentration (IC50) | 520 nM | N.A. | N.A. | Col2 cell | CVCL_D645 | [651] |
| Half Maximal Inhibitory Concentration (IC50) | 520 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [451] |
| Half Maximal Inhibitory Concentration (IC50) | 520.2 nM | N.A. | N.A. | LoVo cell | CVCL_0399 | [648] |
| Half Maximal Inhibitory Concentration (IC50) | 530 nM | N.A. | N.A. | RD cell | CVCL_1649 | [652] |
| Half Maximal Inhibitory Concentration (IC50) | 530 nM | N.A. | N.A. | BALB/3T3 cell | CVCL_0184 | [444] |
| Half Maximal Inhibitory Concentration (IC50) | 530 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [653] |
| Half Maximal Inhibitory Concentration (IC50) | 530 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [654] |
| Half Maximal Inhibitory Concentration (IC50) | 532 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [655] |
| Half Maximal Inhibitory Concentration (IC50) | 540 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [537] |
| Half Maximal Inhibitory Concentration (IC50) | 540 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [656] |
| Half Maximal Inhibitory Concentration (IC50) | 550 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [657] |
| Half Maximal Inhibitory Concentration (IC50) | 550 nM | N.A. | N.A. | KB cell | CVCL_0372 | [658] |
| Half Maximal Inhibitory Concentration (IC50) | 550 nM | N.A. | N.A. | MDA-MB-435 cell | CVCL_0417 | [509] |
| Half Maximal Inhibitory Concentration (IC50) | 560 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [659] |
| Half Maximal Inhibitory Concentration (IC50) | 570 nM | N.A. | N.A. | SMMC-7721 cell | CVCL_0534 | [660] |
| Half Maximal Inhibitory Concentration (IC50) | 570 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [661] |
| Half Maximal Inhibitory Concentration (IC50) | 580 nM | N.A. | N.A. | MCF-10A cell | CVCL_0598 | [572] |
| Half Maximal Inhibitory Concentration (IC50) | 580 nM | N.A. | N.A. | Bel-7402 cell | CVCL_5492 | [662] |
| Half Maximal Inhibitory Concentration (IC50) | 580 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [623] |
| Half Maximal Inhibitory Concentration (IC50) | 590 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [570] |
| Half Maximal Inhibitory Concentration (IC50) | 590 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [578] |
| Half Maximal Inhibitory Concentration (IC50) | 590 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [548] |
| Half Maximal Inhibitory Concentration (IC50) | 590 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [663] |
| Half Maximal Inhibitory Concentration (IC50) | 600 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [664] |
| Half Maximal Inhibitory Concentration (IC50) | 600 nM | N.A. | N.A. | A2780 cell | CVCL_0134 | [665] |
| Half Maximal Inhibitory Concentration (IC50) | 600 nM | N.A. | N.A. | NCI-H292 cell | CVCL_0455 | [666] |
| Half Maximal Inhibitory Concentration (IC50) | 600 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [667] |
| Half Maximal Inhibitory Concentration (IC50) | 600 nM | N.A. | N.A. | ZR-75-1 cell | CVCL_0588 | [641] |
| Half Maximal Inhibitory Concentration (IC50) | 600 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [668] |
| Half Maximal Inhibitory Concentration (IC50) | 600 nM | N.A. | N.A. | SK-MEL-28 cell | CVCL_0526 | [669] |
| Half Maximal Inhibitory Concentration (IC50) | 600 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [670] |
| Half Maximal Inhibitory Concentration (IC50) | 600 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [557] |
| Half Maximal Inhibitory Concentration (IC50) | 600 nM | N.A. | N.A. | U-251MG cell | CVCL_0021 | [329] |
| Half Maximal Inhibitory Concentration (IC50) | 606 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [655] |
| Half Maximal Inhibitory Concentration (IC50) | 610 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [649] |
| Half Maximal Inhibitory Concentration (IC50) | 620 nM | N.A. | N.A. | NCI-H358 cell | CVCL_1559 | [567] |
| Half Maximal Inhibitory Concentration (IC50) | 620 nM | N.A. | N.A. | WiDr cell | CVCL_2760 | [671] |
| Half Maximal Inhibitory Concentration (IC50) | 620 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [672] |
| Half Maximal Inhibitory Concentration (IC50) | 630 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [673] |
| Half Maximal Inhibitory Concentration (IC50) | 630 nM | N.A. | N.A. | PA-1 cell | CVCL_0479 | [517] |
| Half Maximal Inhibitory Concentration (IC50) | 630 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [674] |
| Half Maximal Inhibitory Concentration (IC50) | 630 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [578] |
| Half Maximal Inhibitory Concentration (IC50) | 650 nM | N.A. | N.A. | U2OS cell | CVCL_0042 | [675] |
| Half Maximal Inhibitory Concentration (IC50) | 650 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [676] |
| Half Maximal Inhibitory Concentration (IC50) | 650 nM | N.A. | N.A. | KB cell | CVCL_0372 | [613] |
| Half Maximal Inhibitory Concentration (IC50) | 650 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [677] |
| Half Maximal Inhibitory Concentration (IC50) | 660 nM | N.A. | N.A. | L929 cell | CVCL_0462 | [562] |
| Half Maximal Inhibitory Concentration (IC50) | 670 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [640] |
| Half Maximal Inhibitory Concentration (IC50) | 680 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [678] |
| Half Maximal Inhibitory Concentration (IC50) | 680 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [679] |
| Half Maximal Inhibitory Concentration (IC50) | 690 nM | N.A. | N.A. | NIH3T3 cell | CVCL_0594 | [439] |
| Half Maximal Inhibitory Concentration (IC50) | 690 nM | N.A. | N.A. | SNU-638 cell | CVCL_0102 | [651] |
| Half Maximal Inhibitory Concentration (IC50) | 690 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [680] |
| Half Maximal Inhibitory Concentration (IC50) | 693.1 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [448] |
| Half Maximal Inhibitory Concentration (IC50) | 699 nM | N.A. | N.A. | WI-38 VA13 cell | CVCL_2759 | [681] |
| Half Maximal Inhibitory Concentration (IC50) | 700 nM | N.A. | N.A. | ACHN cell | CVCL_1067 | [437] |
| Half Maximal Inhibitory Concentration (IC50) | 700 nM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [682] |
| Half Maximal Inhibitory Concentration (IC50) | 700 nM | N.A. | N.A. | COLO205 cell | CVCL_F402 | [683] |
| Half Maximal Inhibitory Concentration (IC50) | 700 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [684] |
| Half Maximal Inhibitory Concentration (IC50) | 700 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [685] |
| Half Maximal Inhibitory Concentration (IC50) | 700 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [686] |
| Half Maximal Inhibitory Concentration (IC50) | 710 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [687] |
| Half Maximal Inhibitory Concentration (IC50) | 720 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [639] |
| Half Maximal Inhibitory Concentration (IC50) | 720 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [688] |
| Half Maximal Inhibitory Concentration (IC50) | 730 nM | N.A. | N.A. | PANC-1 cell | CVCL_0480 | [689] |
| Half Maximal Inhibitory Concentration (IC50) | 730 nM | N.A. | N.A. | SMMC-7721 cell | CVCL_0534 | [618] |
| Half Maximal Inhibitory Concentration (IC50) | 740 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [603] |
| Half Maximal Inhibitory Concentration (IC50) | 740 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [690] |
| Half Maximal Inhibitory Concentration (IC50) | 750 nM | N.A. | N.A. | HEK-293T cell | CVCL_0063 | [630] |
| Half Maximal Inhibitory Concentration (IC50) | 750 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [464] |
| Half Maximal Inhibitory Concentration (IC50) | 750 nM | N.A. | N.A. | SF-295 cell | CVCL_1690 | [691] |
| Half Maximal Inhibitory Concentration (IC50) | 750 nM | N.A. | N.A. | A-375 cell | CVCL_0132 | [659] |
| Half Maximal Inhibitory Concentration (IC50) | 760 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [692] |
| Half Maximal Inhibitory Concentration (IC50) | 760 nM | N.A. | N.A. | LAMA-84 cell | CVCL_0388 | [693] |
| Half Maximal Inhibitory Concentration (IC50) | 770 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [694] |
| Half Maximal Inhibitory Concentration (IC50) | 770 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [695] |
| Half Maximal Inhibitory Concentration (IC50) | 770 nM | N.A. | N.A. | SW620 cell | CVCL_0547 | [696] |
| Half Maximal Inhibitory Concentration (IC50) | 800 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [668] |
| Half Maximal Inhibitory Concentration (IC50) | 800 nM | N.A. | N.A. | WI-38 cell | CVCL_0579 | [584] |
| Half Maximal Inhibitory Concentration (IC50) | 800 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [697] |
| Half Maximal Inhibitory Concentration (IC50) | 800 nM | N.A. | N.A. | HCT-8 cell | CVCL_2478 | [698] |
| Half Maximal Inhibitory Concentration (IC50) | 800 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [699] |
| Half Maximal Inhibitory Concentration (IC50) | 800 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [664] |
| Half Maximal Inhibitory Concentration (IC50) | 800 nM | N.A. | N.A. | SNB-19 cell | CVCL_0535 | [669] |
| Half Maximal Inhibitory Concentration (IC50) | 800 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [700] |
| Half Maximal Inhibitory Concentration (IC50) | 800 nM | N.A. | N.A. | U-937 cell | CVCL_0007 | [701] |
| Half Maximal Inhibitory Concentration (IC50) | 820 nM | N.A. | N.A. | M14 cell | CVCL_1395 | [413] |
| Half Maximal Inhibitory Concentration (IC50) | 820 nM | N.A. | N.A. | KB cell | CVCL_0372 | [473] |
| Half Maximal Inhibitory Concentration (IC50) | 820 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [676] |
| Half Maximal Inhibitory Concentration (IC50) | 820 nM | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [525] |
| Half Maximal Inhibitory Concentration (IC50) | 830 nM | N.A. | N.A. | P388 cell | CVCL_7222 | [702] |
| Half Maximal Inhibitory Concentration (IC50) | 830 nM | N.A. | N.A. | P388 cell | CVCL_7222 | [703] |
| Half Maximal Inhibitory Concentration (IC50) | 830 nM | N.A. | N.A. | MDA-MB-435 cell | CVCL_0417 | [704] |
| Half Maximal Inhibitory Concentration (IC50) | 840 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [705] |
| Half Maximal Inhibitory Concentration (IC50) | 840 nM | N.A. | N.A. | KB cell | CVCL_0372 | [706] |
| Half Maximal Inhibitory Concentration (IC50) | 850 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [707] |
| Half Maximal Inhibitory Concentration (IC50) | 850 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [708] |
| Half Maximal Inhibitory Concentration (IC50) | 850 nM | N.A. | N.A. | L02 cell | CVCL_6926 | [627] |
| Half Maximal Inhibitory Concentration (IC50) | 850 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [709] |
| Half Maximal Inhibitory Concentration (IC50) | 860 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [710] |
| Half Maximal Inhibitory Concentration (IC50) | 870 nM | N.A. | N.A. | NCI-H69 cell | CVCL_1579 | [567] |
| Half Maximal Inhibitory Concentration (IC50) | 870 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [690] |
| Half Maximal Inhibitory Concentration (IC50) | 870 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [711] |
| Half Maximal Inhibitory Concentration (IC50) | 880 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [712] |
| Half Maximal Inhibitory Concentration (IC50) | 880 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [690] |
| Half Maximal Inhibitory Concentration (IC50) | 880 nM | N.A. | N.A. | MDA-MB-435 cell | CVCL_0417 | [713] |
| Half Maximal Inhibitory Concentration (IC50) | 890 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [714] |
| Half Maximal Inhibitory Concentration (IC50) | 890 nM | N.A. | N.A. | SH-SY5Y cell | CVCL_0019 | [715] |
| Half Maximal Inhibitory Concentration (IC50) | 900 nM | N.A. | N.A. | BGC-823 cell | CVCL_3360 | [588] |
| Half Maximal Inhibitory Concentration (IC50) | 900 nM | N.A. | N.A. | L1210 cell | CVCL_0382 | [437] |
| Half Maximal Inhibitory Concentration (IC50) | 900 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [716] |
| Half Maximal Inhibitory Concentration (IC50) | 900 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [717] |
| Half Maximal Inhibitory Concentration (IC50) | 900 nM | N.A. | N.A. | KB cell | CVCL_0372 | [718] |
| Half Maximal Inhibitory Concentration (IC50) | 900 nM | N.A. | N.A. | Bel-7402 cell | CVCL_5492 | [719] |
| Half Maximal Inhibitory Concentration (IC50) | 900 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [720] |
| Half Maximal Inhibitory Concentration (IC50) | 900 nM | N.A. | N.A. | SGC-7901 cell | CVCL_0520 | [329] |
| Half Maximal Inhibitory Concentration (IC50) | 900 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [721] |
| Half Maximal Inhibitory Concentration (IC50) | 900 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [722] |
| Half Maximal Inhibitory Concentration (IC50) | 900 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [659] |
| Half Maximal Inhibitory Concentration (IC50) | 910 nM | N.A. | N.A. | KB cell | CVCL_0372 | [723] |
| Half Maximal Inhibitory Concentration (IC50) | 910 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [724] |
| Half Maximal Inhibitory Concentration (IC50) | 912 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [725] |
| Half Maximal Inhibitory Concentration (IC50) | 920 nM | N.A. | N.A. | Vero cell | CVCL_0059 | [561] |
| Half Maximal Inhibitory Concentration (IC50) | 920 nM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [480] |
| Half Maximal Inhibitory Concentration (IC50) | 930 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [674] |
| Half Maximal Inhibitory Concentration (IC50) | 930 nM | N.A. | N.A. | U-937 cell | CVCL_0007 | [669] |
| Half Maximal Inhibitory Concentration (IC50) | 940 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [726] |
| Half Maximal Inhibitory Concentration (IC50) | 940 nM | N.A. | N.A. | MDA-MB-435 cell | CVCL_0417 | [559] |
| Half Maximal Inhibitory Concentration (IC50) | 950 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [727] |
| Half Maximal Inhibitory Concentration (IC50) | 950 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [646] |
| Half Maximal Inhibitory Concentration (IC50) | 960 nM | N.A. | N.A. | MDA-MB-435 cell | CVCL_0417 | [386] |
| Half Maximal Inhibitory Concentration (IC50) | 970 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [728] |
| Half Maximal Inhibitory Concentration (IC50) | 1000 nM | N.A. | N.A. | DU145 cell | CVCL_0105 | [729] |
| Half Maximal Inhibitory Concentration (IC50) | 1000 nM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [730] |
| Half Maximal Inhibitory Concentration (IC50) | 1000 nM | N.A. | N.A. | HEK293 cell | CVCL_0045 | [731] |
| Half Maximal Inhibitory Concentration (IC50) | 1000 nM | N.A. | N.A. | HOP-62 cell | CVCL_1285 | [732] |
| Half Maximal Inhibitory Concentration (IC50) | 1000 nM | N.A. | N.A. | M14 cell | CVCL_1395 | [701] |
| Half Maximal Inhibitory Concentration (IC50) | 1000 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [733] |
| Half Maximal Inhibitory Concentration (IC50) | 1000 nM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [734] |
| Half Maximal Inhibitory Concentration (IC50) | <1000 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [735] |
| Half Maximal Inhibitory Concentration (IC50) | 1000 nM | N.A. | N.A. | HeLa cell | CVCL_0030 | [329] |
| Half Maximal Inhibitory Concentration (IC50) | <1000 nM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [736] |
| Half Maximal Inhibitory Concentration (IC50) | 1000 nM | N.A. | N.A. | LoVo cell | CVCL_0399 | [629] |
| Half Maximal Inhibitory Concentration (IC50) | 1000 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [590] |
| Half Maximal Inhibitory Concentration (IC50) | <1000 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [733] |
| Half Maximal Inhibitory Concentration (IC50) | 1000 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [737] |
| Half Maximal Inhibitory Concentration (IC50) | 1000 nM | N.A. | N.A. | A-375 cell | CVCL_0132 | [738] |
| Half Maximal Inhibitory Concentration (IC50) | 1.006 uM | N.A. | N.A. | HEK293 cell | CVCL_0045 | [739] |
| Half Maximal Inhibitory Concentration (IC50) | 1.02 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [740] |
| Half Maximal Inhibitory Concentration (IC50) | 1.02 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [741] |
| Half Maximal Inhibitory Concentration (IC50) | 1.05 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [566] |
| Half Maximal Inhibitory Concentration (IC50) | 1.05 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [742] |
| Half Maximal Inhibitory Concentration (IC50) | 1.054 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [743] |
| Half Maximal Inhibitory Concentration (IC50) | 1.06 uM | N.A. | N.A. | HEK293 cell | CVCL_0045 | [744] |
| Half Maximal Inhibitory Concentration (IC50) | 1.06 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [745] |
| Half Maximal Inhibitory Concentration (IC50) | 1.07 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [746] |
| Half Maximal Inhibitory Concentration (IC50) | 1.07 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [571] |
| Half Maximal Inhibitory Concentration (IC50) | 1.08 uM | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [727] |
| Half Maximal Inhibitory Concentration (IC50) | 1.089 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [747] |
| Half Maximal Inhibitory Concentration (IC50) | 1.1 uM | N.A. | N.A. | SK-N-SH cell | CVCL_0531 | [748] |
| Half Maximal Inhibitory Concentration (IC50) | 1.1 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [749] |
| Half Maximal Inhibitory Concentration (IC50) | 1.1 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [669] |
| Half Maximal Inhibitory Concentration (IC50) | 1.1 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [724] |
| Half Maximal Inhibitory Concentration (IC50) | 1.117 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [747] |
| Half Maximal Inhibitory Concentration (IC50) | 1.12 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [750] |
| Half Maximal Inhibitory Concentration (IC50) | 1.12 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [751] |
| Half Maximal Inhibitory Concentration (IC50) | 1.15 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [693] |
| Half Maximal Inhibitory Concentration (IC50) | 1.17 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [606] |
| Half Maximal Inhibitory Concentration (IC50) | 1.172 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [752] |
| Half Maximal Inhibitory Concentration (IC50) | 1.18 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [753] |
| Half Maximal Inhibitory Concentration (IC50) | 1.19 uM | N.A. | N.A. | MGC-803 cell | CVCL_5334 | [754] |
| Half Maximal Inhibitory Concentration (IC50) | 1.2 uM | N.A. | N.A. | MCF-10A cell | CVCL_0598 | [755] |
| Half Maximal Inhibitory Concentration (IC50) | 1.2 uM | N.A. | N.A. | Farage cell | CVCL_0214 | [437] |
| Half Maximal Inhibitory Concentration (IC50) | 1.2 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [756] |
| Half Maximal Inhibitory Concentration (IC50) | 1.2 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [735] |
| Half Maximal Inhibitory Concentration (IC50) | 1.2 uM | N.A. | N.A. | AGS cell | CVCL_0139 | [663] |
| Half Maximal Inhibitory Concentration (IC50) | 1.2 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [757] |
| Half Maximal Inhibitory Concentration (IC50) | 1.21 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [758] |
| Half Maximal Inhibitory Concentration (IC50) | 1.21 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [759] |
| Half Maximal Inhibitory Concentration (IC50) | 1.22 uM | N.A. | N.A. | P388 cell | CVCL_7222 | [423] |
| Half Maximal Inhibitory Concentration (IC50) | 1.23 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [707] |
| Half Maximal Inhibitory Concentration (IC50) | 1.246 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [760] |
| Half Maximal Inhibitory Concentration (IC50) | 1.258 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [743] |
| Half Maximal Inhibitory Concentration (IC50) | 1.27 uM | N.A. | N.A. | RD cell | CVCL_1649 | [383] |
| Half Maximal Inhibitory Concentration (IC50) | 1.3 uM | N.A. | N.A. | WI-38 cell | CVCL_0579 | [761] |
| Half Maximal Inhibitory Concentration (IC50) | 1.3 uM | N.A. | N.A. | Ca9-22 cell | CVCL_1102 | [762] |
| Half Maximal Inhibitory Concentration (IC50) | 1.3 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [329] |
| Half Maximal Inhibitory Concentration (IC50) | 1.303 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [763] |
| Half Maximal Inhibitory Concentration (IC50) | 1.32 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [764] |
| Half Maximal Inhibitory Concentration (IC50) | 1.33 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [765] |
| Half Maximal Inhibitory Concentration (IC50) | 1.35 uM | N.A. | N.A. | KB cell | CVCL_0372 | [453] |
| Half Maximal Inhibitory Concentration (IC50) | 1.39 uM | N.A. | N.A. | SMMC-7721 cell | CVCL_0534 | [692] |
| Half Maximal Inhibitory Concentration (IC50) | 1.39 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [688] |
| Half Maximal Inhibitory Concentration (IC50) | 1.4 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [766] |
| Half Maximal Inhibitory Concentration (IC50) | 1.4 uM | N.A. | N.A. | Vero cell | CVCL_0059 | [453] |
| Half Maximal Inhibitory Concentration (IC50) | 1.4 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [767] |
| Half Maximal Inhibitory Concentration (IC50) | 1.4 uM | N.A. | N.A. | KB cell | CVCL_0372 | [679] |
| Half Maximal Inhibitory Concentration (IC50) | 1.4 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [533] |
| Half Maximal Inhibitory Concentration (IC50) | 1.41 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [764] |
| Half Maximal Inhibitory Concentration (IC50) | 1.41 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [768] |
| Half Maximal Inhibitory Concentration (IC50) | 1.43 uM | N.A. | N.A. | A431 cell | CVCL_0037 | [135] |
| Half Maximal Inhibitory Concentration (IC50) | 1.43 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [135] |
| Half Maximal Inhibitory Concentration (IC50) | 1.43 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [769] |
| Half Maximal Inhibitory Concentration (IC50) | 1.46 uM | N.A. | N.A. | LoVo cell | CVCL_0399 | [770] |
| Half Maximal Inhibitory Concentration (IC50) | 1.47 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [771] |
| Half Maximal Inhibitory Concentration (IC50) | 1.5 uM | N.A. | N.A. | SiHa cell | CVCL_0032 | [641] |
| Half Maximal Inhibitory Concentration (IC50) | 1.51 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [772] |
| Half Maximal Inhibitory Concentration (IC50) | 1.54 uM | N.A. | N.A. | MCF-10A cell | CVCL_0598 | [650] |
| Half Maximal Inhibitory Concentration (IC50) | 1.54 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [773] |
| Half Maximal Inhibitory Concentration (IC50) | 1.54 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [707] |
| Half Maximal Inhibitory Concentration (IC50) | 1.546 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [774] |
| Half Maximal Inhibitory Concentration (IC50) | 1.56 uM | N.A. | N.A. | MES-SA cell | CVCL_1404 | [370] |
| Half Maximal Inhibitory Concentration (IC50) | 1.57 uM | N.A. | N.A. | DU145 cell | CVCL_0105 | [688] |
| Half Maximal Inhibitory Concentration (IC50) | 1.57 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [775] |
| Half Maximal Inhibitory Concentration (IC50) | 1.58 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [776] |
| Half Maximal Inhibitory Concentration (IC50) | 1.6 uM | N.A. | N.A. | LLC-PK1 cell | CVCL_0391 | [777] |
| Half Maximal Inhibitory Concentration (IC50) | 1.6 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [778] |
| Half Maximal Inhibitory Concentration (IC50) | 1.6 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [779] |
| Half Maximal Inhibitory Concentration (IC50) | 1.6 uM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [780] |
| Half Maximal Inhibitory Concentration (IC50) | 1.63 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [707] |
| Half Maximal Inhibitory Concentration (IC50) | 1.64 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [781] |
| Half Maximal Inhibitory Concentration (IC50) | 1.664 uM | N.A. | N.A. | PANC-1 cell | CVCL_0480 | [760] |
| Half Maximal Inhibitory Concentration (IC50) | 1.67 uM | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [728] |
| Half Maximal Inhibitory Concentration (IC50) | 1.67 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [386] |
| Half Maximal Inhibitory Concentration (IC50) | 1.67 uM | N.A. | N.A. | MDA-MB-435 cell | CVCL_0417 | [782] |
| Half Maximal Inhibitory Concentration (IC50) | 1.68 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [496] |
| Half Maximal Inhibitory Concentration (IC50) | 1.69 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [783] |
| Half Maximal Inhibitory Concentration (IC50) | 1.69 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [544] |
| Half Maximal Inhibitory Concentration (IC50) | 1.7 uM | N.A. | N.A. | Jurkat cell | CVCL_0065 | [784] |
| Half Maximal Inhibitory Concentration (IC50) | 1.7 uM | N.A. | N.A. | HEL 299 cell | CVCL_2480 | [437] |
| Half Maximal Inhibitory Concentration (IC50) | 1.7 uM | N.A. | N.A. | B16 cell | CVCL_F936 | [785] |
| Half Maximal Inhibitory Concentration (IC50) | 1.7 uM | N.A. | N.A. | L1210 cell | CVCL_0382 | [256] |
| Half Maximal Inhibitory Concentration (IC50) | 1.7 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [786] |
| Half Maximal Inhibitory Concentration (IC50) | 1.7 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [787] |
| Half Maximal Inhibitory Concentration (IC50) | 1.7 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [587] |
| Half Maximal Inhibitory Concentration (IC50) | 1.73 uM | N.A. | N.A. | PANC-1 cell | CVCL_0480 | [788] |
| Half Maximal Inhibitory Concentration (IC50) | 1.73 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [789] |
| Half Maximal Inhibitory Concentration (IC50) | 1.75 uM | N.A. | N.A. | DU145 cell | CVCL_0105 | [783] |
| Half Maximal Inhibitory Concentration (IC50) | 1.76 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [790] |
| Half Maximal Inhibitory Concentration (IC50) | 1.76 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [544] |
| Half Maximal Inhibitory Concentration (IC50) | 1.76 uM | N.A. | N.A. | HCT 15 cell | CVCL_0292 | [731] |
| Half Maximal Inhibitory Concentration (IC50) | 1.78 uM | N.A. | N.A. | HEp-2 cell | CVCL_1906 | [383] |
| Half Maximal Inhibitory Concentration (IC50) | 1.78 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [791] |
| Half Maximal Inhibitory Concentration (IC50) | 1.8 uM | N.A. | N.A. | Capan-1 cell | CVCL_0237 | [256] |
| Half Maximal Inhibitory Concentration (IC50) | 1.8 uM | N.A. | N.A. | KB cell | CVCL_0372 | [792] |
| Half Maximal Inhibitory Concentration (IC50) | 1.8 uM | N.A. | N.A. | THP-1 cell | CVCL_0006 | [793] |
| Half Maximal Inhibitory Concentration (IC50) | 1.807 uM | N.A. | N.A. | DU145 cell | CVCL_0105 | [763] |
| Half Maximal Inhibitory Concentration (IC50) | 1.819 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [794] |
| Half Maximal Inhibitory Concentration (IC50) | 1.82 uM | N.A. | N.A. | T-47D cell | CVCL_0553 | [731] |
| Half Maximal Inhibitory Concentration (IC50) | 1.83 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [795] |
| Half Maximal Inhibitory Concentration (IC50) | 1.86 uM | N.A. | N.A. | DU145 cell | CVCL_0105 | [764] |
| Half Maximal Inhibitory Concentration (IC50) | 1.88 uM | N.A. | N.A. | T-47D cell | CVCL_0553 | [727] |
| Half Maximal Inhibitory Concentration (IC50) | 1.9 uM | N.A. | N.A. | DU145 cell | CVCL_0105 | [700] |
| Half Maximal Inhibitory Concentration (IC50) | 1.9 uM | N.A. | N.A. | MIA PaCa-2 cell | CVCL_0428 | [796] |
| Half Maximal Inhibitory Concentration (IC50) | 1.9 uM | N.A. | N.A. | A2780 cell | CVCL_0134 | [797] |
| Half Maximal Inhibitory Concentration (IC50) | 1.9 uM | N.A. | N.A. | PC-14 cell | CVCL_1640 | [384] |
| Half Maximal Inhibitory Concentration (IC50) | 1.9 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [798] |
| Half Maximal Inhibitory Concentration (IC50) | 1.9 uM | N.A. | N.A. | T-47D cell | CVCL_0553 | [799] |
| Half Maximal Inhibitory Concentration (IC50) | 1.9 uM | N.A. | N.A. | SiHa cell | CVCL_0032 | [800] |
| Half Maximal Inhibitory Concentration (IC50) | 1.91 uM | N.A. | N.A. | DU145 cell | CVCL_0105 | [801] |
| Half Maximal Inhibitory Concentration (IC50) | 1.91 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [764] |
| Half Maximal Inhibitory Concentration (IC50) | 1.92 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [802] |
| Half Maximal Inhibitory Concentration (IC50) | 1.94 uM | N.A. | N.A. | DU145 cell | CVCL_0105 | [674] |
| Half Maximal Inhibitory Concentration (IC50) | 1.945 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [747] |
| Half Maximal Inhibitory Concentration (IC50) | 1.96 uM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [383] |
| Half Maximal Inhibitory Concentration (IC50) | 1.986 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [743] |
| Half Maximal Inhibitory Concentration (IC50) | 1.99 uM | N.A. | N.A. | MDA-MB-453 cell | CVCL_0418 | [801] |
| Half Maximal Inhibitory Concentration (IC50) | 1.995 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [763] |
| Half Maximal Inhibitory Concentration (IC50) | 2 uM | N.A. | N.A. | RD cell | CVCL_1649 | [803] |
| Half Maximal Inhibitory Concentration (IC50) | 2 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [804] |
| Half Maximal Inhibitory Concentration (IC50) | 2 uM | N.A. | N.A. | COLO205 cell | CVCL_F402 | [641] |
| Half Maximal Inhibitory Concentration (IC50) | 2 uM | N.A. | N.A. | Raji cell | CVCL_0511 | [590] |
| Half Maximal Inhibitory Concentration (IC50) | 2 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [805] |
| Half Maximal Inhibitory Concentration (IC50) | 2 uM | N.A. | N.A. | Bcap37 cell | CVCL_0164 | [766] |
| Half Maximal Inhibitory Concentration (IC50) | 2.02 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [661] |
| Half Maximal Inhibitory Concentration (IC50) | 2.03 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [719] |
| Half Maximal Inhibitory Concentration (IC50) | 2.04 uM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [806] |
| Half Maximal Inhibitory Concentration (IC50) | 2.05 uM | N.A. | N.A. | COS-7 cell | CVCL_0224 | [268] |
| Half Maximal Inhibitory Concentration (IC50) | 2.05 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [807] |
| Half Maximal Inhibitory Concentration (IC50) | 2.08 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [808] |
| Half Maximal Inhibitory Concentration (IC50) | 2.089 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [809] |
| Half Maximal Inhibitory Concentration (IC50) | 2.1 uM | N.A. | N.A. | HEK293 cell | CVCL_0045 | [652] |
| Half Maximal Inhibitory Concentration (IC50) | 2.1 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [810] |
| Half Maximal Inhibitory Concentration (IC50) | 2.1 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [811] |
| Half Maximal Inhibitory Concentration (IC50) | 2.1 uM | N.A. | N.A. | A-375 cell | CVCL_0132 | [812] |
| Half Maximal Inhibitory Concentration (IC50) | 2.12 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [721] |
| Half Maximal Inhibitory Concentration (IC50) | 2.18 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [789] |
| Half Maximal Inhibitory Concentration (IC50) | 2.2 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [550] |
| Half Maximal Inhibitory Concentration (IC50) | 2.2 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [813] |
| Half Maximal Inhibitory Concentration (IC50) | 2.21 uM | N.A. | N.A. | V79 cell | CVCL_2234 | [814] |
| Half Maximal Inhibitory Concentration (IC50) | 2.21 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [681] |
| Half Maximal Inhibitory Concentration (IC50) | 2.23 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [790] |
| Half Maximal Inhibitory Concentration (IC50) | 2.24 uM | N.A. | N.A. | SCC-25 cell | CVCL_1682 | [605] |
| Half Maximal Inhibitory Concentration (IC50) | 2.24 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [815] |
| Half Maximal Inhibitory Concentration (IC50) | 2.29 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [816] |
| Half Maximal Inhibitory Concentration (IC50) | 2.3 uM | N.A. | N.A. | IMR-32 cell | CVCL_0346 | [817] |
| Half Maximal Inhibitory Concentration (IC50) | 2.3 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [818] |
| Half Maximal Inhibitory Concentration (IC50) | 2.3 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [786] |
| Half Maximal Inhibitory Concentration (IC50) | 2.3 uM | N.A. | N.A. | SH-SY5Y cell | CVCL_0019 | [819] |
| Half Maximal Inhibitory Concentration (IC50) | 2.33 uM | N.A. | N.A. | BJ cell | CVCL_E483 | [707] |
| Half Maximal Inhibitory Concentration (IC50) | 2.36 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [820] |
| Half Maximal Inhibitory Concentration (IC50) | 2.4 uM | N.A. | N.A. | BEAS-2B cell | CVCL_0168 | [821] |
| Half Maximal Inhibitory Concentration (IC50) | 2.42 uM | N.A. | N.A. | SK-N-SH cell | CVCL_0531 | [564] |
| Half Maximal Inhibitory Concentration (IC50) | 2.43 uM | N.A. | N.A. | SW480 cell | CVCL_0546 | [822] |
| Half Maximal Inhibitory Concentration (IC50) | 2.44 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [579] |
| Half Maximal Inhibitory Concentration (IC50) | 2.45 uM | N.A. | N.A. | DU145 cell | CVCL_0105 | [810] |
| Half Maximal Inhibitory Concentration (IC50) | 2.5 uM | N.A. | N.A. | LNCaP C4-2 cell | CVCL_4782 | [823] |
| Half Maximal Inhibitory Concentration (IC50) | 2.5 uM | N.A. | N.A. | SMMC-7721 cell | CVCL_0534 | [256] |
| Half Maximal Inhibitory Concentration (IC50) | 2.5 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [824] |
| Half Maximal Inhibitory Concentration (IC50) | 2.56 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [825] |
| Half Maximal Inhibitory Concentration (IC50) | 2.57 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [826] |
| Half Maximal Inhibitory Concentration (IC50) | 2.58 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [593] |
| Half Maximal Inhibitory Concentration (IC50) | 2.6 uM | N.A. | N.A. | KB cell | CVCL_0372 | [827] |
| Half Maximal Inhibitory Concentration (IC50) | 2.62 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [828] |
| Half Maximal Inhibitory Concentration (IC50) | 2.63 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [776] |
| Half Maximal Inhibitory Concentration (IC50) | 2.67 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [829] |
| Half Maximal Inhibitory Concentration (IC50) | 2.7 uM | N.A. | N.A. | NIH3T3 cell | CVCL_0594 | [439] |
| Half Maximal Inhibitory Concentration (IC50) | 2.7 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [830] |
| Half Maximal Inhibitory Concentration (IC50) | 2.7 uM | N.A. | N.A. | Caco-2 cell | CVCL_0025 | [830] |
| Half Maximal Inhibitory Concentration (IC50) | 2.71 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [831] |
| Half Maximal Inhibitory Concentration (IC50) | 2.76 uM | N.A. | N.A. | LNCaP cell | CVCL_0395 | [832] |
| Half Maximal Inhibitory Concentration (IC50) | 2.78 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [833] |
| Half Maximal Inhibitory Concentration (IC50) | 2.78 uM | N.A. | N.A. | DLD-1 cell | CVCL_0248 | [711] |
| Half Maximal Inhibitory Concentration (IC50) | 2.8 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [834] |
| Half Maximal Inhibitory Concentration (IC50) | 2.8 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [818] |
| Half Maximal Inhibitory Concentration (IC50) | 2.81 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [835] |
| Half Maximal Inhibitory Concentration (IC50) | 2.81 uM | N.A. | N.A. | MDA-MB-468 cell | CVCL_0419 | [836] |
| Half Maximal Inhibitory Concentration (IC50) | 2.82 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [837] |
| Half Maximal Inhibitory Concentration (IC50) | 2.84 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [838] |
| Half Maximal Inhibitory Concentration (IC50) | 2.85 uM | N.A. | N.A. | SK-N-SH cell | CVCL_0531 | [839] |
| Half Maximal Inhibitory Concentration (IC50) | 2.85 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [840] |
| Half Maximal Inhibitory Concentration (IC50) | 2.87 uM | N.A. | N.A. | Bcap37 cell | CVCL_0164 | [841] |
| Half Maximal Inhibitory Concentration (IC50) | 2.947 uM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [614] |
| Half Maximal Inhibitory Concentration (IC50) | 2.96 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [842] |
| Half Maximal Inhibitory Concentration (IC50) | 2.98 uM | N.A. | N.A. | Raji cell | CVCL_0511 | [843] |
| Half Maximal Inhibitory Concentration (IC50) | 3 uM | N.A. | N.A. | NCI-H69 cell | CVCL_1579 | [590] |
| Half Maximal Inhibitory Concentration (IC50) | 3 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [844] |
| Half Maximal Inhibitory Concentration (IC50) | 3 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [845] |
| Half Maximal Inhibitory Concentration (IC50) | 3 uM | N.A. | N.A. | KB cell | CVCL_0372 | [792] |
| Half Maximal Inhibitory Concentration (IC50) | 3 uM | N.A. | N.A. | M-HeLa cell | CVCL_R965 | [761] |
| Half Maximal Inhibitory Concentration (IC50) | 3 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [844] |
| Half Maximal Inhibitory Concentration (IC50) | 3.1 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [605] |
| Half Maximal Inhibitory Concentration (IC50) | 3.13 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [846] |
| Half Maximal Inhibitory Concentration (IC50) | 3.137 uM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [847] |
| Half Maximal Inhibitory Concentration (IC50) | 3.14 uM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [826] |
| Half Maximal Inhibitory Concentration (IC50) | 3.16 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [807] |
| Half Maximal Inhibitory Concentration (IC50) | 3.16 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [848] |
| Half Maximal Inhibitory Concentration (IC50) | 3.2 uM | N.A. | N.A. | IMR-32 cell | CVCL_0346 | [800] |
| Half Maximal Inhibitory Concentration (IC50) | 3.2 uM | N.A. | N.A. | LNCaP C4-2 cell | CVCL_4782 | [849] |
| Half Maximal Inhibitory Concentration (IC50) | 3.22 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [850] |
| Half Maximal Inhibitory Concentration (IC50) | 3.24 uM | N.A. | N.A. | NIH3T3 cell | CVCL_0594 | [851] |
| Half Maximal Inhibitory Concentration (IC50) | 3.24 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [839] |
| Half Maximal Inhibitory Concentration (IC50) | 3.24 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [828] |
| Half Maximal Inhibitory Concentration (IC50) | 3.27 uM | N.A. | N.A. | DU145 cell | CVCL_0105 | [852] |
| Half Maximal Inhibitory Concentration (IC50) | 3.28 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [853] |
| Half Maximal Inhibitory Concentration (IC50) | 3.3 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [854] |
| Half Maximal Inhibitory Concentration (IC50) | 3.3 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [855] |
| Half Maximal Inhibitory Concentration (IC50) | 3.3 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [856] |
| Half Maximal Inhibitory Concentration (IC50) | 3.39 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [568] |
| Half Maximal Inhibitory Concentration (IC50) | 3.4 uM | N.A. | N.A. | U2OS cell | CVCL_0042 | [588] |
| Half Maximal Inhibitory Concentration (IC50) | 3.4 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [857] |
| Half Maximal Inhibitory Concentration (IC50) | 3.41 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [810] |
| Half Maximal Inhibitory Concentration (IC50) | 3.45 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [858] |
| Half Maximal Inhibitory Concentration (IC50) | 3.5 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [859] |
| Half Maximal Inhibitory Concentration (IC50) | 3.6 uM | N.A. | N.A. | Vero cell | CVCL_0059 | [860] |
| Half Maximal Inhibitory Concentration (IC50) | 3.6 uM | N.A. | N.A. | MGC-803 cell | CVCL_5334 | [699] |
| Half Maximal Inhibitory Concentration (IC50) | 3.61 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [861] |
| Half Maximal Inhibitory Concentration (IC50) | 3.69 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [840] |
| Half Maximal Inhibitory Concentration (IC50) | 3.7 uM | N.A. | N.A. | NIH3T3 cell | CVCL_0594 | [807] |
| Half Maximal Inhibitory Concentration (IC50) | 3.7 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [700] |
| Half Maximal Inhibitory Concentration (IC50) | 3.7 uM | N.A. | N.A. | BT-549 cell | CVCL_1092 | [777] |
| Half Maximal Inhibitory Concentration (IC50) | 3.76 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [862] |
| Half Maximal Inhibitory Concentration (IC50) | 3.8 uM | N.A. | N.A. | COLO205 cell | CVCL_F402 | [863] |
| Half Maximal Inhibitory Concentration (IC50) | 3.86 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [815] |
| Half Maximal Inhibitory Concentration (IC50) | 3.88 uM | N.A. | N.A. | MRC5 cell | CVCL_0440 | [864] |
| Half Maximal Inhibitory Concentration (IC50) | 3.884 uM | N.A. | N.A. | L1210 cell | CVCL_0382 | [419] |
| Half Maximal Inhibitory Concentration (IC50) | 3.89 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [865] |
| Half Maximal Inhibitory Concentration (IC50) | 3.9 uM | N.A. | N.A. | HEK293 cell | CVCL_0045 | [866] |
| Half Maximal Inhibitory Concentration (IC50) | 3.92 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [867] |
| Half Maximal Inhibitory Concentration (IC50) | 3.97 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [268] |
| Half Maximal Inhibitory Concentration (IC50) | 4 uM | N.A. | N.A. | SNU-1 cell | CVCL_0099 | [824] |
| Half Maximal Inhibitory Concentration (IC50) | 4 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [868] |
| Half Maximal Inhibitory Concentration (IC50) | 4.16 uM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [326] |
| Half Maximal Inhibitory Concentration (IC50) | 4.17 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [869] |
| Half Maximal Inhibitory Concentration (IC50) | 4.2 uM | N.A. | N.A. | MES-SA cell | CVCL_1404 | [423] |
| Half Maximal Inhibitory Concentration (IC50) | 4.3 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [857] |
| Half Maximal Inhibitory Concentration (IC50) | 4.4 uM | N.A. | N.A. | SF-295 cell | CVCL_1690 | [669] |
| Half Maximal Inhibitory Concentration (IC50) | 4.5 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [870] |
| Half Maximal Inhibitory Concentration (IC50) | 4.5 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [871] |
| Half Maximal Inhibitory Concentration (IC50) | 4.59 uM | N.A. | N.A. | A549/CDDP cell | CVCL_C5RT | [872] |
| Half Maximal Inhibitory Concentration (IC50) | 4.6 uM | N.A. | N.A. | LNCaP cell | CVCL_0395 | [856] |
| Half Maximal Inhibitory Concentration (IC50) | 4.6 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [873] |
| Half Maximal Inhibitory Concentration (IC50) | 4.78 uM | N.A. | N.A. | SK-MEL-2 cell | CVCL_0069 | [874] |
| Half Maximal Inhibitory Concentration (IC50) | 4.8 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [823] |
| Half Maximal Inhibitory Concentration (IC50) | 4.89 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [852] |
| Half Maximal Inhibitory Concentration (IC50) | 4.91 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [404] |
| Half Maximal Inhibitory Concentration (IC50) | 5 uM | N.A. | N.A. | Vero cell | CVCL_0059 | [351] |
| Half Maximal Inhibitory Concentration (IC50) | 5 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [875] |
| Half Maximal Inhibitory Concentration (IC50) | 5 uM | N.A. | N.A. | Bel-7402 cell | CVCL_5492 | [876] |
| Half Maximal Inhibitory Concentration (IC50) | 5 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [877] |
| Half Maximal Inhibitory Concentration (IC50) | 5.12 uM | N.A. | N.A. | Vero cell | CVCL_0059 | [878] |
| Half Maximal Inhibitory Concentration (IC50) | 5.17 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [879] |
| Half Maximal Inhibitory Concentration (IC50) | 5.18 uM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [880] |
| Half Maximal Inhibitory Concentration (IC50) | 5.2 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [881] |
| Half Maximal Inhibitory Concentration (IC50) | 5.23 uM | N.A. | N.A. | LNCaP cell | CVCL_0395 | [882] |
| Half Maximal Inhibitory Concentration (IC50) | 5.23 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [883] |
| Half Maximal Inhibitory Concentration (IC50) | 5.23 uM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [884] |
| Half Maximal Inhibitory Concentration (IC50) | 5.3 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [538] |
| Half Maximal Inhibitory Concentration (IC50) | 5.3 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [785] |
| Half Maximal Inhibitory Concentration (IC50) | 5.46 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [885] |
| Half Maximal Inhibitory Concentration (IC50) | 5.5 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [886] |
| Half Maximal Inhibitory Concentration (IC50) | 5.51 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [887] |
| Half Maximal Inhibitory Concentration (IC50) | 5.51 uM | N.A. | N.A. | A-375 cell | CVCL_0132 | [745] |
| Half Maximal Inhibitory Concentration (IC50) | 5.57 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [888] |
| Half Maximal Inhibitory Concentration (IC50) | 5.6 uM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [889] |
| Half Maximal Inhibitory Concentration (IC50) | 5.7 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [890] |
| Half Maximal Inhibitory Concentration (IC50) | 5.7 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [891] |
| Half Maximal Inhibitory Concentration (IC50) | 5.8 uM | N.A. | N.A. | KB cell | CVCL_0372 | [876] |
| Half Maximal Inhibitory Concentration (IC50) | 5.96 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [892] |
| Half Maximal Inhibitory Concentration (IC50) | 5.98 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [571] |
| Half Maximal Inhibitory Concentration (IC50) | 6 uM | N.A. | N.A. | DU145 cell | CVCL_0105 | [893] |
| Half Maximal Inhibitory Concentration (IC50) | 6 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [642] |
| Half Maximal Inhibitory Concentration (IC50) | 6 uM | N.A. | N.A. | LoVo cell | CVCL_0399 | [894] |
| Half Maximal Inhibitory Concentration (IC50) | 6.03 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [630] |
| Half Maximal Inhibitory Concentration (IC50) | 6.06 uM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [895] |
| Half Maximal Inhibitory Concentration (IC50) | 6.1 uM | N.A. | N.A. | A498 cell | CVCL_1056 | [896] |
| Half Maximal Inhibitory Concentration (IC50) | 6.1 uM | N.A. | N.A. | HEK293 cell | CVCL_0045 | [886] |
| Half Maximal Inhibitory Concentration (IC50) | 6.1 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [897] |
| Half Maximal Inhibitory Concentration (IC50) | 6.11 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [564] |
| Half Maximal Inhibitory Concentration (IC50) | 6.2 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [898] |
| Half Maximal Inhibitory Concentration (IC50) | <6.25 uM | N.A. | N.A. | hTERT-BJ cell | CVCL_6573 | [899] |
| Half Maximal Inhibitory Concentration (IC50) | <6.25 uM | N.A. | N.A. | TERT-RPE1 cell | CVCL_4388 | [899] |
| Half Maximal Inhibitory Concentration (IC50) | 6.3 uM | N.A. | N.A. | Vero cell | CVCL_0059 | [900] |
| Half Maximal Inhibitory Concentration (IC50) | 6.3 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [256] |
| Half Maximal Inhibitory Concentration (IC50) | 6.46 uM | N.A. | N.A. | LNCaP cell | CVCL_0395 | [630] |
| Half Maximal Inhibitory Concentration (IC50) | 6.6 uM | N.A. | N.A. | Vero cell | CVCL_0059 | [901] |
| Half Maximal Inhibitory Concentration (IC50) | 6.61 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [751] |
| Half Maximal Inhibitory Concentration (IC50) | 6.68 uM | N.A. | N.A. | WI-38 cell | CVCL_0579 | [879] |
| Half Maximal Inhibitory Concentration (IC50) | 6.7 uM | N.A. | N.A. | NCI-ADR-RES cell | CVCL_1452 | [902] |
| Half Maximal Inhibitory Concentration (IC50) | 6.7 uM | N.A. | N.A. | Caco-2 cell | CVCL_0025 | [669] |
| Half Maximal Inhibitory Concentration (IC50) | 6.75 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [903] |
| Half Maximal Inhibitory Concentration (IC50) | 6.75 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [904] |
| Half Maximal Inhibitory Concentration (IC50) | 6.8 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [905] |
| Half Maximal Inhibitory Concentration (IC50) | 7 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [868] |
| Half Maximal Inhibitory Concentration (IC50) | 7.01 uM | N.A. | N.A. | KB cell | CVCL_0372 | [906] |
| Half Maximal Inhibitory Concentration (IC50) | 7.1 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [785] |
| Half Maximal Inhibitory Concentration (IC50) | 7.2 uM | N.A. | N.A. | HEK293 cell | CVCL_0045 | [779] |
| Half Maximal Inhibitory Concentration (IC50) | 7.2 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [907] |
| Half Maximal Inhibitory Concentration (IC50) | 7.2 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [908] |
| Half Maximal Inhibitory Concentration (IC50) | 7.2 uM | N.A. | N.A. | A-375 cell | CVCL_0132 | [846] |
| Half Maximal Inhibitory Concentration (IC50) | 7.3 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [886] |
| Half Maximal Inhibitory Concentration (IC50) | 7.36 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [909] |
| Half Maximal Inhibitory Concentration (IC50) | 7.4 uM | N.A. | N.A. | Vero cell | CVCL_0059 | [910] |
| Half Maximal Inhibitory Concentration (IC50) | 7.47 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [911] |
| Half Maximal Inhibitory Concentration (IC50) | 7.6 uM | N.A. | N.A. | LoVo cell | CVCL_0399 | [450] |
| Half Maximal Inhibitory Concentration (IC50) | 7.88 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [912] |
| Half Maximal Inhibitory Concentration (IC50) | 8 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [913] |
| Half Maximal Inhibitory Concentration (IC50) | 8.1 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [914] |
| Half Maximal Inhibitory Concentration (IC50) | 8.14 uM | N.A. | N.A. | WISH cell | CVCL_1909 | [879] |
| Half Maximal Inhibitory Concentration (IC50) | 8.19 uM | N.A. | N.A. | DU145 cell | CVCL_0105 | [911] |
| Half Maximal Inhibitory Concentration (IC50) | 8.2 uM | N.A. | N.A. | P388/ADR cell | CVCL_IZ75 | [113] |
| Half Maximal Inhibitory Concentration (IC50) | 8.3 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [915] |
| Half Maximal Inhibitory Concentration (IC50) | 8.3 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [916] |
| Half Maximal Inhibitory Concentration (IC50) | 8.46 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [917] |
| Half Maximal Inhibitory Concentration (IC50) | 8.49 uM | N.A. | N.A. | IMR-32 cell | CVCL_0346 | [918] |
| Half Maximal Inhibitory Concentration (IC50) | 8.62 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [832] |
| Half Maximal Inhibitory Concentration (IC50) | 8.66 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [857] |
| Half Maximal Inhibitory Concentration (IC50) | 8.7 uM | N.A. | N.A. | HEp-2 cell | CVCL_1906 | [684] |
| Half Maximal Inhibitory Concentration (IC50) | 8.83 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [919] |
| Half Maximal Inhibitory Concentration (IC50) | 8.87 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [871] |
| Half Maximal Inhibitory Concentration (IC50) | 8.94 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [920] |
| Half Maximal Inhibitory Concentration (IC50) | 9 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [913] |
| Half Maximal Inhibitory Concentration (IC50) | 9.3 uM | N.A. | N.A. | NCI-ADR-RES cell | CVCL_1452 | [439] |
| Half Maximal Inhibitory Concentration (IC50) | 9.58 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [866] |
| Half Maximal Inhibitory Concentration (IC50) | 9.7 uM | N.A. | N.A. | LoVo cell | CVCL_0399 | [397] |
| Half Maximal Inhibitory Concentration (IC50) | 9.74 uM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [921] |
| Half Maximal Inhibitory Concentration (IC50) | 9.8 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [630] |
| Half Maximal Inhibitory Concentration (IC50) | 10 uM | N.A. | N.A. | NCI-ADR-RES cell | CVCL_1452 | [522] |
| Half Maximal Inhibitory Concentration (IC50) | 10 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [922] |
| Half Maximal Inhibitory Concentration (IC50) | >10 uM | N.A. | N.A. | T24 cell | CVCL_0554 | [351] |
| Half Maximal Inhibitory Concentration (IC50) | 10.02 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [923] |
| Half Maximal Inhibitory Concentration (IC50) | 10.15 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [924] |
| Half Maximal Inhibitory Concentration (IC50) | 10.4 uM | N.A. | N.A. | L02 cell | CVCL_6926 | [925] |
| Half Maximal Inhibitory Concentration (IC50) | 10.44 uM | N.A. | N.A. | L02 cell | CVCL_6926 | [926] |
| Half Maximal Inhibitory Concentration (IC50) | 10.6 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [927] |
| Half Maximal Inhibitory Concentration (IC50) | 10.86 uM | N.A. | N.A. | DU145 cell | CVCL_0105 | [832] |
| Half Maximal Inhibitory Concentration (IC50) | 10.9 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [928] |
| Half Maximal Inhibitory Concentration (IC50) | 11 uM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [785] |
| Half Maximal Inhibitory Concentration (IC50) | 11.62 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [929] |
| Half Maximal Inhibitory Concentration (IC50) | 11.67 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [882] |
| Half Maximal Inhibitory Concentration (IC50) | 12 uM | N.A. | N.A. | MES-SA cell | CVCL_1404 | [392] |
| Half Maximal Inhibitory Concentration (IC50) | 12.2 uM | N.A. | N.A. | LoVo cell | CVCL_0399 | [592] |
| Half Maximal Inhibitory Concentration (IC50) | 12.23 uM | N.A. | N.A. | L02 cell | CVCL_6926 | [880] |
| Half Maximal Inhibitory Concentration (IC50) | 12.5 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [930] |
| Half Maximal Inhibitory Concentration (IC50) | 13.1 uM | N.A. | N.A. | LoVo cell | CVCL_0399 | [508] |
| Half Maximal Inhibitory Concentration (IC50) | 13.24 uM | N.A. | N.A. | DU145 cell | CVCL_0105 | [917] |
| Half Maximal Inhibitory Concentration (IC50) | 13.6 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [931] |
| Half Maximal Inhibitory Concentration (IC50) | 14 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [932] |
| Half Maximal Inhibitory Concentration (IC50) | 14.2 uM | N.A. | N.A. | ACHN cell | CVCL_1067 | [933] |
| Half Maximal Inhibitory Concentration (IC50) | 14.3 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [915] |
| Half Maximal Inhibitory Concentration (IC50) | 14.66 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [929] |
| Half Maximal Inhibitory Concentration (IC50) | 14.76 uM | N.A. | N.A. | MRC5 cell | CVCL_0440 | [934] |
| Half Maximal Inhibitory Concentration (IC50) | 14.84 uM | N.A. | N.A. | MRC5 cell | CVCL_0440 | [935] |
| Half Maximal Inhibitory Concentration (IC50) | 15.07 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [936] |
| Half Maximal Inhibitory Concentration (IC50) | 15.65 uM | N.A. | N.A. | DU145 cell | CVCL_0105 | [882] |
| Half Maximal Inhibitory Concentration (IC50) | 15.88 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [937] |
| Half Maximal Inhibitory Concentration (IC50) | 16.2 uM | N.A. | N.A. | MES-SA/Dx5 cell | CVCL_2598 | [524] |
| Half Maximal Inhibitory Concentration (IC50) | 16.31 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [938] |
| Half Maximal Inhibitory Concentration (IC50) | 16.32 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [887] |
| Half Maximal Inhibitory Concentration (IC50) | 17.23 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [939] |
| Half Maximal Inhibitory Concentration (IC50) | 17.54 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [939] |
| Half Maximal Inhibitory Concentration (IC50) | 17.6 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [856] |
| Half Maximal Inhibitory Concentration (IC50) | 17.67 uM | N.A. | N.A. | B16 cell | CVCL_F936 | [940] |
| Half Maximal Inhibitory Concentration (IC50) | 18.23 uM | N.A. | N.A. | MIA PaCa-2 cell | CVCL_0428 | [882] |
| Half Maximal Inhibitory Concentration (IC50) | 18.5 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [941] |
| Half Maximal Inhibitory Concentration (IC50) | 18.56 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [942] |
| Half Maximal Inhibitory Concentration (IC50) | 18.6 uM | N.A. | N.A. | BEAS-2B cell | CVCL_0168 | [821] |
| Half Maximal Inhibitory Concentration (IC50) | 19.01 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [832] |
| Half Maximal Inhibitory Concentration (IC50) | 19.3 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [889] |
| Half Maximal Inhibitory Concentration (IC50) | 19.51 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [943] |
| Half Maximal Inhibitory Concentration (IC50) | >20 uM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [549] |
| Half Maximal Inhibitory Concentration (IC50) | >20 uM | N.A. | N.A. | OVCAR-3 cell | CVCL_0465 | [549] |
| Half Maximal Inhibitory Concentration (IC50) | 20 uM | N.A. | N.A. | Vero cell | CVCL_0059 | [626] |
| Half Maximal Inhibitory Concentration (IC50) | 20.2 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [944] |
| Half Maximal Inhibitory Concentration (IC50) | 20.93 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [832] |
| Half Maximal Inhibitory Concentration (IC50) | 21.2 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [941] |
| Half Maximal Inhibitory Concentration (IC50) | 21.32 uM | N.A. | N.A. | NIH3T3 cell | CVCL_0594 | [849] |
| Half Maximal Inhibitory Concentration (IC50) | 21.5 uM | N.A. | N.A. | B16 cell | CVCL_F936 | [939] |
| Half Maximal Inhibitory Concentration (IC50) | 21.9 uM | N.A. | N.A. | MDA-MB-435 cell | CVCL_0417 | [755] |
| Half Maximal Inhibitory Concentration (IC50) | 23.94 uM | N.A. | N.A. | Vero cell | CVCL_0059 | [407] |
| Half Maximal Inhibitory Concentration (IC50) | 24.27 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [945] |
| Half Maximal Inhibitory Concentration (IC50) | 25.64 uM | N.A. | N.A. | C6 cell | CVCL_0194 | [887] |
| Half Maximal Inhibitory Concentration (IC50) | 26.79 uM | N.A. | N.A. | Caco-2 cell | CVCL_0025 | [946] |
| Half Maximal Inhibitory Concentration (IC50) | 26.81 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [947] |
| Half Maximal Inhibitory Concentration (IC50) | 26.9 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [948] |
| Half Maximal Inhibitory Concentration (IC50) | 29 uM | N.A. | N.A. | NCI-H322M cell | CVCL_1557 | [896] |
| Half Maximal Inhibitory Concentration (IC50) | 32 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [481] |
| Half Maximal Inhibitory Concentration (IC50) | 32.02 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [949] |
| Half Maximal Inhibitory Concentration (IC50) | 33.98 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [950] |
| Half Maximal Inhibitory Concentration (IC50) | 36.3 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [951] |
| Half Maximal Inhibitory Concentration (IC50) | 42.52 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [406] |
| Half Maximal Inhibitory Concentration (IC50) | 44.89 uM | N.A. | N.A. | LoVo cell | CVCL_0399 | [770] |
| Half Maximal Inhibitory Concentration (IC50) | 46.69 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [952] |
| Half Maximal Inhibitory Concentration (IC50) | 50 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [953] |
| Half Maximal Inhibitory Concentration (IC50) | 53.3 uM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [876] |
| Half Maximal Inhibitory Concentration (IC50) | 56.83 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [954] |
| Half Maximal Inhibitory Concentration (IC50) | 62.26 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [882] |
| Half Maximal Inhibitory Concentration (IC50) | 71.8 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [955] |
| Half Maximal Inhibitory Concentration (IC50) | 84.23 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [520] |
| Half Maximal Inhibitory Concentration (IC50) | 90 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [956] |
| Half Maximal Inhibitory Concentration (IC50) | 91 uM | N.A. | N.A. | LoVo cell | CVCL_0399 | [629] |
| Half Maximal Inhibitory Concentration (IC50) | 98.32 uM | N.A. | N.A. | Hs 578T cell | CVCL_0332 | [773] |
| Half Maximal Inhibitory Concentration (IC50) | 100 uM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [957] |
| Half Maximal Inhibitory Concentration (IC50) | >100 uM | N.A. | N.A. | SW1990 cell | CVCL_1723 | [785] |
| Half Maximal Inhibitory Concentration (IC50) | 101 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [958] |
| Half Maximal Inhibitory Concentration (IC50) | 135 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [959] |
| Half Maximal Inhibitory Concentration (IC50) | 148.4 uM | N.A. | N.A. | HEK293 cell | CVCL_0045 | [941] |
| Half Maximal Inhibitory Concentration (IC50) | 163.01 uM | N.A. | N.A. | DLD-1 cell | CVCL_0248 | [268] |
| Half Maximal Inhibitory Concentration (IC50) | 167.5 uM | N.A. | N.A. | AT3B-1 cell | CVCL_3489 | [902] |
| Half Maximal Inhibitory Concentration (IC50) | 200 uM | N.A. | N.A. | HeLa cell | CVCL_0030 | [957] |
| Half Maximal Inhibitory Concentration (IC50) | >400 uM | N.A. | N.A. | KB cell | CVCL_0372 | [456] |
| Half Maximal Inhibitory Concentration (IC50) | >400 uM | N.A. | N.A. | LoVo cell | CVCL_0399 | [456] |
| Half Maximal Inhibitory Concentration (IC50) | 400 uM | N.A. | N.A. | L1210 cell | CVCL_0382 | [957] |
| Half Maximal Lethal Concentration (IC50) | 0.7 ug/mL | N.A. | N.A. | K562 cell | CVCL_0004 | [960] |
| Half Maximal Lethal Concentration (IC50) | 0.8 ug/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [960] |
| Half Maximal Lethal Concentration (IC50) | 60.9 ug/mL | N.A. | N.A. | A498 cell | CVCL_1056 | [961] |
| Half Maximal Lethal Concentration (IC50) | 300 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [200] |
| Half Maximal Lethal Concentration (IC50) | 400 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [962] |
| Half Maximal Lethal Concentration (IC50) | 490 nM | N.A. | N.A. | SK-MEL-5 cell | CVCL_0527 | [141] |
| Half Maximal Lethal Concentration (IC50) | 490 nM | N.A. | N.A. | SK-MEL-28 cell | CVCL_0526 | [146] |
| Half Maximal Lethal Concentration (IC50) | 700 nM | N.A. | N.A. | K562 cell | CVCL_0004 | [963] |
| Half Maximal Lethal Concentration (IC50) | 740 nM | N.A. | N.A. | UACC-62 cell | CVCL_1780 | [152] |
| Half Maximal Lethal Concentration (IC50) | 740 nM | N.A. | N.A. | UACC-257 cell | CVCL_1779 | [146] |
| Half Maximal Lethal Concentration (IC50) | 1.06 uM | N.A. | N.A. | SK-MEL-2 cell | CVCL_0069 | [152] |
| Half Maximal Lethal Concentration (IC50) | 1.1 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [964] |
| Half Maximal Lethal Concentration (IC50) | 1.17 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [965] |
| Half Maximal Lethal Concentration (IC50) | 1.31 uM | N.A. | N.A. | UACC-62 cell | CVCL_1780 | [141] |
| Half Maximal Lethal Concentration (IC50) | 1.56 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [965] |
| Half Maximal Lethal Concentration (IC50) | 1.57 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [150] |
| Half Maximal Lethal Concentration (IC50) | 1.7 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [966] |
| Half Maximal Lethal Concentration (IC50) | 1.8 uM | N.A. | N.A. | BEAS-2B cell | CVCL_0168 | [200] |
| Half Maximal Lethal Concentration (IC50) | 1.9 uM | N.A. | N.A. | A498 cell | CVCL_1056 | [146] |
| Half Maximal Lethal Concentration (IC50) | 2.5 uM | N.A. | N.A. | THP-1 cell | CVCL_0006 | [967] |
| Half Maximal Lethal Concentration (IC50) | 2.8 uM | N.A. | N.A. | NCI-H522 cell | CVCL_1567 | [146] |
| Half Maximal Lethal Concentration (IC50) | 4.69 uM | N.A. | N.A. | RXF 393 cell | CVCL_1673 | [146] |
| Half Maximal Lethal Concentration (IC50) | 8.1 uM | N.A. | N.A. | 184B5 cell | CVCL_4688 | [200] |
| Half Maximal Lethal Concentration (IC50) | 8.15 uM | N.A. | N.A. | SK-MEL-5 cell | CVCL_0527 | [146] |
| Half Maximal Lethal Concentration (IC50) | 9.57 uM | N.A. | N.A. | MDA-MB-435 cell | CVCL_0417 | [146] |
| Half Maximal Lethal Concentration (IC50) | 9.7 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [968] |
| Half Maximal Lethal Concentration (IC50) | 13.15 uM | N.A. | N.A. | NCI-H23 cell | CVCL_1547 | [146] |
| Half Maximal Lethal Concentration (IC50) | 15.92 uM | N.A. | N.A. | SK-MEL-28 cell | CVCL_0526 | [141] |
| Half Maximal Lethal Concentration (IC50) | >17.2 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [190] |
| Half Maximal Lethal Concentration (IC50) | 19.95 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [178] |
| Half Maximal Lethal Concentration (IC50) | 19.95 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [178] |
| Half Maximal Lethal Concentration (IC50) | 19.95 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [178] |
| Half Maximal Lethal Concentration (IC50) | 21.33 uM | N.A. | N.A. | BT-549 cell | CVCL_1092 | [146] |
| Half Maximal Lethal Concentration (IC50) | 21.4 uM | N.A. | N.A. | Hep-G2 cell | CVCL_0027 | [969] |
| Half Maximal Lethal Concentration (IC50) | 21.68 uM | N.A. | N.A. | HCC 2998 cell | CVCL_1266 | [152] |
| Half Maximal Lethal Concentration (IC50) | 25.1 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [200] |
| Half Maximal Lethal Concentration (IC50) | 25.4 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [178] |
| Half Maximal Lethal Concentration (IC50) | 26.1 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [969] |
| Half Maximal Lethal Concentration (IC50) | 26.18 uM | N.A. | N.A. | UO-31 cell | CVCL_1911 | [146] |
| Half Maximal Lethal Concentration (IC50) | 27.23 uM | N.A. | N.A. | SF539 cell | CVCL_1691 | [146] |
| Half Maximal Lethal Concentration (IC50) | 28.3 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [969] |
| Half Maximal Lethal Concentration (IC50) | 30.62 uM | N.A. | N.A. | U-251MG cell | CVCL_0021 | [146] |
| Half Maximal Lethal Concentration (IC50) | 34.75 uM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [146] |
| Half Maximal Lethal Concentration (IC50) | 37.7 uM | N.A. | N.A. | HCT 116 cell | CVCL_0291 | [969] |
| Half Maximal Lethal Concentration (IC50) | 37.9 uM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [148] |
| Half Maximal Lethal Concentration (IC50) | 46 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [148] |
| Half Maximal Lethal Concentration (IC50) | 47.97 uM | N.A. | N.A. | EKVX cell | CVCL_1195 | [146] |
| Half Maximal Lethal Concentration (IC50) | 50.35 uM | N.A. | N.A. | LOX IMVI cell | CVCL_1381 | [141] |
| Half Maximal Lethal Concentration (IC50) | 51.29 uM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [146] |
| Half Maximal Lethal Concentration (IC50) | 58.61 uM | N.A. | N.A. | SW620 cell | CVCL_0547 | [146] |
| Half Maximal Lethal Concentration (IC50) | 64.12 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [146] |
| Half Maximal Lethal Concentration (IC50) | 67.45 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [146] |
| Half Maximal Lethal Concentration (IC50) | 67.61 uM | N.A. | N.A. | HOP-62 cell | CVCL_1285 | [146] |
| Half Maximal Lethal Concentration (IC50) | 67.76 uM | N.A. | N.A. | NCI-H322M cell | CVCL_1557 | [146] |
| Half Maximal Lethal Concentration (IC50) | 72.44 uM | N.A. | N.A. | SN12C cell | CVCL_1705 | [152] |
| Half Maximal Lethal Concentration (IC50) | 84.33 uM | N.A. | N.A. | OVCAR-3 cell | CVCL_0465 | [152] |
| Half Maximal Lethal Concentration (IC50) | 85.7 uM | N.A. | N.A. | Hs 578T cell | CVCL_0332 | [152] |
| Half Maximal Lethal Concentration (IC50) | 86.7 uM | N.A. | N.A. | TK-10 cell | CVCL_1773 | [152] |
| Half Maximal Lethal Concentration (IC50) | 87.1 uM | N.A. | N.A. | PC-3 cell | CVCL_0035 | [146] |
| Half Maximal Lethal Concentration (IC50) | 89.95 uM | N.A. | N.A. | TK-10 cell | CVCL_1773 | [146] |
| Half Maximal Lethal Concentration (IC50) | 92.68 uM | N.A. | N.A. | KM12 cell | CVCL_1331 | [146] |
| Half Maximal Lethal Concentration (IC50) | 100 uM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [146] |
| Half Maximal Lethal Concentration (IC50) | >100 uM | N.A. | N.A. | DU145 cell | CVCL_0105 | [171] |
| Half Maximal Lethal Concentration (IC50) | 100 uM | N.A. | N.A. | DU145 cell | CVCL_0105 | [146] |
| Half Maximal Lethal Concentration (IC50) | 100 uM | N.A. | N.A. | RPMI-8226 cell | CVCL_7353 | [146] |
| Half Maximal Lethal Concentration (IC50) | 100 uM | N.A. | N.A. | SK-OV-3 cell | CVCL_0532 | [146] |
| Half Maximal Lethal Concentration (IC50) | >100 uM | N.A. | N.A. | OVCAR-3 cell | CVCL_0465 | [148] |
| Half Maximal Lethal Concentration (IC50) | 100 uM | N.A. | N.A. | A-549 cell | CVCL_0023 | [146] |
| Half Maximal Lethal Concentration (IC50) | 100 uM | N.A. | N.A. | SR cell | CVCL_1711 | [146] |
| Half Maximal Lethal Concentration (IC50) | 100 uM | N.A. | N.A. | NCI-ADR-RES cell | CVCL_1452 | [146] |
| Half Maximal Lethal Concentration (IC50) | 100 uM | N.A. | N.A. | Caki-1 cell | CVCL_0234 | [146] |
| Half Maximal Lethal Concentration (IC50) | 100 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [146] |
| Half Maximal Lethal dose (LD50) | 0.19 ug/mL | N.A. | N.A. | PC-3 cell | CVCL_0035 | [970] |
| Half Maximal Lethal dose (LD50) | 1.1 uM | N.A. | N.A. | HL-60 cell | CVCL_0002 | [971] |
| Half Maximal Lethal dose (LD50) | 1.2 uM | N.A. | N.A. | AGS cell | CVCL_0139 | [971] |
| Half Maximal Lethal dose (LD50) | 1.8 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [971] |
| Inhibitory Concentration | 25 ng/mL | N.A. | N.A. | A-549 cell | CVCL_0023 | [314] |
| Maximal Effective Concentration (EC50) | 12.1 nM | N.A. | N.A. | WI-38 cell | CVCL_0579 | [396] |
| Median Toxic Dose (TD50) | 14 ng/ml | N.A. | N.A. | OVCAR-3 cell | CVCL_0465 | [256] |
| Tumor Growth Inhibition value (TGI) | 3.3 ug/mL | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [119] |
| Tumor Growth Inhibition value (TGI) | 3.9 ug/mL | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [119] |
| Tumor Growth Inhibition value (TGI) | <10 ug/mL | N.A. | N.A. | NCI-H226 cell | CVCL_1544 | [126] |
| Tumor Growth Inhibition value (TGI) | 32.4 ug/mL | N.A. | N.A. | A498 cell | CVCL_1056 | [961] |
| Tumor Growth Inhibition value (TGI) | 70 nM | N.A. | N.A. | CCRF-CEM cell | CVCL_0207 | [200] |
| Tumor Growth Inhibition value (TGI) | <100 nM | N.A. | N.A. | G-361 cell | CVCL_1220 | [972] |
| Tumor Growth Inhibition value (TGI) | 199.53 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [181] |
| Tumor Growth Inhibition value (TGI) | 200 nM | N.A. | N.A. | A-549 cell | CVCL_0023 | [962] |
| Tumor Growth Inhibition value (TGI) | 210 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [142] |
| Tumor Growth Inhibition value (TGI) | 300 nM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [200] |
| Tumor Growth Inhibition value (TGI) | 300 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [962] |
| Tumor Growth Inhibition value (TGI) | 300 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [962] |
| Tumor Growth Inhibition value (TGI) | 320 nM | N.A. | N.A. | UACC-62 cell | CVCL_1780 | [973] |
| Tumor Growth Inhibition value (TGI) | 400 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [150] |
| Tumor Growth Inhibition value (TGI) | 500 nM | N.A. | N.A. | MDA-MB-231 cell | CVCL_0062 | [190] |
| Tumor Growth Inhibition value (TGI) | 900 nM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [974] |
| Tumor Growth Inhibition value (TGI) | 1.42 uM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [148] |
| Tumor Growth Inhibition value (TGI) | 1.5 uM | N.A. | N.A. | BEAS-2B cell | CVCL_0168 | [200] |
| Tumor Growth Inhibition value (TGI) | 2.0893 uM | N.A. | N.A. | HOP-62 cell | CVCL_1285 | [181] |
| Tumor Growth Inhibition value (TGI) | 2.34423 uM | N.A. | N.A. | A498 cell | CVCL_1056 | [181] |
| Tumor Growth Inhibition value (TGI) | 2.37 uM | N.A. | N.A. | NCI-H460 cell | CVCL_0459 | [973] |
| Tumor Growth Inhibition value (TGI) | 5.01 uM | N.A. | N.A. | K562 cell | CVCL_0004 | [178] |
| Tumor Growth Inhibition value (TGI) | 8.11 uM | N.A. | N.A. | OVCAR-3 cell | CVCL_0465 | [148] |
| Tumor Growth Inhibition value (TGI) | 10.99 uM | N.A. | N.A. | MCF-7 cell | CVCL_0031 | [973] |
| Tumor Growth Inhibition value (TGI) | 16.4 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [973] |
| Tumor Growth Inhibition value (TGI) | 25.4 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [178] |
| Tumor Growth Inhibition value (TGI) | 46 uM | N.A. | N.A. | HT29 cell | CVCL_A8EZ | [148] |
Each Peptide-drug Conjugate Related to This Drug
Full Information of The Activity Data of The PDC(s) Related to This Drug
TH1904 [Preclinical]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [975] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 54 nM | |||
| Administration Time | 12 h | ||||
| MOA of PDC |
Overall, these results indicate that both conjugates alter cell migration in either TNBC or ovarian cancer cell models through a SORT1-dependent mechanism. The effects of TH1902 and TH1904 on cell migration further support the molecular rationale that inhibition of VM activity may result, in part, from the reduction of cancer cell invasive phenotype.
Click to Show/Hide
|
||||
| Description |
TH1904 IC50 value was 54 nM for the number of loops inhibition (Figure 5B).
|
||||
| In Vitro Model | Ovarian clear cell adenocarcinoma | ES-2 cell | CVCL_3509 | ||
(DOX-S)2-S-Pep [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Liver cancer | ||||
| Efficacy Data | Tumor volume | 1500 mm3 | |||
| Administration Time | 24 day | ||||
| Administration Dosage | 6 mg/kg | ||||
| MOA of PDC |
Meanwhile, the thiol groups on Pep were used to covalently link it with a doxorubicin derivative (DOX-SH) and form PDC to kill tumors and inhibit tumor metastasis. Two doxorubicin molecules were linked to one Pep, and the drug loading was as high as 50.17%. The functional PDC molecule was synthesized from DOX-SH and 2,2-dipyridyl disulfide activated peptide (Pep-S-S-Py), and the molecular weight is 2565.98. CSNs were prepared for the effective codelivery of MMPI Pep and DOX to the tumor site. The MPL shell displayed a negative surface charge for prolonged blood circulation, and the PDC core was able to aggregate in the tumor matrix and adhere to the cell membrane. PDC aggregation could constantly release the MMPI peptide and DOX via low concentration and long-term glutathione-mediated reduction conditions in the tumor matrix. DOX can effectively enter the tumor cell and kill them. Meanwhile, MMPI peptide adherence to the cell membrane was able to selectively inhibit the activity of the MMP2 and achieve the effect of inhibiting tumor metastasis. Our study suggests that CSNs are of great potential for treating metastatic tumors.
Click to Show/Hide
|
||||
| Description |
The antitumor activity of CSNs was assessed in BALB/c male nude mice inoculated with high metastatic HCCLM3-LUC cells. As can be seen from Figure 3A,B, DOX caused severe systemic toxicity and a significant weight loss (up to 24.38%) during treatment. In contrast, there was little change in body weight for PDC- and CSNs-treated mice. Tumor progression was greatly suppressed in the CSNs group, as observed by bioluminescence. Tumor tissue images collected on day 27 confirmed that nude mice treated with CSNs bore the smallest tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c male nude mice inoculated with high metastatic HCCLM3-LUC cells. | ||||
| In Vitro Model | Adult hepatocellular carcinoma | HCCLM3-Luc cell | CVCL_6832 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Liver cancer | ||||
| Efficacy Data | Lung metastasis nodules inhibition rates | 81.82% | |||
| Evaluation Method | H&E stain assay | ||||
| Administration Time | 5 week | ||||
| Administration Dosage | 6 mg/kg | ||||
| MOA of PDC |
Meanwhile, the thiol groups on Pep were used to covalently link it with a doxorubicin derivative (DOX-SH) and form PDC to kill tumors and inhibit tumor metastasis. Two doxorubicin molecules were linked to one Pep, and the drug loading was as high as 50.17%. The functional PDC molecule was synthesized from DOX-SH and 2,2-dipyridyl disulfide activated peptide (Pep-S-S-Py), and the molecular weight is 2565.98. CSNs were prepared for the effective codelivery of MMPI Pep and DOX to the tumor site. The MPL shell displayed a negative surface charge for prolonged blood circulation, and the PDC core was able to aggregate in the tumor matrix and adhere to the cell membrane. PDC aggregation could constantly release the MMPI peptide and DOX via low concentration and long-term glutathione-mediated reduction conditions in the tumor matrix. DOX can effectively enter the tumor cell and kill them. Meanwhile, MMPI peptide adherence to the cell membrane was able to selectively inhibit the activity of the MMP2 and achieve the effect of inhibiting tumor metastasis. Our study suggests that CSNs are of great potential for treating metastatic tumors.
Click to Show/Hide
|
||||
| Description |
For the PDC- and CSN-treated groups, the number of lung metastasis nodules was decreased (inhibition rates of 81.82% and 93.18%, respectively), and smaller areas of tumor cells were observed in the H&E-stained images. Nude mice treated with DOX displayed 7.8 nodules, which was not significantly different from what was seen in the PBS group.
|
||||
| In Vivo Model | BALB/c male nude mice inoculated with high metastatic HCCLM3-LUC cells. | ||||
| In Vitro Model | Adult hepatocellular carcinoma | HCCLM3-Luc cell | CVCL_6832 | ||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Liver cancer | ||||
| Efficacy Data | Inhibitory effect | 82.84 ± 2.22% | |||
| Evaluation Method | Wound healing and transwell assays | ||||
| MOA of PDC |
Meanwhile, the thiol groups on Pep were used to covalently link it with a doxorubicin derivative (DOX-SH) and form PDC to kill tumors and inhibit tumor metastasis. Two doxorubicin molecules were linked to one Pep, and the drug loading was as high as 50.17%. The functional PDC molecule was synthesized from DOX-SH and 2,2-dipyridyl disulfide activated peptide (Pep-S-S-Py), and the molecular weight is 2565.98. CSNs were prepared for the effective codelivery of MMPI Pep and DOX to the tumor site. The MPL shell displayed a negative surface charge for prolonged blood circulation, and the PDC core was able to aggregate in the tumor matrix and adhere to the cell membrane. PDC aggregation could constantly release the MMPI peptide and DOX via low concentration and long-term glutathione-mediated reduction conditions in the tumor matrix. DOX can effectively enter the tumor cell and kill them. Meanwhile, MMPI peptide adherence to the cell membrane was able to selectively inhibit the activity of the MMP2 and achieve the effect of inhibiting tumor metastasis. Our study suggests that CSNs are of great potential for treating metastatic tumors.
Click to Show/Hide
|
||||
| Description |
The tumor invasion ability was also inhibited under the action of Pep and the PDC. Pep and PDC treatment resulted in a strong inhibition of invasion ability of SMCC-7721 cells with rates of 79.93 ± 3.86% and 82.84 ± 2.22%, respectively. These results were used to guide follow-up in vivo studies.
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Liver cancer | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) | 20.22 µM | |||
| Evaluation Method | MTT colorimetric assay | ||||
| MOA of PDC |
Meanwhile, the thiol groups on Pep were used to covalently link it with a doxorubicin derivative (DOX-SH) and form PDC to kill tumors and inhibit tumor metastasis. Two doxorubicin molecules were linked to one Pep, and the drug loading was as high as 50.17%. The functional PDC molecule was synthesized from DOX-SH and 2,2-dipyridyl disulfide activated peptide (Pep-S-S-Py), and the molecular weight is 2565.98. CSNs were prepared for the effective codelivery of MMPI Pep and DOX to the tumor site. The MPL shell displayed a negative surface charge for prolonged blood circulation, and the PDC core was able to aggregate in the tumor matrix and adhere to the cell membrane. PDC aggregation could constantly release the MMPI peptide and DOX via low concentration and long-term glutathione-mediated reduction conditions in the tumor matrix. DOX can effectively enter the tumor cell and kill them. Meanwhile, MMPI peptide adherence to the cell membrane was able to selectively inhibit the activity of the MMP2 and achieve the effect of inhibiting tumor metastasis. Our study suggests that CSNs are of great potential for treating metastatic tumors.
Click to Show/Hide
|
||||
| Description |
An MTT colorimetric assay was used to evaluate the in vitro cytotoxicity of materials on SMMC-7721 cells. Within the concentration range of 1.25-100 uM, DOX, DOX-SH, and PDC had a certain killing effect on liver cancer cells. The IC50 values of DOX, DOX-SH, and PDC were 17.62, 14.75, and 20.22 uM, respectively.
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
MPD1 [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [976] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 12.50% | |||
| Administration Time | 30 days | ||||
| Administration Dosage | 10 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BxPC-3 cells (KRAS wild type) xenografted mice. | ||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | BxPC-3 cell | CVCL_0186 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [976] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 69.10% | |||
| Administration Time | 30 days | ||||
| Administration Dosage | 5 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | AsPC-1 cells (G12D KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | AsPC-1 (KRAS G12D) cell | L-929 cell line | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [976] | ||||
| Indication | Lung cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 80.00% | |||
| Administration Time | 30 days | ||||
| Administration Dosage | 5 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | A549 cells (G12S KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Lung adenocarcinoma | A-549 (KRAS G12S) cell | CVCL_0023 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [976] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 86.80% | |||
| Administration Time | 30 days | ||||
| Administration Dosage | 5 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | HCT116 cells (G13D KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Colon carcinoma | HCT 116 (KRAS G13D) cell | CVCL_0291 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [976] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 95.10% | |||
| Administration Time | 30 days | ||||
| Administration Dosage | 10 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | AsPC-1 cells (G12D KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | AsPC-1 (KRAS G12D) cell | L-929 cell line | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [976] | ||||
| Indication | Lung cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 95.10% | |||
| Administration Time | 30 days | ||||
| Administration Dosage | 10 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | A549 cells (G12S KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Lung adenocarcinoma | A-549 (KRAS G12S) cell | CVCL_0023 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [976] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 96.50% | |||
| Administration Time | 30 days | ||||
| Administration Dosage | 5 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | MDA-MB-231 cells (G13D KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (KRAS G13D) cell | CVCL_0062 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 8 Reporting the Activity Data of This PDC | [976] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 97.80% | |||
| Administration Time | 30 days | ||||
| Administration Dosage | 10 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | HCT116 cells (G13D KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Colon carcinoma | HCT 116 (KRAS G13D) cell | CVCL_0291 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 9 Reporting the Activity Data of This PDC | [976] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 99.90% | |||
| Administration Time | 30 days | ||||
| Administration Dosage | 5 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | MIA PaCa-2 cells (G12C KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | MIA PaCa-2 (KRAS G12C) cell | CVCL_0428 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 10 Reporting the Activity Data of This PDC | [976] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 100.00% | |||
| Administration Time | 30 days | ||||
| Administration Dosage | 10 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | MIA PaCa-2 cells (G12C KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | MIA PaCa-2 (KRAS G12C) cell | CVCL_0428 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 11 Reporting the Activity Data of This PDC | [976] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 100.00% | |||
| Administration Time | 30 days | ||||
| Administration Dosage | 10 mg/kg | ||||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | MDA-MB-231 cells (G13D KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (KRAS G13D) cell | CVCL_0062 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 12 Reporting the Activity Data of This PDC | [976] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Albumin uptake rate | 6.67% | |||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | AsPC-1 cells (G12D KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | AsPC-1 (KRAS G12D) cell | L-929 cell line | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 13 Reporting the Activity Data of This PDC | [976] | ||||
| Indication | Lung cancer | ||||
| Efficacy Data | Albumin uptake rate | 12.80% | |||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | A549 cells (G12S KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Lung adenocarcinoma | A-549 (KRAS G12S) cell | CVCL_0023 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 14 Reporting the Activity Data of This PDC | [976] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Albumin uptake rate | 51.57% | |||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | MDA-MB-231 cells (G13D KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 (KRAS G13D) cell | CVCL_0062 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 15 Reporting the Activity Data of This PDC | [976] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Albumin uptake rate | 52.30% | |||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | HCT116 cells (G13D KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Colon carcinoma | HCT 116 (KRAS G13D) cell | CVCL_0291 | ||
| Half life period | 8.51 ± 0.50 h | ||||
| Experiment 16 Reporting the Activity Data of This PDC | [976] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Albumin uptake rate | 68.43% | |||
| MOA of PDC |
To address these challenges, we developed a novel peptide-drug conjugate (PDC) to target pan-KRAS mutant cancers by exploiting enhanced albumin metabolism in KRAS mutant cancer cells .Such enhanced albumin metabolism is particularly found in cancer cells with oncogenic hypermutations in the RAS-PI3K signaling pathway, which are associated with the proliferation and survival of cancer cells. Particularly, Ras hyperactivated cancer cells in various solid tumors use macropinocytosis as a nutrient scavenging source for intracellular uptake of extracellular proteins, including albumin. Recent studies evidenced that the Ras superfamily of small guanosine triphosphatases (GTPases) including Rac, Cdc42, Arf6, and Rab5 are known stimulating factors or receptors for promoting membrane ruffle formation via actin polymerization as well as vacuolization of macropinosome. However, this altered mechanism can be taken advantage of as a potential drug delivery route in targeting RAS-transformed cancer cells. For this study, we adopted a previously developed albumin-binding caspase-3-cleavable peptide-doxorubicin conjugate (MPD1). In contrast to cytostatic small molecule inhibitors, MPD1 uses a cytotoxic anti-cancer agent (doxorubicin) as its warhead to capitalize on its potency to directly kill cancer cells non-selectively. More specifically, the albumin-bound MPD1 is intended to be delivered into KRAS mutant cancer cells through enhanced macropinocytosis and subsequently degraded by lysosomal enzymes to release the cytotoxic payload, which can induce apoptosis within albumin-engulfing cancer cells. Furthermore, albumin metabolism-induced apoptotic cells release caspase-3 to activate unabsorbed extracellular albumin-bound MPD1 through the cleavage of DEVD peptide to free doxorubicin, which induces the subsequent apoptosis of neighboring cancer cells in a non-selective manner.
Click to Show/Hide
|
||||
| Description |
The in vivo anti-cancer activity of MPD1 was evaluated in MIA PaCa-2- and BxPC-3-xenografted mice. When the average tumor volume reached 200 mm3, mice were treated with 5 or 10 mg/kg of MPD1 via intravenous administration every other day for 4 weeks. MPD1 demonstrated potent anti-cancer activity, yielding 100% and 113% TGI for 5 and 10 mg/kg, respectively, compared to the control group in MIA PaCa-2 tumor model (30-day tumor volume [mm3]: 5 mg/kg, 268.48 ± 135.66, P < 0.0001; 10 mg/kg, 46.19 ± 45.92, P < 0.0001). However, when BxPC-3-xenografted mice were treated with the same doses of MPD1, no therapeutic efficacy was observed (30-day tumor volume [mm3]: 5 mg/kg, 1728.68 ± 311.91, P = 0.77; 10 mg/kg, 1221.27 ± 306.77, P = 0.36). There were no noticeable body weight changes or obvious abnormalities in heart, kidney, liver, and spleen in histological assessment indicating that MPD1 was tolerable up to 10 mg/kg when administered 14 times in both xenograft models. Immunohistochemical analysis of the caspase-3 expression and TUNEL staining of MIA PaCa-2 and BxPC-3 tumors from MPD1-treated mice confirmed that MPD1 caused a substantial degree of apoptosis and caspase-3 upregulation only in MIA PaCa-2. In contrast, an increased dose of 10 mg/kg of MPD1 did not show upregulated apoptotic events or caspase-3 expression in BxPC-3 tumors.
Click to Show/Hide
|
||||
| In Vivo Model | MIA PaCa-2 cells (G12C KRAS mutation) xenografted mice model. | ||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | MIA PaCa-2 (KRAS G12C) cell | CVCL_0428 | ||
| Half life period | 8.51 ± 0.50 h | ||||
DOXO-EMCH-(RNWELRLK-PEG4)2 [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) | 51.1 ± 3.1% | |||
| Administration Time | 14 days | ||||
| Administration Dosage | 5 mg/kg | ||||
| MOA of PDC |
Tumor-targeting peptide-drug conjugates (PDCs) have become a focus of research in recent years. However, due to the instability of peptides and their short in vivo effective half-life, they have limited clinical application. Herein, we propose a new DOX PDC based on a homodimer HER-2-targeting peptide and acid-sensitive hydrazone bond, which could enhance the anti-tumor effect of DOX and reduce systemic toxicities. The PDC could accurately deliver DOX into HER2-positive SKBR-3 cells, with it showing 2.9 times higher cellular uptake than free DOX and enhanced cytotoxicity with respect to IC50of 140 nM (vs. 410 nM for free DOX). In vitro assays showed that the PDC had high cellular internalization efficiency and cytotoxicity. In vivo anti-tumor experiments indicated that the PDC could significantly inhibit the growth of HER2-positive breast cancer xenografts in mice and reduce the side effects of DOX. In summary, we constructed a novel PDC molecule targeting HER2-positive tumors, which may overcome some deficiencies of DOX in breast cancer therapy.
Click to Show/Hide
|
||||
| Description |
In vivo anti-tumor studies were evaluated by using SKBR-3 xenografted (BALB/c nude) mice treated with the PDC, free DOX, or saline. Figure 5a demonstrates that the PDC had a much more powerful anti-tumor effect than free DOX, reducing tumor growth by 51.1 ± 3.1% on day 14 post treatment, while free DOX only achieved a 23.13 ± 2.4% reduction. Additionally, the PDC had a significantly higher tumor weight inhibition of 57.5 ± 3.4% compared to free DOX.
Click to Show/Hide
|
||||
| In Vivo Model | SKBR-3 cells female BALB/c mice xenograft model. | ||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 140 nM | |||
| Evaluation Method | CCK8 assay | ||||
| Administration Time | 24 h | ||||
| MOA of PDC |
Tumor-targeting peptide-drug conjugates (PDCs) have become a focus of research in recent years. However, due to the instability of peptides and their short in vivo effective half-life, they have limited clinical application. Herein, we propose a new DOX PDC based on a homodimer HER-2-targeting peptide and acid-sensitive hydrazone bond, which could enhance the anti-tumor effect of DOX and reduce systemic toxicities. The PDC could accurately deliver DOX into HER2-positive SKBR-3 cells, with it showing 2.9 times higher cellular uptake than free DOX and enhanced cytotoxicity with respect to IC50of 140 nM (vs. 410 nM for free DOX). In vitro assays showed that the PDC had high cellular internalization efficiency and cytotoxicity. In vivo anti-tumor experiments indicated that the PDC could significantly inhibit the growth of HER2-positive breast cancer xenografts in mice and reduce the side effects of DOX. In summary, we constructed a novel PDC molecule targeting HER2-positive tumors, which may overcome some deficiencies of DOX in breast cancer therapy.
Click to Show/Hide
|
||||
| Description |
Tumor-targeting peptide-drug conjugates (PDCs) have become a focus of research in recent years. However, due to the instability of peptides and their short in vivo effective half-life, they have limited clinical application. Herein, we propose a new DOX PDC based on a homodimer HER-2-targeting peptide and acid-sensitive hydrazone bond, which could enhance the anti-tumor effect of DOX and reduce systemic toxicities. The PDC could accurately deliver DOX into HER2-positive SKBR-3 cells, with it showing 2.9 times higher cellular uptake than free DOX and enhanced cytotoxicity with respect to IC50of 140 nM (vs. 410 nM for free DOX). In vitro assays showed that the PDC had high cellular internalization efficiency and cytotoxicity. In vivo anti-tumor experiments indicated that the PDC could significantly inhibit the growth of HER2-positive breast cancer xenografts in mice and reduce the side effects of DOX. In summary, we constructed a novel PDC molecule targeting HER2-positive tumors, which may overcome some deficiencies of DOX in breast cancer therapy.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
SMAC-FRRG-DOX [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [977] | ||||
| Indication | Drug-resistant cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 76.00% | |||
| Administration Dosage | 1 mg/kg of DOX | ||||
| MOA of PDC |
These results suggest that the DD-NPs could improve the in vivo bioavailability and cancer targeting efficiency of SMAC via their stable nanoparticle-derived EPR effect, leading to effective IAPs inhibition in targeted tumor tissues.
|
||||
| Description |
Finally, the tumor volumes were successfully suppressed from 76% to 89% when the intravenous dose of DD-NPs increased from 1 mg/kg of DOX to 5 mg/kg of DOX.
|
||||
| In Vivo Model | MCF-7 tumor-bearing mice. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [977] | ||||
| Indication | Drug-resistant cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 89%-99% | |||
| Administration Dosage | 5 mg/kg | ||||
| MOA of PDC |
These results suggest that the DD-NPs could improve the in vivo bioavailability and cancer targeting efficiency of SMAC via their stable nanoparticle-derived EPR effect, leading to effective IAPs inhibition in targeted tumor tissues.
|
||||
| Description |
Finally, the tumor volumes were successfully suppressed from 76% to 89% when the intravenous dose of DD-NPs increased from 1 mg/kg of DOX to 5 mg/kg of DOX.
|
||||
| In Vivo Model | MCF-7 tumor-bearing mice. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [977] | ||||
| Indication | Drug-resistant cancer | ||||
| Efficacy Data | Tumer volume | 10 mm3 | |||
| Administration Time | 21 days | ||||
| MOA of PDC |
These results suggest that the DD-NPs could improve the in vivo bioavailability and cancer targeting efficiency of SMAC via their stable nanoparticle-derived EPR effect, leading to effective IAPs inhibition in targeted tumor tissues.
|
||||
| In Vivo Model | MCF-7 tumor-bearing mice. | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [977] | ||||
| Indication | Drug-resistant cancer | ||||
| Efficacy Data | Body weight | 23.5g | |||
| Administration Time | 13 days | ||||
| MOA of PDC |
These results suggest that the DD-NPs could improve the in vivo bioavailability and cancer targeting efficiency of SMAC via their stable nanoparticle-derived EPR effect, leading to effective IAPs inhibition in targeted tumor tissues.
|
||||
| In Vivo Model | MCF-7 tumor-bearing mice. | ||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [977] | ||||
| Indication | Drug-resistant cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 7.53 μM | |||
| MOA of PDC |
These results suggest that the DD-NPs could improve the in vivo bioavailability and cancer targeting efficiency of SMAC via their stable nanoparticle-derived EPR effect, leading to effective IAPs inhibition in targeted tumor tissues.
|
||||
| Description |
When both cells were treated with different concentrations of DD-NPs (0-186 μg/ml), the cytotoxicity was induced only in MCF-7, showing similar cytotoxicity with free DOX at a high concentration (Fig. 2h). However, DD-NPs did not exhibit significant cytotoxicity in H9C2 whereas free DOX showed similar cytotoxicity when treated to MCF-7 (Fig. 2i).
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [977] | ||||
| Indication | Drug-resistant cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 9.2 μM | |||
| Administration Time | 48 h | ||||
| MOA of PDC |
These results suggest that the DD-NPs could improve the in vivo bioavailability and cancer targeting efficiency of SMAC via their stable nanoparticle-derived EPR effect, leading to effective IAPs inhibition in targeted tumor tissues.
|
||||
| Description |
However, DD-NP-treated group showed similar IC50 in wild-MCF-7 (7.53 μM) and ADR-MCF-7 (9.2 μM), due to the synergetic pro-apoptotic effect of SMAC molecules in DD-NPs (Fig. 3g).
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7/ADR cell | CVCL_0031 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [977] | ||||
| Indication | Drug-resistant cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | >> 200 μM | |||
| MOA of PDC |
These results suggest that the DD-NPs could improve the in vivo bioavailability and cancer targeting efficiency of SMAC via their stable nanoparticle-derived EPR effect, leading to effective IAPs inhibition in targeted tumor tissues.
|
||||
| Description |
When both cells were treated with different concentrations of DD-NPs (0-186 μg/ml), the cytotoxicity was induced only in MCF-7, showing similar cytotoxicity with free DOX at a high concentration (Fig. 2h). However, DD-NPs did not exhibit significant cytotoxicity in H9C2 whereas free DOX showed similar cytotoxicity when treated to MCF-7 (Fig. 2i).
Click to Show/Hide
|
||||
| In Vitro Model | Normal | H9c2 cell | CVCL_0286 | ||
DOX-ApoA-1 mimic peptide [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [978] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) | 83% | |||
| Administration Time | 15 days | ||||
| MOA of PDC |
Herein, we report a dual-functional sHDL (DP-sHDL) that is co-assembled from DOX-ApoA1 mimetic peptide conjugate, PSB-603 (an A2BR inhibitor), phospholipid and cholesterol oleate (CO) through a microfluidic-based methodology. We expect that DP-sHDL could accumulate in tumor, where they can target cancerous cells, CAF and tumor-associated macrophages (TAMs). The DOX would induce the ICD of cancer cells and promote DC maturation and T lymphocyte infiltration and activity. PSB-603 is expected to block the A2BR, which would down-regulate CD73 and thus ADO, leading to a decrease in the intratumoral densities of several immunosuppressive cells. By simultaneously activating anti-tumor immunity and relieving immunosuppressive microenvironment, DP-sHDL may inhibit TNBC tumor growth and prolong the survival of animals, which could be further improved when used in combination with an immune checkpoint inhibitor.
Click to Show/Hide
|
||||
| Description |
Given the potent effects of DP-sHDL in boosting anti-tumor immunity and relieving immunosuppressive microenvironment, its efficacy in treating TNBC model mice was evaluated. Through a 3-dose regime, DP-sHDL inhibited the growth of tumors by 84% and significantly reduced tumor burden. In contrast, DOX was less effective and caused a progressive decline in body weight. Further biochemical assays revealed that the levels of AST, CREA, CK and LDH in DOX-treated mice were significantly higher than those receiving PBS. Accordingly, inflammatory cell infiltration, cell necrosis and local swelling were observed in the heart, liver and kidneys, respectively, in the DOX-injection group. With the same regime, no visible damage was monitored in the major organs in DP-sHDL group. Based on these findings, the long-term efficacy of D-sHDL, P-sHDL and DP-sHDL were further evaluated. DP-sHDL retarded the tumor growth, and prolonged the median survival time (MST) from 18 days (PBS-treated mice) to 35 days, without body weight loss. These results proved that DP-sHDL could reduce the toxicity of DOX and effectively inhibit tumor growth.
Click to Show/Hide
|
||||
| In Vivo Model | Female BALB/c mice murine 4T1 breast cancer cells xenograft tumor models. | ||||
| In Vitro Model | Breast cancer | Murine 4T1 breast cancer cell | Homo sapiens | ||
FDPC-NPs [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [979] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Tumer volume | 200 mm3 | |||
| Administration Time | 13 days | ||||
| Administration Dosage | 10 mg DOX/kg | ||||
| MOA of PDC |
In this work, we reported doxorubicin-peptide conjugates (DPCs) with an extracellular tumor acid-responsive sphere-fiber transformation for enhanced residence in tumors. As illustrated in Scheme Scheme1,1, the chemotherapy drug doxorubicin (DOX) was coupled with a peptide (KIGLFRWR) to design a DPC molecule with assembly ability. First, the DPCs, driven by hydrophobic forces from the hydrophobic drug DOX and the IGL fragment, can form spherical DPC nanoparticles (DPC-NPs). Then, along with hydrogen bond between peptides, the aromatic amino acids F and W give the DPC-NPs the ability of self-assembly to DPC-nanofibers (DPC-NFs) due to π-π stacking. The step-by-step assembly process provides opportunities for morphological transformation control. To meet the particle size requirements for intravenous injection, the acid-responsive material 2,3-dimethylmaleic anhydride grafted polylysine, named the functional polylysine graft (FPG), was designed as a shielding layer for DPC-NPs and formed functional doxorubicin-peptide conjugate nanoparticles (FDPC-NPs) by an electrostatic interaction to avoid π-π stacking interactions and hydrogen bond between the DPC-NPs. Therefore, the FDPC-NPs could maintain an appropriate size in blood vessels until entering the tumor stroma by the EPR effect. When the FDPC-NPs passed through the blood vessel and entered the weakly acidic microenvironment of the tumor, the surface potential of the shield was reversed from negative to positive because of acid-sensitive 2,3-dimethylmaleic groups on the FPG. Therefore, FPG would separate from the DPC-NPs because of the mutual repulsion effect from the like charges. Then, DPC-NPs self-assembled into DPC-NFs, thereby staying in the tumor region for a long time. After that, the fibers degraded gradually and free drug penetrated into tumor cells, exerting sustained anti-tumor effect. This study is original and provides new ideas for the design of targeted and long-acting drug delivery systems for tumor therapy.
Click to Show/Hide
|
||||
| Description |
Results showed that DOX solution, DOX-liposomes and FDPC-NPs displayed significant therapeutic effects against tumors (P < 0.001). Particularly, DOX-liposomes and FDPC-NPs behaved better due to the EPR effect.
|
||||
| In Vivo Model | H22 hepatocarcinoma tumor-bearing mouse. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [979] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Percent survival | 95% | |||
| Administration Time | 13 days | ||||
| Administration Dosage | 10 mg DOX/kg | ||||
| MOA of PDC |
In this work, we reported doxorubicin-peptide conjugates (DPCs) with an extracellular tumor acid-responsive sphere-fiber transformation for enhanced residence in tumors. As illustrated in Scheme Scheme1,1, the chemotherapy drug doxorubicin (DOX) was coupled with a peptide (KIGLFRWR) to design a DPC molecule with assembly ability. First, the DPCs, driven by hydrophobic forces from the hydrophobic drug DOX and the IGL fragment, can form spherical DPC nanoparticles (DPC-NPs). Then, along with hydrogen bond between peptides, the aromatic amino acids F and W give the DPC-NPs the ability of self-assembly to DPC-nanofibers (DPC-NFs) due to π-π stacking. The step-by-step assembly process provides opportunities for morphological transformation control. To meet the particle size requirements for intravenous injection, the acid-responsive material 2,3-dimethylmaleic anhydride grafted polylysine, named the functional polylysine graft (FPG), was designed as a shielding layer for DPC-NPs and formed functional doxorubicin-peptide conjugate nanoparticles (FDPC-NPs) by an electrostatic interaction to avoid π-π stacking interactions and hydrogen bond between the DPC-NPs. Therefore, the FDPC-NPs could maintain an appropriate size in blood vessels until entering the tumor stroma by the EPR effect. When the FDPC-NPs passed through the blood vessel and entered the weakly acidic microenvironment of the tumor, the surface potential of the shield was reversed from negative to positive because of acid-sensitive 2,3-dimethylmaleic groups on the FPG. Therefore, FPG would separate from the DPC-NPs because of the mutual repulsion effect from the like charges. Then, DPC-NPs self-assembled into DPC-NFs, thereby staying in the tumor region for a long time. After that, the fibers degraded gradually and free drug penetrated into tumor cells, exerting sustained anti-tumor effect. This study is original and provides new ideas for the design of targeted and long-acting drug delivery systems for tumor therapy.
Click to Show/Hide
|
||||
| Description |
Moreover, based on the survivorship curves, treatment with FDPC-NPs remarkably promoted the survival rate of tumor-bearing mice, which furtherly confirmed the therapeutic effect and biological safety of FDPC-NPs.
|
||||
| In Vivo Model | H22 hepatocarcinoma tumor-bearing mouse. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [979] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Body weight | 32g | |||
| Administration Time | 13 days | ||||
| Administration Dosage | 10 mg DOX/kg | ||||
| MOA of PDC |
In this work, we reported doxorubicin-peptide conjugates (DPCs) with an extracellular tumor acid-responsive sphere-fiber transformation for enhanced residence in tumors. As illustrated in Scheme Scheme1,1, the chemotherapy drug doxorubicin (DOX) was coupled with a peptide (KIGLFRWR) to design a DPC molecule with assembly ability. First, the DPCs, driven by hydrophobic forces from the hydrophobic drug DOX and the IGL fragment, can form spherical DPC nanoparticles (DPC-NPs). Then, along with hydrogen bond between peptides, the aromatic amino acids F and W give the DPC-NPs the ability of self-assembly to DPC-nanofibers (DPC-NFs) due to π-π stacking. The step-by-step assembly process provides opportunities for morphological transformation control. To meet the particle size requirements for intravenous injection, the acid-responsive material 2,3-dimethylmaleic anhydride grafted polylysine, named the functional polylysine graft (FPG), was designed as a shielding layer for DPC-NPs and formed functional doxorubicin-peptide conjugate nanoparticles (FDPC-NPs) by an electrostatic interaction to avoid π-π stacking interactions and hydrogen bond between the DPC-NPs. Therefore, the FDPC-NPs could maintain an appropriate size in blood vessels until entering the tumor stroma by the EPR effect. When the FDPC-NPs passed through the blood vessel and entered the weakly acidic microenvironment of the tumor, the surface potential of the shield was reversed from negative to positive because of acid-sensitive 2,3-dimethylmaleic groups on the FPG. Therefore, FPG would separate from the DPC-NPs because of the mutual repulsion effect from the like charges. Then, DPC-NPs self-assembled into DPC-NFs, thereby staying in the tumor region for a long time. After that, the fibers degraded gradually and free drug penetrated into tumor cells, exerting sustained anti-tumor effect. This study is original and provides new ideas for the design of targeted and long-acting drug delivery systems for tumor therapy.
Click to Show/Hide
|
||||
| Description |
On the contrary, the administration of DOX-liposomes and FDPC-NPs barely influenced the body weights of the model mice, revealing the safety of DOX-liposomes and FDPC-NPs.
|
||||
| In Vivo Model | H22 hepatocarcinoma tumor-bearing mouse. | ||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [979] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 5.896 μg/mL | |||
| MOA of PDC |
In this work, we reported doxorubicin-peptide conjugates (DPCs) with an extracellular tumor acid-responsive sphere-fiber transformation for enhanced residence in tumors. As illustrated in Scheme Scheme1,1, the chemotherapy drug doxorubicin (DOX) was coupled with a peptide (KIGLFRWR) to design a DPC molecule with assembly ability. First, the DPCs, driven by hydrophobic forces from the hydrophobic drug DOX and the IGL fragment, can form spherical DPC nanoparticles (DPC-NPs). Then, along with hydrogen bond between peptides, the aromatic amino acids F and W give the DPC-NPs the ability of self-assembly to DPC-nanofibers (DPC-NFs) due to π-π stacking. The step-by-step assembly process provides opportunities for morphological transformation control. To meet the particle size requirements for intravenous injection, the acid-responsive material 2,3-dimethylmaleic anhydride grafted polylysine, named the functional polylysine graft (FPG), was designed as a shielding layer for DPC-NPs and formed functional doxorubicin-peptide conjugate nanoparticles (FDPC-NPs) by an electrostatic interaction to avoid π-π stacking interactions and hydrogen bond between the DPC-NPs. Therefore, the FDPC-NPs could maintain an appropriate size in blood vessels until entering the tumor stroma by the EPR effect. When the FDPC-NPs passed through the blood vessel and entered the weakly acidic microenvironment of the tumor, the surface potential of the shield was reversed from negative to positive because of acid-sensitive 2,3-dimethylmaleic groups on the FPG. Therefore, FPG would separate from the DPC-NPs because of the mutual repulsion effect from the like charges. Then, DPC-NPs self-assembled into DPC-NFs, thereby staying in the tumor region for a long time. After that, the fibers degraded gradually and free drug penetrated into tumor cells, exerting sustained anti-tumor effect. This study is original and provides new ideas for the design of targeted and long-acting drug delivery systems for tumor therapy.
Click to Show/Hide
|
||||
| Description |
Both the peptide and FPG exhibited no obvious cytotoxicity, while FDPC-NPs and DOX displayed cytotoxicity against tumor cells (IC50 DOX = 2.965 μg/mL; IC50 FDPC-NPs = 5.896 μg/mL) (Figure (Figure6A).6A).
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [979] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability | 10.00% | |||
| Evaluation Method | MTT assay | ||||
| Administration Dosage | 50 μg/ml | ||||
| MOA of PDC |
In this work, we reported doxorubicin-peptide conjugates (DPCs) with an extracellular tumor acid-responsive sphere-fiber transformation for enhanced residence in tumors. As illustrated in Scheme Scheme1,1, the chemotherapy drug doxorubicin (DOX) was coupled with a peptide (KIGLFRWR) to design a DPC molecule with assembly ability. First, the DPCs, driven by hydrophobic forces from the hydrophobic drug DOX and the IGL fragment, can form spherical DPC nanoparticles (DPC-NPs). Then, along with hydrogen bond between peptides, the aromatic amino acids F and W give the DPC-NPs the ability of self-assembly to DPC-nanofibers (DPC-NFs) due to π-π stacking. The step-by-step assembly process provides opportunities for morphological transformation control. To meet the particle size requirements for intravenous injection, the acid-responsive material 2,3-dimethylmaleic anhydride grafted polylysine, named the functional polylysine graft (FPG), was designed as a shielding layer for DPC-NPs and formed functional doxorubicin-peptide conjugate nanoparticles (FDPC-NPs) by an electrostatic interaction to avoid π-π stacking interactions and hydrogen bond between the DPC-NPs. Therefore, the FDPC-NPs could maintain an appropriate size in blood vessels until entering the tumor stroma by the EPR effect. When the FDPC-NPs passed through the blood vessel and entered the weakly acidic microenvironment of the tumor, the surface potential of the shield was reversed from negative to positive because of acid-sensitive 2,3-dimethylmaleic groups on the FPG. Therefore, FPG would separate from the DPC-NPs because of the mutual repulsion effect from the like charges. Then, DPC-NPs self-assembled into DPC-NFs, thereby staying in the tumor region for a long time. After that, the fibers degraded gradually and free drug penetrated into tumor cells, exerting sustained anti-tumor effect. This study is original and provides new ideas for the design of targeted and long-acting drug delivery systems for tumor therapy.
Click to Show/Hide
|
||||
| Description |
Both the peptide and FPG exhibited no obvious cytotoxicity, while FDPC-NPs and DOX displayed cytotoxicity against tumor cells (IC50 DOX = 2.965 μg/mL; IC50 FDPC-NPs = 5.896 μg/mL) (Figure (Figure6A).6A).
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [979] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability | 25.00% | |||
| Evaluation Method | MTT assay | ||||
| Administration Dosage | 25 μg/ml | ||||
| MOA of PDC |
In this work, we reported doxorubicin-peptide conjugates (DPCs) with an extracellular tumor acid-responsive sphere-fiber transformation for enhanced residence in tumors. As illustrated in Scheme Scheme1,1, the chemotherapy drug doxorubicin (DOX) was coupled with a peptide (KIGLFRWR) to design a DPC molecule with assembly ability. First, the DPCs, driven by hydrophobic forces from the hydrophobic drug DOX and the IGL fragment, can form spherical DPC nanoparticles (DPC-NPs). Then, along with hydrogen bond between peptides, the aromatic amino acids F and W give the DPC-NPs the ability of self-assembly to DPC-nanofibers (DPC-NFs) due to π-π stacking. The step-by-step assembly process provides opportunities for morphological transformation control. To meet the particle size requirements for intravenous injection, the acid-responsive material 2,3-dimethylmaleic anhydride grafted polylysine, named the functional polylysine graft (FPG), was designed as a shielding layer for DPC-NPs and formed functional doxorubicin-peptide conjugate nanoparticles (FDPC-NPs) by an electrostatic interaction to avoid π-π stacking interactions and hydrogen bond between the DPC-NPs. Therefore, the FDPC-NPs could maintain an appropriate size in blood vessels until entering the tumor stroma by the EPR effect. When the FDPC-NPs passed through the blood vessel and entered the weakly acidic microenvironment of the tumor, the surface potential of the shield was reversed from negative to positive because of acid-sensitive 2,3-dimethylmaleic groups on the FPG. Therefore, FPG would separate from the DPC-NPs because of the mutual repulsion effect from the like charges. Then, DPC-NPs self-assembled into DPC-NFs, thereby staying in the tumor region for a long time. After that, the fibers degraded gradually and free drug penetrated into tumor cells, exerting sustained anti-tumor effect. This study is original and provides new ideas for the design of targeted and long-acting drug delivery systems for tumor therapy.
Click to Show/Hide
|
||||
| Description |
Both the peptide and FPG exhibited no obvious cytotoxicity, while FDPC-NPs and DOX displayed cytotoxicity against tumor cells (IC50 DOX = 2.965 μg/mL; IC50 FDPC-NPs = 5.896 μg/mL) (Figure (Figure6A).6A).
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [979] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability | 40.00% | |||
| Evaluation Method | MTT assay | ||||
| Administration Dosage | 10 μg/ml | ||||
| MOA of PDC |
In this work, we reported doxorubicin-peptide conjugates (DPCs) with an extracellular tumor acid-responsive sphere-fiber transformation for enhanced residence in tumors. As illustrated in Scheme Scheme1,1, the chemotherapy drug doxorubicin (DOX) was coupled with a peptide (KIGLFRWR) to design a DPC molecule with assembly ability. First, the DPCs, driven by hydrophobic forces from the hydrophobic drug DOX and the IGL fragment, can form spherical DPC nanoparticles (DPC-NPs). Then, along with hydrogen bond between peptides, the aromatic amino acids F and W give the DPC-NPs the ability of self-assembly to DPC-nanofibers (DPC-NFs) due to π-π stacking. The step-by-step assembly process provides opportunities for morphological transformation control. To meet the particle size requirements for intravenous injection, the acid-responsive material 2,3-dimethylmaleic anhydride grafted polylysine, named the functional polylysine graft (FPG), was designed as a shielding layer for DPC-NPs and formed functional doxorubicin-peptide conjugate nanoparticles (FDPC-NPs) by an electrostatic interaction to avoid π-π stacking interactions and hydrogen bond between the DPC-NPs. Therefore, the FDPC-NPs could maintain an appropriate size in blood vessels until entering the tumor stroma by the EPR effect. When the FDPC-NPs passed through the blood vessel and entered the weakly acidic microenvironment of the tumor, the surface potential of the shield was reversed from negative to positive because of acid-sensitive 2,3-dimethylmaleic groups on the FPG. Therefore, FPG would separate from the DPC-NPs because of the mutual repulsion effect from the like charges. Then, DPC-NPs self-assembled into DPC-NFs, thereby staying in the tumor region for a long time. After that, the fibers degraded gradually and free drug penetrated into tumor cells, exerting sustained anti-tumor effect. This study is original and provides new ideas for the design of targeted and long-acting drug delivery systems for tumor therapy.
Click to Show/Hide
|
||||
| Description |
Both the peptide and FPG exhibited no obvious cytotoxicity, while FDPC-NPs and DOX displayed cytotoxicity against tumor cells (IC50 DOX = 2.965 μg/mL; IC50 FDPC-NPs = 5.896 μg/mL) (Figure (Figure6A).6A).
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [979] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability | 50.00% | |||
| Evaluation Method | MTT assay | ||||
| Administration Dosage | 5 μg/ml | ||||
| MOA of PDC |
In this work, we reported doxorubicin-peptide conjugates (DPCs) with an extracellular tumor acid-responsive sphere-fiber transformation for enhanced residence in tumors. As illustrated in Scheme Scheme1,1, the chemotherapy drug doxorubicin (DOX) was coupled with a peptide (KIGLFRWR) to design a DPC molecule with assembly ability. First, the DPCs, driven by hydrophobic forces from the hydrophobic drug DOX and the IGL fragment, can form spherical DPC nanoparticles (DPC-NPs). Then, along with hydrogen bond between peptides, the aromatic amino acids F and W give the DPC-NPs the ability of self-assembly to DPC-nanofibers (DPC-NFs) due to π-π stacking. The step-by-step assembly process provides opportunities for morphological transformation control. To meet the particle size requirements for intravenous injection, the acid-responsive material 2,3-dimethylmaleic anhydride grafted polylysine, named the functional polylysine graft (FPG), was designed as a shielding layer for DPC-NPs and formed functional doxorubicin-peptide conjugate nanoparticles (FDPC-NPs) by an electrostatic interaction to avoid π-π stacking interactions and hydrogen bond between the DPC-NPs. Therefore, the FDPC-NPs could maintain an appropriate size in blood vessels until entering the tumor stroma by the EPR effect. When the FDPC-NPs passed through the blood vessel and entered the weakly acidic microenvironment of the tumor, the surface potential of the shield was reversed from negative to positive because of acid-sensitive 2,3-dimethylmaleic groups on the FPG. Therefore, FPG would separate from the DPC-NPs because of the mutual repulsion effect from the like charges. Then, DPC-NPs self-assembled into DPC-NFs, thereby staying in the tumor region for a long time. After that, the fibers degraded gradually and free drug penetrated into tumor cells, exerting sustained anti-tumor effect. This study is original and provides new ideas for the design of targeted and long-acting drug delivery systems for tumor therapy.
Click to Show/Hide
|
||||
| Description |
Both the peptide and FPG exhibited no obvious cytotoxicity, while FDPC-NPs and DOX displayed cytotoxicity against tumor cells (IC50 DOX = 2.965 μg/mL; IC50 FDPC-NPs = 5.896 μg/mL) (Figure (Figure6A).6A).
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [979] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability | 78.00% | |||
| Evaluation Method | MTT assay | ||||
| Administration Dosage | 2.5 μg/ml | ||||
| MOA of PDC |
In this work, we reported doxorubicin-peptide conjugates (DPCs) with an extracellular tumor acid-responsive sphere-fiber transformation for enhanced residence in tumors. As illustrated in Scheme Scheme1,1, the chemotherapy drug doxorubicin (DOX) was coupled with a peptide (KIGLFRWR) to design a DPC molecule with assembly ability. First, the DPCs, driven by hydrophobic forces from the hydrophobic drug DOX and the IGL fragment, can form spherical DPC nanoparticles (DPC-NPs). Then, along with hydrogen bond between peptides, the aromatic amino acids F and W give the DPC-NPs the ability of self-assembly to DPC-nanofibers (DPC-NFs) due to π-π stacking. The step-by-step assembly process provides opportunities for morphological transformation control. To meet the particle size requirements for intravenous injection, the acid-responsive material 2,3-dimethylmaleic anhydride grafted polylysine, named the functional polylysine graft (FPG), was designed as a shielding layer for DPC-NPs and formed functional doxorubicin-peptide conjugate nanoparticles (FDPC-NPs) by an electrostatic interaction to avoid π-π stacking interactions and hydrogen bond between the DPC-NPs. Therefore, the FDPC-NPs could maintain an appropriate size in blood vessels until entering the tumor stroma by the EPR effect. When the FDPC-NPs passed through the blood vessel and entered the weakly acidic microenvironment of the tumor, the surface potential of the shield was reversed from negative to positive because of acid-sensitive 2,3-dimethylmaleic groups on the FPG. Therefore, FPG would separate from the DPC-NPs because of the mutual repulsion effect from the like charges. Then, DPC-NPs self-assembled into DPC-NFs, thereby staying in the tumor region for a long time. After that, the fibers degraded gradually and free drug penetrated into tumor cells, exerting sustained anti-tumor effect. This study is original and provides new ideas for the design of targeted and long-acting drug delivery systems for tumor therapy.
Click to Show/Hide
|
||||
| Description |
Both the peptide and FPG exhibited no obvious cytotoxicity, while FDPC-NPs and DOX displayed cytotoxicity against tumor cells (IC50 DOX = 2.965 μg/mL; IC50 FDPC-NPs = 5.896 μg/mL) (Figure (Figure6A).6A).
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [979] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability | 85.00% | |||
| Evaluation Method | MTT assay | ||||
| Administration Dosage | 1 μg/ml | ||||
| MOA of PDC |
In this work, we reported doxorubicin-peptide conjugates (DPCs) with an extracellular tumor acid-responsive sphere-fiber transformation for enhanced residence in tumors. As illustrated in Scheme Scheme1,1, the chemotherapy drug doxorubicin (DOX) was coupled with a peptide (KIGLFRWR) to design a DPC molecule with assembly ability. First, the DPCs, driven by hydrophobic forces from the hydrophobic drug DOX and the IGL fragment, can form spherical DPC nanoparticles (DPC-NPs). Then, along with hydrogen bond between peptides, the aromatic amino acids F and W give the DPC-NPs the ability of self-assembly to DPC-nanofibers (DPC-NFs) due to π-π stacking. The step-by-step assembly process provides opportunities for morphological transformation control. To meet the particle size requirements for intravenous injection, the acid-responsive material 2,3-dimethylmaleic anhydride grafted polylysine, named the functional polylysine graft (FPG), was designed as a shielding layer for DPC-NPs and formed functional doxorubicin-peptide conjugate nanoparticles (FDPC-NPs) by an electrostatic interaction to avoid π-π stacking interactions and hydrogen bond between the DPC-NPs. Therefore, the FDPC-NPs could maintain an appropriate size in blood vessels until entering the tumor stroma by the EPR effect. When the FDPC-NPs passed through the blood vessel and entered the weakly acidic microenvironment of the tumor, the surface potential of the shield was reversed from negative to positive because of acid-sensitive 2,3-dimethylmaleic groups on the FPG. Therefore, FPG would separate from the DPC-NPs because of the mutual repulsion effect from the like charges. Then, DPC-NPs self-assembled into DPC-NFs, thereby staying in the tumor region for a long time. After that, the fibers degraded gradually and free drug penetrated into tumor cells, exerting sustained anti-tumor effect. This study is original and provides new ideas for the design of targeted and long-acting drug delivery systems for tumor therapy.
Click to Show/Hide
|
||||
| Description |
Both the peptide and FPG exhibited no obvious cytotoxicity, while FDPC-NPs and DOX displayed cytotoxicity against tumor cells (IC50 DOX = 2.965 μg/mL; IC50 FDPC-NPs = 5.896 μg/mL) (Figure (Figure6A).6A).
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [979] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability | 98.00% | |||
| Evaluation Method | MTT assay | ||||
| Administration Dosage | 0.1 μg/ml | ||||
| MOA of PDC |
In this work, we reported doxorubicin-peptide conjugates (DPCs) with an extracellular tumor acid-responsive sphere-fiber transformation for enhanced residence in tumors. As illustrated in Scheme Scheme1,1, the chemotherapy drug doxorubicin (DOX) was coupled with a peptide (KIGLFRWR) to design a DPC molecule with assembly ability. First, the DPCs, driven by hydrophobic forces from the hydrophobic drug DOX and the IGL fragment, can form spherical DPC nanoparticles (DPC-NPs). Then, along with hydrogen bond between peptides, the aromatic amino acids F and W give the DPC-NPs the ability of self-assembly to DPC-nanofibers (DPC-NFs) due to π-π stacking. The step-by-step assembly process provides opportunities for morphological transformation control. To meet the particle size requirements for intravenous injection, the acid-responsive material 2,3-dimethylmaleic anhydride grafted polylysine, named the functional polylysine graft (FPG), was designed as a shielding layer for DPC-NPs and formed functional doxorubicin-peptide conjugate nanoparticles (FDPC-NPs) by an electrostatic interaction to avoid π-π stacking interactions and hydrogen bond between the DPC-NPs. Therefore, the FDPC-NPs could maintain an appropriate size in blood vessels until entering the tumor stroma by the EPR effect. When the FDPC-NPs passed through the blood vessel and entered the weakly acidic microenvironment of the tumor, the surface potential of the shield was reversed from negative to positive because of acid-sensitive 2,3-dimethylmaleic groups on the FPG. Therefore, FPG would separate from the DPC-NPs because of the mutual repulsion effect from the like charges. Then, DPC-NPs self-assembled into DPC-NFs, thereby staying in the tumor region for a long time. After that, the fibers degraded gradually and free drug penetrated into tumor cells, exerting sustained anti-tumor effect. This study is original and provides new ideas for the design of targeted and long-acting drug delivery systems for tumor therapy.
Click to Show/Hide
|
||||
| Description |
Both the peptide and FPG exhibited no obvious cytotoxicity, while FDPC-NPs and DOX displayed cytotoxicity against tumor cells (IC50 DOX = 2.965 μg/mL; IC50 FDPC-NPs = 5.896 μg/mL) (Figure (Figure6A).6A).
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [979] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Apotosis rate | 22.00% | |||
| Administration Dosage | 2 μM | ||||
| MOA of PDC |
In this work, we reported doxorubicin-peptide conjugates (DPCs) with an extracellular tumor acid-responsive sphere-fiber transformation for enhanced residence in tumors. As illustrated in Scheme Scheme1,1, the chemotherapy drug doxorubicin (DOX) was coupled with a peptide (KIGLFRWR) to design a DPC molecule with assembly ability. First, the DPCs, driven by hydrophobic forces from the hydrophobic drug DOX and the IGL fragment, can form spherical DPC nanoparticles (DPC-NPs). Then, along with hydrogen bond between peptides, the aromatic amino acids F and W give the DPC-NPs the ability of self-assembly to DPC-nanofibers (DPC-NFs) due to π-π stacking. The step-by-step assembly process provides opportunities for morphological transformation control. To meet the particle size requirements for intravenous injection, the acid-responsive material 2,3-dimethylmaleic anhydride grafted polylysine, named the functional polylysine graft (FPG), was designed as a shielding layer for DPC-NPs and formed functional doxorubicin-peptide conjugate nanoparticles (FDPC-NPs) by an electrostatic interaction to avoid π-π stacking interactions and hydrogen bond between the DPC-NPs. Therefore, the FDPC-NPs could maintain an appropriate size in blood vessels until entering the tumor stroma by the EPR effect. When the FDPC-NPs passed through the blood vessel and entered the weakly acidic microenvironment of the tumor, the surface potential of the shield was reversed from negative to positive because of acid-sensitive 2,3-dimethylmaleic groups on the FPG. Therefore, FPG would separate from the DPC-NPs because of the mutual repulsion effect from the like charges. Then, DPC-NPs self-assembled into DPC-NFs, thereby staying in the tumor region for a long time. After that, the fibers degraded gradually and free drug penetrated into tumor cells, exerting sustained anti-tumor effect. This study is original and provides new ideas for the design of targeted and long-acting drug delivery systems for tumor therapy.
Click to Show/Hide
|
||||
| Description |
Additionally, the results from flow cytometry with annexin-V-FITC/PI double staining showed that FDPC-NPs and DOX significantly increased the proportion of apoptotic cells in a concentration-dependent manner, and the two groups exhibited similar cytotoxicity.
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [979] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Apotosis rate | 30.00% | |||
| Administration Dosage | 10 μM | ||||
| MOA of PDC |
In this work, we reported doxorubicin-peptide conjugates (DPCs) with an extracellular tumor acid-responsive sphere-fiber transformation for enhanced residence in tumors. As illustrated in Scheme Scheme1,1, the chemotherapy drug doxorubicin (DOX) was coupled with a peptide (KIGLFRWR) to design a DPC molecule with assembly ability. First, the DPCs, driven by hydrophobic forces from the hydrophobic drug DOX and the IGL fragment, can form spherical DPC nanoparticles (DPC-NPs). Then, along with hydrogen bond between peptides, the aromatic amino acids F and W give the DPC-NPs the ability of self-assembly to DPC-nanofibers (DPC-NFs) due to π-π stacking. The step-by-step assembly process provides opportunities for morphological transformation control. To meet the particle size requirements for intravenous injection, the acid-responsive material 2,3-dimethylmaleic anhydride grafted polylysine, named the functional polylysine graft (FPG), was designed as a shielding layer for DPC-NPs and formed functional doxorubicin-peptide conjugate nanoparticles (FDPC-NPs) by an electrostatic interaction to avoid π-π stacking interactions and hydrogen bond between the DPC-NPs. Therefore, the FDPC-NPs could maintain an appropriate size in blood vessels until entering the tumor stroma by the EPR effect. When the FDPC-NPs passed through the blood vessel and entered the weakly acidic microenvironment of the tumor, the surface potential of the shield was reversed from negative to positive because of acid-sensitive 2,3-dimethylmaleic groups on the FPG. Therefore, FPG would separate from the DPC-NPs because of the mutual repulsion effect from the like charges. Then, DPC-NPs self-assembled into DPC-NFs, thereby staying in the tumor region for a long time. After that, the fibers degraded gradually and free drug penetrated into tumor cells, exerting sustained anti-tumor effect. This study is original and provides new ideas for the design of targeted and long-acting drug delivery systems for tumor therapy.
Click to Show/Hide
|
||||
| Description |
Additionally, the results from flow cytometry with annexin-V-FITC/PI double staining showed that FDPC-NPs and DOX significantly increased the proportion of apoptotic cells in a concentration-dependent manner, and the two groups exhibited similar cytotoxicity.
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [979] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Apotosis rate | 40.00% | |||
| Administration Dosage | 20 μM | ||||
| MOA of PDC |
In this work, we reported doxorubicin-peptide conjugates (DPCs) with an extracellular tumor acid-responsive sphere-fiber transformation for enhanced residence in tumors. As illustrated in Scheme Scheme1,1, the chemotherapy drug doxorubicin (DOX) was coupled with a peptide (KIGLFRWR) to design a DPC molecule with assembly ability. First, the DPCs, driven by hydrophobic forces from the hydrophobic drug DOX and the IGL fragment, can form spherical DPC nanoparticles (DPC-NPs). Then, along with hydrogen bond between peptides, the aromatic amino acids F and W give the DPC-NPs the ability of self-assembly to DPC-nanofibers (DPC-NFs) due to π-π stacking. The step-by-step assembly process provides opportunities for morphological transformation control. To meet the particle size requirements for intravenous injection, the acid-responsive material 2,3-dimethylmaleic anhydride grafted polylysine, named the functional polylysine graft (FPG), was designed as a shielding layer for DPC-NPs and formed functional doxorubicin-peptide conjugate nanoparticles (FDPC-NPs) by an electrostatic interaction to avoid π-π stacking interactions and hydrogen bond between the DPC-NPs. Therefore, the FDPC-NPs could maintain an appropriate size in blood vessels until entering the tumor stroma by the EPR effect. When the FDPC-NPs passed through the blood vessel and entered the weakly acidic microenvironment of the tumor, the surface potential of the shield was reversed from negative to positive because of acid-sensitive 2,3-dimethylmaleic groups on the FPG. Therefore, FPG would separate from the DPC-NPs because of the mutual repulsion effect from the like charges. Then, DPC-NPs self-assembled into DPC-NFs, thereby staying in the tumor region for a long time. After that, the fibers degraded gradually and free drug penetrated into tumor cells, exerting sustained anti-tumor effect. This study is original and provides new ideas for the design of targeted and long-acting drug delivery systems for tumor therapy.
Click to Show/Hide
|
||||
| Description |
Additionally, the results from flow cytometry with annexin-V-FITC/PI double staining showed that FDPC-NPs and DOX significantly increased the proportion of apoptotic cells in a concentration-dependent manner, and the two groups exhibited similar cytotoxicity.
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
HA@PDC-DOX2 [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [980] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Tumer volume | 550 mm3 | |||
| Administration Time | 18 days | ||||
| Administration Dosage | 11 mg/kg | ||||
| MOA of PDC |
In this study, we designed and synthesized a novel peptide-drug conjugate (PDC-DOX2), in which two doxorubicin (DOX) molecules are covalently linked to a modified peptide with two carboxyl groups (Pep-AA). In neutral aqueous solution, PDC-DOX2 can self-assemble into stable spherical micelles due to hydrophilic-hydrophobic interactions. The sphere morphology can provide for the feasibility of intravenous injections of such peptide drug conjugates. PDC-DOX2 nanomicelles are stable spherical structures under neutral conditions, while they aggregate with decreased pH values. The pH value affected the assembly performance of PDC-DOX2 to a certain extent. With a decrease in pH (from a neutral to an acid environment), the morphology transforms from independent nanomicelles to slightly aggregated micelles and then to very aggregate micelles with diameters of nearly 3000 nm. The surfaces of PDC-DOX2 micelles were positively charged due to the lysine and arginine residues in the peptides. To avoid being engulfed by macrophages in plasma and prolong their blood circulation time, we further coated the positively charged micelles with a negatively charged natural polysaccharide shell, hyaluronic acid (HA), to form core-shell structure nanomedicine HA@PDC-DOX2. HA has various advantages, such as biodegradability, non-inflammatory, and non-immunogenicity. In addition, HA-coated nanomicelles allow for enhanced targeting in cancer therapy because HA can interact with overexpressed receptors in cancer cells, such as cluster determinant 44 (CD44), receptor for hyaluronic acid mediated motility (RHAMM) and intercellular adhesion molecule 1 (ICAM-1). Particularly, we found that the amount of HA influences the properties of HA@PDC-DOX2. The particle size of HA@PDC-DOX2 decreases with increasing HA content. The amount of HA can regulate the particle size, and HA@PDC-DOX2 become more stable in solution due to eliminating electrostatic repulsion of PDC-DOX2. The schematic mechanism of HA@PDC-DOX2 is shown in Scheme 1. First, PDC-DOX2 self-assembles into nanomicelles in neutral aqueous solution. Then, HA@PDC-DOX2 is constructed by negative HA shells and positively PDC-DOX2 cores. HA@PDC-DOX2 can deliver DOX into tumor sites via passive and active targeting effects. The core-shell structure HA@PDC-DOX2 nanomedicine showed better treatment effects on hepatocellular carcinoma, compared with PDC-DOX2 micelles and free DOX.
Click to Show/Hide
|
||||
| Description |
For the control group receiving PBS injections, the tumor volume expanded rapidly, whereas the tumor growth of the group receiving free DOX, PDC-DOX2, and HA@PDC-DOX2 could be suppressed to some degree. Among them, the inhibition in the HA@PDC-DOX2 group was the most obvious.
|
||||
| In Vivo Model | H22 tumor-bearing C57BL/6 mice. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [980] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Body weight | 19g | |||
| Administration Time | 18 days | ||||
| Administration Dosage | 11 mg/kg | ||||
| MOA of PDC |
In this study, we designed and synthesized a novel peptide-drug conjugate (PDC-DOX2), in which two doxorubicin (DOX) molecules are covalently linked to a modified peptide with two carboxyl groups (Pep-AA). In neutral aqueous solution, PDC-DOX2 can self-assemble into stable spherical micelles due to hydrophilic-hydrophobic interactions. The sphere morphology can provide for the feasibility of intravenous injections of such peptide drug conjugates. PDC-DOX2 nanomicelles are stable spherical structures under neutral conditions, while they aggregate with decreased pH values. The pH value affected the assembly performance of PDC-DOX2 to a certain extent. With a decrease in pH (from a neutral to an acid environment), the morphology transforms from independent nanomicelles to slightly aggregated micelles and then to very aggregate micelles with diameters of nearly 3000 nm. The surfaces of PDC-DOX2 micelles were positively charged due to the lysine and arginine residues in the peptides. To avoid being engulfed by macrophages in plasma and prolong their blood circulation time, we further coated the positively charged micelles with a negatively charged natural polysaccharide shell, hyaluronic acid (HA), to form core-shell structure nanomedicine HA@PDC-DOX2. HA has various advantages, such as biodegradability, non-inflammatory, and non-immunogenicity. In addition, HA-coated nanomicelles allow for enhanced targeting in cancer therapy because HA can interact with overexpressed receptors in cancer cells, such as cluster determinant 44 (CD44), receptor for hyaluronic acid mediated motility (RHAMM) and intercellular adhesion molecule 1 (ICAM-1). Particularly, we found that the amount of HA influences the properties of HA@PDC-DOX2. The particle size of HA@PDC-DOX2 decreases with increasing HA content. The amount of HA can regulate the particle size, and HA@PDC-DOX2 become more stable in solution due to eliminating electrostatic repulsion of PDC-DOX2. The schematic mechanism of HA@PDC-DOX2 is shown in Scheme 1. First, PDC-DOX2 self-assembles into nanomicelles in neutral aqueous solution. Then, HA@PDC-DOX2 is constructed by negative HA shells and positively PDC-DOX2 cores. HA@PDC-DOX2 can deliver DOX into tumor sites via passive and active targeting effects. The core-shell structure HA@PDC-DOX2 nanomedicine showed better treatment effects on hepatocellular carcinoma, compared with PDC-DOX2 micelles and free DOX.
Click to Show/Hide
|
||||
| Description |
Body weight changes in all of the C57BL/6 mice in treatment groups, presented steady decreases.
|
||||
| In Vivo Model | H22 tumor-bearing C57BL/6 mice. | ||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [980] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Cell viability | 40% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 4 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| MOA of PDC |
In this study, we designed and synthesized a novel peptide-drug conjugate (PDC-DOX2), in which two doxorubicin (DOX) molecules are covalently linked to a modified peptide with two carboxyl groups (Pep-AA). In neutral aqueous solution, PDC-DOX2 can self-assemble into stable spherical micelles due to hydrophilic-hydrophobic interactions. The sphere morphology can provide for the feasibility of intravenous injections of such peptide drug conjugates. PDC-DOX2 nanomicelles are stable spherical structures under neutral conditions, while they aggregate with decreased pH values. The pH value affected the assembly performance of PDC-DOX2 to a certain extent. With a decrease in pH (from a neutral to an acid environment), the morphology transforms from independent nanomicelles to slightly aggregated micelles and then to very aggregate micelles with diameters of nearly 3000 nm. The surfaces of PDC-DOX2 micelles were positively charged due to the lysine and arginine residues in the peptides. To avoid being engulfed by macrophages in plasma and prolong their blood circulation time, we further coated the positively charged micelles with a negatively charged natural polysaccharide shell, hyaluronic acid (HA), to form core-shell structure nanomedicine HA@PDC-DOX2. HA has various advantages, such as biodegradability, non-inflammatory, and non-immunogenicity. In addition, HA-coated nanomicelles allow for enhanced targeting in cancer therapy because HA can interact with overexpressed receptors in cancer cells, such as cluster determinant 44 (CD44), receptor for hyaluronic acid mediated motility (RHAMM) and intercellular adhesion molecule 1 (ICAM-1). Particularly, we found that the amount of HA influences the properties of HA@PDC-DOX2. The particle size of HA@PDC-DOX2 decreases with increasing HA content. The amount of HA can regulate the particle size, and HA@PDC-DOX2 become more stable in solution due to eliminating electrostatic repulsion of PDC-DOX2. The schematic mechanism of HA@PDC-DOX2 is shown in Scheme 1. First, PDC-DOX2 self-assembles into nanomicelles in neutral aqueous solution. Then, HA@PDC-DOX2 is constructed by negative HA shells and positively PDC-DOX2 cores. HA@PDC-DOX2 can deliver DOX into tumor sites via passive and active targeting effects. The core-shell structure HA@PDC-DOX2 nanomedicine showed better treatment effects on hepatocellular carcinoma, compared with PDC-DOX2 micelles and free DOX.
Click to Show/Hide
|
||||
| Description |
All of the samples inhibited tumor cell activity in a dose-dependent manner (0.1-50 μg/mL).The antitumor activity of PDC-DOX2 and HA@PDC-DOX2 was lower than that of free DOX (IC50 of DOX: 3.102 μg/mL, IC50 of PDC-DOX2: 7.449 μg/mL, IC50 of HA@PDC-DOX2: 24.05 μg/mL).
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [980] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Cell viability | 50% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 4 h | ||||
| Administration Dosage | 25 μg/ml | ||||
| MOA of PDC |
In this study, we designed and synthesized a novel peptide-drug conjugate (PDC-DOX2), in which two doxorubicin (DOX) molecules are covalently linked to a modified peptide with two carboxyl groups (Pep-AA). In neutral aqueous solution, PDC-DOX2 can self-assemble into stable spherical micelles due to hydrophilic-hydrophobic interactions. The sphere morphology can provide for the feasibility of intravenous injections of such peptide drug conjugates. PDC-DOX2 nanomicelles are stable spherical structures under neutral conditions, while they aggregate with decreased pH values. The pH value affected the assembly performance of PDC-DOX2 to a certain extent. With a decrease in pH (from a neutral to an acid environment), the morphology transforms from independent nanomicelles to slightly aggregated micelles and then to very aggregate micelles with diameters of nearly 3000 nm. The surfaces of PDC-DOX2 micelles were positively charged due to the lysine and arginine residues in the peptides. To avoid being engulfed by macrophages in plasma and prolong their blood circulation time, we further coated the positively charged micelles with a negatively charged natural polysaccharide shell, hyaluronic acid (HA), to form core-shell structure nanomedicine HA@PDC-DOX2. HA has various advantages, such as biodegradability, non-inflammatory, and non-immunogenicity. In addition, HA-coated nanomicelles allow for enhanced targeting in cancer therapy because HA can interact with overexpressed receptors in cancer cells, such as cluster determinant 44 (CD44), receptor for hyaluronic acid mediated motility (RHAMM) and intercellular adhesion molecule 1 (ICAM-1). Particularly, we found that the amount of HA influences the properties of HA@PDC-DOX2. The particle size of HA@PDC-DOX2 decreases with increasing HA content. The amount of HA can regulate the particle size, and HA@PDC-DOX2 become more stable in solution due to eliminating electrostatic repulsion of PDC-DOX2. The schematic mechanism of HA@PDC-DOX2 is shown in Scheme 1. First, PDC-DOX2 self-assembles into nanomicelles in neutral aqueous solution. Then, HA@PDC-DOX2 is constructed by negative HA shells and positively PDC-DOX2 cores. HA@PDC-DOX2 can deliver DOX into tumor sites via passive and active targeting effects. The core-shell structure HA@PDC-DOX2 nanomedicine showed better treatment effects on hepatocellular carcinoma, compared with PDC-DOX2 micelles and free DOX.
Click to Show/Hide
|
||||
| Description |
All of the samples inhibited tumor cell activity in a dose-dependent manner (0.1-50 μg/mL).The antitumor activity of PDC-DOX2 and HA@PDC-DOX2 was lower than that of free DOX (IC50 of DOX: 3.102 μg/mL, IC50 of PDC-DOX2: 7.449 μg/mL, IC50 of HA@PDC-DOX2: 24.05 μg/mL).
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [980] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Cell viability | 60% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 4 h | ||||
| Administration Dosage | 10 μg/ml | ||||
| MOA of PDC |
In this study, we designed and synthesized a novel peptide-drug conjugate (PDC-DOX2), in which two doxorubicin (DOX) molecules are covalently linked to a modified peptide with two carboxyl groups (Pep-AA). In neutral aqueous solution, PDC-DOX2 can self-assemble into stable spherical micelles due to hydrophilic-hydrophobic interactions. The sphere morphology can provide for the feasibility of intravenous injections of such peptide drug conjugates. PDC-DOX2 nanomicelles are stable spherical structures under neutral conditions, while they aggregate with decreased pH values. The pH value affected the assembly performance of PDC-DOX2 to a certain extent. With a decrease in pH (from a neutral to an acid environment), the morphology transforms from independent nanomicelles to slightly aggregated micelles and then to very aggregate micelles with diameters of nearly 3000 nm. The surfaces of PDC-DOX2 micelles were positively charged due to the lysine and arginine residues in the peptides. To avoid being engulfed by macrophages in plasma and prolong their blood circulation time, we further coated the positively charged micelles with a negatively charged natural polysaccharide shell, hyaluronic acid (HA), to form core-shell structure nanomedicine HA@PDC-DOX2. HA has various advantages, such as biodegradability, non-inflammatory, and non-immunogenicity. In addition, HA-coated nanomicelles allow for enhanced targeting in cancer therapy because HA can interact with overexpressed receptors in cancer cells, such as cluster determinant 44 (CD44), receptor for hyaluronic acid mediated motility (RHAMM) and intercellular adhesion molecule 1 (ICAM-1). Particularly, we found that the amount of HA influences the properties of HA@PDC-DOX2. The particle size of HA@PDC-DOX2 decreases with increasing HA content. The amount of HA can regulate the particle size, and HA@PDC-DOX2 become more stable in solution due to eliminating electrostatic repulsion of PDC-DOX2. The schematic mechanism of HA@PDC-DOX2 is shown in Scheme 1. First, PDC-DOX2 self-assembles into nanomicelles in neutral aqueous solution. Then, HA@PDC-DOX2 is constructed by negative HA shells and positively PDC-DOX2 cores. HA@PDC-DOX2 can deliver DOX into tumor sites via passive and active targeting effects. The core-shell structure HA@PDC-DOX2 nanomedicine showed better treatment effects on hepatocellular carcinoma, compared with PDC-DOX2 micelles and free DOX.
Click to Show/Hide
|
||||
| Description |
All of the samples inhibited tumor cell activity in a dose-dependent manner (0.1-50 μg/mL).The antitumor activity of PDC-DOX2 and HA@PDC-DOX2 was lower than that of free DOX (IC50 of DOX: 3.102 μg/mL, IC50 of PDC-DOX2: 7.449 μg/mL, IC50 of HA@PDC-DOX2: 24.05 μg/mL).
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [980] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Cell viability | 70% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 4 h | ||||
| Administration Dosage | 5 μg/ml | ||||
| MOA of PDC |
In this study, we designed and synthesized a novel peptide-drug conjugate (PDC-DOX2), in which two doxorubicin (DOX) molecules are covalently linked to a modified peptide with two carboxyl groups (Pep-AA). In neutral aqueous solution, PDC-DOX2 can self-assemble into stable spherical micelles due to hydrophilic-hydrophobic interactions. The sphere morphology can provide for the feasibility of intravenous injections of such peptide drug conjugates. PDC-DOX2 nanomicelles are stable spherical structures under neutral conditions, while they aggregate with decreased pH values. The pH value affected the assembly performance of PDC-DOX2 to a certain extent. With a decrease in pH (from a neutral to an acid environment), the morphology transforms from independent nanomicelles to slightly aggregated micelles and then to very aggregate micelles with diameters of nearly 3000 nm. The surfaces of PDC-DOX2 micelles were positively charged due to the lysine and arginine residues in the peptides. To avoid being engulfed by macrophages in plasma and prolong their blood circulation time, we further coated the positively charged micelles with a negatively charged natural polysaccharide shell, hyaluronic acid (HA), to form core-shell structure nanomedicine HA@PDC-DOX2. HA has various advantages, such as biodegradability, non-inflammatory, and non-immunogenicity. In addition, HA-coated nanomicelles allow for enhanced targeting in cancer therapy because HA can interact with overexpressed receptors in cancer cells, such as cluster determinant 44 (CD44), receptor for hyaluronic acid mediated motility (RHAMM) and intercellular adhesion molecule 1 (ICAM-1). Particularly, we found that the amount of HA influences the properties of HA@PDC-DOX2. The particle size of HA@PDC-DOX2 decreases with increasing HA content. The amount of HA can regulate the particle size, and HA@PDC-DOX2 become more stable in solution due to eliminating electrostatic repulsion of PDC-DOX2. The schematic mechanism of HA@PDC-DOX2 is shown in Scheme 1. First, PDC-DOX2 self-assembles into nanomicelles in neutral aqueous solution. Then, HA@PDC-DOX2 is constructed by negative HA shells and positively PDC-DOX2 cores. HA@PDC-DOX2 can deliver DOX into tumor sites via passive and active targeting effects. The core-shell structure HA@PDC-DOX2 nanomedicine showed better treatment effects on hepatocellular carcinoma, compared with PDC-DOX2 micelles and free DOX.
Click to Show/Hide
|
||||
| Description |
All of the samples inhibited tumor cell activity in a dose-dependent manner (0.1-50 μg/mL).The antitumor activity of PDC-DOX2 and HA@PDC-DOX2 was lower than that of free DOX (IC50 of DOX: 3.102 μg/mL, IC50 of PDC-DOX2: 7.449 μg/mL, IC50 of HA@PDC-DOX2: 24.05 μg/mL).
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [980] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Cell viability | 72% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 4 h | ||||
| Administration Dosage | 2.5 μg/ml | ||||
| MOA of PDC |
In this study, we designed and synthesized a novel peptide-drug conjugate (PDC-DOX2), in which two doxorubicin (DOX) molecules are covalently linked to a modified peptide with two carboxyl groups (Pep-AA). In neutral aqueous solution, PDC-DOX2 can self-assemble into stable spherical micelles due to hydrophilic-hydrophobic interactions. The sphere morphology can provide for the feasibility of intravenous injections of such peptide drug conjugates. PDC-DOX2 nanomicelles are stable spherical structures under neutral conditions, while they aggregate with decreased pH values. The pH value affected the assembly performance of PDC-DOX2 to a certain extent. With a decrease in pH (from a neutral to an acid environment), the morphology transforms from independent nanomicelles to slightly aggregated micelles and then to very aggregate micelles with diameters of nearly 3000 nm. The surfaces of PDC-DOX2 micelles were positively charged due to the lysine and arginine residues in the peptides. To avoid being engulfed by macrophages in plasma and prolong their blood circulation time, we further coated the positively charged micelles with a negatively charged natural polysaccharide shell, hyaluronic acid (HA), to form core-shell structure nanomedicine HA@PDC-DOX2. HA has various advantages, such as biodegradability, non-inflammatory, and non-immunogenicity. In addition, HA-coated nanomicelles allow for enhanced targeting in cancer therapy because HA can interact with overexpressed receptors in cancer cells, such as cluster determinant 44 (CD44), receptor for hyaluronic acid mediated motility (RHAMM) and intercellular adhesion molecule 1 (ICAM-1). Particularly, we found that the amount of HA influences the properties of HA@PDC-DOX2. The particle size of HA@PDC-DOX2 decreases with increasing HA content. The amount of HA can regulate the particle size, and HA@PDC-DOX2 become more stable in solution due to eliminating electrostatic repulsion of PDC-DOX2. The schematic mechanism of HA@PDC-DOX2 is shown in Scheme 1. First, PDC-DOX2 self-assembles into nanomicelles in neutral aqueous solution. Then, HA@PDC-DOX2 is constructed by negative HA shells and positively PDC-DOX2 cores. HA@PDC-DOX2 can deliver DOX into tumor sites via passive and active targeting effects. The core-shell structure HA@PDC-DOX2 nanomedicine showed better treatment effects on hepatocellular carcinoma, compared with PDC-DOX2 micelles and free DOX.
Click to Show/Hide
|
||||
| Description |
All of the samples inhibited tumor cell activity in a dose-dependent manner (0.1-50 μg/mL).The antitumor activity of PDC-DOX2 and HA@PDC-DOX2 was lower than that of free DOX (IC50 of DOX: 3.102 μg/mL, IC50 of PDC-DOX2: 7.449 μg/mL, IC50 of HA@PDC-DOX2: 24.05 μg/mL).
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [980] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Cell viability | 75% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 4 h | ||||
| Administration Dosage | 1 μg/ml | ||||
| MOA of PDC |
In this study, we designed and synthesized a novel peptide-drug conjugate (PDC-DOX2), in which two doxorubicin (DOX) molecules are covalently linked to a modified peptide with two carboxyl groups (Pep-AA). In neutral aqueous solution, PDC-DOX2 can self-assemble into stable spherical micelles due to hydrophilic-hydrophobic interactions. The sphere morphology can provide for the feasibility of intravenous injections of such peptide drug conjugates. PDC-DOX2 nanomicelles are stable spherical structures under neutral conditions, while they aggregate with decreased pH values. The pH value affected the assembly performance of PDC-DOX2 to a certain extent. With a decrease in pH (from a neutral to an acid environment), the morphology transforms from independent nanomicelles to slightly aggregated micelles and then to very aggregate micelles with diameters of nearly 3000 nm. The surfaces of PDC-DOX2 micelles were positively charged due to the lysine and arginine residues in the peptides. To avoid being engulfed by macrophages in plasma and prolong their blood circulation time, we further coated the positively charged micelles with a negatively charged natural polysaccharide shell, hyaluronic acid (HA), to form core-shell structure nanomedicine HA@PDC-DOX2. HA has various advantages, such as biodegradability, non-inflammatory, and non-immunogenicity. In addition, HA-coated nanomicelles allow for enhanced targeting in cancer therapy because HA can interact with overexpressed receptors in cancer cells, such as cluster determinant 44 (CD44), receptor for hyaluronic acid mediated motility (RHAMM) and intercellular adhesion molecule 1 (ICAM-1). Particularly, we found that the amount of HA influences the properties of HA@PDC-DOX2. The particle size of HA@PDC-DOX2 decreases with increasing HA content. The amount of HA can regulate the particle size, and HA@PDC-DOX2 become more stable in solution due to eliminating electrostatic repulsion of PDC-DOX2. The schematic mechanism of HA@PDC-DOX2 is shown in Scheme 1. First, PDC-DOX2 self-assembles into nanomicelles in neutral aqueous solution. Then, HA@PDC-DOX2 is constructed by negative HA shells and positively PDC-DOX2 cores. HA@PDC-DOX2 can deliver DOX into tumor sites via passive and active targeting effects. The core-shell structure HA@PDC-DOX2 nanomedicine showed better treatment effects on hepatocellular carcinoma, compared with PDC-DOX2 micelles and free DOX.
Click to Show/Hide
|
||||
| Description |
All of the samples inhibited tumor cell activity in a dose-dependent manner (0.1-50 μg/mL).The antitumor activity of PDC-DOX2 and HA@PDC-DOX2 was lower than that of free DOX (IC50 of DOX: 3.102 μg/mL, IC50 of PDC-DOX2: 7.449 μg/mL, IC50 of HA@PDC-DOX2: 24.05 μg/mL).
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [980] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Cell viability | 78% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 4 h | ||||
| Administration Dosage | 0.1 μg/ml | ||||
| MOA of PDC |
In this study, we designed and synthesized a novel peptide-drug conjugate (PDC-DOX2), in which two doxorubicin (DOX) molecules are covalently linked to a modified peptide with two carboxyl groups (Pep-AA). In neutral aqueous solution, PDC-DOX2 can self-assemble into stable spherical micelles due to hydrophilic-hydrophobic interactions. The sphere morphology can provide for the feasibility of intravenous injections of such peptide drug conjugates. PDC-DOX2 nanomicelles are stable spherical structures under neutral conditions, while they aggregate with decreased pH values. The pH value affected the assembly performance of PDC-DOX2 to a certain extent. With a decrease in pH (from a neutral to an acid environment), the morphology transforms from independent nanomicelles to slightly aggregated micelles and then to very aggregate micelles with diameters of nearly 3000 nm. The surfaces of PDC-DOX2 micelles were positively charged due to the lysine and arginine residues in the peptides. To avoid being engulfed by macrophages in plasma and prolong their blood circulation time, we further coated the positively charged micelles with a negatively charged natural polysaccharide shell, hyaluronic acid (HA), to form core-shell structure nanomedicine HA@PDC-DOX2. HA has various advantages, such as biodegradability, non-inflammatory, and non-immunogenicity. In addition, HA-coated nanomicelles allow for enhanced targeting in cancer therapy because HA can interact with overexpressed receptors in cancer cells, such as cluster determinant 44 (CD44), receptor for hyaluronic acid mediated motility (RHAMM) and intercellular adhesion molecule 1 (ICAM-1). Particularly, we found that the amount of HA influences the properties of HA@PDC-DOX2. The particle size of HA@PDC-DOX2 decreases with increasing HA content. The amount of HA can regulate the particle size, and HA@PDC-DOX2 become more stable in solution due to eliminating electrostatic repulsion of PDC-DOX2. The schematic mechanism of HA@PDC-DOX2 is shown in Scheme 1. First, PDC-DOX2 self-assembles into nanomicelles in neutral aqueous solution. Then, HA@PDC-DOX2 is constructed by negative HA shells and positively PDC-DOX2 cores. HA@PDC-DOX2 can deliver DOX into tumor sites via passive and active targeting effects. The core-shell structure HA@PDC-DOX2 nanomedicine showed better treatment effects on hepatocellular carcinoma, compared with PDC-DOX2 micelles and free DOX.
Click to Show/Hide
|
||||
| Description |
All of the samples inhibited tumor cell activity in a dose-dependent manner (0.1-50 μg/mL).The antitumor activity of PDC-DOX2 and HA@PDC-DOX2 was lower than that of free DOX (IC50 of DOX: 3.102 μg/mL, IC50 of PDC-DOX2: 7.449 μg/mL, IC50 of HA@PDC-DOX2: 24.05 μg/mL).
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
PDC-DOX2 [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [980] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Tumer volume | 580 mm3 | |||
| Administration Time | 18 days | ||||
| Administration Dosage | 11 mg/kg | ||||
| MOA of PDC |
In this study, we designed and synthesized a novel peptide-drug conjugate (PDC-DOX2), in which two doxorubicin (DOX) molecules are covalently linked to a modified peptide with two carboxyl groups (Pep-AA). In neutral aqueous solution, PDC-DOX2 can self-assemble into stable spherical micelles due to hydrophilic-hydrophobic interactions. The sphere morphology can provide for the feasibility of intravenous injections of such peptide drug conjugates. PDC-DOX2 nanomicelles are stable spherical structures under neutral conditions, while they aggregate with decreased pH values. The pH value affected the assembly performance of PDC-DOX2 to a certain extent. With a decrease in pH (from a neutral to an acid environment), the morphology transforms from independent nanomicelles to slightly aggregated micelles and then to very aggregate micelles with diameters of nearly 3000 nm. The surfaces of PDC-DOX2 micelles were positively charged due to the lysine and arginine residues in the peptides. To avoid being engulfed by macrophages in plasma and prolong their blood circulation time, we further coated the positively charged micelles with a negatively charged natural polysaccharide shell, hyaluronic acid (HA), to form core-shell structure nanomedicine HA@PDC-DOX2. HA has various advantages, such as biodegradability, non-inflammatory, and non-immunogenicity. In addition, HA-coated nanomicelles allow for enhanced targeting in cancer therapy because HA can interact with overexpressed receptors in cancer cells, such as cluster determinant 44 (CD44), receptor for hyaluronic acid mediated motility (RHAMM) and intercellular adhesion molecule 1 (ICAM-1). Particularly, we found that the amount of HA influences the properties of HA@PDC-DOX2. The particle size of HA@PDC-DOX2 decreases with increasing HA content. The amount of HA can regulate the particle size, and HA@PDC-DOX2 become more stable in solution due to eliminating electrostatic repulsion of PDC-DOX2. The schematic mechanism of HA@PDC-DOX2 is shown in Scheme 1. First, PDC-DOX2 self-assembles into nanomicelles in neutral aqueous solution. Then, HA@PDC-DOX2 is constructed by negative HA shells and positively PDC-DOX2 cores. HA@PDC-DOX2 can deliver DOX into tumor sites via passive and active targeting effects. The core-shell structure HA@PDC-DOX2 nanomedicine showed better treatment effects on hepatocellular carcinoma, compared with PDC-DOX2 micelles and free DOX.
Click to Show/Hide
|
||||
| Description |
For the control group receiving PBS injections, the tumor volume expanded rapidly, whereas the tumor growth of the group receiving free DOX, PDC-DOX2, and HA@PDC-DOX2 could be suppressed to some degree. Among them, the inhibition in the HA@PDC-DOX2 group was the most obvious.
|
||||
| In Vivo Model | H22 tumor-bearing C57BL/6 mice. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [980] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Body weight | 19g | |||
| Administration Time | 18 days | ||||
| Administration Dosage | 11 mg/kg | ||||
| MOA of PDC |
In this study, we designed and synthesized a novel peptide-drug conjugate (PDC-DOX2), in which two doxorubicin (DOX) molecules are covalently linked to a modified peptide with two carboxyl groups (Pep-AA). In neutral aqueous solution, PDC-DOX2 can self-assemble into stable spherical micelles due to hydrophilic-hydrophobic interactions. The sphere morphology can provide for the feasibility of intravenous injections of such peptide drug conjugates. PDC-DOX2 nanomicelles are stable spherical structures under neutral conditions, while they aggregate with decreased pH values. The pH value affected the assembly performance of PDC-DOX2 to a certain extent. With a decrease in pH (from a neutral to an acid environment), the morphology transforms from independent nanomicelles to slightly aggregated micelles and then to very aggregate micelles with diameters of nearly 3000 nm. The surfaces of PDC-DOX2 micelles were positively charged due to the lysine and arginine residues in the peptides. To avoid being engulfed by macrophages in plasma and prolong their blood circulation time, we further coated the positively charged micelles with a negatively charged natural polysaccharide shell, hyaluronic acid (HA), to form core-shell structure nanomedicine HA@PDC-DOX2. HA has various advantages, such as biodegradability, non-inflammatory, and non-immunogenicity. In addition, HA-coated nanomicelles allow for enhanced targeting in cancer therapy because HA can interact with overexpressed receptors in cancer cells, such as cluster determinant 44 (CD44), receptor for hyaluronic acid mediated motility (RHAMM) and intercellular adhesion molecule 1 (ICAM-1). Particularly, we found that the amount of HA influences the properties of HA@PDC-DOX2. The particle size of HA@PDC-DOX2 decreases with increasing HA content. The amount of HA can regulate the particle size, and HA@PDC-DOX2 become more stable in solution due to eliminating electrostatic repulsion of PDC-DOX2. The schematic mechanism of HA@PDC-DOX2 is shown in Scheme 1. First, PDC-DOX2 self-assembles into nanomicelles in neutral aqueous solution. Then, HA@PDC-DOX2 is constructed by negative HA shells and positively PDC-DOX2 cores. HA@PDC-DOX2 can deliver DOX into tumor sites via passive and active targeting effects. The core-shell structure HA@PDC-DOX2 nanomedicine showed better treatment effects on hepatocellular carcinoma, compared with PDC-DOX2 micelles and free DOX.
Click to Show/Hide
|
||||
| Description |
Body weight changes in all of the C57BL/6 mice in treatment groups, presented steady decreases.
|
||||
| In Vivo Model | H22 tumor-bearing C57BL/6 mice. | ||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [980] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Cell viability | 38% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 4 h | ||||
| Administration Dosage | 50 μg/ml | ||||
| MOA of PDC |
In this study, we designed and synthesized a novel peptide-drug conjugate (PDC-DOX2), in which two doxorubicin (DOX) molecules are covalently linked to a modified peptide with two carboxyl groups (Pep-AA). In neutral aqueous solution, PDC-DOX2 can self-assemble into stable spherical micelles due to hydrophilic-hydrophobic interactions. The sphere morphology can provide for the feasibility of intravenous injections of such peptide drug conjugates. PDC-DOX2 nanomicelles are stable spherical structures under neutral conditions, while they aggregate with decreased pH values. The pH value affected the assembly performance of PDC-DOX2 to a certain extent. With a decrease in pH (from a neutral to an acid environment), the morphology transforms from independent nanomicelles to slightly aggregated micelles and then to very aggregate micelles with diameters of nearly 3000 nm. The surfaces of PDC-DOX2 micelles were positively charged due to the lysine and arginine residues in the peptides. To avoid being engulfed by macrophages in plasma and prolong their blood circulation time, we further coated the positively charged micelles with a negatively charged natural polysaccharide shell, hyaluronic acid (HA), to form core-shell structure nanomedicine HA@PDC-DOX2. HA has various advantages, such as biodegradability, non-inflammatory, and non-immunogenicity. In addition, HA-coated nanomicelles allow for enhanced targeting in cancer therapy because HA can interact with overexpressed receptors in cancer cells, such as cluster determinant 44 (CD44), receptor for hyaluronic acid mediated motility (RHAMM) and intercellular adhesion molecule 1 (ICAM-1). Particularly, we found that the amount of HA influences the properties of HA@PDC-DOX2. The particle size of HA@PDC-DOX2 decreases with increasing HA content. The amount of HA can regulate the particle size, and HA@PDC-DOX2 become more stable in solution due to eliminating electrostatic repulsion of PDC-DOX2. The schematic mechanism of HA@PDC-DOX2 is shown in Scheme 1. First, PDC-DOX2 self-assembles into nanomicelles in neutral aqueous solution. Then, HA@PDC-DOX2 is constructed by negative HA shells and positively PDC-DOX2 cores. HA@PDC-DOX2 can deliver DOX into tumor sites via passive and active targeting effects. The core-shell structure HA@PDC-DOX2 nanomedicine showed better treatment effects on hepatocellular carcinoma, compared with PDC-DOX2 micelles and free DOX.
Click to Show/Hide
|
||||
| Description |
All of the samples inhibited tumor cell activity in a dose-dependent manner (0.1-50 μg/mL).The antitumor activity of PDC-DOX2 and HA@PDC-DOX2 was lower than that of free DOX (IC50 of DOX: 3.102 μg/mL, IC50 of PDC-DOX2: 7.449 μg/mL, IC50 of HA@PDC-DOX2: 24.05 μg/mL).
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [980] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Cell viability | 40% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 4 h | ||||
| Administration Dosage | 25 μg/ml | ||||
| MOA of PDC |
In this study, we designed and synthesized a novel peptide-drug conjugate (PDC-DOX2), in which two doxorubicin (DOX) molecules are covalently linked to a modified peptide with two carboxyl groups (Pep-AA). In neutral aqueous solution, PDC-DOX2 can self-assemble into stable spherical micelles due to hydrophilic-hydrophobic interactions. The sphere morphology can provide for the feasibility of intravenous injections of such peptide drug conjugates. PDC-DOX2 nanomicelles are stable spherical structures under neutral conditions, while they aggregate with decreased pH values. The pH value affected the assembly performance of PDC-DOX2 to a certain extent. With a decrease in pH (from a neutral to an acid environment), the morphology transforms from independent nanomicelles to slightly aggregated micelles and then to very aggregate micelles with diameters of nearly 3000 nm. The surfaces of PDC-DOX2 micelles were positively charged due to the lysine and arginine residues in the peptides. To avoid being engulfed by macrophages in plasma and prolong their blood circulation time, we further coated the positively charged micelles with a negatively charged natural polysaccharide shell, hyaluronic acid (HA), to form core-shell structure nanomedicine HA@PDC-DOX2. HA has various advantages, such as biodegradability, non-inflammatory, and non-immunogenicity. In addition, HA-coated nanomicelles allow for enhanced targeting in cancer therapy because HA can interact with overexpressed receptors in cancer cells, such as cluster determinant 44 (CD44), receptor for hyaluronic acid mediated motility (RHAMM) and intercellular adhesion molecule 1 (ICAM-1). Particularly, we found that the amount of HA influences the properties of HA@PDC-DOX2. The particle size of HA@PDC-DOX2 decreases with increasing HA content. The amount of HA can regulate the particle size, and HA@PDC-DOX2 become more stable in solution due to eliminating electrostatic repulsion of PDC-DOX2. The schematic mechanism of HA@PDC-DOX2 is shown in Scheme 1. First, PDC-DOX2 self-assembles into nanomicelles in neutral aqueous solution. Then, HA@PDC-DOX2 is constructed by negative HA shells and positively PDC-DOX2 cores. HA@PDC-DOX2 can deliver DOX into tumor sites via passive and active targeting effects. The core-shell structure HA@PDC-DOX2 nanomedicine showed better treatment effects on hepatocellular carcinoma, compared with PDC-DOX2 micelles and free DOX.
Click to Show/Hide
|
||||
| Description |
All of the samples inhibited tumor cell activity in a dose-dependent manner (0.1-50 μg/mL).The antitumor activity of PDC-DOX2 and HA@PDC-DOX2 was lower than that of free DOX (IC50 of DOX: 3.102 μg/mL, IC50 of PDC-DOX2: 7.449 μg/mL, IC50 of HA@PDC-DOX2: 24.05 μg/mL).
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [980] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Cell viability | 48% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 4 h | ||||
| Administration Dosage | 10 μg/ml | ||||
| MOA of PDC |
In this study, we designed and synthesized a novel peptide-drug conjugate (PDC-DOX2), in which two doxorubicin (DOX) molecules are covalently linked to a modified peptide with two carboxyl groups (Pep-AA). In neutral aqueous solution, PDC-DOX2 can self-assemble into stable spherical micelles due to hydrophilic-hydrophobic interactions. The sphere morphology can provide for the feasibility of intravenous injections of such peptide drug conjugates. PDC-DOX2 nanomicelles are stable spherical structures under neutral conditions, while they aggregate with decreased pH values. The pH value affected the assembly performance of PDC-DOX2 to a certain extent. With a decrease in pH (from a neutral to an acid environment), the morphology transforms from independent nanomicelles to slightly aggregated micelles and then to very aggregate micelles with diameters of nearly 3000 nm. The surfaces of PDC-DOX2 micelles were positively charged due to the lysine and arginine residues in the peptides. To avoid being engulfed by macrophages in plasma and prolong their blood circulation time, we further coated the positively charged micelles with a negatively charged natural polysaccharide shell, hyaluronic acid (HA), to form core-shell structure nanomedicine HA@PDC-DOX2. HA has various advantages, such as biodegradability, non-inflammatory, and non-immunogenicity. In addition, HA-coated nanomicelles allow for enhanced targeting in cancer therapy because HA can interact with overexpressed receptors in cancer cells, such as cluster determinant 44 (CD44), receptor for hyaluronic acid mediated motility (RHAMM) and intercellular adhesion molecule 1 (ICAM-1). Particularly, we found that the amount of HA influences the properties of HA@PDC-DOX2. The particle size of HA@PDC-DOX2 decreases with increasing HA content. The amount of HA can regulate the particle size, and HA@PDC-DOX2 become more stable in solution due to eliminating electrostatic repulsion of PDC-DOX2. The schematic mechanism of HA@PDC-DOX2 is shown in Scheme 1. First, PDC-DOX2 self-assembles into nanomicelles in neutral aqueous solution. Then, HA@PDC-DOX2 is constructed by negative HA shells and positively PDC-DOX2 cores. HA@PDC-DOX2 can deliver DOX into tumor sites via passive and active targeting effects. The core-shell structure HA@PDC-DOX2 nanomedicine showed better treatment effects on hepatocellular carcinoma, compared with PDC-DOX2 micelles and free DOX.
Click to Show/Hide
|
||||
| Description |
All of the samples inhibited tumor cell activity in a dose-dependent manner (0.1-50 μg/mL).The antitumor activity of PDC-DOX2 and HA@PDC-DOX2 was lower than that of free DOX (IC50 of DOX: 3.102 μg/mL, IC50 of PDC-DOX2: 7.449 μg/mL, IC50 of HA@PDC-DOX2: 24.05 μg/mL).
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [980] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Cell viability | 50% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 4 h | ||||
| Administration Dosage | 5 μg/ml | ||||
| MOA of PDC |
In this study, we designed and synthesized a novel peptide-drug conjugate (PDC-DOX2), in which two doxorubicin (DOX) molecules are covalently linked to a modified peptide with two carboxyl groups (Pep-AA). In neutral aqueous solution, PDC-DOX2 can self-assemble into stable spherical micelles due to hydrophilic-hydrophobic interactions. The sphere morphology can provide for the feasibility of intravenous injections of such peptide drug conjugates. PDC-DOX2 nanomicelles are stable spherical structures under neutral conditions, while they aggregate with decreased pH values. The pH value affected the assembly performance of PDC-DOX2 to a certain extent. With a decrease in pH (from a neutral to an acid environment), the morphology transforms from independent nanomicelles to slightly aggregated micelles and then to very aggregate micelles with diameters of nearly 3000 nm. The surfaces of PDC-DOX2 micelles were positively charged due to the lysine and arginine residues in the peptides. To avoid being engulfed by macrophages in plasma and prolong their blood circulation time, we further coated the positively charged micelles with a negatively charged natural polysaccharide shell, hyaluronic acid (HA), to form core-shell structure nanomedicine HA@PDC-DOX2. HA has various advantages, such as biodegradability, non-inflammatory, and non-immunogenicity. In addition, HA-coated nanomicelles allow for enhanced targeting in cancer therapy because HA can interact with overexpressed receptors in cancer cells, such as cluster determinant 44 (CD44), receptor for hyaluronic acid mediated motility (RHAMM) and intercellular adhesion molecule 1 (ICAM-1). Particularly, we found that the amount of HA influences the properties of HA@PDC-DOX2. The particle size of HA@PDC-DOX2 decreases with increasing HA content. The amount of HA can regulate the particle size, and HA@PDC-DOX2 become more stable in solution due to eliminating electrostatic repulsion of PDC-DOX2. The schematic mechanism of HA@PDC-DOX2 is shown in Scheme 1. First, PDC-DOX2 self-assembles into nanomicelles in neutral aqueous solution. Then, HA@PDC-DOX2 is constructed by negative HA shells and positively PDC-DOX2 cores. HA@PDC-DOX2 can deliver DOX into tumor sites via passive and active targeting effects. The core-shell structure HA@PDC-DOX2 nanomedicine showed better treatment effects on hepatocellular carcinoma, compared with PDC-DOX2 micelles and free DOX.
Click to Show/Hide
|
||||
| Description |
All of the samples inhibited tumor cell activity in a dose-dependent manner (0.1-50 μg/mL).The antitumor activity of PDC-DOX2 and HA@PDC-DOX2 was lower than that of free DOX (IC50 of DOX: 3.102 μg/mL, IC50 of PDC-DOX2: 7.449 μg/mL, IC50 of HA@PDC-DOX2: 24.05 μg/mL).
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [980] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Cell viability | 58% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 4 h | ||||
| Administration Dosage | 2.5 μg/ml | ||||
| MOA of PDC |
In this study, we designed and synthesized a novel peptide-drug conjugate (PDC-DOX2), in which two doxorubicin (DOX) molecules are covalently linked to a modified peptide with two carboxyl groups (Pep-AA). In neutral aqueous solution, PDC-DOX2 can self-assemble into stable spherical micelles due to hydrophilic-hydrophobic interactions. The sphere morphology can provide for the feasibility of intravenous injections of such peptide drug conjugates. PDC-DOX2 nanomicelles are stable spherical structures under neutral conditions, while they aggregate with decreased pH values. The pH value affected the assembly performance of PDC-DOX2 to a certain extent. With a decrease in pH (from a neutral to an acid environment), the morphology transforms from independent nanomicelles to slightly aggregated micelles and then to very aggregate micelles with diameters of nearly 3000 nm. The surfaces of PDC-DOX2 micelles were positively charged due to the lysine and arginine residues in the peptides. To avoid being engulfed by macrophages in plasma and prolong their blood circulation time, we further coated the positively charged micelles with a negatively charged natural polysaccharide shell, hyaluronic acid (HA), to form core-shell structure nanomedicine HA@PDC-DOX2. HA has various advantages, such as biodegradability, non-inflammatory, and non-immunogenicity. In addition, HA-coated nanomicelles allow for enhanced targeting in cancer therapy because HA can interact with overexpressed receptors in cancer cells, such as cluster determinant 44 (CD44), receptor for hyaluronic acid mediated motility (RHAMM) and intercellular adhesion molecule 1 (ICAM-1). Particularly, we found that the amount of HA influences the properties of HA@PDC-DOX2. The particle size of HA@PDC-DOX2 decreases with increasing HA content. The amount of HA can regulate the particle size, and HA@PDC-DOX2 become more stable in solution due to eliminating electrostatic repulsion of PDC-DOX2. The schematic mechanism of HA@PDC-DOX2 is shown in Scheme 1. First, PDC-DOX2 self-assembles into nanomicelles in neutral aqueous solution. Then, HA@PDC-DOX2 is constructed by negative HA shells and positively PDC-DOX2 cores. HA@PDC-DOX2 can deliver DOX into tumor sites via passive and active targeting effects. The core-shell structure HA@PDC-DOX2 nanomedicine showed better treatment effects on hepatocellular carcinoma, compared with PDC-DOX2 micelles and free DOX.
Click to Show/Hide
|
||||
| Description |
All of the samples inhibited tumor cell activity in a dose-dependent manner (0.1-50 μg/mL).The antitumor activity of PDC-DOX2 and HA@PDC-DOX2 was lower than that of free DOX (IC50 of DOX: 3.102 μg/mL, IC50 of PDC-DOX2: 7.449 μg/mL, IC50 of HA@PDC-DOX2: 24.05 μg/mL).
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [980] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Cell viability | 63% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 4 h | ||||
| Administration Dosage | 1 μg/ml | ||||
| MOA of PDC |
In this study, we designed and synthesized a novel peptide-drug conjugate (PDC-DOX2), in which two doxorubicin (DOX) molecules are covalently linked to a modified peptide with two carboxyl groups (Pep-AA). In neutral aqueous solution, PDC-DOX2 can self-assemble into stable spherical micelles due to hydrophilic-hydrophobic interactions. The sphere morphology can provide for the feasibility of intravenous injections of such peptide drug conjugates. PDC-DOX2 nanomicelles are stable spherical structures under neutral conditions, while they aggregate with decreased pH values. The pH value affected the assembly performance of PDC-DOX2 to a certain extent. With a decrease in pH (from a neutral to an acid environment), the morphology transforms from independent nanomicelles to slightly aggregated micelles and then to very aggregate micelles with diameters of nearly 3000 nm. The surfaces of PDC-DOX2 micelles were positively charged due to the lysine and arginine residues in the peptides. To avoid being engulfed by macrophages in plasma and prolong their blood circulation time, we further coated the positively charged micelles with a negatively charged natural polysaccharide shell, hyaluronic acid (HA), to form core-shell structure nanomedicine HA@PDC-DOX2. HA has various advantages, such as biodegradability, non-inflammatory, and non-immunogenicity. In addition, HA-coated nanomicelles allow for enhanced targeting in cancer therapy because HA can interact with overexpressed receptors in cancer cells, such as cluster determinant 44 (CD44), receptor for hyaluronic acid mediated motility (RHAMM) and intercellular adhesion molecule 1 (ICAM-1). Particularly, we found that the amount of HA influences the properties of HA@PDC-DOX2. The particle size of HA@PDC-DOX2 decreases with increasing HA content. The amount of HA can regulate the particle size, and HA@PDC-DOX2 become more stable in solution due to eliminating electrostatic repulsion of PDC-DOX2. The schematic mechanism of HA@PDC-DOX2 is shown in Scheme 1. First, PDC-DOX2 self-assembles into nanomicelles in neutral aqueous solution. Then, HA@PDC-DOX2 is constructed by negative HA shells and positively PDC-DOX2 cores. HA@PDC-DOX2 can deliver DOX into tumor sites via passive and active targeting effects. The core-shell structure HA@PDC-DOX2 nanomedicine showed better treatment effects on hepatocellular carcinoma, compared with PDC-DOX2 micelles and free DOX.
Click to Show/Hide
|
||||
| Description |
All of the samples inhibited tumor cell activity in a dose-dependent manner (0.1-50 μg/mL).The antitumor activity of PDC-DOX2 and HA@PDC-DOX2 was lower than that of free DOX (IC50 of DOX: 3.102 μg/mL, IC50 of PDC-DOX2: 7.449 μg/mL, IC50 of HA@PDC-DOX2: 24.05 μg/mL).
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [980] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Cell viability | 84% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 4 h | ||||
| Administration Dosage | 0.1 μg/ml | ||||
| MOA of PDC |
In this study, we designed and synthesized a novel peptide-drug conjugate (PDC-DOX2), in which two doxorubicin (DOX) molecules are covalently linked to a modified peptide with two carboxyl groups (Pep-AA). In neutral aqueous solution, PDC-DOX2 can self-assemble into stable spherical micelles due to hydrophilic-hydrophobic interactions. The sphere morphology can provide for the feasibility of intravenous injections of such peptide drug conjugates. PDC-DOX2 nanomicelles are stable spherical structures under neutral conditions, while they aggregate with decreased pH values. The pH value affected the assembly performance of PDC-DOX2 to a certain extent. With a decrease in pH (from a neutral to an acid environment), the morphology transforms from independent nanomicelles to slightly aggregated micelles and then to very aggregate micelles with diameters of nearly 3000 nm. The surfaces of PDC-DOX2 micelles were positively charged due to the lysine and arginine residues in the peptides. To avoid being engulfed by macrophages in plasma and prolong their blood circulation time, we further coated the positively charged micelles with a negatively charged natural polysaccharide shell, hyaluronic acid (HA), to form core-shell structure nanomedicine HA@PDC-DOX2. HA has various advantages, such as biodegradability, non-inflammatory, and non-immunogenicity. In addition, HA-coated nanomicelles allow for enhanced targeting in cancer therapy because HA can interact with overexpressed receptors in cancer cells, such as cluster determinant 44 (CD44), receptor for hyaluronic acid mediated motility (RHAMM) and intercellular adhesion molecule 1 (ICAM-1). Particularly, we found that the amount of HA influences the properties of HA@PDC-DOX2. The particle size of HA@PDC-DOX2 decreases with increasing HA content. The amount of HA can regulate the particle size, and HA@PDC-DOX2 become more stable in solution due to eliminating electrostatic repulsion of PDC-DOX2. The schematic mechanism of HA@PDC-DOX2 is shown in Scheme 1. First, PDC-DOX2 self-assembles into nanomicelles in neutral aqueous solution. Then, HA@PDC-DOX2 is constructed by negative HA shells and positively PDC-DOX2 cores. HA@PDC-DOX2 can deliver DOX into tumor sites via passive and active targeting effects. The core-shell structure HA@PDC-DOX2 nanomedicine showed better treatment effects on hepatocellular carcinoma, compared with PDC-DOX2 micelles and free DOX.
Click to Show/Hide
|
||||
| Description |
All of the samples inhibited tumor cell activity in a dose-dependent manner (0.1-50 μg/mL).The antitumor activity of PDC-DOX2 and HA@PDC-DOX2 was lower than that of free DOX (IC50 of DOX: 3.102 μg/mL, IC50 of PDC-DOX2: 7.449 μg/mL, IC50 of HA@PDC-DOX2: 24.05 μg/mL).
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
TPP-DOX-AP2H [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [981] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Average tumor volume shrunk | 55% | |||
| Administration Time | 18 days | ||||
| Administration Dosage | 20 µM | ||||
| MOA of PDC |
Initiated by the interaction between AP2H and membrane-anchored LAPTM4B protein, PDC specifically recognized and bound cancer cells. The receptor-mediated endocytic pathway then led to the delivery of PDC to lysosomes. The low pH environment in lysosomes efficiently cut the hydrazone bond and released TPP-DOX. The successful escape from lysosomes enabled further transportation of TPP-DOX to mitochondria directed by targeting group TPP. Because TPP-DOX is highly active in producing ROS and mitochondria are prone to oxidative damage, the disruption in the electron transport chain and ATP synthesis finally lead to mitochondrial dysfunction and cell death. With dual-targeting ability, PDC can effectively bypass the interaction with P-gp. Meanwhile, due to the energy-dependent expression feature of P-gp, the damage to mitochondria inhibit P-gp expression, thus inhibit the efflux. (9) The targeted and mitochondria interrupted cell damaging pathway allow PDC to exert toxicity both wild type and drug resistant
Click to Show/Hide
|
||||
| Description |
The average tumor volume shrunk by 55% at day 18 compared with that of the control group (Figure 4c).
|
||||
| In Vivo Model | HepG2 tumor xenograft model. | ||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [981] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 15 µM | |||
| MOA of PDC |
Initiated by the interaction between AP2H and membrane-anchored LAPTM4B protein, PDC specifically recognized and bound cancer cells. The receptor-mediated endocytic pathway then led to the delivery of PDC to lysosomes. The low pH environment in lysosomes efficiently cut the hydrazone bond and released TPP-DOX. The successful escape from lysosomes enabled further transportation of TPP-DOX to mitochondria directed by targeting group TPP. Because TPP-DOX is highly active in producing ROS and mitochondria are prone to oxidative damage, the disruption in the electron transport chain and ATP synthesis finally lead to mitochondrial dysfunction and cell death. With dual-targeting ability, PDC can effectively bypass the interaction with P-gp. Meanwhile, due to the energy-dependent expression feature of P-gp, the damage to mitochondria inhibit P-gp expression, thus inhibit the efflux. (9) The targeted and mitochondria interrupted cell damaging pathway allow PDC to exert toxicity both wild type and drug resistant
Click to Show/Hide
|
||||
| Description |
For PDC, its IC50 values against MCF-7/WT (15 uM) and MCF-7/ADR (18 uM) are almost the same, confirming its effectiveness in bypass drug resistance.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [981] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 18 µM | |||
| MOA of PDC |
Initiated by the interaction between AP2H and membrane-anchored LAPTM4B protein, PDC specifically recognized and bound cancer cells. The receptor-mediated endocytic pathway then led to the delivery of PDC to lysosomes. The low pH environment in lysosomes efficiently cut the hydrazone bond and released TPP-DOX. The successful escape from lysosomes enabled further transportation of TPP-DOX to mitochondria directed by targeting group TPP. Because TPP-DOX is highly active in producing ROS and mitochondria are prone to oxidative damage, the disruption in the electron transport chain and ATP synthesis finally lead to mitochondrial dysfunction and cell death. With dual-targeting ability, PDC can effectively bypass the interaction with P-gp. Meanwhile, due to the energy-dependent expression feature of P-gp, the damage to mitochondria inhibit P-gp expression, thus inhibit the efflux. (9) The targeted and mitochondria interrupted cell damaging pathway allow PDC to exert toxicity both wild type and drug resistant
Click to Show/Hide
|
||||
| Description |
For PDC, its IC50 values against MCF-7/WT (15 uM) and MCF-7/ADR (18 uM) are almost the same, confirming its effectiveness in bypass drug resistance.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF7/ADR cell | CVCL_0031 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [981] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 17% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 20 µM | ||||
| MOA of PDC |
Initiated by the interaction between AP2H and membrane-anchored LAPTM4B protein, PDC specifically recognized and bound cancer cells. The receptor-mediated endocytic pathway then led to the delivery of PDC to lysosomes. The low pH environment in lysosomes efficiently cut the hydrazone bond and released TPP-DOX. The successful escape from lysosomes enabled further transportation of TPP-DOX to mitochondria directed by targeting group TPP. Because TPP-DOX is highly active in producing ROS and mitochondria are prone to oxidative damage, the disruption in the electron transport chain and ATP synthesis finally lead to mitochondrial dysfunction and cell death. With dual-targeting ability, PDC can effectively bypass the interaction with P-gp. Meanwhile, due to the energy-dependent expression feature of P-gp, the damage to mitochondria inhibit P-gp expression, thus inhibit the efflux. (9) The targeted and mitochondria interrupted cell damaging pathway allow PDC to exert toxicity both wild type and drug resistant
Click to Show/Hide
|
||||
| Description |
PDC killed most HepG2 cells with a high efficiency of 83% and left HEK293 cells unaffected.
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [981] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 40% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 20 µM | ||||
| MOA of PDC |
Initiated by the interaction between AP2H and membrane-anchored LAPTM4B protein, PDC specifically recognized and bound cancer cells. The receptor-mediated endocytic pathway then led to the delivery of PDC to lysosomes. The low pH environment in lysosomes efficiently cut the hydrazone bond and released TPP-DOX. The successful escape from lysosomes enabled further transportation of TPP-DOX to mitochondria directed by targeting group TPP. Because TPP-DOX is highly active in producing ROS and mitochondria are prone to oxidative damage, the disruption in the electron transport chain and ATP synthesis finally lead to mitochondrial dysfunction and cell death. With dual-targeting ability, PDC can effectively bypass the interaction with P-gp. Meanwhile, due to the energy-dependent expression feature of P-gp, the damage to mitochondria inhibit P-gp expression, thus inhibit the efflux. (9) The targeted and mitochondria interrupted cell damaging pathway allow PDC to exert toxicity both wild type and drug resistant
Click to Show/Hide
|
||||
| Description |
PDC maintained almost the same cytotoxicity against MCF-7/ADR cells and MCF-7/WT cells
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [981] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 45% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 20 µM | ||||
| MOA of PDC |
Initiated by the interaction between AP2H and membrane-anchored LAPTM4B protein, PDC specifically recognized and bound cancer cells. The receptor-mediated endocytic pathway then led to the delivery of PDC to lysosomes. The low pH environment in lysosomes efficiently cut the hydrazone bond and released TPP-DOX. The successful escape from lysosomes enabled further transportation of TPP-DOX to mitochondria directed by targeting group TPP. Because TPP-DOX is highly active in producing ROS and mitochondria are prone to oxidative damage, the disruption in the electron transport chain and ATP synthesis finally lead to mitochondrial dysfunction and cell death. With dual-targeting ability, PDC can effectively bypass the interaction with P-gp. Meanwhile, due to the energy-dependent expression feature of P-gp, the damage to mitochondria inhibit P-gp expression, thus inhibit the efflux. (9) The targeted and mitochondria interrupted cell damaging pathway allow PDC to exert toxicity both wild type and drug resistant
Click to Show/Hide
|
||||
| Description |
PDC maintained almost the same cytotoxicity against MCF-7/ADR cells and MCF-7/WT cells
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF7/ADR cell | CVCL_0031 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [981] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 100% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 20 µM | ||||
| MOA of PDC |
Initiated by the interaction between AP2H and membrane-anchored LAPTM4B protein, PDC specifically recognized and bound cancer cells. The receptor-mediated endocytic pathway then led to the delivery of PDC to lysosomes. The low pH environment in lysosomes efficiently cut the hydrazone bond and released TPP-DOX. The successful escape from lysosomes enabled further transportation of TPP-DOX to mitochondria directed by targeting group TPP. Because TPP-DOX is highly active in producing ROS and mitochondria are prone to oxidative damage, the disruption in the electron transport chain and ATP synthesis finally lead to mitochondrial dysfunction and cell death. With dual-targeting ability, PDC can effectively bypass the interaction with P-gp. Meanwhile, due to the energy-dependent expression feature of P-gp, the damage to mitochondria inhibit P-gp expression, thus inhibit the efflux. (9) The targeted and mitochondria interrupted cell damaging pathway allow PDC to exert toxicity both wild type and drug resistant
Click to Show/Hide
|
||||
| Description |
PDC killed most HepG2 cells with a high efficiency of 83% and left HEK293 cells unaffected.
|
||||
| In Vitro Model | Normal | HEK293 cell | CVCL_0045 | ||
DOXKGFRWR [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [982] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Tumor volume | 376 mm3 | |||
| Description |
The antitumor efficacy of the DOX-KGFRWR nanofiber was superior to all other treatments, with a final tumor volume of 376 mm3.
|
||||
| In Vivo Model | SMMC7721 tumorbearing mice. | ||||
| Half life period | 24.52±13.17 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [982] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Percent survival | 50% | |||
| Administration Time | 40 days | ||||
| Description |
DOX-KGFRWR has significantly prolonged survival rates.
|
||||
| In Vivo Model | SMMC7721 tumorbearing mice. | ||||
| Half life period | 24.52±13.17 h | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [982] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Percent survival | > 40 days | |||
| Administration Time | 30 days | ||||
| Description |
DOX-KGFRWR has significantly prolonged survival rates.
|
||||
| In Vivo Model | SMMC7721 tumorbearing mice. | ||||
| Half life period | 24.52±13.17 h | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [982] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 5.27 µM | |||
| Description |
The IC50 values for KGFRWR, DOX, and DOX-KGFRWR against the MMP2 enzyme were 14.19, 8.68, and 5.27 10-6 m, respectively.
|
||||
| In Vivo Model | SMMC7721 pulmonary metastatic mouse model. | ||||
| Half life period | 24.52±13.17 h | ||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [982] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Migration rates | 19.90% | |||
| Description |
Cells treated with DOX and DOX-KGFRWR exhibited markedly decreased migration, with migration rates of 39.4% and 19.9%, respectively, compared with those in the control, indicating that DOX-KGWRFR exerted a stronger inhibiting effect on migration than DOX.
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Half life period | 24.52±13.17 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [982] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 34.55 µg mL-1 | |||
| Evaluation Method | MTT assay | ||||
| Description |
The IC50 values for DOX-KGFRWR is 34.55 μg mL-1.
|
||||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Half life period | 24.52±13.17 h | ||||
ABD-RPARPAR-DOXO [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [983] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Retarded the tumor growth rate | 47.09% | |||
| Administration Time | 20 days | ||||
| Administration Dosage | 3.0 mg/kg doxorubicin equivalent | ||||
| Description |
The doxorubicin, DOXO-EMCH, ABD-RGDK-DOXO, and ABD-RPARPAR-DOXO retarded the tumor growth rate by 24.28, 25.67, 47.78, and 47.09% on day 20, respectively.
|
||||
| In Vivo Model | Tumor-bearing BALB/c nude mice. | ||||
| Half life period | 23.88 ± 0.94 h | ||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [983] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 11.21 ± 4.54 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Description |
Whereas conjugation of a peptide would hinder the passive diffusion route, resulting in higher IC50 values for the peptide-DOX-conjugates (9.27 ± 3.56 uM for ABD-RGDK-DOXO and 11.21 ± 4.54 uM for ABD-RPARPAR-DOXO, respectively).
|
||||
| In Vitro Model | Lung adenocarcinoma | A-549 cell | CVCL_0023 | ||
| Half life period | 23.88 ± 0.94 h | ||||
ABD-RGDK-DOXO [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [983] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Retarded the tumor growth rate | 47.78% | |||
| Administration Time | 20 days | ||||
| Administration Dosage | 3.0 mg/kg doxorubicin equivalent | ||||
| Description |
The doxorubicin, DOXO-EMCH, ABD-RGDK-DOXO, and ABD-RPARPAR-DOXO retarded the tumor growth rate by 24.28, 25.67, 47.78, and 47.09% on day 20, respectively.
|
||||
| In Vivo Model | Tumor-bearing BALB/c nude mice. | ||||
| Half life period | 23.67 ± 0.35 h | ||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [983] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 9.27 ± 3.56 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Description |
Whereas conjugation of a peptide would hinder the passive diffusion route, resulting in higher IC50 values for the peptide-DOX-conjugates (9.27 ± 3.56 uM for ABD-RGDK-DOXO and 11.21 ± 4.54 uM for ABD-RPARPAR-DOXO, respectively).
|
||||
| In Vitro Model | Lung adenocarcinoma | A-549 cell | CVCL_0023 | ||
| Half life period | 23.67 ± 0.35 h | ||||
E1-3 doxorubicin [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [984] | ||||
| Indication | Medulloblastoma | ||||
| Efficacy Data | Blood-brain barrier permeability | 8 | |||
| Administration Time | 30 min | ||||
| Description |
Notably, its permeability efficiency was significantly higher compared to free doxorubicin (5) (36.93 ± 0.7 uM and 28.93 ± 0.2 uM, respectively, p < 0.001) at 120 min post-treatment (Figure 8D).
|
||||
| In Vivo Model | Blood brain barrier model. | ||||
| In Vitro Model | Normal | HBEC-5i cell | CVCL_4D10 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [984] | ||||
| Indication | Medulloblastoma | ||||
| Efficacy Data | Blood-brain barrier permeability | 20 | |||
| Administration Time | 60 min | ||||
| Description |
Notably, its permeability efficiency was significantly higher compared to free doxorubicin (5) (36.93 ± 0.7 uM and 28.93 ± 0.2 uM, respectively, p < 0.001) at 120 min post-treatment (Figure 8D).
|
||||
| In Vivo Model | Blood brain barrier model. | ||||
| In Vitro Model | Normal | HBEC-5i cell | CVCL_4D10 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [984] | ||||
| Indication | Medulloblastoma | ||||
| Efficacy Data | Blood-brain barrier permeability | 37 | |||
| Administration Time | 120 min | ||||
| Description |
Notably, its permeability efficiency was significantly higher compared to free doxorubicin (5) (36.93 ± 0.7 uM and 28.93 ± 0.2 uM, respectively, p < 0.001) at 120 min post-treatment (Figure 8D).
|
||||
| In Vivo Model | Blood brain barrier model. | ||||
| In Vitro Model | Normal | HBEC-5i cell | CVCL_4D10 | ||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [984] | ||||
| Indication | Medulloblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 25 ± 1.22 nM | |||
| Administration Time | 72 h | ||||
| Description |
This was confirmed with E1-7 doxorubicin conjugate (4) displaying a 5-fold reduction in cytotoxicity compared to E1-3 doxorubicin conjugate (3) (IC50 values of 130 ± 1.27 nM and 25 ± 1.22 nM, respectively) and 14-fold reduction in cytotoxicity compared to free doxorubicin (5) (IC50 values of 130 ± 1.27 nM and 8.8 ± 1.31 nM, respectively) (Figure 7).
Click to Show/Hide
|
||||
| In Vitro Model | Medulloblastoma | Medulloblastoma cell | Homo sapiens | ||
| Experiment 2 Reporting the Activity Data of This PDC | [984] | ||||
| Indication | Medulloblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 842.0 ± 1.10 nM | |||
| Administration Time | 72 h | ||||
| Description |
E1-3 doxorubicin conjugate had a pronounced reduction in cytotoxicity (>72-fold reduction, IC50 value of 10754 ± 1.38 nM) compared to free doxorubicin (IC50 value of 148 ± 1.15 nM) in human fibroblasts. E1-7 doxorubicin was also able to reduce the cytotoxicity of doxorubicin on fibroblasts but not to the same degree as the E1-3 doxorubicin conjugate. E1-3 doxorubicin conjugate (3) also had reduced cytotoxicity compared to free doxorubicin (>7.4-fold reduction, IC50 values of 842 ± 1.10 nM and 113 ± 1.14 nM, respectively) in primary cultures of human astrocytes, a major cell type located in the brain and spinal cord.
Click to Show/Hide
|
||||
| In Vitro Model | Glioma | Brain astrocytes | Homo sapiens | ||
| Experiment 3 Reporting the Activity Data of This PDC | [984] | ||||
| Indication | Medulloblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 10754 ± 1.38 nM | |||
| Administration Time | 72 h | ||||
| Description |
E1-3 doxorubicin conjugate had a pronounced reduction in cytotoxicity (>72-fold reduction, IC50 value of 10754 ± 1.38 nM) compared to free doxorubicin (IC50 value of 148 ± 1.15 nM) in human fibroblasts. E1-7 doxorubicin was also able to reduce the cytotoxicity of doxorubicin on fibroblasts but not to the same degree as the E1-3 doxorubicin conjugate. E1-3 doxorubicin conjugate (3) also had reduced cytotoxicity compared to free doxorubicin (>7.4-fold reduction, IC50 values of 842 ± 1.10 nM and 113 ± 1.14 nM, respectively) in primary cultures of human astrocytes, a major cell type located in the brain and spinal cord.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MRC-5 cell | CVCL_0440 | ||
Peptide 18-4 analog doxorubicin conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [985] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) | 75% | |||
| Administration Time | 30 days | ||||
| Administration Dosage | 2.5 mg DOX equivalent/kg | ||||
| MOA of PDC |
Keratin 1 (K1) is a novel receptor, present on the surface of cancer cells (breast and neuroblastoma) and cells that have undergone oxidative stress, that is being used for targeted drug delivery. We showed that K1 is present on the surface of MCF-7 breast cancer cells, and a comparison of the total K1 levels in cell lysates using Western blot showed that cancer cells (MCF-7 and MDA-MB-435) have a much higher expression of K1 compared to non-cancerous breast tissue derived epithelial (MCF-10A) cells. We engineered peptides, such as linear 18-4 and cyclic analogues, for specific uptake by breast cancer cells (MCF-7 and MDA-MB-231) via cell surface K1 mediated endocytosis. Further, K1 targeting linear peptide 18-4 was used to synthesize four peptide-doxorubicin conjugates with different linker chemistries, such as ester, amide, succinimidyl thioether, and hydrazone. We showed specific uptake of the targeted PDCs via receptor mediated endocytosis in MCF-7 and MDA-MB-435-MDR cancer cells. The PDCs with K1 targeting peptide 18-4 were more cytotoxic to TNBC cells (IC50 1.2-4.7 μM) compared to non-cancerous human mammary epithelial MCF-10A cells (IC50 15.1-38.6 μM), while free drug (doxorubicin) was equally cytotoxic to both cancer and non-cancerous cells (IC50 0.24-1.5 μM). To explore the in vivo efficacy and evaluate the potential of K1 targeting PDC for TNBC treatment, we report here the antitumor activity of one of these peptide-doxorubicin conjugates, where the peptide (18-4) is conjugated to Dox via an acid-sensitive N-acyl hydrazone linker in a mouse model for TNBC. TNBC MDA-MB-231 cells were subcutaneously injected into female NOD/SCID mice to generate TNBC cell-derived xenograft models. Mice treated with the conjugate showed better efficacy, pharmacokinetics, and safety profile compared to the Dox treated mice, supporting the future clinical development of K1 targeted PDCs for treatment of TNBC.
Click to Show/Hide
|
||||
| Description |
After the tumor xenografts reached a volume of around 100150 mm3, mice were randomized into three groups (n = 7), namely, saline (negative control), free doxorubicin (positive control), and hydrazone PDC. A low dose of 2.5 mg/kg Dox or 2.5 mg/kg Dox equivalent for PDC was chosen to study the antitumor efficacy in vivo. Mice were intravenously administered treatment by tail vein every seventh day for six doses. Compared to the saline group, the PDC reduced tumor growth significantly (3.8 times) on day 35 after treatment, whereas the reduction of tumor growth after free Dox treatment was 2.5 times, suggesting the PDC, at the same equivalent dose, was more potent than the free Dox. In addition, the mice treated with PDC remained in overall good health condition, as evidenced by the general appearance, behavior, diet consumption, and body weight. On day 32 during the treatment period, there were no significant differences observed between the PDC and saline groups in the average body weight (p > 0.05). However, the mice treated with Dox showed significant body weight loss (reduced by 11.2%) compared to the PDC group. Twenty-four hours after the last treatment with PDC or free Dox, mice were euthanized. Mice treated with saline were euthanized on day 32 because of the tumor size per IACUC policy and the conditions for euthanasia. Tumor and other major tissues were collected and weighted for further analysis. The mice with PDC treatment exhibited a greater reduction (three times reduction compared to saline) of tumor weight compared to that of free Dox treated (two times reduction compared to saline).
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [991] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.2 µM | |||
| Evaluation Method | MTT assay | ||||
| Description |
The results showed that the cytotoxicity of both conjugates 1 and 2 (IC50 = 1.3 and 2.2 uM, respectively), as well as the free Dox (IC50 = 1.5 uM) on MDA-MB-231 breast cancer cell line, were in the low micromolar range (Figure 4). For the breast cancer cell line MDA-MB-468, the free Dox (IC50 = 0.35 uM) was slightly more toxic compared to conjugates 1 (4.7 uM) and 2 (1.2 uM). For the non-cancerous cell line MCF 10A, the free Dox was highly toxic (IC50 = 0.24 uM) whereas conjugates 1 and 2 displayed much-reduced toxicity (IC50 = 38.6 and 15.1 uM, respectively).
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-468 cell | CVCL_0419 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [991] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 2.2 µM | |||
| Evaluation Method | MTT assay | ||||
| Description |
The results showed that the cytotoxicity of both conjugates 1 and 2 (IC50 = 1.3 and 2.2 uM, respectively), as well as the free Dox (IC50 = 1.5 uM) on MDA-MB-231 breast cancer cell line, were in the low micromolar range (Figure 4). For the breast cancer cell line MDA-MB-468, the free Dox (IC50 = 0.35 uM) was slightly more toxic compared to conjugates 1 (4.7 uM) and 2 (1.2 uM). For the non-cancerous cell line MCF 10A, the free Dox was highly toxic (IC50 = 0.24 uM) whereas conjugates 1 and 2 displayed much-reduced toxicity (IC50 = 38.6 and 15.1 uM, respectively).
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [991] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 15.1 µM | |||
| Evaluation Method | MTT assay | ||||
| Description |
The results showed that the cytotoxicity of both conjugates 1 and 2 (IC50 = 1.3 and 2.2 uM, respectively), as well as the free Dox (IC50 = 1.5 uM) on MDA-MB-231 breast cancer cell line, were in the low micromolar range (Figure 4). For the breast cancer cell line MDA-MB-468, the free Dox (IC50 = 0.35 uM) was slightly more toxic compared to conjugates 1 (4.7 uM) and 2 (1.2 uM). For the non-cancerous cell line MCF 10A, the free Dox was highly toxic (IC50 = 0.24 uM) whereas conjugates 1 and 2 displayed much-reduced toxicity (IC50 = 38.6 and 15.1 uM, respectively).
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
DPV1047-E-Dox [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Relative tumor volume | 29.50% | |||
| Administration Time | Three injections per week for 3 weeks | ||||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
The conjugates 8a-c all showed activity; the most active was 8a. Compound 12a showed no activity and confirmed the in vitro data. In a second experiment, conjugates 9c, 9b, and 8a were compared. Compound 9c was completely inactive and 9b was only partially active, which correlated with the reduction of in vitro cytotoxicity of both 9c and 9b compared to 8a observed in the HCT116 model. The in vivo evaluation of the Vectocell-doxorubicin conjugates confirmed that 8a (15 μmol/kg) is the optimal conjugate with a T/C value of 29.5% (Table 1), whereas T/C obtained with the other conjugates ranged from 34.1% (8c) to 86.2% (9c). Moreover, in this experiment, the conjugate 8a (15 μmol/kg) showed better efficacy than doxorubicin (10, 6.5 μmol/kg: doxorubucin's maximal tolerated dose, MTD), with a T/C of 49.6%. It should be noted that it is possible to administer 8a at twice the MTD of doxorubicin (10), demonstrating that 8a is less toxic and more active than 10. For this reason 8a was therefore selected for further preclinical evaluation in both doxorubicin-sensitive and -resistant models.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT 116 cell | CVCL_0291 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 4 μM | |||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
In the case of Vp followed by DPV1047-E-Dox (8a) treatment, only a moderate increase in sensitivity was observed, with only a 3.6-5-fold increase in activity. The difference between doxorubicin (10) and DPV1047-E-Dox (8a) cytotoxicity following Vp treatment suggests that DPV1047-E-Dox (8a) is a poor substrate for Pgp-mediated cell extrusion, which could result in an increase in the intracellular concentration of the therapeutic molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT 116 cell | CVCL_0291 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 5 μM | |||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
In the case of Vp followed by DPV1047-E-Dox (8a) treatment, only a moderate increase in sensitivity was observed, with only a 3.6-5-fold increase in activity. The difference between doxorubicin (10) and DPV1047-E-Dox (8a) cytotoxicity following Vp treatment suggests that DPV1047-E-Dox (8a) is a poor substrate for Pgp-mediated cell extrusion, which could result in an increase in the intracellular concentration of the therapeutic molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT 116 cell | CVCL_0291 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 6 μM | |||
| Administration Time | Three injections per week for 3 weeks | ||||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
The Vectocell peptides alone showed no cytotoxic activity (data not shown). The Vectocell-doxorubicin conjugates 8a-c showed no loss of cytotoxicity compared to free doxorubicin. Compounds 9b and 9c showed a loss of activity in the HCT116 model but not in the MCF-7/6 cell model, compared to free doxorubicin (10). In contrast, the compounds 12a and 12b showed a significant loss of cytotoxicity in both tumor cell lines (loss of 1-2 logs in IC50). The in vitro data, therefore, suggest that the ester (8a-c) and thioether (9b and 9c) bonds are optimal for Vectocell-doxorubicin conjugate activity and that conjugation to the 3 position via an amide bond (12a and 12b) partially inactivates doxorubicin.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT 116 cell | CVCL_0291 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 11 μM | |||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
In the case of Vp followed by DPV1047-E-Dox (8a) treatment, only a moderate increase in sensitivity was observed, with only a 3.6-5-fold increase in activity. The difference between doxorubicin (10) and DPV1047-E-Dox (8a) cytotoxicity following Vp treatment suggests that DPV1047-E-Dox (8a) is a poor substrate for Pgp-mediated cell extrusion, which could result in an increase in the intracellular concentration of the therapeutic molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT 116 cell | CVCL_0291 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 22 μM | |||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
In the case of Vp followed by DPV1047-E-Dox (8a) treatment, only a moderate increase in sensitivity was observed, with only a 3.6-5-fold increase in activity. The difference between doxorubicin (10) and DPV1047-E-Dox (8a) cytotoxicity following Vp treatment suggests that DPV1047-E-Dox (8a) is a poor substrate for Pgp-mediated cell extrusion, which could result in an increase in the intracellular concentration of the therapeutic molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Uterine sarcoma | MES-SA/Dx5 cell | CVCL_2598 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 29 μM | |||
| Administration Time | Three injections per week for 3 weeks | ||||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
The Vectocell peptides alone showed no cytotoxic activity (data not shown). The Vectocell-doxorubicin conjugates 8a-c showed no loss of cytotoxicity compared to free doxorubicin. Compounds 9b and 9c showed a loss of activity in the HCT116 model but not in the MCF-7/6 cell model, compared to free doxorubicin (10). In contrast, the compounds 12a and 12b showed a significant loss of cytotoxicity in both tumor cell lines (loss of 1-2 logs in IC50). The in vitro data, therefore, suggest that the ester (8a-c) and thioether (9b and 9c) bonds are optimal for Vectocell-doxorubicin conjugate activity and that conjugation to the 3 position via an amide bond (12a and 12b) partially inactivates doxorubicin.
Click to Show/Hide
|
||||
| In Vitro Model | Breast cancer | MCF-7/6 cell | CVCL_W972 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 50 μM | |||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
In these resistant cell lines, DPV1047-E-Dox (8a) always showed better antiproliferative activity than doxorubicin. In MCF7-Adr and MES-SA-dx5 cells, which express high levels of Pgp, the enhanced efficacy of DPV1047-E-Dox (8a) was highly significant compared to that of doxorubicin alone (p < 0.001).
|
||||
| In Vitro Model | Colon adenocarcinoma | HCT 15 cell | CVCL_0292 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 50 μM | |||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
In the case of Vp followed by DPV1047-E-Dox (8a) treatment, only a moderate increase in sensitivity was observed, with only a 3.6-5-fold increase in activity. The difference between doxorubicin (10) and DPV1047-E-Dox (8a) cytotoxicity following Vp treatment suggests that DPV1047-E-Dox (8a) is a poor substrate for Pgp-mediated cell extrusion, which could result in an increase in the intracellular concentration of the therapeutic molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT 116 cell | CVCL_0291 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 52 μM | |||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
In the case of Vp followed by DPV1047-E-Dox (8a) treatment, only a moderate increase in sensitivity was observed, with only a 3.6-5-fold increase in activity. The difference between doxorubicin (10) and DPV1047-E-Dox (8a) cytotoxicity following Vp treatment suggests that DPV1047-E-Dox (8a) is a poor substrate for Pgp-mediated cell extrusion, which could result in an increase in the intracellular concentration of the therapeutic molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7/ADR cell | CVCL_0031 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 79 μM | |||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
In the case of Vp followed by DPV1047-E-Dox (8a) treatment, only a moderate increase in sensitivity was observed, with only a 3.6-5-fold increase in activity. The difference between doxorubicin (10) and DPV1047-E-Dox (8a) cytotoxicity following Vp treatment suggests that DPV1047-E-Dox (8a) is a poor substrate for Pgp-mediated cell extrusion, which could result in an increase in the intracellular concentration of the therapeutic molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Uterine sarcoma | MES-SA/Dx5 cell | CVCL_2598 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 81 μM | |||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
In these resistant cell lines, DPV1047-E-Dox (8a) always showed better antiproliferative activity than doxorubicin. In MCF7-Adr and MES-SA-dx5 cells, which express high levels of Pgp, the enhanced efficacy of DPV1047-E-Dox (8a) was highly significant compared to that of doxorubicin alone (p < 0.001).
|
||||
| In Vitro Model | Uterine sarcoma | MES-SA/Dx5 cell | CVCL_2598 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 133 μM | |||
| Administration Time | Three injections per week for 3 weeks | ||||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
These experiments showed that it is possible to overcome the doxorubicin-resistant phenotype by conjugation of doxorubicin to Vectocell peptides. Vectocell peptides DPV1047 (8a and 12a) and DPV10 (8b and 9b) are able to inhibit the doxorubicin- resistant phenotype of MCF7-Adr cells. However, the compounds 8a and 9b exhibited the greatest cytotoxic activity in the doxorubicin-resistant cell model, although 9b showed a loss of cytotoxic activity in the HCT116 in vitro model (Table 1). The in vitro data therefore suggested that the optimal conjugate for both doxorubicin-sensitive and -resistant models was 8a.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7/ADR cell | CVCL_0031 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 260 μM | |||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
In these resistant cell lines, DPV1047-E-Dox (8a) always showed better antiproliferative activity than doxorubicin. In MCF7-Adr and MES-SA-dx5 cells, which express high levels of Pgp, the enhanced efficacy of DPV1047-E-Dox (8a) was highly significant compared to that of doxorubicin alone (p < 0.001).
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7/ADR cell | CVCL_0031 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 260 μM | |||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
In the case of Vp followed by DPV1047-E-Dox (8a) treatment, only a moderate increase in sensitivity was observed, with only a 3.6-5-fold increase in activity. The difference between doxorubicin (10) and DPV1047-E-Dox (8a) cytotoxicity following Vp treatment suggests that DPV1047-E-Dox (8a) is a poor substrate for Pgp-mediated cell extrusion, which could result in an increase in the intracellular concentration of the therapeutic molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7/ADR cell | CVCL_0031 | ||
DPV7b-E-Dox [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Relative tumor volume | 34.10% | |||
| Administration Time | Three injections per week for 3 weeks | ||||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
The conjugates 8a-c all showed activity; the most active was 8a. Compound 12a showed no activity and confirmed the in vitro data. In a second experiment, conjugates 9c, 9b, and 8a were compared. Compound 9c was completely inactive and 9b was only partially active, which correlated with the reduction of in vitro cytotoxicity of both 9c and 9b compared to 8a observed in the HCT116 model. The in vivo evaluation of the Vectocell-doxorubicin conjugates confirmed that 8a (15 μmol/kg) is the optimal conjugate with a T/C value of 29.5% (Table 1), whereas T/C obtained with the other conjugates ranged from 34.1% (8c) to 86.2% (9c). Moreover, in this experiment, the conjugate 8a (15 μmol/kg) showed better efficacy than doxorubicin (10, 6.5 μmol/kg: doxorubucin's maximal tolerated dose, MTD), with a T/C of 49.6%. It should be noted that it is possible to administer 8a at twice the MTD of doxorubicin (10), demonstrating that 8a is less toxic and more active than 10. For this reason 8a was therefore selected for further preclinical evaluation in both doxorubicin-sensitive and -resistant models.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT 116 cell | CVCL_0291 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 5 μM | |||
| Administration Time | Three injections per week for 3 weeks | ||||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
The Vectocell peptides alone showed no cytotoxic activity (data not shown). The Vectocell-doxorubicin conjugates 8a-c showed no loss of cytotoxicity compared to free doxorubicin. Compounds 9b and 9c showed a loss of activity in the HCT116 model but not in the MCF-7/6 cell model, compared to free doxorubicin (10). In contrast, the compounds 12a and 12b showed a significant loss of cytotoxicity in both tumor cell lines (loss of 1-2 logs in IC50). The in vitro data, therefore, suggest that the ester (8a-c) and thioether (9b and 9c) bonds are optimal for Vectocell-doxorubicin conjugate activity and that conjugation to the 3 position via an amide bond (12a and 12b) partially inactivates doxorubicin.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT 116 cell | CVCL_0291 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 10 μM | |||
| Administration Time | Three injections per week for 3 weeks | ||||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
The Vectocell peptides alone showed no cytotoxic activity (data not shown). The Vectocell-doxorubicin conjugates 8a-c showed no loss of cytotoxicity compared to free doxorubicin. Compounds 9b and 9c showed a loss of activity in the HCT116 model but not in the MCF-7/6 cell model, compared to free doxorubicin (10). In contrast, the compounds 12a and 12b showed a significant loss of cytotoxicity in both tumor cell lines (loss of 1-2 logs in IC50). The in vitro data, therefore, suggest that the ester (8a-c) and thioether (9b and 9c) bonds are optimal for Vectocell-doxorubicin conjugate activity and that conjugation to the 3 position via an amide bond (12a and 12b) partially inactivates doxorubicin.
Click to Show/Hide
|
||||
| In Vitro Model | Breast cancer | MCF-7/6 cell | CVCL_W972 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 1000 μM | |||
| Administration Time | Three injections per week for 3 weeks | ||||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
These experiments showed that it is possible to overcome the doxorubicin-resistant phenotype by conjugation of doxorubicin to Vectocell peptides. Vectocell peptides DPV1047 (8a and 12a) and DPV10 (8b and 9b) are able to inhibit the doxorubicin- resistant phenotype of MCF7-Adr cells. However, the compounds 8a and 9b exhibited the greatest cytotoxic activity in the doxorubicin-resistant cell model, although 9b showed a loss of cytotoxic activity in the HCT116 in vitro model (Table 1). The in vitro data therefore suggested that the optimal conjugate for both doxorubicin-sensitive and -resistant models was 8a.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7/ADR cell | CVCL_0031 | ||
DPV10-E-Dox [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Relative tumor volume | 39% | |||
| Administration Time | Three injections per week for 3 weeks | ||||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
The conjugates 8a-c all showed activity; the most active was 8a. Compound 12a showed no activity and confirmed the in vitro data. In a second experiment, conjugates 9c, 9b, and 8a were compared. Compound 9c was completely inactive and 9b was only partially active, which correlated with the reduction of in vitro cytotoxicity of both 9c and 9b compared to 8a observed in the HCT116 model. The in vivo evaluation of the Vectocell-doxorubicin conjugates confirmed that 8a (15 μmol/kg) is the optimal conjugate with a T/C value of 29.5% (Table 1), whereas T/C obtained with the other conjugates ranged from 34.1% (8c) to 86.2% (9c). Moreover, in this experiment, the conjugate 8a (15 μmol/kg) showed better efficacy than doxorubicin (10, 6.5 μmol/kg: doxorubucin's maximal tolerated dose, MTD), with a T/C of 49.6%. It should be noted that it is possible to administer 8a at twice the MTD of doxorubicin (10), demonstrating that 8a is less toxic and more active than 10. For this reason 8a was therefore selected for further preclinical evaluation in both doxorubicin-sensitive and -resistant models.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT 116 cell | CVCL_0291 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 2 μM | |||
| Administration Time | Three injections per week for 3 weeks | ||||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
The Vectocell peptides alone showed no cytotoxic activity (data not shown). The Vectocell-doxorubicin conjugates 8a-c showed no loss of cytotoxicity compared to free doxorubicin. Compounds 9b and 9c showed a loss of activity in the HCT116 model but not in the MCF-7/6 cell model, compared to free doxorubicin (10). In contrast, the compounds 12a and 12b showed a significant loss of cytotoxicity in both tumor cell lines (loss of 1-2 logs in IC50). The in vitro data, therefore, suggest that the ester (8a-c) and thioether (9b and 9c) bonds are optimal for Vectocell-doxorubicin conjugate activity and that conjugation to the 3 position via an amide bond (12a and 12b) partially inactivates doxorubicin.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT 116 cell | CVCL_0291 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 9 μM | |||
| Administration Time | Three injections per week for 3 weeks | ||||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
The Vectocell peptides alone showed no cytotoxic activity (data not shown). The Vectocell-doxorubicin conjugates 8a-c showed no loss of cytotoxicity compared to free doxorubicin. Compounds 9b and 9c showed a loss of activity in the HCT116 model but not in the MCF-7/6 cell model, compared to free doxorubicin (10). In contrast, the compounds 12a and 12b showed a significant loss of cytotoxicity in both tumor cell lines (loss of 1-2 logs in IC50). The in vitro data, therefore, suggest that the ester (8a-c) and thioether (9b and 9c) bonds are optimal for Vectocell-doxorubicin conjugate activity and that conjugation to the 3 position via an amide bond (12a and 12b) partially inactivates doxorubicin.
Click to Show/Hide
|
||||
| In Vitro Model | Breast cancer | MCF-7/6 cell | CVCL_W972 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 532 μM | |||
| Administration Time | Three injections per week for 3 weeks | ||||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
These experiments showed that it is possible to overcome the doxorubicin-resistant phenotype by conjugation of doxorubicin to Vectocell peptides. Vectocell peptides DPV1047 (8a and 12a) and DPV10 (8b and 9b) are able to inhibit the doxorubicin- resistant phenotype of MCF7-Adr cells. However, the compounds 8a and 9b exhibited the greatest cytotoxic activity in the doxorubicin-resistant cell model, although 9b showed a loss of cytotoxic activity in the HCT116 in vitro model (Table 1). The in vitro data therefore suggested that the optimal conjugate for both doxorubicin-sensitive and -resistant models was 8a.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7/ADR cell | CVCL_0031 | ||
DPV10-TE-Dox [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Relative tumor volume | 55.80% | |||
| Administration Time | Three injections per week for 3 weeks | ||||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
The conjugates 8a-c all showed activity; the most active was 8a. Compound 12a showed no activity and confirmed the in vitro data. In a second experiment, conjugates 9c, 9b, and 8a were compared. Compound 9c was completely inactive and 9b was only partially active, which correlated with the reduction of in vitro cytotoxicity of both 9c and 9b compared to 8a observed in the HCT116 model. The in vivo evaluation of the Vectocell-doxorubicin conjugates confirmed that 8a (15 μmol/kg) is the optimal conjugate with a T/C value of 29.5% (Table 1), whereas T/C obtained with the other conjugates ranged from 34.1% (8c) to 86.2% (9c). Moreover, in this experiment, the conjugate 8a (15 μmol/kg) showed better efficacy than doxorubicin (10, 6.5 μmol/kg: doxorubucin's maximal tolerated dose, MTD), with a T/C of 49.6%. It should be noted that it is possible to administer 8a at twice the MTD of doxorubicin (10), demonstrating that 8a is less toxic and more active than 10. For this reason 8a was therefore selected for further preclinical evaluation in both doxorubicin-sensitive and -resistant models.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT 116 cell | CVCL_0291 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 37 μM | |||
| Administration Time | Three injections per week for 3 weeks | ||||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
The Vectocell peptides alone showed no cytotoxic activity (data not shown). The Vectocell-doxorubicin conjugates 8a-c showed no loss of cytotoxicity compared to free doxorubicin. Compounds 9b and 9c showed a loss of activity in the HCT116 model but not in the MCF-7/6 cell model, compared to free doxorubicin (10). In contrast, the compounds 12a and 12b showed a significant loss of cytotoxicity in both tumor cell lines (loss of 1-2 logs in IC50). The in vitro data, therefore, suggest that the ester (8a-c) and thioether (9b and 9c) bonds are optimal for Vectocell-doxorubicin conjugate activity and that conjugation to the 3 position via an amide bond (12a and 12b) partially inactivates doxorubicin.
Click to Show/Hide
|
||||
| In Vitro Model | Breast cancer | MCF-7/6 cell | CVCL_W972 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 121 μM | |||
| Administration Time | Three injections per week for 3 weeks | ||||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
These experiments showed that it is possible to overcome the doxorubicin-resistant phenotype by conjugation of doxorubicin to Vectocell peptides. Vectocell peptides DPV1047 (8a and 12a) and DPV10 (8b and 9b) are able to inhibit the doxorubicin- resistant phenotype of MCF7-Adr cells. However, the compounds 8a and 9b exhibited the greatest cytotoxic activity in the doxorubicin-resistant cell model, although 9b showed a loss of cytotoxic activity in the HCT116 in vitro model (Table 1). The in vitro data therefore suggested that the optimal conjugate for both doxorubicin-sensitive and -resistant models was 8a.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7/ADR cell | CVCL_0031 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 153 μM | |||
| Administration Time | Three injections per week for 3 weeks | ||||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
The Vectocell peptides alone showed no cytotoxic activity (data not shown). The Vectocell-doxorubicin conjugates 8a-c showed no loss of cytotoxicity compared to free doxorubicin. Compounds 9b and 9c showed a loss of activity in the HCT116 model but not in the MCF-7/6 cell model, compared to free doxorubicin (10). In contrast, the compounds 12a and 12b showed a significant loss of cytotoxicity in both tumor cell lines (loss of 1-2 logs in IC50). The in vitro data, therefore, suggest that the ester (8a-c) and thioether (9b and 9c) bonds are optimal for Vectocell-doxorubicin conjugate activity and that conjugation to the 3 position via an amide bond (12a and 12b) partially inactivates doxorubicin.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT 116 cell | CVCL_0291 | ||
DPV1047-A-Dox [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Relative tumor volume | 79.70% | |||
| Administration Time | Three injections per week for 3 weeks | ||||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
The conjugates 8a-c all showed activity; the most active was 8a. Compound 12a showed no activity and confirmed the in vitro data. In a second experiment, conjugates 9c, 9b, and 8a were compared. Compound 9c was completely inactive and 9b was only partially active, which correlated with the reduction of in vitro cytotoxicity of both 9c and 9b compared to 8a observed in the HCT116 model. The in vivo evaluation of the Vectocell-doxorubicin conjugates confirmed that 8a (15 μmol/kg) is the optimal conjugate with a T/C value of 29.5% (Table 1), whereas T/C obtained with the other conjugates ranged from 34.1% (8c) to 86.2% (9c). Moreover, in this experiment, the conjugate 8a (15 μmol/kg) showed better efficacy than doxorubicin (10, 6.5 μmol/kg: doxorubucin's maximal tolerated dose, MTD), with a T/C of 49.6%. It should be noted that it is possible to administer 8a at twice the MTD of doxorubicin (10), demonstrating that 8a is less toxic and more active than 10. For this reason 8a was therefore selected for further preclinical evaluation in both doxorubicin-sensitive and -resistant models.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT 116 cell | CVCL_0291 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | . Not detected | |||
| Administration Time | Three injections per week for 3 weeks | ||||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
The Vectocell peptides alone showed no cytotoxic activity (data not shown). The Vectocell-doxorubicin conjugates 8a-c showed no loss of cytotoxicity compared to free doxorubicin. Compounds 9b and 9c showed a loss of activity in the HCT116 model but not in the MCF-7/6 cell model, compared to free doxorubicin (10). In contrast, the compounds 12a and 12b showed a significant loss of cytotoxicity in both tumor cell lines (loss of 1-2 logs in IC50). The in vitro data, therefore, suggest that the ester (8a-c) and thioether (9b and 9c) bonds are optimal for Vectocell-doxorubicin conjugate activity and that conjugation to the 3 position via an amide bond (12a and 12b) partially inactivates doxorubicin.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT 116 cell | CVCL_0291 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 108 μM | |||
| Administration Time | Three injections per week for 3 weeks | ||||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
The Vectocell peptides alone showed no cytotoxic activity (data not shown). The Vectocell-doxorubicin conjugates 8a-c showed no loss of cytotoxicity compared to free doxorubicin. Compounds 9b and 9c showed a loss of activity in the HCT116 model but not in the MCF-7/6 cell model, compared to free doxorubicin (10). In contrast, the compounds 12a and 12b showed a significant loss of cytotoxicity in both tumor cell lines (loss of 1-2 logs in IC50). The in vitro data, therefore, suggest that the ester (8a-c) and thioether (9b and 9c) bonds are optimal for Vectocell-doxorubicin conjugate activity and that conjugation to the 3 position via an amide bond (12a and 12b) partially inactivates doxorubicin.
Click to Show/Hide
|
||||
| In Vitro Model | Breast cancer | MCF-7/6 cell | CVCL_W972 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 500 μM | |||
| Administration Time | Three injections per week for 3 weeks | ||||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
These experiments showed that it is possible to overcome the doxorubicin-resistant phenotype by conjugation of doxorubicin to Vectocell peptides. Vectocell peptides DPV1047 (8a and 12a) and DPV10 (8b and 9b) are able to inhibit the doxorubicin- resistant phenotype of MCF7-Adr cells. However, the compounds 8a and 9b exhibited the greatest cytotoxic activity in the doxorubicin-resistant cell model, although 9b showed a loss of cytotoxic activity in the HCT116 in vitro model (Table 1). The in vitro data therefore suggested that the optimal conjugate for both doxorubicin-sensitive and -resistant models was 8a.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7/ADR cell | CVCL_0031 | ||
DPV7b-TE-Dox [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Relative tumor volume | 86.20% | |||
| Administration Time | Three injections per week for 3 weeks | ||||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
The conjugates 8a-c all showed activity; the most active was 8a. Compound 12a showed no activity and confirmed the in vitro data. In a second experiment, conjugates 9c, 9b, and 8a were compared. Compound 9c was completely inactive and 9b was only partially active, which correlated with the reduction of in vitro cytotoxicity of both 9c and 9b compared to 8a observed in the HCT116 model. The in vivo evaluation of the Vectocell-doxorubicin conjugates confirmed that 8a (15 μmol/kg) is the optimal conjugate with a T/C value of 29.5% (Table 1), whereas T/C obtained with the other conjugates ranged from 34.1% (8c) to 86.2% (9c). Moreover, in this experiment, the conjugate 8a (15 μmol/kg) showed better efficacy than doxorubicin (10, 6.5 μmol/kg: doxorubucin's maximal tolerated dose, MTD), with a T/C of 49.6%. It should be noted that it is possible to administer 8a at twice the MTD of doxorubicin (10), demonstrating that 8a is less toxic and more active than 10. For this reason 8a was therefore selected for further preclinical evaluation in both doxorubicin-sensitive and -resistant models.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT 116 cell | CVCL_0291 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 17 μM | |||
| Administration Time | Three injections per week for 3 weeks | ||||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
The Vectocell peptides alone showed no cytotoxic activity (data not shown). The Vectocell-doxorubicin conjugates 8a-c showed no loss of cytotoxicity compared to free doxorubicin. Compounds 9b and 9c showed a loss of activity in the HCT116 model but not in the MCF-7/6 cell model, compared to free doxorubicin (10). In contrast, the compounds 12a and 12b showed a significant loss of cytotoxicity in both tumor cell lines (loss of 1-2 logs in IC50). The in vitro data, therefore, suggest that the ester (8a-c) and thioether (9b and 9c) bonds are optimal for Vectocell-doxorubicin conjugate activity and that conjugation to the 3 position via an amide bond (12a and 12b) partially inactivates doxorubicin.
Click to Show/Hide
|
||||
| In Vitro Model | Breast cancer | MCF-7/6 cell | CVCL_W972 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 30 μM | |||
| Administration Time | Three injections per week for 3 weeks | ||||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
The Vectocell peptides alone showed no cytotoxic activity (data not shown). The Vectocell-doxorubicin conjugates 8a-c showed no loss of cytotoxicity compared to free doxorubicin. Compounds 9b and 9c showed a loss of activity in the HCT116 model but not in the MCF-7/6 cell model, compared to free doxorubicin (10). In contrast, the compounds 12a and 12b showed a significant loss of cytotoxicity in both tumor cell lines (loss of 1-2 logs in IC50). The in vitro data, therefore, suggest that the ester (8a-c) and thioether (9b and 9c) bonds are optimal for Vectocell-doxorubicin conjugate activity and that conjugation to the 3 position via an amide bond (12a and 12b) partially inactivates doxorubicin.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT 116 cell | CVCL_0291 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 862 μM | |||
| Administration Time | Three injections per week for 3 weeks | ||||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
These experiments showed that it is possible to overcome the doxorubicin-resistant phenotype by conjugation of doxorubicin to Vectocell peptides. Vectocell peptides DPV1047 (8a and 12a) and DPV10 (8b and 9b) are able to inhibit the doxorubicin- resistant phenotype of MCF7-Adr cells. However, the compounds 8a and 9b exhibited the greatest cytotoxic activity in the doxorubicin-resistant cell model, although 9b showed a loss of cytotoxic activity in the HCT116 in vitro model (Table 1). The in vitro data therefore suggested that the optimal conjugate for both doxorubicin-sensitive and -resistant models was 8a.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7/ADR cell | CVCL_0031 | ||
LT7-SS-DOX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Proliferation inhibitory activity | 41.00% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Both conjugates exhibited targeted antiproliferative effects on TfR overexpressed tumor cells and little toxicity to TfR low-expressed normal cells compared with free DOX. Moreover, the DT7-SS-DOX conjugate possessed higher serum stability, more sustained reduction-triggered drug release characteristics, and stronger in vitro antiproliferative activity as compared to LT7-SS-DOX.
Click to Show/Hide
|
||||
| Description |
Even at an equal DOX concentration of 40 μM, the cell viability of the three types of tumor cells after exposure to this conjugate for 48 h were 41.0%, 61.5%, and 67.2%, respectively.
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Proliferation inhibitory activity | 61.50% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Both conjugates exhibited targeted antiproliferative effects on TfR overexpressed tumor cells and little toxicity to TfR low-expressed normal cells compared with free DOX. Moreover, the DT7-SS-DOX conjugate possessed higher serum stability, more sustained reduction-triggered drug release characteristics, and stronger in vitro antiproliferative activity as compared to LT7-SS-DOX.
Click to Show/Hide
|
||||
| Description |
Even at an equal DOX concentration of 40 μM, the cell viability of the three types of tumor cells after exposure to this conjugate for 48 h were 41.0%, 61.5%, and 67.2%, respectively.
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Lung adenocarcinoma | ||||
| Efficacy Data | Proliferation inhibitory activity | 67.20% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Both conjugates exhibited targeted antiproliferative effects on TfR overexpressed tumor cells and little toxicity to TfR low-expressed normal cells compared with free DOX. Moreover, the DT7-SS-DOX conjugate possessed higher serum stability, more sustained reduction-triggered drug release characteristics, and stronger in vitro antiproliferative activity as compared to LT7-SS-DOX.
Click to Show/Hide
|
||||
| Description |
Even at an equal DOX concentration of 40 μM, the cell viability of the three types of tumor cells after exposure to this conjugate for 48 h were 41.0%, 61.5%, and 67.2%, respectively.
|
||||
| In Vitro Model | Lung adenocarcinoma | A-549 cell | CVCL_0023 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Proliferation inhibitory activity | 73.10% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Both conjugates exhibited targeted antiproliferative effects on TfR overexpressed tumor cells and little toxicity to TfR low-expressed normal cells compared with free DOX. Moreover, the DT7-SS-DOX conjugate possessed higher serum stability, more sustained reduction-triggered drug release characteristics, and stronger in vitro antiproliferative activity as compared to LT7-SS-DOX.
Click to Show/Hide
|
||||
| Description |
The proliferation inhibitory activity of LT7-SS-DOX was the weakest among the three drugs because the cell viabilities of U87, HepG2, and A549 cells after incubation with LT7-SS-DOX (equal DOX concentration of 20 μM) for 48 h were 95.1%, 73.1%, and 83.2%, respectively.
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Lung adenocarcinoma | ||||
| Efficacy Data | Proliferation inhibitory activity | 83.20% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Both conjugates exhibited targeted antiproliferative effects on TfR overexpressed tumor cells and little toxicity to TfR low-expressed normal cells compared with free DOX. Moreover, the DT7-SS-DOX conjugate possessed higher serum stability, more sustained reduction-triggered drug release characteristics, and stronger in vitro antiproliferative activity as compared to LT7-SS-DOX.
Click to Show/Hide
|
||||
| Description |
The proliferation inhibitory activity of LT7-SS-DOX was the weakest among the three drugs because the cell viabilities of U87, HepG2, and A549 cells after incubation with LT7-SS-DOX (equal DOX concentration of 20 μM) for 48 h were 95.1%, 73.1%, and 83.2%, respectively.
|
||||
| In Vitro Model | Lung adenocarcinoma | A-549 cell | CVCL_0023 | ||
DT7-SS-DOX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Proliferation inhibitory activity | 95.10% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Both conjugates exhibited targeted antiproliferative effects on TfR overexpressed tumor cells and little toxicity to TfR low-expressed normal cells compared with free DOX. Moreover, the DT7-SS-DOX conjugate possessed higher serum stability, more sustained reduction-triggered drug release characteristics, and stronger in vitro antiproliferative activity as compared to LT7-SS-DOX.
Click to Show/Hide
|
||||
| Description |
The proliferation inhibitory activity of LT7-SS-DOX was the weakest among the three drugs because the cell viabilities of U87, HepG2, and A549 cells after incubation with LT7-SS-DOX (equal DOX concentration of 20 μM) for 48 h were 95.1%, 73.1%, and 83.2%, respectively.
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 5.70 ± 0.22 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Both conjugates exhibited targeted antiproliferative effects on TfR overexpressed tumor cells and little toxicity to TfR low-expressed normal cells compared with free DOX. Moreover, the DT7-SS-DOX conjugate possessed higher serum stability, more sustained reduction-triggered drug release characteristics, and stronger in vitro antiproliferative activity as compared to LT7-SS-DOX.
Click to Show/Hide
|
||||
| Description |
DT7-SS-DOX exhibited good in vitro antiproliferative activity against the three tumor cell lines, with IC50 values of 5.70 ± 0.22 μM (U87), 7.01 ± 1.64 μM (HepG2), and 20.61 ± 4.81 μM (A549), respectively.
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Hepatoblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 7.01 ± 1.64 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Both conjugates exhibited targeted antiproliferative effects on TfR overexpressed tumor cells and little toxicity to TfR low-expressed normal cells compared with free DOX. Moreover, the DT7-SS-DOX conjugate possessed higher serum stability, more sustained reduction-triggered drug release characteristics, and stronger in vitro antiproliferative activity as compared to LT7-SS-DOX.
Click to Show/Hide
|
||||
| Description |
DT7-SS-DOX exhibited good in vitro antiproliferative activity against the three tumor cell lines, with IC50 values of 5.70 ± 0.22 μM (U87), 7.01 ± 1.64 μM (HepG2), and 20.61 ± 4.81 μM (A549), respectively.
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Lung adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 20.61 ± 4.81 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Both conjugates exhibited targeted antiproliferative effects on TfR overexpressed tumor cells and little toxicity to TfR low-expressed normal cells compared with free DOX. Moreover, the DT7-SS-DOX conjugate possessed higher serum stability, more sustained reduction-triggered drug release characteristics, and stronger in vitro antiproliferative activity as compared to LT7-SS-DOX.
Click to Show/Hide
|
||||
| Description |
DT7-SS-DOX exhibited good in vitro antiproliferative activity against the three tumor cell lines, with IC50 values of 5.70 ± 0.22 μM (U87), 7.01 ± 1.64 μM (HepG2), and 20.61 ± 4.81 μM (A549), respectively.
|
||||
| In Vitro Model | Lung adenocarcinoma | A-549 cell | CVCL_0023 | ||
MAHNP-Dox conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [987] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 110.1 ± 12.7 nM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
You et al. developed a unique approach for the treatment of human epidermal growth factor receptor 2 (HER2)-positive breast cancer. A peptide fragment from the heavy chain 3 of the full-length antibody trastuzumab was obtained and termed AHNP. The 12-mer AHNP binds the extracellular domain of HER2 with high affinity and displays similar potency as trastuzumab. The peptide mimetic AHNP was then conjugated via an MMP-2 sensitive linker with doxorubicin (Dox) to form MAHNP-Dox conjugate essentially using ester/amide bonds.
Click to Show/Hide
|
||||
| Description |
Conjugate MAHNP-Dox was compared to Dox using HER2 positive breast cancer cell lines BT474 and SKBR3. Cellular toxicity analysis showed that MAHNP-Dox was more potent than free Dox, as indicated by lower IC50 values for both cell lines. The BT474 and SKBR3 cell lines had an IC50 of 746.8 ± 81.5 nM and 110.1 ± 12.7 nM for MAHNP-DOX and 2075.0 ± 368.0 nM and 172.9 ± 19.2 nM for Dox, respectively.
Click to Show/Hide
|
||||
| Experiment 2 Reporting the Activity Data of This PDC | [987] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 746.8 ± 81.5 nM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
You et al. developed a unique approach for the treatment of human epidermal growth factor receptor 2 (HER2)-positive breast cancer. A peptide fragment from the heavy chain 3 of the full-length antibody trastuzumab was obtained and termed AHNP. The 12-mer AHNP binds the extracellular domain of HER2 with high affinity and displays similar potency as trastuzumab. The peptide mimetic AHNP was then conjugated via an MMP-2 sensitive linker with doxorubicin (Dox) to form MAHNP-Dox conjugate essentially using ester/amide bonds.
Click to Show/Hide
|
||||
| Description |
Conjugate MAHNP-Dox was compared to Dox using HER2 positive breast cancer cell lines BT474 and SKBR3. Cellular toxicity analysis showed that MAHNP-Dox was more potent than free Dox, as indicated by lower IC50 values for both cell lines. The BT474 and SKBR3 cell lines had an IC50 of 746.8 ± 81.5 nM and 110.1 ± 12.7 nM for MAHNP-DOX and 2075.0 ± 368.0 nM and 172.9 ± 19.2 nM for Dox, respectively.
Click to Show/Hide
|
||||
E1-7 doxorubicin [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [984] | ||||
| Indication | Medulloblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 130 ± 1.27 nM | |||
| Administration Time | 72 h | ||||
| Description |
This was confirmed with E1-7 doxorubicin conjugate (4) displaying a 5-fold reduction in cytotoxicity compared to E1-3 doxorubicin conjugate (3) (IC50 values of 130 ± 1.27 nM and 25 ± 1.22 nM, respectively) and 14-fold reduction in cytotoxicity compared to free doxorubicin (5) (IC50 values of 130 ± 1.27 nM and 8.8 ± 1.31 nM, respectively) (Figure 7).
Click to Show/Hide
|
||||
| In Vitro Model | Medulloblastoma | Medulloblastoma cell | Homo sapiens | ||
DPV10-A-Dox [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | . Not detected | |||
| Administration Time | Three injections per week for 3 weeks | ||||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
The conjugates 8a-c all showed activity; the most active was 8a. Compound 12a showed no activity and confirmed the in vitro data. In a second experiment, conjugates 9c, 9b, and 8a were compared. Compound 9c was completely inactive and 9b was only partially active, which correlated with the reduction of in vitro cytotoxicity of both 9c and 9b compared to 8a observed in the HCT116 model. The in vivo evaluation of the Vectocell-doxorubicin conjugates confirmed that 8a (15 μmol/kg) is the optimal conjugate with a T/C value of 29.5% (Table 1), whereas T/C obtained with the other conjugates ranged from 34.1% (8c) to 86.2% (9c). Moreover, in this experiment, the conjugate 8a (15 μmol/kg) showed better efficacy than doxorubicin (10, 6.5 μmol/kg: doxorubucin's maximal tolerated dose, MTD), with a T/C of 49.6%. It should be noted that it is possible to administer 8a at twice the MTD of doxorubicin (10), demonstrating that 8a is less toxic and more active than 10. For this reason 8a was therefore selected for further preclinical evaluation in both doxorubicin-sensitive and -resistant models.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT 116 cell | CVCL_0291 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 114 μM | |||
| Administration Time | Three injections per week for 3 weeks | ||||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
The Vectocell peptides alone showed no cytotoxic activity (data not shown). The Vectocell-doxorubicin conjugates 8a-c showed no loss of cytotoxicity compared to free doxorubicin. Compounds 9b and 9c showed a loss of activity in the HCT116 model but not in the MCF-7/6 cell model, compared to free doxorubicin (10). In contrast, the compounds 12a and 12b showed a significant loss of cytotoxicity in both tumor cell lines (loss of 1-2 logs in IC50). The in vitro data, therefore, suggest that the ester (8a-c) and thioether (9b and 9c) bonds are optimal for Vectocell-doxorubicin conjugate activity and that conjugation to the 3 position via an amide bond (12a and 12b) partially inactivates doxorubicin.
Click to Show/Hide
|
||||
| In Vitro Model | Breast cancer | MCF-7/6 cell | CVCL_W972 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 200 μM | |||
| Administration Time | Three injections per week for 3 weeks | ||||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
The Vectocell peptides alone showed no cytotoxic activity (data not shown). The Vectocell-doxorubicin conjugates 8a-c showed no loss of cytotoxicity compared to free doxorubicin. Compounds 9b and 9c showed a loss of activity in the HCT116 model but not in the MCF-7/6 cell model, compared to free doxorubicin (10). In contrast, the compounds 12a and 12b showed a significant loss of cytotoxicity in both tumor cell lines (loss of 1-2 logs in IC50). The in vitro data, therefore, suggest that the ester (8a-c) and thioether (9b and 9c) bonds are optimal for Vectocell-doxorubicin conjugate activity and that conjugation to the 3 position via an amide bond (12a and 12b) partially inactivates doxorubicin.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT 116 cell | CVCL_0291 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [986] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 625 μM | |||
| Administration Time | Three injections per week for 3 weeks | ||||
| MOA of PDC |
The conjugation of Vectocell peptides to cytotoxic molecules can modify the in vivo distribution of the therapeutic molecules, improving pharmacokinetic properties and/or reducing systemic toxicity by driving tissue and intracellular delivery.2,5 In addition, conjugation of Vectocell peptides to small molecules/drugs may also provide a means of inhibiting the extracellular export of the therapeutic agents by proteins involved in multidrug resistance (MDR). Multidrug resistance can seriously limit cancer chemotherapy treatment, for example, through the overexpression of membrane transporters that mediate unidirectional energy-dependent drug efflux, thus reducing intracellular drug levels. These membrane transporters are normally expressed in high levels within cells involved in detoxification, such as the liver, kidney, and colon. Tumors arising from these cells are often resistant to chemotherapy treatment from their onset, while other tumors can acquire resistance through the induction of MDR transport proteins during treatment. Many inhibitors of MDR transporters have been identified. However, these inhibitors also interfere with the clearance of the cytotoxic drug, resulting in elevated plasma concentrations of the cytotoxic agent and associated toxicity. An alternative approach is to circumvent rather than to directly inhibit MDR mechanisms, by developing anticancer therapies that are not substrates for extracellular export. Doxorubicin, an anthracycline antibiotic, remains among the most widely used cytotoxic agents for the treatment of a broad spectrum of cancers, including breast, stomach, non-Hodgkin's lymphoma, and bladder cancer. As with many cytotoxic drugs, doxorubicin has severe short- and long-term side effects, in this case mostly associated with bone marrow and myocardial cell toxicity. Cardiotoxicity limits the cumulative dosage of doxorubicin to 500-600 mg/m2, which may be a dose at which tumor is still responding to treatment but for which no further doxorubicin treatment can be given. Another drawback of doxorubicin is the emergence of drug resistance that results in the reduction of the intracellular concentration of doxorubicin. The present study aimed to generate novel peptidic-doxorubicin conjugates by use of three Vectocell peptides that differ in terms of their charge, size, and intracellular delivery characteristics and to assess their ability to enhance the therapeutic potential of doxorubicin and to prevent the appearance of MDR. Different conjugation sites and linkers of different stabilities were used to generate Vectocell-doxorubicin conjugates in order to evaluate their effect on efficacy. Chemical routes were developed to allow the conjugation of doxorubicin to Vectocell peptides through ester, thioether, and amide chemical linkers. The ester and thioether involved carbon 14 of doxorubicin, and the amide carbon 3. In vitro and in vivo characterization has defined the optimal conjugate-linker combination that significantly increases efficacy above unconjugated doxorubicin in both doxorubicin-sensitive and -resistant models. The data presented therefore provide in vivo proof of concept for the use of Vectocell peptides to improve the therapeutic index of doxorubicin, and potentially many other cytotoxic or small molecule anticancer drugs.
Click to Show/Hide
|
||||
| Description |
These experiments showed that it is possible to overcome the doxorubicin-resistant phenotype by conjugation of doxorubicin to Vectocell peptides. Vectocell peptides DPV1047 (8a and 12a) and DPV10 (8b and 9b) are able to inhibit the doxorubicin- resistant phenotype of MCF7-Adr cells. However, the compounds 8a and 9b exhibited the greatest cytotoxic activity in the doxorubicin-resistant cell model, although 9b showed a loss of cytotoxic activity in the HCT116 in vitro model (Table 1). The in vitro data therefore suggested that the optimal conjugate for both doxorubicin-sensitive and -resistant models was 8a.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7/ADR cell | CVCL_0031 | ||
[8Lys(Dox-O-glut)]-GnRH-III [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [988] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.1 ± 0.1 µM | |||
| Administration Time | 72 h | ||||
| MOA of PDC |
The efficacy of current chemotherapeutic treatments against most solid tumors is limited by their systemic toxicity, which is partly associated with the cytotoxic properties of agents such as docetaxel or doxorubicin. To avoid or minimize adverse effects from chemotherapeutic molecules, a promising targeted approach is through peptide-drug conjugates (PDCs) that link anticancer molecules to peptides designed to interact with receptors highly expressed on cancer cells, and which can mediate the molecules rapid internalization within those cells. One such receptor is sortilin (SORT1), also known as neurotensin receptor3, a membranebound receptor that belongs to the VPS10P family of receptors. TH19P01 peptide was recently designed to target and exploit SORT1s ligand internalization function. Studies have confirmed that both TH1902 (a docetaxel-TH19P01 conjugate) and TH1904 (a doxorubicin-TH19P01 conjugate) require a SORT1-dependent mechanism of action to exert anticancer activities. In recent preclinical studies performed in immunocompromised animal models, which are unable to produce mature T-cells, TH1902 was effective against several human SORT1-positive xenograft models including triple-negative breast cancer (TNBC), ovarian cancer, and endometrial cancer.
Click to Show/Hide
|
||||
| Description |
The main drug-related toxic side effect of anthracyclines is cardiotoxicity, leading to cardiomyopathy and heart failure. Comparative experiments to determine the cardiotoxicity of peptide-anthracycline conjugates with different linkages might be informative for the other conjugates with different types of drugs as well. For this purpose, human cardiomyocytes (HCM) and human umbilical vein endothelial cells (HUVEC) were used as models. The long-term (0-72 h) cytotoxic effect of sixteen GnRH-based conjugates containing Dox and Dau was determined by real-time impedimetric sensing using the xCELLigence SP system (ACEA Biosciences, San Diego, CA, USA) [75]. The results indicated that the ester-linked GnRH-Dox conjugates, including Zoptarelin Doxorubicin, showed significant toxicity at 100 nM and 1 uM, which was remarkably pronounced on the HCM cells. The cytotoxic effect was comparable to that of the free drug, especially at the highest concentration. In contrast, the conjugates with oxime-linked Dau showed no or only a minor toxicity on both the cell lines (Table 2). These data confirm that the linkage between the payload and homing peptide has a significant influence on early drug release and, consequently, an undesired toxic side effect. We may also conclude that the search for more suitable homing peptides might be more important than the application of cleavable bonds between the drug and the peptide to develop efficient DDSs.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [988] | ||||
| Indication | Colon cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 2.4 ± 0.2 µM | |||
| Administration Time | 72 h | ||||
| MOA of PDC |
The crucial steps during the synthesis of aminooxyacetylated peptides are the incorporation of aminooxyacetic acid (Aoa), including a protecting group, and the final cleavage and the working-up procedure of the Aoa-containing peptide derivatives. In most cases, Boc-protected Aoa is attached to a peptide chain in the last step of solid-phase peptide synthesis. It has been observed that over-acylation (additional Boc-Aoa-OH connected to the Aoa moiety) may occur as the main side reaction. Many approaches have been investigated to overcome this problem, including carbodiimide-mediated one-pot acylation without a base or the application of Boc-Aoa-OSu active ester as an acylating agent, as well as the use of a high excess (8 equiv) of Boc-Aoa-OH and coupling agents for a short acylation time (10 min). Nevertheless, the coupling of the diBoc-protected Aoa derivative has proved to be the best solution. However, the aminooxyacetyl moiety is very sensitive to molecules containing carbonyl groups, with the partial impact of the peptide sequence. Therefore, the free NH2-O-R group reacts often with these compounds during the working-up procedure after the final cleavage of Aoa-modified peptides from the resin. These carbonyl group-containing derivatives might come from the plastic tubes or residues of acetone used in a laboratory. This cannot be prevented even by using diBoc-protected Aoa or working in argon. We found a highly sensitive peptide to this side reaction; the synthesis of a somatostatin analog developed in Schallys laboratory elongated with Aoa (H-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2) was unsuccessful. After several trials to optimize the reaction conditions, we elaborated the following procedure: The semi-protected peptide H-D-Phe-Cys-Tyr-D-Trp-Lys(Dde)-Val-Cys-Thr-NH2 was cleaved from Rink Amide MBHA resin and reacted with Boc-Aoa-OPcp to incorporate Aoa into the N-terminus in a solution. After the efficient coupling reaction, the Dde-protecting group was removed with 2% hydrazine in DMF. Surprisingly, during the cleavage of Dde, a cyclic peptide also formed (Boc-Aoa-D-Phe-c[Cys-Tyr-D-Trp-Lys-Val-Cys]-Thr-NH2). The Boc group was cleaved in 95% TFA solution in the presence of 10 equiv-free Aoa as a carbonyl capture reagent that could prevent the reaction of the peptide with any carbonyl derivative. The crude product was purified by RP-HPLC, and the solvent was evaporated, followed by direct ligation to daunomycin (Dau) in 0.2 M NaOAc solution at pH 5. This procedure proved to be very efficient to prepare oxime-linked Dau-peptide conjugates.
Click to Show/Hide
|
||||
| Description |
The main drug-related toxic side effect of anthracyclines is cardiotoxicity, leading to cardiomyopathy and heart failure. Comparative experiments to determine the cardiotoxicity of peptide-anthracycline conjugates with different linkages might be informative for the other conjugates with different types of drugs as well. For this purpose, human cardiomyocytes (HCM) and human umbilical vein endothelial cells (HUVEC) were used as models. The long-term (0-72 h) cytotoxic effect of sixteen GnRH-based conjugates containing Dox and Dau was determined by real-time impedimetric sensing using the xCELLigence SP system (ACEA Biosciences, San Diego, CA, USA) [75]. The results indicated that the ester-linked GnRH-Dox conjugates, including Zoptarelin Doxorubicin, showed significant toxicity at 100 nM and 1 uM, which was remarkably pronounced on the HCM cells. The cytotoxic effect was comparable to that of the free drug, especially at the highest concentration. In contrast, the conjugates with oxime-linked Dau showed no or only a minor toxicity on both the cell lines (Table 2). These data confirm that the linkage between the payload and homing peptide has a significant influence on early drug release and, consequently, an undesired toxic side effect. We may also conclude that the search for more suitable homing peptides might be more important than the application of cleavable bonds between the drug and the peptide to develop efficient DDSs.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
pHA-AOHX-VAP-DOX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Glioma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.26 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
The existence of the blood-brain barrier (BBB) and blood-brain tumor barrier (BBTB) greatly limits the application of chemotherapy in glioma. To address this challenge, an optimal drug delivery system must efficiently cross the BBB/BBTB and specifically deliver therapeutic drugs into glioma cells while minimizing systemic toxicity. Here we demonstrated that glucose-regulated protein 78 (GRP78) and dopamine receptor D2 were highly expressed in patient-derived glioma tissues, and dopamine receptors were highly expressed on the BBB. Subsequently, we synthesized a novel Y-shaped peptide and compared the effects of different linkers on the receptor affinity and targeting ability of the peptide. A peptide-drug conjugate (pHA-AOHX-VAP-doxorubicin conjugate, pHA-AOHX-VAP-DOX) with a better affinity for glioma cells and higher solubility was derived for glioma treatment. pHA-AOHX-VAP-DOX could cross both BBB and BBTB via dopamine receptor and GRP78 receptor, and finally target glioma cells, significantly prolonging the survival time of nude mice bearing intracranial glioma. Furthermore, pHA-AOHX-VAP-DOX significantly reduced the toxicity of DOX and increased the maximum tolerated dose (MTD). Collectively, this work paves a new avenue for overcoming multiple barriers and effectively delivering chemotherapeutic agents to glioma cells while providing key evidence to identify potential receptors for glioma-targeted drug delivery.
Click to Show/Hide
|
||||
| Description |
The coupling of peptides with chemotherapeutic drugs often increased the solubility of the drugs, so the solubility of peptide-drug conjugates was determined, and the results were shown in Fig. S9. DOX free base exhibited poor solubility, with a solubility of approximately 0.22 ± 0.03 mg/mL in PBS. However, upon formation of a peptide-drug conjugate, the hydrophilicity of the peptide significantly enhanced the solubility of DOX. The solubility of pHA-AHX-VAP-DOX and pHA-AOHX-VAP-DOX drastically increased to 7.09 ± 0.15 mg/mL and 17.29 ± 0.43 mg/mL, which was 32-fold and 78-fold higher than that of the DOX free base, respectively. The improved solubility performance was consistent with their LogP values predicted by the ALOGPS 2.1 program.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Glioma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.32 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
The existence of the blood-brain barrier (BBB) and blood-brain tumor barrier (BBTB) greatly limits the application of chemotherapy in glioma. To address this challenge, an optimal drug delivery system must efficiently cross the BBB/BBTB and specifically deliver therapeutic drugs into glioma cells while minimizing systemic toxicity. Here we demonstrated that glucose-regulated protein 78 (GRP78) and dopamine receptor D2 were highly expressed in patient-derived glioma tissues, and dopamine receptors were highly expressed on the BBB. Subsequently, we synthesized a novel Y-shaped peptide and compared the effects of different linkers on the receptor affinity and targeting ability of the peptide. A peptide-drug conjugate (pHA-AOHX-VAP-doxorubicin conjugate, pHA-AOHX-VAP-DOX) with a better affinity for glioma cells and higher solubility was derived for glioma treatment. pHA-AOHX-VAP-DOX could cross both BBB and BBTB via dopamine receptor and GRP78 receptor, and finally target glioma cells, significantly prolonging the survival time of nude mice bearing intracranial glioma. Furthermore, pHA-AOHX-VAP-DOX significantly reduced the toxicity of DOX and increased the maximum tolerated dose (MTD). Collectively, this work paves a new avenue for overcoming multiple barriers and effectively delivering chemotherapeutic agents to glioma cells while providing key evidence to identify potential receptors for glioma-targeted drug delivery.
Click to Show/Hide
|
||||
| Description |
The coupling of peptides with chemotherapeutic drugs often increased the solubility of the drugs, so the solubility of peptide-drug conjugates was determined, and the results were shown in Fig. S9. DOX free base exhibited poor solubility, with a solubility of approximately 0.22 ± 0.03 mg/mL in PBS. However, upon formation of a peptide-drug conjugate, the hydrophilicity of the peptide significantly enhanced the solubility of DOX. The solubility of pHA-AHX-VAP-DOX and pHA-AOHX-VAP-DOX drastically increased to 7.09 ± 0.15 mg/mL and 17.29 ± 0.43 mg/mL, which was 32-fold and 78-fold higher than that of the DOX free base, respectively. The improved solubility performance was consistent with their LogP values predicted by the ALOGPS 2.1 program.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cell | Homo sapiens | ||
pHA-AHX-VAP-DOX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Glioma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.56 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
The existence of the blood-brain barrier (BBB) and blood-brain tumor barrier (BBTB) greatly limits the application of chemotherapy in glioma. To address this challenge, an optimal drug delivery system must efficiently cross the BBB/BBTB and specifically deliver therapeutic drugs into glioma cells while minimizing systemic toxicity. Here we demonstrated that glucose-regulated protein 78 (GRP78) and dopamine receptor D2 were highly expressed in patient-derived glioma tissues, and dopamine receptors were highly expressed on the BBB. Subsequently, we synthesized a novel Y-shaped peptide and compared the effects of different linkers on the receptor affinity and targeting ability of the peptide. A peptide-drug conjugate (pHA-AOHX-VAP-doxorubicin conjugate, pHA-AOHX-VAP-DOX) with a better affinity for glioma cells and higher solubility was derived for glioma treatment. pHA-AOHX-VAP-DOX could cross both BBB and BBTB via dopamine receptor and GRP78 receptor, and finally target glioma cells, significantly prolonging the survival time of nude mice bearing intracranial glioma. Furthermore, pHA-AOHX-VAP-DOX significantly reduced the toxicity of DOX and increased the maximum tolerated dose (MTD). Collectively, this work paves a new avenue for overcoming multiple barriers and effectively delivering chemotherapeutic agents to glioma cells while providing key evidence to identify potential receptors for glioma-targeted drug delivery.
Click to Show/Hide
|
||||
| Description |
The coupling of peptides with chemotherapeutic drugs often increased the solubility of the drugs, so the solubility of peptide-drug conjugates was determined, and the results were shown in Fig. S9. DOX free base exhibited poor solubility, with a solubility of approximately 0.22 ± 0.03 mg/mL in PBS. However, upon formation of a peptide-drug conjugate, the hydrophilicity of the peptide significantly enhanced the solubility of DOX. The solubility of pHA-AHX-VAP-DOX and pHA-AOHX-VAP-DOX drastically increased to 7.09 ± 0.15 mg/mL and 17.29 ± 0.43 mg/mL, which was 32-fold and 78-fold higher than that of the DOX free base, respectively. The improved solubility performance was consistent with their LogP values predicted by the ALOGPS 2.1 program.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cell | Homo sapiens | ||
| Experiment 2 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Glioma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.62 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
The existence of the blood-brain barrier (BBB) and blood-brain tumor barrier (BBTB) greatly limits the application of chemotherapy in glioma. To address this challenge, an optimal drug delivery system must efficiently cross the BBB/BBTB and specifically deliver therapeutic drugs into glioma cells while minimizing systemic toxicity. Here we demonstrated that glucose-regulated protein 78 (GRP78) and dopamine receptor D2 were highly expressed in patient-derived glioma tissues, and dopamine receptors were highly expressed on the BBB. Subsequently, we synthesized a novel Y-shaped peptide and compared the effects of different linkers on the receptor affinity and targeting ability of the peptide. A peptide-drug conjugate (pHA-AOHX-VAP-doxorubicin conjugate, pHA-AOHX-VAP-DOX) with a better affinity for glioma cells and higher solubility was derived for glioma treatment. pHA-AOHX-VAP-DOX could cross both BBB and BBTB via dopamine receptor and GRP78 receptor, and finally target glioma cells, significantly prolonging the survival time of nude mice bearing intracranial glioma. Furthermore, pHA-AOHX-VAP-DOX significantly reduced the toxicity of DOX and increased the maximum tolerated dose (MTD). Collectively, this work paves a new avenue for overcoming multiple barriers and effectively delivering chemotherapeutic agents to glioma cells while providing key evidence to identify potential receptors for glioma-targeted drug delivery.
Click to Show/Hide
|
||||
| Description |
The coupling of peptides with chemotherapeutic drugs often increased the solubility of the drugs, so the solubility of peptide-drug conjugates was determined, and the results were shown in Fig. S9. DOX free base exhibited poor solubility, with a solubility of approximately 0.22 ± 0.03 mg/mL in PBS. However, upon formation of a peptide-drug conjugate, the hydrophilicity of the peptide significantly enhanced the solubility of DOX. The solubility of pHA-AHX-VAP-DOX and pHA-AOHX-VAP-DOX drastically increased to 7.09 ± 0.15 mg/mL and 17.29 ± 0.43 mg/mL, which was 32-fold and 78-fold higher than that of the DOX free base, respectively. The improved solubility performance was consistent with their LogP values predicted by the ALOGPS 2.1 program.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
GE11-DOX conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [989] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.87 µM | |||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [989] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 3.66 µM | |||
| In Vitro Model | Invasive breast carcinoma of no special type | EGFR-overexpressing MCF-7 cell | CVCL_0031 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [989] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 32.2µM | |||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
Peptide 18-4 doxorubicin conjugate 1 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [990] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 0.9 ± 0.07 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Here we report the design and synthesis of two new peptide-Dox conjugates (1 and 2) for the specific delivery of Dox to the breast cancer cells and the ability to overcome P-glycoprotein multidrug resistance pathway in both drug-sensitive and drug-resistant cancer cells. Peptide-Dox conjugates were evaluated for DOX release in human serum, intracellular delivery compared to free Dox in three cancerous cells (MCF-7, MDA-MB-435, and MDA-MB-435-MDR) and two noncancerous cell lines (HUVEC and MCF-10A), and cytotoxicity compared to free Dox. Results show that both the peptide-Dox conjugates (1 and 2) enter sensitive and resistant cell lines with minimal uptake in normal cells compared to free Dox. Cellular uptake is most likely mediated by a cell specific receptor, as the amount of internalized conjugates significantly decreased in the presence of excess free peptide. Importantly, conjugate 1 is equally cytotoxic as Dox in drug sensitive breast cancer cells and 4 times more potent than free Dox in Dox resistant cell line. Overall, the peptide-Dox ester conjugate 1 showed better breast targeting efficacy than the amide conjugate 2, most likely due to the slow release of Dox from the stable amide linkage.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity experiment was done by incubating the cells with different treatments for 48 h. The results show that conjugate 1 is quite similar to free Dox for toxicity to MCF-7 and MDA-MB-435 cancer cells. In contrast, conjugate 2 was ?20 times less cytotoxic to breast cancerous cells compared to free Dox. This could be attributed to the stability of the amide conjugate 2 inside the cells. Furthermore, the cytotoxicity of conjugate 1 in doxorubicin-resistant cell model MDA-MB-435-MDR (IC50 = 5.4 μM) is 4 times more than free Dox (IC50 = 22 μM). In normal cells (HUVEC and MCF-10A) the two conjugates were 3540 times less toxic compared to breast cancer cells, whereas free Dox was equally cytotoxic (equal IC50) to breast cancer cells and noncancerous cells. Overall these results provide clear evidence that of the two conjugates (conjugate 1 and 2), conjugate 1 has optimal characteristics for specific Dox targeting to breast cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [990] | ||||
| Indication | Amelanotic melanoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 1.5 ± 0.09 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Here we report the design and synthesis of two new peptide-Dox conjugates (1 and 2) for the specific delivery of Dox to the breast cancer cells and the ability to overcome P-glycoprotein multidrug resistance pathway in both drug-sensitive and drug-resistant cancer cells. Peptide-Dox conjugates were evaluated for DOX release in human serum, intracellular delivery compared to free Dox in three cancerous cells (MCF-7, MDA-MB-435, and MDA-MB-435-MDR) and two noncancerous cell lines (HUVEC and MCF-10A), and cytotoxicity compared to free Dox. Results show that both the peptide-Dox conjugates (1 and 2) enter sensitive and resistant cell lines with minimal uptake in normal cells compared to free Dox. Cellular uptake is most likely mediated by a cell specific receptor, as the amount of internalized conjugates significantly decreased in the presence of excess free peptide. Importantly, conjugate 1 is equally cytotoxic as Dox in drug sensitive breast cancer cells and 4 times more potent than free Dox in Dox resistant cell line. Overall, the peptide-Dox ester conjugate 1 showed better breast targeting efficacy than the amide conjugate 2, most likely due to the slow release of Dox from the stable amide linkage.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity experiment was done by incubating the cells with different treatments for 48 h. The results show that conjugate 1 is quite similar to free Dox for toxicity to MCF-7 and MDA-MB-435 cancer cells. In contrast, conjugate 2 was ?20 times less cytotoxic to breast cancerous cells compared to free Dox. This could be attributed to the stability of the amide conjugate 2 inside the cells. Furthermore, the cytotoxicity of conjugate 1 in doxorubicin-resistant cell model MDA-MB-435-MDR (IC50 = 5.4 μM) is 4 times more than free Dox (IC50 = 22 μM). In normal cells (HUVEC and MCF-10A) the two conjugates were 3540 times less toxic compared to breast cancer cells, whereas free Dox was equally cytotoxic (equal IC50) to breast cancer cells and noncancerous cells. Overall these results provide clear evidence that of the two conjugates (conjugate 1 and 2), conjugate 1 has optimal characteristics for specific Dox targeting to breast cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | MDA-MB-435 cell | CVCL_0417 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [990] | ||||
| Indication | Amelanotic melanoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 5.4 ± 0.62 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Here we report the design and synthesis of two new peptide-Dox conjugates (1 and 2) for the specific delivery of Dox to the breast cancer cells and the ability to overcome P-glycoprotein multidrug resistance pathway in both drug-sensitive and drug-resistant cancer cells. Peptide-Dox conjugates were evaluated for DOX release in human serum, intracellular delivery compared to free Dox in three cancerous cells (MCF-7, MDA-MB-435, and MDA-MB-435-MDR) and two noncancerous cell lines (HUVEC and MCF-10A), and cytotoxicity compared to free Dox. Results show that both the peptide-Dox conjugates (1 and 2) enter sensitive and resistant cell lines with minimal uptake in normal cells compared to free Dox. Cellular uptake is most likely mediated by a cell specific receptor, as the amount of internalized conjugates significantly decreased in the presence of excess free peptide. Importantly, conjugate 1 is equally cytotoxic as Dox in drug sensitive breast cancer cells and 4 times more potent than free Dox in Dox resistant cell line. Overall, the peptide-Dox ester conjugate 1 showed better breast targeting efficacy than the amide conjugate 2, most likely due to the slow release of Dox from the stable amide linkage.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity experiment was done by incubating the cells with different treatments for 48 h. The results show that conjugate 1 is quite similar to free Dox for toxicity to MCF-7 and MDA-MB-435 cancer cells. In contrast, conjugate 2 was ?20 times less cytotoxic to breast cancerous cells compared to free Dox. This could be attributed to the stability of the amide conjugate 2 inside the cells. Furthermore, the cytotoxicity of conjugate 1 in doxorubicin-resistant cell model MDA-MB-435-MDR (IC50 = 5.4 μM) is 4 times more than free Dox (IC50 = 22 μM). In normal cells (HUVEC and MCF-10A) the two conjugates were 3540 times less toxic compared to breast cancer cells, whereas free Dox was equally cytotoxic (equal IC50) to breast cancer cells and noncancerous cells. Overall these results provide clear evidence that of the two conjugates (conjugate 1 and 2), conjugate 1 has optimal characteristics for specific Dox targeting to breast cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | MDA-MB-435 cell | CVCL_0417 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [990] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 35.1 ± 2.2 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Here we report the design and synthesis of two new peptide-Dox conjugates (1 and 2) for the specific delivery of Dox to the breast cancer cells and the ability to overcome P-glycoprotein multidrug resistance pathway in both drug-sensitive and drug-resistant cancer cells. Peptide-Dox conjugates were evaluated for DOX release in human serum, intracellular delivery compared to free Dox in three cancerous cells (MCF-7, MDA-MB-435, and MDA-MB-435-MDR) and two noncancerous cell lines (HUVEC and MCF-10A), and cytotoxicity compared to free Dox. Results show that both the peptide-Dox conjugates (1 and 2) enter sensitive and resistant cell lines with minimal uptake in normal cells compared to free Dox. Cellular uptake is most likely mediated by a cell specific receptor, as the amount of internalized conjugates significantly decreased in the presence of excess free peptide. Importantly, conjugate 1 is equally cytotoxic as Dox in drug sensitive breast cancer cells and 4 times more potent than free Dox in Dox resistant cell line. Overall, the peptide-Dox ester conjugate 1 showed better breast targeting efficacy than the amide conjugate 2, most likely due to the slow release of Dox from the stable amide linkage.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity experiment was done by incubating the cells with different treatments for 48 h. The results show that conjugate 1 is quite similar to free Dox for toxicity to MCF-7 and MDA-MB-435 cancer cells. In contrast, conjugate 2 was ?20 times less cytotoxic to breast cancerous cells compared to free Dox. This could be attributed to the stability of the amide conjugate 2 inside the cells. Furthermore, the cytotoxicity of conjugate 1 in doxorubicin-resistant cell model MDA-MB-435-MDR (IC50 = 5.4 μM) is 4 times more than free Dox (IC50 = 22 μM). In normal cells (HUVEC and MCF-10A) the two conjugates were 3540 times less toxic compared to breast cancer cells, whereas free Dox was equally cytotoxic (equal IC50) to breast cancer cells and noncancerous cells. Overall these results provide clear evidence that of the two conjugates (conjugate 1 and 2), conjugate 1 has optimal characteristics for specific Dox targeting to breast cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [990] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 42.3 ± 2.4 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Here we report the design and synthesis of two new peptide-Dox conjugates (1 and 2) for the specific delivery of Dox to the breast cancer cells and the ability to overcome P-glycoprotein multidrug resistance pathway in both drug-sensitive and drug-resistant cancer cells. Peptide-Dox conjugates were evaluated for DOX release in human serum, intracellular delivery compared to free Dox in three cancerous cells (MCF-7, MDA-MB-435, and MDA-MB-435-MDR) and two noncancerous cell lines (HUVEC and MCF-10A), and cytotoxicity compared to free Dox. Results show that both the peptide-Dox conjugates (1 and 2) enter sensitive and resistant cell lines with minimal uptake in normal cells compared to free Dox. Cellular uptake is most likely mediated by a cell specific receptor, as the amount of internalized conjugates significantly decreased in the presence of excess free peptide. Importantly, conjugate 1 is equally cytotoxic as Dox in drug sensitive breast cancer cells and 4 times more potent than free Dox in Dox resistant cell line. Overall, the peptide-Dox ester conjugate 1 showed better breast targeting efficacy than the amide conjugate 2, most likely due to the slow release of Dox from the stable amide linkage.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity experiment was done by incubating the cells with different treatments for 48 h. The results show that conjugate 1 is quite similar to free Dox for toxicity to MCF-7 and MDA-MB-435 cancer cells. In contrast, conjugate 2 was ?20 times less cytotoxic to breast cancerous cells compared to free Dox. This could be attributed to the stability of the amide conjugate 2 inside the cells. Furthermore, the cytotoxicity of conjugate 1 in doxorubicin-resistant cell model MDA-MB-435-MDR (IC50 = 5.4 μM) is 4 times more than free Dox (IC50 = 22 μM). In normal cells (HUVEC and MCF-10A) the two conjugates were 3540 times less toxic compared to breast cancer cells, whereas free Dox was equally cytotoxic (equal IC50) to breast cancer cells and noncancerous cells. Overall these results provide clear evidence that of the two conjugates (conjugate 1 and 2), conjugate 1 has optimal characteristics for specific Dox targeting to breast cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cell | Homo sapiens | ||
Peptide 18-4-doxorubicin [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [991] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.3 µM | |||
| Evaluation Method | MTT assay | ||||
| Description |
The results showed that the cytotoxicity of both conjugates 1 and 2 (IC50 = 1.3 and 2.2 uM, respectively), as well as the free Dox (IC50 = 1.5 uM) on MDA-MB-231 breast cancer cell line, were in the low micromolar range (Figure 4). For the breast cancer cell line MDA-MB-468, the free Dox (IC50 = 0.35 uM) was slightly more toxic compared to conjugates 1 (4.7 uM) and 2 (1.2 uM). For the non-cancerous cell line MCF 10A, the free Dox was highly toxic (IC50 = 0.24 uM) whereas conjugates 1 and 2 displayed much-reduced toxicity (IC50 = 38.6 and 15.1 uM, respectively).
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [992] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.3 ± 0.2µM | |||
| MOA of PDC |
We engineered peptides that bind to cell-surface K1 and are internalized by breast cancer cells via cell-surface K1 receptor-mediated endocytosis. Peptide 18-4 (WxEAAYQrFL), with two D-amino acids, is a second-generation breast cancer cell-targeting peptide that is proteolytically stable (100% intact up to 24 h when incubated with human serum or liver homogenate from mice) and has shown high specific uptake by breast cancer cells and minimal/no binding to non-cancerous cells. Affinity purification of breast cancer cell lysates using the immobilized peptide, followed by liquid chromatography-tandem mass spectrometry and proteomics were used to identify K1 as the novel target for peptide 18-4 in cancer cells. Further, we showed that the uptake of the peptide by the cancer cells is dependent on K1 expression.
Click to Show/Hide
|
||||
| Description |
Several PDCs have been prepared with peptide 18-4 and doxorubicin (Dox) using different linker chemistries such as ester, amide, succinimidyl thioether, or acyl hydrazone. In vitro results showed that these PDCs were highly specific toward breast cancer cells. PDCs displayed similar toxicity as free Dox toward the breast cancer cells and several-fold (7-40 times) less toxicity toward the non-cancerous cells such as MCF-10A and human umbilical vein endothelial cells (HUVECs). A peptide 18-4-Dox conjugate with amide/succinimidyl thioether linkage showed high selective toxicity toward triple negative breast cancer (TNBC) cell lines, i.e., MDA-MB-231 cells (IC50 1.3 ± 0.2 uM) and MDA-MB-468 cells (IC50 4.7 ± 0.3 uM) compared to the normal breast MCF10A cells (IC50 38.6 ± 1.1 uM). The linkage between the drug and the peptide was stable as the degradation half-life of peptide 18-4-Dox conjugate in the presence of human serum was found to be ˜18 h. Herein, we describe the first in vivo evidence for improved efficacy of this PDC targeting K1 receptor in an orthotopic TNBC mouse model. We also show a higher accumulation of PDC in TNBC tumors in mice, in accord with K1 overexpression in tumor over non-tumor tissues in MDA-MB-231 xenografted mice.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [991] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 4.7 µM | |||
| Evaluation Method | MTT assay | ||||
| Description |
The results showed that the cytotoxicity of both conjugates 1 and 2 (IC50 = 1.3 and 2.2 uM, respectively), as well as the free Dox (IC50 = 1.5 uM) on MDA-MB-231 breast cancer cell line, were in the low micromolar range (Figure 4). For the breast cancer cell line MDA-MB-468, the free Dox (IC50 = 0.35 uM) was slightly more toxic compared to conjugates 1 (4.7 uM) and 2 (1.2 uM). For the non-cancerous cell line MCF 10A, the free Dox was highly toxic (IC50 = 0.24 uM) whereas conjugates 1 and 2 displayed much-reduced toxicity (IC50 = 38.6 and 15.1 uM, respectively).
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-468 cell | CVCL_0419 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [992] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 4.7 ± 0.3µM | |||
| MOA of PDC |
We engineered peptides that bind to cell-surface K1 and are internalized by breast cancer cells via cell-surface K1 receptor-mediated endocytosis. Peptide 18-4 (WxEAAYQrFL), with two D-amino acids, is a second-generation breast cancer cell-targeting peptide that is proteolytically stable (100% intact up to 24 h when incubated with human serum or liver homogenate from mice) and has shown high specific uptake by breast cancer cells and minimal/no binding to non-cancerous cells. Affinity purification of breast cancer cell lysates using the immobilized peptide, followed by liquid chromatography-tandem mass spectrometry and proteomics were used to identify K1 as the novel target for peptide 18-4 in cancer cells. Further, we showed that the uptake of the peptide by the cancer cells is dependent on K1 expression.
Click to Show/Hide
|
||||
| Description |
Several PDCs have been prepared with peptide 18-4 and doxorubicin (Dox) using different linker chemistries such as ester, amide, succinimidyl thioether, or acyl hydrazone. In vitro results showed that these PDCs were highly specific toward breast cancer cells. PDCs displayed similar toxicity as free Dox toward the breast cancer cells and several-fold (7-40 times) less toxicity toward the non-cancerous cells such as MCF-10A and human umbilical vein endothelial cells (HUVECs). A peptide 18-4-Dox conjugate with amide/succinimidyl thioether linkage showed high selective toxicity toward triple negative breast cancer (TNBC) cell lines, i.e., MDA-MB-231 cells (IC50 1.3 ± 0.2 uM) and MDA-MB-468 cells (IC50 4.7 ± 0.3 uM) compared to the normal breast MCF10A cells (IC50 38.6 ± 1.1 uM). The linkage between the drug and the peptide was stable as the degradation half-life of peptide 18-4-Dox conjugate in the presence of human serum was found to be ˜18 h. Herein, we describe the first in vivo evidence for improved efficacy of this PDC targeting K1 receptor in an orthotopic TNBC mouse model. We also show a higher accumulation of PDC in TNBC tumors in mice, in accord with K1 overexpression in tumor over non-tumor tissues in MDA-MB-231 xenografted mice.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-468 cell | CVCL_0419 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [991] | ||||
| Indication | Triple-negative breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 38.6 µM | |||
| Evaluation Method | MTT assay | ||||
| Description |
The results showed that the cytotoxicity of both conjugates 1 and 2 (IC50 = 1.3 and 2.2 uM, respectively), as well as the free Dox (IC50 = 1.5 uM) on MDA-MB-231 breast cancer cell line, were in the low micromolar range (Figure 4). For the breast cancer cell line MDA-MB-468, the free Dox (IC50 = 0.35 uM) was slightly more toxic compared to conjugates 1 (4.7 uM) and 2 (1.2 uM). For the non-cancerous cell line MCF 10A, the free Dox was highly toxic (IC50 = 0.24 uM) whereas conjugates 1 and 2 displayed much-reduced toxicity (IC50 = 38.6 and 15.1 uM, respectively).
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [992] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 38.6 ± 1.1µM | |||
| MOA of PDC |
We engineered peptides that bind to cell-surface K1 and are internalized by breast cancer cells via cell-surface K1 receptor-mediated endocytosis. Peptide 18-4 (WxEAAYQrFL), with two D-amino acids, is a second-generation breast cancer cell-targeting peptide that is proteolytically stable (100% intact up to 24 h when incubated with human serum or liver homogenate from mice) and has shown high specific uptake by breast cancer cells and minimal/no binding to non-cancerous cells. Affinity purification of breast cancer cell lysates using the immobilized peptide, followed by liquid chromatography-tandem mass spectrometry and proteomics were used to identify K1 as the novel target for peptide 18-4 in cancer cells. Further, we showed that the uptake of the peptide by the cancer cells is dependent on K1 expression.
Click to Show/Hide
|
||||
| Description |
Several PDCs have been prepared with peptide 18-4 and doxorubicin (Dox) using different linker chemistries such as ester, amide, succinimidyl thioether, or acyl hydrazone. In vitro results showed that these PDCs were highly specific toward breast cancer cells. PDCs displayed similar toxicity as free Dox toward the breast cancer cells and several-fold (7-40 times) less toxicity toward the non-cancerous cells such as MCF-10A and human umbilical vein endothelial cells (HUVECs). A peptide 18-4-Dox conjugate with amide/succinimidyl thioether linkage showed high selective toxicity toward triple negative breast cancer (TNBC) cell lines, i.e., MDA-MB-231 cells (IC50 1.3 ± 0.2 uM) and MDA-MB-468 cells (IC50 4.7 ± 0.3 uM) compared to the normal breast MCF10A cells (IC50 38.6 ± 1.1 uM). The linkage between the drug and the peptide was stable as the degradation half-life of peptide 18-4-Dox conjugate in the presence of human serum was found to be ˜18 h. Herein, we describe the first in vivo evidence for improved efficacy of this PDC targeting K1 receptor in an orthotopic TNBC mouse model. We also show a higher accumulation of PDC in TNBC tumors in mice, in accord with K1 overexpression in tumor over non-tumor tissues in MDA-MB-231 xenografted mice.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
Peptide-drug conjugate 10 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Neuroblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 2.47 ± 0.91 µM | |||
| Evaluation Method | CellTiter-Glo luminescent cell viability assay | ||||
| Administration Time | 72 h | ||||
| Description |
The cytotoxic effect of the peptide-drug conjugate on Kelly-WT cells was comparable to that of 9, but not as strong as the one of the native drug. Contrary to the other utilized substances, the antiproliferative action of the bioconjugate in wild-type and drug-resistant cells was nearly the same. In comparison with doxorubicin the cytotoxicity of 10 against Kelly-ADR cells was increased by a factor of 3 according to the obtained IC50 values. Even the unmodified dimer had roughly the same cytotoxic effect on Kelly-ADR cells as doxorubicin.
Click to Show/Hide
|
||||
| In Vitro Model | Neuroblastoma | Kelly-WT cell | CVCL_2092 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Neuroblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 2.76 ± 0.33 µM | |||
| Evaluation Method | CellTiter-Glo luminescent cell viability assay | ||||
| Administration Time | 72 h | ||||
| Description |
The cytotoxic effect of the peptide-drug conjugate on Kelly-WT cells was comparable to that of 9, but not as strong as the one of the native drug. Contrary to the other utilized substances, the antiproliferative action of the bioconjugate in wild-type and drug-resistant cells was nearly the same. In comparison with doxorubicin the cytotoxicity of 10 against Kelly-ADR cells was increased by a factor of 3 according to the obtained IC50 values. Even the unmodified dimer had roughly the same cytotoxic effect on Kelly-ADR cells as doxorubicin.
Click to Show/Hide
|
||||
| In Vitro Model | Neuroblastoma | Kelly-ADR cell | CVCL_2092 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Neuroblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 3.66 ± 0.77 µM | |||
| Evaluation Method | CellTiter-Glo luminescent cell viability assay | ||||
| Administration Time | 72 h | ||||
| Description |
The obtained IC50 values for 9 (6.73 ± 2.44 uM) and 10 (3.66 ± 0.77 uM) were higher than for doxorubicin (0.90 ± 0.12 uM).
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
RGD-GFLG-DOX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [10] | ||||
| Indication | Lung cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 4 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 36 h | ||||
| MOA of PDC |
In conclusion, we developed a robust and regioselective rhodium-catalyzed methodology for C(7)-H Trp maleimidation. This reaction served as an efficient tool for peptide/drug modification, ligation, and particularly peptide cyclization, confirming its promising potential in pharmaceutical chemistry and drug synthesis. Notably, this catalytical system is not limited by the Trp position in the peptides. We also demonstrated that tryptophan-substituted maleimide could be used as an effective click functional group to rapidly react with sulfhydryl groups. Moreover, the introduced N-pivaloyl directing group and protecting groups of the peptides could be removed in a single step, providing a more convenient approach compared to the previous methods, which require multi-step removal of the corresponding directing groups and peptide protection groups. Additionally, cyclic peptide 10a exhibited excellent binding affinity to integrin vβ3, indicating its good drug-like properties. With rational design, RGD- GFLG -DOX, which is a stapled PDC, displayed higher selectivity, stronger binding affinity and better cell penetrability than the more commonly used DOX. The proposed strategy for rapid preparation of stapled peptides is expected to further improve PDC formulation.
Click to Show/Hide
|
||||
| Description |
Doxorubicin (DOX) is one of the most effective anticancer drugs and has been successfully used in clinical practice. However, DOX cannot differentiate between cancer cells and normal cells, which may induce unwanted side effects and severe toxicity. Compared with traditional small-molecule anticancer drugs, the peptide-drug conjugates (PDCs) have enhanced targeting specificity and water solubility. Based on these advantages, to further explore the function of 10a, we designed and prepared a anticancer PDC drug compound RGD-GFLG-DOX containing the tetrapeptide linker Gly-Phe-Leu-Gly, which can be cleaved in presence of cathepsin B, a highly upregulated enzyme in malignant tumors, to release the drug. RGD-GFLG was synthesized as a control. The inhibitory effects of RGD-GFLG-DOX on cancer cell lines were assessed using cytotoxicity assay. Specifically, the effects of RGD-GFLG-DOX were evaluated on integrin v3-positive cancer cell lines, including A549 and U87MG cells, integrin v3-negative cancer cell lines such as HeLa and MCF-7 cells, as well as normal cell lines, namely LO2 and MIHA cells. RGD-GFLG-DOX exhibited a lower cytotoxicity on HeLa, MCF-7, LO2 and MIHA cells, but a stronger cytotoxicity than DOX on A549 and U87MG cells. For comparison, RGD-GFLG demonstrated minimal cytotoxicity. In addition, the cytotoxicity of RGD-GFLG-DOX with various concentrations (0-40 uM) on A549 and U87MG cells was studied. The results showed that the cytotoxicity of RGD-GFLG-DOX on A549 and U87MG cells was dose-dependent. These indicate that RGD-GFLG-DOX has a good specificity and inhibitory activity toward integrin v3-overexpressed A549 and U87MG cells.
Click to Show/Hide
|
||||
| In Vitro Model | Lung adenocarcinoma | A-549 cell | CVCL_0023 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [10] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 4 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 36 h | ||||
| MOA of PDC |
In conclusion, we developed a robust and regioselective rhodium-catalyzed methodology for C(7)-H Trp maleimidation. This reaction served as an efficient tool for peptide/drug modification, ligation, and particularly peptide cyclization, confirming its promising potential in pharmaceutical chemistry and drug synthesis. Notably, this catalytical system is not limited by the Trp position in the peptides. We also demonstrated that tryptophan-substituted maleimide could be used as an effective click functional group to rapidly react with sulfhydryl groups. Moreover, the introduced N-pivaloyl directing group and protecting groups of the peptides could be removed in a single step, providing a more convenient approach compared to the previous methods, which require multi-step removal of the corresponding directing groups and peptide protection groups. Additionally, cyclic peptide 10a exhibited excellent binding affinity to integrin vβ3, indicating its good drug-like properties. With rational design, RGD- GFLG -DOX, which is a stapled PDC, displayed higher selectivity, stronger binding affinity and better cell penetrability than the more commonly used DOX. The proposed strategy for rapid preparation of stapled peptides is expected to further improve PDC formulation.
Click to Show/Hide
|
||||
| Description |
Doxorubicin (DOX) is one of the most effective anticancer drugs and has been successfully used in clinical practice. However, DOX cannot differentiate between cancer cells and normal cells, which may induce unwanted side effects and severe toxicity. Compared with traditional small-molecule anticancer drugs, the peptide-drug conjugates (PDCs) have enhanced targeting specificity and water solubility. Based on these advantages, to further explore the function of 10a, we designed and prepared a anticancer PDC drug compound RGD-GFLG-DOX containing the tetrapeptide linker Gly-Phe-Leu-Gly, which can be cleaved in presence of cathepsin B, a highly upregulated enzyme in malignant tumors, to release the drug. RGD-GFLG was synthesized as a control. The inhibitory effects of RGD-GFLG-DOX on cancer cell lines were assessed using cytotoxicity assay. Specifically, the effects of RGD-GFLG-DOX were evaluated on integrin v3-positive cancer cell lines, including A549 and U87MG cells, integrin v3-negative cancer cell lines such as HeLa and MCF-7 cells, as well as normal cell lines, namely LO2 and MIHA cells. RGD-GFLG-DOX exhibited a lower cytotoxicity on HeLa, MCF-7, LO2 and MIHA cells, but a stronger cytotoxicity than DOX on A549 and U87MG cells. For comparison, RGD-GFLG demonstrated minimal cytotoxicity. In addition, the cytotoxicity of RGD-GFLG-DOX with various concentrations (0-40 uM) on A549 and U87MG cells was studied. The results showed that the cytotoxicity of RGD-GFLG-DOX on A549 and U87MG cells was dose-dependent. These indicate that RGD-GFLG-DOX has a good specificity and inhibitory activity toward integrin v3-overexpressed A549 and U87MG cells.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [10] | ||||
| Indication | Cervical carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 40 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 36 h | ||||
| MOA of PDC |
In conclusion, we developed a robust and regioselective rhodium-catalyzed methodology for C(7)-H Trp maleimidation. This reaction served as an efficient tool for peptide/drug modification, ligation, and particularly peptide cyclization, confirming its promising potential in pharmaceutical chemistry and drug synthesis. Notably, this catalytical system is not limited by the Trp position in the peptides. We also demonstrated that tryptophan-substituted maleimide could be used as an effective click functional group to rapidly react with sulfhydryl groups. Moreover, the introduced N-pivaloyl directing group and protecting groups of the peptides could be removed in a single step, providing a more convenient approach compared to the previous methods, which require multi-step removal of the corresponding directing groups and peptide protection groups. Additionally, cyclic peptide 10a exhibited excellent binding affinity to integrin vβ3, indicating its good drug-like properties. With rational design, RGD- GFLG -DOX, which is a stapled PDC, displayed higher selectivity, stronger binding affinity and better cell penetrability than the more commonly used DOX. The proposed strategy for rapid preparation of stapled peptides is expected to further improve PDC formulation.
Click to Show/Hide
|
||||
| Description |
Doxorubicin (DOX) is one of the most effective anticancer drugs and has been successfully used in clinical practice. However, DOX cannot differentiate between cancer cells and normal cells, which may induce unwanted side effects and severe toxicity. Compared with traditional small-molecule anticancer drugs, the peptide-drug conjugates (PDCs) have enhanced targeting specificity and water solubility. Based on these advantages, to further explore the function of 10a, we designed and prepared a anticancer PDC drug compound RGD-GFLG-DOX containing the tetrapeptide linker Gly-Phe-Leu-Gly, which can be cleaved in presence of cathepsin B, a highly upregulated enzyme in malignant tumors, to release the drug. RGD-GFLG was synthesized as a control. The inhibitory effects of RGD-GFLG-DOX on cancer cell lines were assessed using cytotoxicity assay. Specifically, the effects of RGD-GFLG-DOX were evaluated on integrin v3-positive cancer cell lines, including A549 and U87MG cells, integrin v3-negative cancer cell lines such as HeLa and MCF-7 cells, as well as normal cell lines, namely LO2 and MIHA cells. RGD-GFLG-DOX exhibited a lower cytotoxicity on HeLa, MCF-7, LO2 and MIHA cells, but a stronger cytotoxicity than DOX on A549 and U87MG cells. For comparison, RGD-GFLG demonstrated minimal cytotoxicity. In addition, the cytotoxicity of RGD-GFLG-DOX with various concentrations (0-40 uM) on A549 and U87MG cells was studied. The results showed that the cytotoxicity of RGD-GFLG-DOX on A549 and U87MG cells was dose-dependent. These indicate that RGD-GFLG-DOX has a good specificity and inhibitory activity toward integrin v3-overexpressed A549 and U87MG cells.
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [10] | ||||
| Indication | Liver cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 40 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 36 h | ||||
| MOA of PDC |
In conclusion, we developed a robust and regioselective rhodium-catalyzed methodology for C(7)-H Trp maleimidation. This reaction served as an efficient tool for peptide/drug modification, ligation, and particularly peptide cyclization, confirming its promising potential in pharmaceutical chemistry and drug synthesis. Notably, this catalytical system is not limited by the Trp position in the peptides. We also demonstrated that tryptophan-substituted maleimide could be used as an effective click functional group to rapidly react with sulfhydryl groups. Moreover, the introduced N-pivaloyl directing group and protecting groups of the peptides could be removed in a single step, providing a more convenient approach compared to the previous methods, which require multi-step removal of the corresponding directing groups and peptide protection groups. Additionally, cyclic peptide 10a exhibited excellent binding affinity to integrin vβ3, indicating its good drug-like properties. With rational design, RGD- GFLG -DOX, which is a stapled PDC, displayed higher selectivity, stronger binding affinity and better cell penetrability than the more commonly used DOX. The proposed strategy for rapid preparation of stapled peptides is expected to further improve PDC formulation.
Click to Show/Hide
|
||||
| Description |
Doxorubicin (DOX) is one of the most effective anticancer drugs and has been successfully used in clinical practice. However, DOX cannot differentiate between cancer cells and normal cells, which may induce unwanted side effects and severe toxicity. Compared with traditional small-molecule anticancer drugs, the peptide-drug conjugates (PDCs) have enhanced targeting specificity and water solubility. Based on these advantages, to further explore the function of 10a, we designed and prepared a anticancer PDC drug compound RGD-GFLG-DOX containing the tetrapeptide linker Gly-Phe-Leu-Gly, which can be cleaved in presence of cathepsin B, a highly upregulated enzyme in malignant tumors, to release the drug. RGD-GFLG was synthesized as a control. The inhibitory effects of RGD-GFLG-DOX on cancer cell lines were assessed using cytotoxicity assay. Specifically, the effects of RGD-GFLG-DOX were evaluated on integrin v3-positive cancer cell lines, including A549 and U87MG cells, integrin v3-negative cancer cell lines such as HeLa and MCF-7 cells, as well as normal cell lines, namely LO2 and MIHA cells. RGD-GFLG-DOX exhibited a lower cytotoxicity on HeLa, MCF-7, LO2 and MIHA cells, but a stronger cytotoxicity than DOX on A549 and U87MG cells. For comparison, RGD-GFLG demonstrated minimal cytotoxicity. In addition, the cytotoxicity of RGD-GFLG-DOX with various concentrations (0-40 uM) on A549 and U87MG cells was studied. The results showed that the cytotoxicity of RGD-GFLG-DOX on A549 and U87MG cells was dose-dependent. These indicate that RGD-GFLG-DOX has a good specificity and inhibitory activity toward integrin v3-overexpressed A549 and U87MG cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MIHA cell | CVCL_SA11 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [10] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 40 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 36 h | ||||
| MOA of PDC |
In conclusion, we developed a robust and regioselective rhodium-catalyzed methodology for C(7)-H Trp maleimidation. This reaction served as an efficient tool for peptide/drug modification, ligation, and particularly peptide cyclization, confirming its promising potential in pharmaceutical chemistry and drug synthesis. Notably, this catalytical system is not limited by the Trp position in the peptides. We also demonstrated that tryptophan-substituted maleimide could be used as an effective click functional group to rapidly react with sulfhydryl groups. Moreover, the introduced N-pivaloyl directing group and protecting groups of the peptides could be removed in a single step, providing a more convenient approach compared to the previous methods, which require multi-step removal of the corresponding directing groups and peptide protection groups. Additionally, cyclic peptide 10a exhibited excellent binding affinity to integrin vβ3, indicating its good drug-like properties. With rational design, RGD- GFLG -DOX, which is a stapled PDC, displayed higher selectivity, stronger binding affinity and better cell penetrability than the more commonly used DOX. The proposed strategy for rapid preparation of stapled peptides is expected to further improve PDC formulation.
Click to Show/Hide
|
||||
| Description |
Doxorubicin (DOX) is one of the most effective anticancer drugs and has been successfully used in clinical practice. However, DOX cannot differentiate between cancer cells and normal cells, which may induce unwanted side effects and severe toxicity. Compared with traditional small-molecule anticancer drugs, the peptide-drug conjugates (PDCs) have enhanced targeting specificity and water solubility. Based on these advantages, to further explore the function of 10a, we designed and prepared a anticancer PDC drug compound RGD-GFLG-DOX containing the tetrapeptide linker Gly-Phe-Leu-Gly, which can be cleaved in presence of cathepsin B, a highly upregulated enzyme in malignant tumors, to release the drug. RGD-GFLG was synthesized as a control. The inhibitory effects of RGD-GFLG-DOX on cancer cell lines were assessed using cytotoxicity assay. Specifically, the effects of RGD-GFLG-DOX were evaluated on integrin v3-positive cancer cell lines, including A549 and U87MG cells, integrin v3-negative cancer cell lines such as HeLa and MCF-7 cells, as well as normal cell lines, namely LO2 and MIHA cells. RGD-GFLG-DOX exhibited a lower cytotoxicity on HeLa, MCF-7, LO2 and MIHA cells, but a stronger cytotoxicity than DOX on A549 and U87MG cells. For comparison, RGD-GFLG demonstrated minimal cytotoxicity. In addition, the cytotoxicity of RGD-GFLG-DOX with various concentrations (0-40 uM) on A549 and U87MG cells was studied. The results showed that the cytotoxicity of RGD-GFLG-DOX on A549 and U87MG cells was dose-dependent. These indicate that RGD-GFLG-DOX has a good specificity and inhibitory activity toward integrin v3-overexpressed A549 and U87MG cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [10] | ||||
| Indication | Liver cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 40 µM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 36 h | ||||
| MOA of PDC |
In conclusion, we developed a robust and regioselective rhodium-catalyzed methodology for C(7)-H Trp maleimidation. This reaction served as an efficient tool for peptide/drug modification, ligation, and particularly peptide cyclization, confirming its promising potential in pharmaceutical chemistry and drug synthesis. Notably, this catalytical system is not limited by the Trp position in the peptides. We also demonstrated that tryptophan-substituted maleimide could be used as an effective click functional group to rapidly react with sulfhydryl groups. Moreover, the introduced N-pivaloyl directing group and protecting groups of the peptides could be removed in a single step, providing a more convenient approach compared to the previous methods, which require multi-step removal of the corresponding directing groups and peptide protection groups. Additionally, cyclic peptide 10a exhibited excellent binding affinity to integrin vβ3, indicating its good drug-like properties. With rational design, RGD- GFLG -DOX, which is a stapled PDC, displayed higher selectivity, stronger binding affinity and better cell penetrability than the more commonly used DOX. The proposed strategy for rapid preparation of stapled peptides is expected to further improve PDC formulation.
Click to Show/Hide
|
||||
| Description |
Doxorubicin (DOX) is one of the most effective anticancer drugs and has been successfully used in clinical practice. However, DOX cannot differentiate between cancer cells and normal cells, which may induce unwanted side effects and severe toxicity. Compared with traditional small-molecule anticancer drugs, the peptide-drug conjugates (PDCs) have enhanced targeting specificity and water solubility. Based on these advantages, to further explore the function of 10a, we designed and prepared a anticancer PDC drug compound RGD-GFLG-DOX containing the tetrapeptide linker Gly-Phe-Leu-Gly, which can be cleaved in presence of cathepsin B, a highly upregulated enzyme in malignant tumors, to release the drug. RGD-GFLG was synthesized as a control. The inhibitory effects of RGD-GFLG-DOX on cancer cell lines were assessed using cytotoxicity assay. Specifically, the effects of RGD-GFLG-DOX were evaluated on integrin v3-positive cancer cell lines, including A549 and U87MG cells, integrin v3-negative cancer cell lines such as HeLa and MCF-7 cells, as well as normal cell lines, namely LO2 and MIHA cells. RGD-GFLG-DOX exhibited a lower cytotoxicity on HeLa, MCF-7, LO2 and MIHA cells, but a stronger cytotoxicity than DOX on A549 and U87MG cells. For comparison, RGD-GFLG demonstrated minimal cytotoxicity. In addition, the cytotoxicity of RGD-GFLG-DOX with various concentrations (0-40 uM) on A549 and U87MG cells was studied. The results showed that the cytotoxicity of RGD-GFLG-DOX on A549 and U87MG cells was dose-dependent. These indicate that RGD-GFLG-DOX has a good specificity and inhibitory activity toward integrin v3-overexpressed A549 and U87MG cells.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | LO #2 cell | CVCL_C7SD | ||
BP9a-SS-DOX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [262] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 6.21 ± 1.12 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Description |
The BP9a-SS-DOX conjugate exhibited impressed antiproliferative activity against HepG2 cells with an IC50 value of 6.21 ± 1.12 uM which was lower than that of free DOX.
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
DOXoxmCPP [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [993] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 11.4 μM | |||
| Description |
Doxorubicin binds with high affinity to DNA by intercalation, which leads to DNA damage and subsequent growth inhibition44,45. We performed MTT assays in order to determine the viability of the tumor cells and additionally to assess the proliferative potential of the cells after drug treatment. MCF-7 and HT-29 cells were incubated with doxorubicin and the peptide conjugates at various concentrations in the range of 0.1 and 50 μmfor 72 h (in a serum-containing medium), and the results were expressed as IC50-values (Table 1).
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [993] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 27 μM | |||
| Description |
Doxorubicin binds with high affinity to DNA by intercalation, which leads to DNA damage and subsequent growth inhibition44,45. We performed MTT assays in order to determine the viability of the tumor cells and additionally to assess the proliferative potential of the cells after drug treatment. MCF-7 and HT-29 cells were incubated with doxorubicin and the peptide conjugates at various concentrations in the range of 0.1 and 50 μmfor 72 h (in a serum-containing medium), and the results were expressed as IC50-values (Table 1).
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
BP9a-DOX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [994] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 18.5 ± 3.77 µM | |||
| Evaluation Method | CCK-8 assay | ||||
| Description |
Its antiproliferative activity against HepG2 cells (IC50 18.5 ± 3.77 uM) was lower than that of free DOX.
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
Peptide 18-4 doxorubicin conjugate 2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [990] | ||||
| Indication | Amelanotic melanoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 18.6 ± 2.5 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Here we report the design and synthesis of two new peptide-Dox conjugates (1 and 2) for the specific delivery of Dox to the breast cancer cells and the ability to overcome P-glycoprotein multidrug resistance pathway in both drug-sensitive and drug-resistant cancer cells. Peptide-Dox conjugates were evaluated for DOX release in human serum, intracellular delivery compared to free Dox in three cancerous cells (MCF-7, MDA-MB-435, and MDA-MB-435-MDR) and two noncancerous cell lines (HUVEC and MCF-10A), and cytotoxicity compared to free Dox. Results show that both the peptide-Dox conjugates (1 and 2) enter sensitive and resistant cell lines with minimal uptake in normal cells compared to free Dox. Cellular uptake is most likely mediated by a cell specific receptor, as the amount of internalized conjugates significantly decreased in the presence of excess free peptide. Importantly, conjugate 1 is equally cytotoxic as Dox in drug sensitive breast cancer cells and 4 times more potent than free Dox in Dox resistant cell line. Overall, the peptide-Dox ester conjugate 1 showed better breast targeting efficacy than the amide conjugate 2, most likely due to the slow release of Dox from the stable amide linkage.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity experiment was done by incubating the cells with different treatments for 48 h. The results show that conjugate 1 is quite similar to free Dox for toxicity to MCF-7 and MDA-MB-435 cancer cells. In contrast, conjugate 2 was ?20 times less cytotoxic to breast cancerous cells compared to free Dox. This could be attributed to the stability of the amide conjugate 2 inside the cells. Furthermore, the cytotoxicity of conjugate 1 in doxorubicin-resistant cell model MDA-MB-435-MDR (IC50 = 5.4 μM) is 4 times more than free Dox (IC50 = 22 μM). In normal cells (HUVEC and MCF-10A) the two conjugates were 3540 times less toxic compared to breast cancer cells, whereas free Dox was equally cytotoxic (equal IC50) to breast cancer cells and noncancerous cells. Overall these results provide clear evidence that of the two conjugates (conjugate 1 and 2), conjugate 1 has optimal characteristics for specific Dox targeting to breast cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | MDA-MB-435 cell | CVCL_0417 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [990] | ||||
| Indication | Amelanotic melanoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 19.7 ± 3.1 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Here we report the design and synthesis of two new peptide-Dox conjugates (1 and 2) for the specific delivery of Dox to the breast cancer cells and the ability to overcome P-glycoprotein multidrug resistance pathway in both drug-sensitive and drug-resistant cancer cells. Peptide-Dox conjugates were evaluated for DOX release in human serum, intracellular delivery compared to free Dox in three cancerous cells (MCF-7, MDA-MB-435, and MDA-MB-435-MDR) and two noncancerous cell lines (HUVEC and MCF-10A), and cytotoxicity compared to free Dox. Results show that both the peptide-Dox conjugates (1 and 2) enter sensitive and resistant cell lines with minimal uptake in normal cells compared to free Dox. Cellular uptake is most likely mediated by a cell specific receptor, as the amount of internalized conjugates significantly decreased in the presence of excess free peptide. Importantly, conjugate 1 is equally cytotoxic as Dox in drug sensitive breast cancer cells and 4 times more potent than free Dox in Dox resistant cell line. Overall, the peptide-Dox ester conjugate 1 showed better breast targeting efficacy than the amide conjugate 2, most likely due to the slow release of Dox from the stable amide linkage.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity experiment was done by incubating the cells with different treatments for 48 h. The results show that conjugate 1 is quite similar to free Dox for toxicity to MCF-7 and MDA-MB-435 cancer cells. In contrast, conjugate 2 was ?20 times less cytotoxic to breast cancerous cells compared to free Dox. This could be attributed to the stability of the amide conjugate 2 inside the cells. Furthermore, the cytotoxicity of conjugate 1 in doxorubicin-resistant cell model MDA-MB-435-MDR (IC50 = 5.4 μM) is 4 times more than free Dox (IC50 = 22 μM). In normal cells (HUVEC and MCF-10A) the two conjugates were 3540 times less toxic compared to breast cancer cells, whereas free Dox was equally cytotoxic (equal IC50) to breast cancer cells and noncancerous cells. Overall these results provide clear evidence that of the two conjugates (conjugate 1 and 2), conjugate 1 has optimal characteristics for specific Dox targeting to breast cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Amelanotic melanoma | MDA-MB-435 cell | CVCL_0417 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [990] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 40.5 ± 4.3 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Here we report the design and synthesis of two new peptide-Dox conjugates (1 and 2) for the specific delivery of Dox to the breast cancer cells and the ability to overcome P-glycoprotein multidrug resistance pathway in both drug-sensitive and drug-resistant cancer cells. Peptide-Dox conjugates were evaluated for DOX release in human serum, intracellular delivery compared to free Dox in three cancerous cells (MCF-7, MDA-MB-435, and MDA-MB-435-MDR) and two noncancerous cell lines (HUVEC and MCF-10A), and cytotoxicity compared to free Dox. Results show that both the peptide-Dox conjugates (1 and 2) enter sensitive and resistant cell lines with minimal uptake in normal cells compared to free Dox. Cellular uptake is most likely mediated by a cell specific receptor, as the amount of internalized conjugates significantly decreased in the presence of excess free peptide. Importantly, conjugate 1 is equally cytotoxic as Dox in drug sensitive breast cancer cells and 4 times more potent than free Dox in Dox resistant cell line. Overall, the peptide-Dox ester conjugate 1 showed better breast targeting efficacy than the amide conjugate 2, most likely due to the slow release of Dox from the stable amide linkage.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity experiment was done by incubating the cells with different treatments for 48 h. The results show that conjugate 1 is quite similar to free Dox for toxicity to MCF-7 and MDA-MB-435 cancer cells. In contrast, conjugate 2 was ?20 times less cytotoxic to breast cancerous cells compared to free Dox. This could be attributed to the stability of the amide conjugate 2 inside the cells. Furthermore, the cytotoxicity of conjugate 1 in doxorubicin-resistant cell model MDA-MB-435-MDR (IC50 = 5.4 μM) is 4 times more than free Dox (IC50 = 22 μM). In normal cells (HUVEC and MCF-10A) the two conjugates were 3540 times less toxic compared to breast cancer cells, whereas free Dox was equally cytotoxic (equal IC50) to breast cancer cells and noncancerous cells. Overall these results provide clear evidence that of the two conjugates (conjugate 1 and 2), conjugate 1 has optimal characteristics for specific Dox targeting to breast cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [990] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 50.9 ± 3.2 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Here we report the design and synthesis of two new peptide-Dox conjugates (1 and 2) for the specific delivery of Dox to the breast cancer cells and the ability to overcome P-glycoprotein multidrug resistance pathway in both drug-sensitive and drug-resistant cancer cells. Peptide-Dox conjugates were evaluated for DOX release in human serum, intracellular delivery compared to free Dox in three cancerous cells (MCF-7, MDA-MB-435, and MDA-MB-435-MDR) and two noncancerous cell lines (HUVEC and MCF-10A), and cytotoxicity compared to free Dox. Results show that both the peptide-Dox conjugates (1 and 2) enter sensitive and resistant cell lines with minimal uptake in normal cells compared to free Dox. Cellular uptake is most likely mediated by a cell specific receptor, as the amount of internalized conjugates significantly decreased in the presence of excess free peptide. Importantly, conjugate 1 is equally cytotoxic as Dox in drug sensitive breast cancer cells and 4 times more potent than free Dox in Dox resistant cell line. Overall, the peptide-Dox ester conjugate 1 showed better breast targeting efficacy than the amide conjugate 2, most likely due to the slow release of Dox from the stable amide linkage.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity experiment was done by incubating the cells with different treatments for 48 h. The results show that conjugate 1 is quite similar to free Dox for toxicity to MCF-7 and MDA-MB-435 cancer cells. In contrast, conjugate 2 was ?20 times less cytotoxic to breast cancerous cells compared to free Dox. This could be attributed to the stability of the amide conjugate 2 inside the cells. Furthermore, the cytotoxicity of conjugate 1 in doxorubicin-resistant cell model MDA-MB-435-MDR (IC50 = 5.4 μM) is 4 times more than free Dox (IC50 = 22 μM). In normal cells (HUVEC and MCF-10A) the two conjugates were 3540 times less toxic compared to breast cancer cells, whereas free Dox was equally cytotoxic (equal IC50) to breast cancer cells and noncancerous cells. Overall these results provide clear evidence that of the two conjugates (conjugate 1 and 2), conjugate 1 has optimal characteristics for specific Dox targeting to breast cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cell | Homo sapiens | ||
| Experiment 5 Reporting the Activity Data of This PDC | [990] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 191 ± 2.8 μM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| MOA of PDC |
Here we report the design and synthesis of two new peptide-Dox conjugates (1 and 2) for the specific delivery of Dox to the breast cancer cells and the ability to overcome P-glycoprotein multidrug resistance pathway in both drug-sensitive and drug-resistant cancer cells. Peptide-Dox conjugates were evaluated for DOX release in human serum, intracellular delivery compared to free Dox in three cancerous cells (MCF-7, MDA-MB-435, and MDA-MB-435-MDR) and two noncancerous cell lines (HUVEC and MCF-10A), and cytotoxicity compared to free Dox. Results show that both the peptide-Dox conjugates (1 and 2) enter sensitive and resistant cell lines with minimal uptake in normal cells compared to free Dox. Cellular uptake is most likely mediated by a cell specific receptor, as the amount of internalized conjugates significantly decreased in the presence of excess free peptide. Importantly, conjugate 1 is equally cytotoxic as Dox in drug sensitive breast cancer cells and 4 times more potent than free Dox in Dox resistant cell line. Overall, the peptide-Dox ester conjugate 1 showed better breast targeting efficacy than the amide conjugate 2, most likely due to the slow release of Dox from the stable amide linkage.
Click to Show/Hide
|
||||
| Description |
The cytotoxicity experiment was done by incubating the cells with different treatments for 48 h. The results show that conjugate 1 is quite similar to free Dox for toxicity to MCF-7 and MDA-MB-435 cancer cells. In contrast, conjugate 2 was ?20 times less cytotoxic to breast cancerous cells compared to free Dox. This could be attributed to the stability of the amide conjugate 2 inside the cells. Furthermore, the cytotoxicity of conjugate 1 in doxorubicin-resistant cell model MDA-MB-435-MDR (IC50 = 5.4 μM) is 4 times more than free Dox (IC50 = 22 μM). In normal cells (HUVEC and MCF-10A) the two conjugates were 3540 times less toxic compared to breast cancer cells, whereas free Dox was equally cytotoxic (equal IC50) to breast cancer cells and noncancerous cells. Overall these results provide clear evidence that of the two conjugates (conjugate 1 and 2), conjugate 1 has optimal characteristics for specific Dox targeting to breast cancer cells.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
DOXoxmCPPamph [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [993] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 19 μM | |||
| Description |
Doxorubicin binds with high affinity to DNA by intercalation, which leads to DNA damage and subsequent growth inhibition44,45. We performed MTT assays in order to determine the viability of the tumor cells and additionally to assess the proliferative potential of the cells after drug treatment. MCF-7 and HT-29 cells were incubated with doxorubicin and the peptide conjugates at various concentrations in the range of 0.1 and 50 μmfor 72 h (in a serum-containing medium), and the results were expressed as IC50-values (Table 1).
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
| Experiment 2 Reporting the Activity Data of This PDC | [993] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 24.7 μM | |||
| Description |
Doxorubicin binds with high affinity to DNA by intercalation, which leads to DNA damage and subsequent growth inhibition44,45. We performed MTT assays in order to determine the viability of the tumor cells and additionally to assess the proliferative potential of the cells after drug treatment. MCF-7 and HT-29 cells were incubated with doxorubicin and the peptide conjugates at various concentrations in the range of 0.1 and 50 μmfor 72 h (in a serum-containing medium), and the results were expressed as IC50-values (Table 1).
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
Octreotide doxorubicin conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [995] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 27.14 ± 2.47 μM | |||
| Evaluation Method | CellTiter-Glo cell proliferation assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
In the present work, we introduce a new approach to overcome all the aforementioned limitations. The cytotoxic drug doxorubicin is coupled to the tumor-targeting vector octreotide via a disulfide-intercalating cross-linking reagent. On the one hand, this reagent creates an oxime bond with the drug, and on the other hand, two disulfides with octreotide to keep the cyclic structure of the peptide. The combination of a hydrolytically stable oxime bond and disulfides leads to the formation of a novel bioconjugate superior to any previous anticancer drug-somatostatin analog hybrid as it allows the efficient release of the toxic cargo within the reducing environment of cancer cells. The versatility of the linker molecule described here will enable its future application not only in targeted drug delivery, but also in the chemical modification of therapeutic proteins.
Click to Show/Hide
|
||||
| Description |
We selected cells, where doxorubicin is typically applied and which overexpress somatostatin receptors, like the human pancreatic carcinoma cell line MIA PaCa-2 or MCF-7. Doxorubicins antiproliferative action on MIA PaCa-2 after 72 h incubation with the drug was as expected very strong (IC50 = 0.80 ± 0.13 μM), whereas octreotide was not able to inhibit cell growth by 50% up to a concentration of 150 μM. The cytotoxic effects of the conjugate expressed, as half maximal inhibitory concentration was much stronger compared to the precursor peptide, but, nevertheless, lower than that of doxorubicin (IC50 = 31.50 ± 1.74 μM). This characteristic feature of 12 is attributed to the different cellular uptake mechanisms of the substances. Doxorubicin is taken up quickly by passive diffusion, while octreotide enters cells by receptor-mediated endocytosis. Furthermore, it needs to be considered that the conjugate releases after cleavage by glutathione a doxorubicin derivative, which is still carrying a small cross-linker residue. Previous studies have shown that these types of molecules are, nevertheless, capable of successfully interacting with DNA, to mediate their cytotoxic properties, but to a lesser extent.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [995] | ||||
| Indication | Pancreatic carcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 31.50 ± 1.74 μM | |||
| Evaluation Method | CellTiter-Glo cell proliferation assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
In the present work, we introduce a new approach to overcome all the aforementioned limitations. The cytotoxic drug doxorubicin is coupled to the tumor-targeting vector octreotide via a disulfide-intercalating cross-linking reagent. On the one hand, this reagent creates an oxime bond with the drug, and on the other hand, two disulfides with octreotide to keep the cyclic structure of the peptide. The combination of a hydrolytically stable oxime bond and disulfides leads to the formation of a novel bioconjugate superior to any previous anticancer drug-somatostatin analog hybrid as it allows the efficient release of the toxic cargo within the reducing environment of cancer cells. The versatility of the linker molecule described here will enable its future application not only in targeted drug delivery, but also in the chemical modification of therapeutic proteins.
Click to Show/Hide
|
||||
| Description |
We selected cells, where doxorubicin is typically applied and which overexpress somatostatin receptors, like the human pancreatic carcinoma cell line MIA PaCa-2 or MCF-7. Doxorubicins antiproliferative action on MIA PaCa-2 after 72 h incubation with the drug was as expected very strong (IC50 = 0.80 ± 0.13 μM), whereas octreotide was not able to inhibit cell growth by 50% up to a concentration of 150 μM. The cytotoxic effects of the conjugate expressed, as half maximal inhibitory concentration was much stronger compared to the precursor peptide, but, nevertheless, lower than that of doxorubicin (IC50 = 31.50 ± 1.74 μM). This characteristic feature of 12 is attributed to the different cellular uptake mechanisms of the substances. Doxorubicin is taken up quickly by passive diffusion, while octreotide enters cells by receptor-mediated endocytosis. Furthermore, it needs to be considered that the conjugate releases after cleavage by glutathione a doxorubicin derivative, which is still carrying a small cross-linker residue. Previous studies have shown that these types of molecules are, nevertheless, capable of successfully interacting with DNA, to mediate their cytotoxic properties, but to a lesser extent.
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | MIA PaCa-2 cell | CVCL_0428 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [995] | ||||
| Indication | Pancreatic adenocarcinoma | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 48.90 ± 5.40 μM | |||
| Evaluation Method | CellTiter-Glo cell proliferation assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
In the present work, we introduce a new approach to overcome all the aforementioned limitations. The cytotoxic drug doxorubicin is coupled to the tumor-targeting vector octreotide via a disulfide-intercalating cross-linking reagent. On the one hand, this reagent creates an oxime bond with the drug, and on the other hand, two disulfides with octreotide to keep the cyclic structure of the peptide. The combination of a hydrolytically stable oxime bond and disulfides leads to the formation of a novel bioconjugate superior to any previous anticancer drug-somatostatin analog hybrid as it allows the efficient release of the toxic cargo within the reducing environment of cancer cells. The versatility of the linker molecule described here will enable its future application not only in targeted drug delivery, but also in the chemical modification of therapeutic proteins.
Click to Show/Hide
|
||||
| Description |
We selected cells, where doxorubicin is typically applied and which overexpress somatostatin receptors, like the human pancreatic carcinoma cell line MIA PaCa-2 or MCF-7. Doxorubicins antiproliferative action on MIA PaCa-2 after 72 h incubation with the drug was as expected very strong (IC50 = 0.80 ± 0.13 μM), whereas octreotide was not able to inhibit cell growth by 50% up to a concentration of 150 μM. The cytotoxic effects of the conjugate expressed, as half maximal inhibitory concentration was much stronger compared to the precursor peptide, but, nevertheless, lower than that of doxorubicin (IC50 = 31.50 ± 1.74 μM). This characteristic feature of 12 is attributed to the different cellular uptake mechanisms of the substances. Doxorubicin is taken up quickly by passive diffusion, while octreotide enters cells by receptor-mediated endocytosis. Furthermore, it needs to be considered that the conjugate releases after cleavage by glutathione a doxorubicin derivative, which is still carrying a small cross-linker residue. Previous studies have shown that these types of molecules are, nevertheless, capable of successfully interacting with DNA, to mediate their cytotoxic properties, but to a lesser extent.
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | Capan-1 cell | CVCL_0237 | ||
pHLIP-SS-DOX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [996] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 400 µM | |||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
Dox-GLRKRLRKFRNKIKK [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Cervical carcinoma | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) | 14.47 µM | |||
| Administration Time | 24 h | ||||
| MOA of PDC |
Herein, the design and synthesis of peptide-drug conjugates (PDCs) including different variants of the cell-penetrating peptide sC18 is presented. We first generated a series of novel sequence mutants of sC18 having either amino acid deletions and/or substitutions, and then tested their biological activity. The effects of histidine substituents were found to be not meaningful for sC18 uptake and cell selectivity. Moreover, building a nearly perfect amphipathic structure within a shortened sC18 derivative provided a peptide that was highly membrane-active, but also too cytotoxic. As a result, the most promising analog was sC18E, which stands out due to its higher uptake efficacy compared to parent sC18. In the last set of experiments, we let the peptides react with the cytotoxic drug doxorubicin by Thiol-Michael addition to form novel PDCs. Our results indicate that sC18E could be a more efficient drug carrier than parent sC18 for biomedical applications. However, cellular uptake using endocytosis and resulting entrapment of cargo inside vesicles is still a major critical step to overcome in CPP-containing peptide-drug development.
Click to Show/Hide
|
||||
| Description |
First, we probed the novel PDCs in non-cancerous human foreskin fibroblasts (HFF-1 cells). After 24 h treatment with different concentrations of PDCs, HFF-1 cells were still viable, while after adding doxorubicin viability was decreased up to 72%. In comparison, when we elucidated PDC activity in HeLa cells and exposed them for 24 h to various concentrations of the conjugates (2.5-70 μM), all of the PDCs, as well as free Dox, exhibited EC50 values in the lower micromolar range (PDC-1: 15.34 μM, PDC-2: 14.47 μM, PDC-3: 27.01 μM, Dox: 6.78 μM data not shown). The higher activity compared to HFF cells might be attributed to the fact that the PDCs were internalized to far less of an extent into the non-cancerous cell line. This observation might be advantageous and could reflect some selectivity of the more basic and positively charged peptides towards cancerous cells. We also noted that the obtained EC50 values somehow agreed with the results of the former assays. For example, sC18E was taken up to a significantly higher extent compared to sC18* and should, therefore, exhibit higher activity, e.g., drug delivery. However, surprisingly, the EC50 values of the PDCs containing sC18 and sC18E were quite similar, although sC18E significantly outcompeted sC18 in internalization activity.
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
Dox-GLRKRLRKFRNKIKEK [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Cervical carcinoma | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) | 15.34 µM | |||
| Administration Time | 24 h | ||||
| MOA of PDC |
Herein, the design and synthesis of peptide-drug conjugates (PDCs) including different variants of the cell-penetrating peptide sC18 is presented. We first generated a series of novel sequence mutants of sC18 having either amino acid deletions and/or substitutions, and then tested their biological activity. The effects of histidine substituents were found to be not meaningful for sC18 uptake and cell selectivity. Moreover, building a nearly perfect amphipathic structure within a shortened sC18 derivative provided a peptide that was highly membrane-active, but also too cytotoxic. As a result, the most promising analog was sC18E, which stands out due to its higher uptake efficacy compared to parent sC18. In the last set of experiments, we let the peptides react with the cytotoxic drug doxorubicin by Thiol-Michael addition to form novel PDCs. Our results indicate that sC18δE could be a more efficient drug carrier than parent sC18 for biomedical applications. However, cellular uptake using endocytosis and resulting entrapment of cargo inside vesicles is still a major critical step to overcome in CPP-containing peptide-drug development.
Click to Show/Hide
|
||||
| Description |
First, we probed the novel PDCs in non-cancerous human foreskin fibroblasts (HFF-1 cells). After 24 h treatment with different concentrations of PDCs, HFF-1 cells were still viable, while after adding doxorubicin viability was decreased up to 72%. In comparison, when we elucidated PDC activity in HeLa cells and exposed them for 24 h to various concentrations of the conjugates (2.5-70 μM), all of the PDCs, as well as free Dox, exhibited EC50 values in the lower micromolar range (PDC-1: 15.34 μM, PDC-2: 14.47 μM, PDC-3: 27.01 μM, Dox: 6.78 μM data not shown). The higher activity compared to HFF cells might be attributed to the fact that the PDCs were internalized to far less of an extent into the non-cancerous cell line. This observation might be advantageous and could reflect some selectivity of the more basic and positively charged peptides towards cancerous cells. We also noted that the obtained EC50 values somehow agreed with the results of the former assays. For example, sC18E was taken up to a significantly higher extent compared to sC18* and should, therefore, exhibit higher activity, e.g., drug delivery. However, surprisingly, the EC50 values of the PDCs containing sC18 and sC18E were quite similar, although sC18E significantly outcompeted sC18 in internalization activity.
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
Dox-GLRKRLRKFRNK [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Cervical carcinoma | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) | 27.01 µM | |||
| Administration Time | 24 h | ||||
| MOA of PDC |
Herein, the design and synthesis of peptide-drug conjugates (PDCs) including different variants of the cell-penetrating peptide sC18 is presented. We first generated a series of novel sequence mutants of sC18 having either amino acid deletions and/or substitutions, and then tested their biological activity. The effects of histidine substituents were found to be not meaningful for sC18 uptake and cell selectivity. Moreover, building a nearly perfect amphipathic structure within a shortened sC18 derivative provided a peptide that was highly membrane-active, but also too cytotoxic. As a result, the most promising analog was sC18E, which stands out due to its higher uptake efficacy compared to parent sC18. In the last set of experiments, we let the peptides react with the cytotoxic drug doxorubicin by Thiol-Michael addition to form novel PDCs. Our results indicate that sC18E could be a more efficient drug carrier than parent sC18 for biomedical applications. However, cellular uptake using endocytosis and resulting entrapment of cargo inside vesicles is still a major critical step to overcome in CPP-containing peptide-drug development.
Click to Show/Hide
|
||||
| Description |
First, we probed the novel PDCs in non-cancerous human foreskin fibroblasts (HFF-1 cells). After 24 h treatment with different concentrations of PDCs, HFF-1 cells were still viable, while after adding doxorubicin viability was decreased up to 72%. In comparison, when we elucidated PDC activity in HeLa cells and exposed them for 24 h to various concentrations of the conjugates (2.5-70 μM), all of the PDCs, as well as free Dox, exhibited EC50 values in the lower micromolar range (PDC-1: 15.34 μM, PDC-2: 14.47 μM, PDC-3: 27.01 μM, Dox: 6.78 μM data not shown). The higher activity compared to HFF cells might be attributed to the fact that the PDCs were internalized to far less of an extent into the non-cancerous cell line. This observation might be advantageous and could reflect some selectivity of the more basic and positively charged peptides towards cancerous cells. We also noted that the obtained EC50 values somehow agreed with the results of the former assays. For example, sC18E was taken up to a significantly higher extent compared to sC18* and should, therefore, exhibit higher activity, e.g., drug delivery. However, surprisingly, the EC50 values of the PDCs containing sC18 and sC18E were quite similar, although sC18E significantly outcompeted sC18 in internalization activity.
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
rL-A9-DOX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell viability | 7% | |||
| Evaluation Method | Flow cytometry assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 15 µM | ||||
| MOA of PDC |
The peptide A9 has been reported in the literature to exhibit high affinity and specificity towards the HER2 receptor. In our previous report, we observed the retro variant of A9 peptide, rL-A9, to be a promising molecule for targeting HER2-expressing breast cancer cells. The present study, thus aimed at designing and synthesis of a peptide-drug conjugate by linking the rL-A9 peptide with DOX. A linker, N-succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate (SMCC) was utilized for conjugation of DOX at one end and the peptide at the other end. The N-hydroxysuccinimidyl ester group of SMCC was conjugated with the amine (-NH2) group of DOX resulting in the formation of amide bond. The thiol (-SH) functionality was introduced by coupling cysteine amino acid at the N-terminus of the rL-A9 peptide for covalent linkage with the maleimide group of SMCC. Successful synthesis of the conjugate was confirmed by MALDI-TOF mass spectrometry. Cytotoxicity, cellular uptake and internalization of the peptide, drug and peptide-drug conjugate were assessed in SKOV3 cells using flow cytometry and confocal fluorescence microscopy.
Click to Show/Hide
|
||||
| Description |
Analysis of dot-plots suggests that the rL-A9 peptide alone does not exert any significant cytotoxic effects on either HER2-positive, SKOV3 cells or HER2-negative, MDA-MB-231 cells at any of the investigated concentrations. In contrast, incubation of drug DOX with either of the cells resulted in enhanced cell death with a negligible viable population even at lower concentrations. However, it was observed that the peptide-drug conjugate, rL-A9-DOX had a concentration-dependent impact on cell death (SKOV3 cells). Notably, nearly half of the cell population died at 3.75 uM. At a concentration of 15 uM, there were only 5% viable SKOV3 cells. The higher percent of the viable cell population was observed at corresponding concentrations in the case of incubation of rL-A9-DOX with HER2-negative, MDA-MB-231 cells. Comparative data of viability in two different cell lines is presented in Figure 6 for the peptide-drug conjugate, rL-A9-DOX.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell viability | 13% | |||
| Evaluation Method | Flow cytometry assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 15 µM | ||||
| MOA of PDC |
The peptide A9 has been reported in the literature to exhibit high affinity and specificity towards the HER2 receptor. In our previous report, we observed the retro variant of A9 peptide, rL-A9, to be a promising molecule for targeting HER2-expressing breast cancer cells. The present study, thus aimed at designing and synthesis of a peptide-drug conjugate by linking the rL-A9 peptide with DOX. A linker, N-succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate (SMCC) was utilized for conjugation of DOX at one end and the peptide at the other end. The N-hydroxysuccinimidyl ester group of SMCC was conjugated with the amine (-NH2) group of DOX resulting in the formation of amide bond. The thiol (-SH) functionality was introduced by coupling cysteine amino acid at the N-terminus of the rL-A9 peptide for covalent linkage with the maleimide group of SMCC. Successful synthesis of the conjugate was confirmed by MALDI-TOF mass spectrometry. Cytotoxicity, cellular uptake and internalization of the peptide, drug and peptide-drug conjugate were assessed in SKOV3 cells using flow cytometry and confocal fluorescence microscopy.
Click to Show/Hide
|
||||
| Description |
Analysis of dot-plots suggests that the rL-A9 peptide alone does not exert any significant cytotoxic effects on either HER2-positive, SKOV3 cells or HER2-negative, MDA-MB-231 cells at any of the investigated concentrations. In contrast, incubation of drug DOX with either of the cells resulted in enhanced cell death with a negligible viable population even at lower concentrations. However, it was observed that the peptide-drug conjugate, rL-A9-DOX had a concentration-dependent impact on cell death (SKOV3 cells). Notably, nearly half of the cell population died at 3.75 uM. At a concentration of 15 uM, there were only 5% viable SKOV3 cells. The higher percent of the viable cell population was observed at corresponding concentrations in the case of incubation of rL-A9-DOX with HER2-negative, MDA-MB-231 cells. Comparative data of viability in two different cell lines is presented in Figure 6 for the peptide-drug conjugate, rL-A9-DOX.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell viability | 26% | |||
| Evaluation Method | Flow cytometry assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 7.5 µM | ||||
| MOA of PDC |
The peptide A9 has been reported in the literature to exhibit high affinity and specificity towards the HER2 receptor. In our previous report, we observed the retro variant of A9 peptide, rL-A9, to be a promising molecule for targeting HER2-expressing breast cancer cells. The present study, thus aimed at designing and synthesis of a peptide-drug conjugate by linking the rL-A9 peptide with DOX. A linker, N-succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate (SMCC) was utilized for conjugation of DOX at one end and the peptide at the other end. The N-hydroxysuccinimidyl ester group of SMCC was conjugated with the amine (-NH2) group of DOX resulting in the formation of amide bond. The thiol (-SH) functionality was introduced by coupling cysteine amino acid at the N-terminus of the rL-A9 peptide for covalent linkage with the maleimide group of SMCC. Successful synthesis of the conjugate was confirmed by MALDI-TOF mass spectrometry. Cytotoxicity, cellular uptake and internalization of the peptide, drug and peptide-drug conjugate were assessed in SKOV3 cells using flow cytometry and confocal fluorescence microscopy.
Click to Show/Hide
|
||||
| Description |
Analysis of dot-plots suggests that the rL-A9 peptide alone does not exert any significant cytotoxic effects on either HER2-positive, SKOV3 cells or HER2-negative, MDA-MB-231 cells at any of the investigated concentrations. In contrast, incubation of drug DOX with either of the cells resulted in enhanced cell death with a negligible viable population even at lower concentrations. However, it was observed that the peptide-drug conjugate, rL-A9-DOX had a concentration-dependent impact on cell death (SKOV3 cells). Notably, nearly half of the cell population died at 3.75 uM. At a concentration of 15 uM, there were only 5% viable SKOV3 cells. The higher percent of the viable cell population was observed at corresponding concentrations in the case of incubation of rL-A9-DOX with HER2-negative, MDA-MB-231 cells. Comparative data of viability in two different cell lines is presented in Figure 6 for the peptide-drug conjugate, rL-A9-DOX.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell viability | 33% | |||
| Evaluation Method | Flow cytometry assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 7.5 µM | ||||
| MOA of PDC |
The peptide A9 has been reported in the literature to exhibit high affinity and specificity towards the HER2 receptor. In our previous report, we observed the retro variant of A9 peptide, rL-A9, to be a promising molecule for targeting HER2-expressing breast cancer cells. The present study, thus aimed at designing and synthesis of a peptide-drug conjugate by linking the rL-A9 peptide with DOX. A linker, N-succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate (SMCC) was utilized for conjugation of DOX at one end and the peptide at the other end. The N-hydroxysuccinimidyl ester group of SMCC was conjugated with the amine (-NH2) group of DOX resulting in the formation of amide bond. The thiol (-SH) functionality was introduced by coupling cysteine amino acid at the N-terminus of the rL-A9 peptide for covalent linkage with the maleimide group of SMCC. Successful synthesis of the conjugate was confirmed by MALDI-TOF mass spectrometry. Cytotoxicity, cellular uptake and internalization of the peptide, drug and peptide-drug conjugate were assessed in SKOV3 cells using flow cytometry and confocal fluorescence microscopy.
Click to Show/Hide
|
||||
| Description |
Analysis of dot-plots suggests that the rL-A9 peptide alone does not exert any significant cytotoxic effects on either HER2-positive, SKOV3 cells or HER2-negative, MDA-MB-231 cells at any of the investigated concentrations. In contrast, incubation of drug DOX with either of the cells resulted in enhanced cell death with a negligible viable population even at lower concentrations. However, it was observed that the peptide-drug conjugate, rL-A9-DOX had a concentration-dependent impact on cell death (SKOV3 cells). Notably, nearly half of the cell population died at 3.75 uM. At a concentration of 15 uM, there were only 5% viable SKOV3 cells. The higher percent of the viable cell population was observed at corresponding concentrations in the case of incubation of rL-A9-DOX with HER2-negative, MDA-MB-231 cells. Comparative data of viability in two different cell lines is presented in Figure 6 for the peptide-drug conjugate, rL-A9-DOX.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell viability | 49% | |||
| Evaluation Method | Flow cytometry assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 3.75 µM | ||||
| MOA of PDC |
The peptide A9 has been reported in the literature to exhibit high affinity and specificity towards the HER2 receptor. In our previous report, we observed the retro variant of A9 peptide, rL-A9, to be a promising molecule for targeting HER2-expressing breast cancer cells. The present study, thus aimed at designing and synthesis of a peptide-drug conjugate by linking the rL-A9 peptide with DOX. A linker, N-succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate (SMCC) was utilized for conjugation of DOX at one end and the peptide at the other end. The N-hydroxysuccinimidyl ester group of SMCC was conjugated with the amine (-NH2) group of DOX resulting in the formation of amide bond. The thiol (-SH) functionality was introduced by coupling cysteine amino acid at the N-terminus of the rL-A9 peptide for covalent linkage with the maleimide group of SMCC. Successful synthesis of the conjugate was confirmed by MALDI-TOF mass spectrometry. Cytotoxicity, cellular uptake and internalization of the peptide, drug and peptide-drug conjugate were assessed in SKOV3 cells using flow cytometry and confocal fluorescence microscopy.
Click to Show/Hide
|
||||
| Description |
Analysis of dot-plots suggests that the rL-A9 peptide alone does not exert any significant cytotoxic effects on either HER2-positive, SKOV3 cells or HER2-negative, MDA-MB-231 cells at any of the investigated concentrations. In contrast, incubation of drug DOX with either of the cells resulted in enhanced cell death with a negligible viable population even at lower concentrations. However, it was observed that the peptide-drug conjugate, rL-A9-DOX had a concentration-dependent impact on cell death (SKOV3 cells). Notably, nearly half of the cell population died at 3.75 uM. At a concentration of 15 uM, there were only 5% viable SKOV3 cells. The higher percent of the viable cell population was observed at corresponding concentrations in the case of incubation of rL-A9-DOX with HER2-negative, MDA-MB-231 cells. Comparative data of viability in two different cell lines is presented in Figure 6 for the peptide-drug conjugate, rL-A9-DOX.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell viability | 67% | |||
| Evaluation Method | Flow cytometry assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 1.87 µM | ||||
| MOA of PDC |
The peptide A9 has been reported in the literature to exhibit high affinity and specificity towards the HER2 receptor. In our previous report, we observed the retro variant of A9 peptide, rL-A9, to be a promising molecule for targeting HER2-expressing breast cancer cells. The present study, thus aimed at designing and synthesis of a peptide-drug conjugate by linking the rL-A9 peptide with DOX. A linker, N-succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate (SMCC) was utilized for conjugation of DOX at one end and the peptide at the other end. The N-hydroxysuccinimidyl ester group of SMCC was conjugated with the amine (-NH2) group of DOX resulting in the formation of amide bond. The thiol (-SH) functionality was introduced by coupling cysteine amino acid at the N-terminus of the rL-A9 peptide for covalent linkage with the maleimide group of SMCC. Successful synthesis of the conjugate was confirmed by MALDI-TOF mass spectrometry. Cytotoxicity, cellular uptake and internalization of the peptide, drug and peptide-drug conjugate were assessed in SKOV3 cells using flow cytometry and confocal fluorescence microscopy.
Click to Show/Hide
|
||||
| Description |
Analysis of dot-plots suggests that the rL-A9 peptide alone does not exert any significant cytotoxic effects on either HER2-positive, SKOV3 cells or HER2-negative, MDA-MB-231 cells at any of the investigated concentrations. In contrast, incubation of drug DOX with either of the cells resulted in enhanced cell death with a negligible viable population even at lower concentrations. However, it was observed that the peptide-drug conjugate, rL-A9-DOX had a concentration-dependent impact on cell death (SKOV3 cells). Notably, nearly half of the cell population died at 3.75 uM. At a concentration of 15 uM, there were only 5% viable SKOV3 cells. The higher percent of the viable cell population was observed at corresponding concentrations in the case of incubation of rL-A9-DOX with HER2-negative, MDA-MB-231 cells. Comparative data of viability in two different cell lines is presented in Figure 6 for the peptide-drug conjugate, rL-A9-DOX.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell viability | 87% | |||
| Evaluation Method | Flow cytometry assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 3.75 µM | ||||
| MOA of PDC |
The peptide A9 has been reported in the literature to exhibit high affinity and specificity towards the HER2 receptor. In our previous report, we observed the retro variant of A9 peptide, rL-A9, to be a promising molecule for targeting HER2-expressing breast cancer cells. The present study, thus aimed at designing and synthesis of a peptide-drug conjugate by linking the rL-A9 peptide with DOX. A linker, N-succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate (SMCC) was utilized for conjugation of DOX at one end and the peptide at the other end. The N-hydroxysuccinimidyl ester group of SMCC was conjugated with the amine (-NH2) group of DOX resulting in the formation of amide bond. The thiol (-SH) functionality was introduced by coupling cysteine amino acid at the N-terminus of the rL-A9 peptide for covalent linkage with the maleimide group of SMCC. Successful synthesis of the conjugate was confirmed by MALDI-TOF mass spectrometry. Cytotoxicity, cellular uptake and internalization of the peptide, drug and peptide-drug conjugate were assessed in SKOV3 cells using flow cytometry and confocal fluorescence microscopy.
Click to Show/Hide
|
||||
| Description |
Analysis of dot-plots suggests that the rL-A9 peptide alone does not exert any significant cytotoxic effects on either HER2-positive, SKOV3 cells or HER2-negative, MDA-MB-231 cells at any of the investigated concentrations. In contrast, incubation of drug DOX with either of the cells resulted in enhanced cell death with a negligible viable population even at lower concentrations. However, it was observed that the peptide-drug conjugate, rL-A9-DOX had a concentration-dependent impact on cell death (SKOV3 cells). Notably, nearly half of the cell population died at 3.75 uM. At a concentration of 15 uM, there were only 5% viable SKOV3 cells. The higher percent of the viable cell population was observed at corresponding concentrations in the case of incubation of rL-A9-DOX with HER2-negative, MDA-MB-231 cells. Comparative data of viability in two different cell lines is presented in Figure 6 for the peptide-drug conjugate, rL-A9-DOX.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell viability | 91% | |||
| Evaluation Method | Flow cytometry assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 0.94 µM | ||||
| MOA of PDC |
The peptide A9 has been reported in the literature to exhibit high affinity and specificity towards the HER2 receptor. In our previous report, we observed the retro variant of A9 peptide, rL-A9, to be a promising molecule for targeting HER2-expressing breast cancer cells. The present study, thus aimed at designing and synthesis of a peptide-drug conjugate by linking the rL-A9 peptide with DOX. A linker, N-succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate (SMCC) was utilized for conjugation of DOX at one end and the peptide at the other end. The N-hydroxysuccinimidyl ester group of SMCC was conjugated with the amine (-NH2) group of DOX resulting in the formation of amide bond. The thiol (-SH) functionality was introduced by coupling cysteine amino acid at the N-terminus of the rL-A9 peptide for covalent linkage with the maleimide group of SMCC. Successful synthesis of the conjugate was confirmed by MALDI-TOF mass spectrometry. Cytotoxicity, cellular uptake and internalization of the peptide, drug and peptide-drug conjugate were assessed in SKOV3 cells using flow cytometry and confocal fluorescence microscopy.
Click to Show/Hide
|
||||
| Description |
Analysis of dot-plots suggests that the rL-A9 peptide alone does not exert any significant cytotoxic effects on either HER2-positive, SKOV3 cells or HER2-negative, MDA-MB-231 cells at any of the investigated concentrations. In contrast, incubation of drug DOX with either of the cells resulted in enhanced cell death with a negligible viable population even at lower concentrations. However, it was observed that the peptide-drug conjugate, rL-A9-DOX had a concentration-dependent impact on cell death (SKOV3 cells). Notably, nearly half of the cell population died at 3.75 uM. At a concentration of 15 uM, there were only 5% viable SKOV3 cells. The higher percent of the viable cell population was observed at corresponding concentrations in the case of incubation of rL-A9-DOX with HER2-negative, MDA-MB-231 cells. Comparative data of viability in two different cell lines is presented in Figure 6 for the peptide-drug conjugate, rL-A9-DOX.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell viability | 94% | |||
| Evaluation Method | Flow cytometry assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 1.87 µM | ||||
| MOA of PDC |
The peptide A9 has been reported in the literature to exhibit high affinity and specificity towards the HER2 receptor. In our previous report, we observed the retro variant of A9 peptide, rL-A9, to be a promising molecule for targeting HER2-expressing breast cancer cells. The present study, thus aimed at designing and synthesis of a peptide-drug conjugate by linking the rL-A9 peptide with DOX. A linker, N-succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate (SMCC) was utilized for conjugation of DOX at one end and the peptide at the other end. The N-hydroxysuccinimidyl ester group of SMCC was conjugated with the amine (-NH2) group of DOX resulting in the formation of amide bond. The thiol (-SH) functionality was introduced by coupling cysteine amino acid at the N-terminus of the rL-A9 peptide for covalent linkage with the maleimide group of SMCC. Successful synthesis of the conjugate was confirmed by MALDI-TOF mass spectrometry. Cytotoxicity, cellular uptake and internalization of the peptide, drug and peptide-drug conjugate were assessed in SKOV3 cells using flow cytometry and confocal fluorescence microscopy.
Click to Show/Hide
|
||||
| Description |
Analysis of dot-plots suggests that the rL-A9 peptide alone does not exert any significant cytotoxic effects on either HER2-positive, SKOV3 cells or HER2-negative, MDA-MB-231 cells at any of the investigated concentrations. In contrast, incubation of drug DOX with either of the cells resulted in enhanced cell death with a negligible viable population even at lower concentrations. However, it was observed that the peptide-drug conjugate, rL-A9-DOX had a concentration-dependent impact on cell death (SKOV3 cells). Notably, nearly half of the cell population died at 3.75 uM. At a concentration of 15 uM, there were only 5% viable SKOV3 cells. The higher percent of the viable cell population was observed at corresponding concentrations in the case of incubation of rL-A9-DOX with HER2-negative, MDA-MB-231 cells. Comparative data of viability in two different cell lines is presented in Figure 6 for the peptide-drug conjugate, rL-A9-DOX.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell viability | 95% | |||
| Evaluation Method | Flow cytometry assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 0.47 µM | ||||
| MOA of PDC |
The peptide A9 has been reported in the literature to exhibit high affinity and specificity towards the HER2 receptor. In our previous report, we observed the retro variant of A9 peptide, rL-A9, to be a promising molecule for targeting HER2-expressing breast cancer cells. The present study, thus aimed at designing and synthesis of a peptide-drug conjugate by linking the rL-A9 peptide with DOX. A linker, N-succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate (SMCC) was utilized for conjugation of DOX at one end and the peptide at the other end. The N-hydroxysuccinimidyl ester group of SMCC was conjugated with the amine (-NH2) group of DOX resulting in the formation of amide bond. The thiol (-SH) functionality was introduced by coupling cysteine amino acid at the N-terminus of the rL-A9 peptide for covalent linkage with the maleimide group of SMCC. Successful synthesis of the conjugate was confirmed by MALDI-TOF mass spectrometry. Cytotoxicity, cellular uptake and internalization of the peptide, drug and peptide-drug conjugate were assessed in SKOV3 cells using flow cytometry and confocal fluorescence microscopy.
Click to Show/Hide
|
||||
| Description |
Analysis of dot-plots suggests that the rL-A9 peptide alone does not exert any significant cytotoxic effects on either HER2-positive, SKOV3 cells or HER2-negative, MDA-MB-231 cells at any of the investigated concentrations. In contrast, incubation of drug DOX with either of the cells resulted in enhanced cell death with a negligible viable population even at lower concentrations. However, it was observed that the peptide-drug conjugate, rL-A9-DOX had a concentration-dependent impact on cell death (SKOV3 cells). Notably, nearly half of the cell population died at 3.75 uM. At a concentration of 15 uM, there were only 5% viable SKOV3 cells. The higher percent of the viable cell population was observed at corresponding concentrations in the case of incubation of rL-A9-DOX with HER2-negative, MDA-MB-231 cells. Comparative data of viability in two different cell lines is presented in Figure 6 for the peptide-drug conjugate, rL-A9-DOX.
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian serous cystadenocarcinoma | SK-OV-3 cell | CVCL_0532 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell viability | 96% | |||
| Evaluation Method | Flow cytometry assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 0.94 µM | ||||
| MOA of PDC |
The peptide A9 has been reported in the literature to exhibit high affinity and specificity towards the HER2 receptor. In our previous report, we observed the retro variant of A9 peptide, rL-A9, to be a promising molecule for targeting HER2-expressing breast cancer cells. The present study, thus aimed at designing and synthesis of a peptide-drug conjugate by linking the rL-A9 peptide with DOX. A linker, N-succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate (SMCC) was utilized for conjugation of DOX at one end and the peptide at the other end. The N-hydroxysuccinimidyl ester group of SMCC was conjugated with the amine (-NH2) group of DOX resulting in the formation of amide bond. The thiol (-SH) functionality was introduced by coupling cysteine amino acid at the N-terminus of the rL-A9 peptide for covalent linkage with the maleimide group of SMCC. Successful synthesis of the conjugate was confirmed by MALDI-TOF mass spectrometry. Cytotoxicity, cellular uptake and internalization of the peptide, drug and peptide-drug conjugate were assessed in SKOV3 cells using flow cytometry and confocal fluorescence microscopy.
Click to Show/Hide
|
||||
| Description |
Analysis of dot-plots suggests that the rL-A9 peptide alone does not exert any significant cytotoxic effects on either HER2-positive, SKOV3 cells or HER2-negative, MDA-MB-231 cells at any of the investigated concentrations. In contrast, incubation of drug DOX with either of the cells resulted in enhanced cell death with a negligible viable population even at lower concentrations. However, it was observed that the peptide-drug conjugate, rL-A9-DOX had a concentration-dependent impact on cell death (SKOV3 cells). Notably, nearly half of the cell population died at 3.75 uM. At a concentration of 15 uM, there were only 5% viable SKOV3 cells. The higher percent of the viable cell population was observed at corresponding concentrations in the case of incubation of rL-A9-DOX with HER2-negative, MDA-MB-231 cells. Comparative data of viability in two different cell lines is presented in Figure 6 for the peptide-drug conjugate, rL-A9-DOX.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell viability | 97% | |||
| Evaluation Method | Flow cytometry assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 0.47 µM | ||||
| MOA of PDC |
The peptide A9 has been reported in the literature to exhibit high affinity and specificity towards the HER2 receptor. In our previous report, we observed the retro variant of A9 peptide, rL-A9, to be a promising molecule for targeting HER2-expressing breast cancer cells. The present study, thus aimed at designing and synthesis of a peptide-drug conjugate by linking the rL-A9 peptide with DOX. A linker, N-succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate (SMCC) was utilized for conjugation of DOX at one end and the peptide at the other end. The N-hydroxysuccinimidyl ester group of SMCC was conjugated with the amine (-NH2) group of DOX resulting in the formation of amide bond. The thiol (-SH) functionality was introduced by coupling cysteine amino acid at the N-terminus of the rL-A9 peptide for covalent linkage with the maleimide group of SMCC. Successful synthesis of the conjugate was confirmed by MALDI-TOF mass spectrometry. Cytotoxicity, cellular uptake and internalization of the peptide, drug and peptide-drug conjugate were assessed in SKOV3 cells using flow cytometry and confocal fluorescence microscopy.
Click to Show/Hide
|
||||
| Description |
Analysis of dot-plots suggests that the rL-A9 peptide alone does not exert any significant cytotoxic effects on either HER2-positive, SKOV3 cells or HER2-negative, MDA-MB-231 cells at any of the investigated concentrations. In contrast, incubation of drug DOX with either of the cells resulted in enhanced cell death with a negligible viable population even at lower concentrations. However, it was observed that the peptide-drug conjugate, rL-A9-DOX had a concentration-dependent impact on cell death (SKOV3 cells). Notably, nearly half of the cell population died at 3.75 uM. At a concentration of 15 uM, there were only 5% viable SKOV3 cells. The higher percent of the viable cell population was observed at corresponding concentrations in the case of incubation of rL-A9-DOX with HER2-negative, MDA-MB-231 cells. Comparative data of viability in two different cell lines is presented in Figure 6 for the peptide-drug conjugate, rL-A9-DOX.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
[d-Cys6-des-Gly10-Pro9-NHEt]-GnRH-Dox [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [997] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 10% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 100 µM | ||||
| Description |
The results showed that all the conjugates had lower antiproliferative effects than Dox at the tested concentrations (Figure 2A); this may be related to inefficient release of Dox from the conjugate caused by the relative stable thioether bond linkage between Dox-SMP and GnRH analog. The antiproliferative effects of the three conjugates were close at 25, 50, and 75 uM. While at 100 uM, conjugate II exhibited higher inhibitory effect than that of I and III.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 7.45 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [997] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 28% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 75 µM | ||||
| MOA of PDC |
Although RNT with 177Lu-DOTATATE/PSMA is known as a novel and effective therapy option for cancer that significantly improves the quality of life and survival of patients, it may have acute or chronic side effects. Therefore, any method that can ameliorate these side effects is useful in the RNT process. For this purpose, a few clinical studies have reported that antioxidants as free radical scavengers such as amifostine and vitamins C and E can reduce radioiodine-related side effects, particularly in salivary glands in thyroid cancer patients.
Click to Show/Hide
|
||||
| Description |
The results showed that all the conjugates had lower antiproliferative effects than Dox at the tested concentrations (Figure 2A); this may be related to inefficient release of Dox from the conjugate caused by the relative stable thioether bond linkage between Dox-SMP and GnRH analog. The antiproliferative effects of the three conjugates were close at 25, 50, and 75 uM. While at 100 uM, conjugate II exhibited higher inhibitory effect than that of I and III.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 7.45 h | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [997] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 45% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 50 µM | ||||
| Description |
The results showed that all the conjugates had lower antiproliferative effects than Dox at the tested concentrations (Figure 2A); this may be related to inefficient release of Dox from the conjugate caused by the relative stable thioether bond linkage between Dox-SMP and GnRH analog. The antiproliferative effects of the three conjugates were close at 25, 50, and 75 uM. While at 100 uM, conjugate II exhibited higher inhibitory effect than that of I and III.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 7.45 h | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [997] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 48% | |||
| Administration Time | 48 h | ||||
| Administration Dosage | 100 µM; with 10- µM leuprolide for 2 hours | ||||
| Description |
The combinations of conjugate II and leuprolide exhibited lower antiproliferative efficacy on MCF-7 cells than conjugate II individually at all the tested concentrations (Figure 3A).
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 7.45 h | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [997] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 50% | |||
| Administration Time | 48 h | ||||
| Administration Dosage | 75 µM; with 10- µM leuprolide for 2 hours | ||||
| MOA of PDC |
Vitamin C as a water-soluble vitamin is the reduced form of ascorbic acid. No significant adverse effect of taking high doses of vitamin C (over 2000 mg/day) has been reported due to the water-soluble feature of vitamin C. Vitamin C directly reacts with hydroxy, alkoxyl, and lipid peroxyl radicals and converts them to alcohol, water, and hydroperoxide lipid, respectively. It has been shown that taking vitamin C before radioiodine therapy can ameliorate the oxidative stress effect of radioiodine. The radioprotective effects of vitamin C are mainly due to its free radical scavenging activity.
Click to Show/Hide
|
||||
| Description |
The combinations of conjugate II and leuprolide exhibited lower antiproliferative efficacy on MCF-7 cells than conjugate II individually at all the tested concentrations (Figure 3A).
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 7.45 h | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [997] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 52% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 25 µM | ||||
| Description |
The results showed that all the conjugates had lower antiproliferative effects than Dox at the tested concentrations (Figure 2A); this may be related to inefficient release of Dox from the conjugate caused by the relative stable thioether bond linkage between Dox-SMP and GnRH analog. The antiproliferative effects of the three conjugates were close at 25, 50, and 75 uM. While at 100 uM, conjugate II exhibited higher inhibitory effect than that of I and III.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 7.45 h | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [997] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 72% | |||
| Administration Time | 48 h | ||||
| Administration Dosage | 50 µM; with 10- µM leuprolide for 2 hours | ||||
| Description |
The combinations of conjugate II and leuprolide exhibited lower antiproliferative efficacy on MCF-7 cells than conjugate II individually at all the tested concentrations (Figure 3A).
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 7.45 h | ||||
| Experiment 8 Reporting the Activity Data of This PDC | [997] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 75% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 100 µM | ||||
| Description |
However, by linking to [d-Cys6-des-Gly10-Pro9-NHEt]-GnRH, the cytotoxicity of Dox against 3T3 cells was reduced because the cell viability was over 87% after treatment with conjugate II in 25, 50, and 75 uM of equivalent concentration of Dox. Even at 100 uM, conjugate II maintained the cell viability to about 74% (Figure 2B).
|
||||
| In Vitro Model | Normal | NIH 3T3 cell | CVCL_0594 | ||
| Half life period | 7.45 h | ||||
| Experiment 9 Reporting the Activity Data of This PDC | [997] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 90% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 75 µM | ||||
| MOA of PDC |
In biological systems, antioxidants such as catalase, superoxide dismutase, glutathione peroxidase, and glutathione reductase are responsible for the elimination or reduction of the adverse effects of ROS, that is, they prevent or reduce ROS generation. Dietary antioxidants, such as vitamins E, A, and C, and anthocyanins and polyphenols have a role in the protection of cells against ROS damage.
Click to Show/Hide
|
||||
| Description |
However, by linking to [d-Cys6-des-Gly10-Pro9-NHEt]-GnRH, the cytotoxicity of Dox against 3T3 cells was reduced because the cell viability was over 87% after treatment with conjugate II in 25, 50, and 75 uM of equivalent concentration of Dox. Even at 100 uM, conjugate II maintained the cell viability to about 74% (Figure 2B).
|
||||
| In Vitro Model | Normal | NIH 3T3 cell | CVCL_0594 | ||
| Half life period | 7.45 h | ||||
| Experiment 10 Reporting the Activity Data of This PDC | [997] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 92% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 50 µM | ||||
| Description |
However, by linking to [d-Cys6-des-Gly10-Pro9-NHEt]-GnRH, the cytotoxicity of Dox against 3T3 cells was reduced because the cell viability was over 87% after treatment with conjugate II in 25, 50, and 75 uM of equivalent concentration of Dox. Even at 100 uM, conjugate II maintained the cell viability to about 74% (Figure 2B).
|
||||
| In Vitro Model | Normal | NIH 3T3 cell | CVCL_0594 | ||
| Half life period | 7.45 h | ||||
| Experiment 11 Reporting the Activity Data of This PDC | [997] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 98% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 25 µM | ||||
| Description |
However, by linking to [d-Cys6-des-Gly10-Pro9-NHEt]-GnRH, the cytotoxicity of Dox against 3T3 cells was reduced because the cell viability was over 87% after treatment with conjugate II in 25, 50, and 75 uM of equivalent concentration of Dox. Even at 100 uM, conjugate II maintained the cell viability to about 74% (Figure 2B).
|
||||
| In Vitro Model | Normal | NIH 3T3 cell | CVCL_0594 | ||
| Half life period | 7.45 h | ||||
| Experiment 12 Reporting the Activity Data of This PDC | [997] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 108% | |||
| Administration Time | 48 h | ||||
| Administration Dosage | 25 µM; with 10- µM leuprolide for 2 hours | ||||
| Description |
The combinations of conjugate II and leuprolide exhibited lower antiproliferative efficacy on MCF-7 cells than conjugate II individually at all the tested concentrations (Figure 3A).
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 7.45 h | ||||
pHLIP-PAMAM-DOX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 15% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 10 µM | ||||
| Description |
The cytotoxicity data for the pHLIP-PAMAM-DOX conjugate mirrored quite closely that of the pHLIP-S-S-DOX conjugate, particularly at the higher concentrations (>1.25 μM). However, at lower pHLIP concentrations (0.16 μM-0.63 μM), the PAMAM conjugate exhibited higher cytotoxicity than the single DOX conjugate (˜up to 17% higher toxicity).
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 20% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 2.5 µM | ||||
| Description |
The cytotoxicity data for the pHLIP-PAMAM-DOX conjugate mirrored quite closely that of the pHLIP-S-S-DOX conjugate, particularly at the higher concentrations (>1.25 μM). However, at lower pHLIP concentrations (0.16 μM-0.63 μM), the PAMAM conjugate exhibited higher cytotoxicity than the single DOX conjugate (˜up to 17% higher toxicity).
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 20% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 5.0 µM | ||||
| Description |
The cytotoxicity data for the pHLIP-PAMAM-DOX conjugate mirrored quite closely that of the pHLIP-S-S-DOX conjugate, particularly at the higher concentrations (>1.25 μM). However, at lower pHLIP concentrations (0.16 μM-0.63 μM), the PAMAM conjugate exhibited higher cytotoxicity than the single DOX conjugate (˜up to 17% higher toxicity).
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 90% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 0.16 µM | ||||
| Description |
The cytotoxicity data for the pHLIP-PAMAM-DOX conjugate mirrored quite closely that of the pHLIP-S-S-DOX conjugate, particularly at the higher concentrations (>1.25 μM). However, at lower pHLIP concentrations (0.16 μM-0.63 μM), the PAMAM conjugate exhibited higher cytotoxicity than the single DOX conjugate (˜up to 17% higher toxicity).
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 90% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 0.31 µM | ||||
| Description |
The cytotoxicity data for the pHLIP-PAMAM-DOX conjugate mirrored quite closely that of the pHLIP-S-S-DOX conjugate, particularly at the higher concentrations (>1.25 μM). However, at lower pHLIP concentrations (0.16 μM-0.63 μM), the PAMAM conjugate exhibited higher cytotoxicity than the single DOX conjugate (˜up to 17% higher toxicity).
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 90% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 1.3 µM | ||||
| Description |
The cytotoxicity data for the pHLIP-PAMAM-DOX conjugate mirrored quite closely that of the pHLIP-S-S-DOX conjugate, particularly at the higher concentrations (>1.25 μM). However, at lower pHLIP concentrations (0.16 μM-0.63 μM), the PAMAM conjugate exhibited higher cytotoxicity than the single DOX conjugate (˜up to 17% higher toxicity).
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 95% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 0.63 µM | ||||
| Description |
The cytotoxicity data for the pHLIP-PAMAM-DOX conjugate mirrored quite closely that of the pHLIP-S-S-DOX conjugate, particularly at the higher concentrations (>1.25 μM). However, at lower pHLIP concentrations (0.16 μM-0.63 μM), the PAMAM conjugate exhibited higher cytotoxicity than the single DOX conjugate (˜up to 17% higher toxicity).
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 98% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 10 µM | ||||
| Description |
The cytotoxicity data for the pHLIP-PAMAM-DOX conjugate mirrored quite closely that of the pHLIP-S-S-DOX conjugate, particularly at the higher concentrations (>1.25 μM). However, at lower pHLIP concentrations (0.16 μM-0.63 μM), the PAMAM conjugate exhibited higher cytotoxicity than the single DOX conjugate (˜up to 17% higher toxicity).
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 100% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 2.5 µM | ||||
| Description |
The cytotoxicity data for the pHLIP-PAMAM-DOX conjugate mirrored quite closely that of the pHLIP-S-S-DOX conjugate, particularly at the higher concentrations (>1.25 μM). However, at lower pHLIP concentrations (0.16 μM-0.63 μM), the PAMAM conjugate exhibited higher cytotoxicity than the single DOX conjugate (˜up to 17% higher toxicity).
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 100% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 5.0 µM | ||||
| Description |
The cytotoxicity data for the pHLIP-PAMAM-DOX conjugate mirrored quite closely that of the pHLIP-S-S-DOX conjugate, particularly at the higher concentrations (>1.25 μM). However, at lower pHLIP concentrations (0.16 μM-0.63 μM), the PAMAM conjugate exhibited higher cytotoxicity than the single DOX conjugate (˜up to 17% higher toxicity).
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
pHLIP-S-S-DOX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 18% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 10 µM | ||||
| Description |
HeLa cells treated with pHLIP-S-S-DOX at low pH showed significant toxicity (˜50% viability at the lowest concentration tested (0.16 μM pHLIP)) which, in this construct, corresponds to the DOX concentration. Cellular viability tracked inversely with increasing pHLIP conjugate concentration, peaking at ˜18% viability at 16 μM.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 20% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 1.3 µM | ||||
| Description |
HeLa cells treated with pHLIP-S-S-DOX at low pH showed significant toxicity (˜50% viability at the lowest concentration tested (0.16 μM pHLIP)) which, in this construct, corresponds to the DOX concentration. Cellular viability tracked inversely with increasing pHLIP conjugate concentration, peaking at ˜18% viability at 13 μM.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 22% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 2.5 µM | ||||
| Description |
HeLa cells treated with pHLIP-S-S-DOX at low pH showed significant toxicity (˜50% viability at the lowest concentration tested (0.16 μM pHLIP)) which, in this construct, corresponds to the DOX concentration. Cellular viability tracked inversely with increasing pHLIP conjugate concentration, peaking at ˜18% viability at 14 μM.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 22% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 5.0 µM | ||||
| Description |
HeLa cells treated with pHLIP-S-S-DOX at low pH showed significant toxicity (˜50% viability at the lowest concentration tested (0.16 μM pHLIP)) which, in this construct, corresponds to the DOX concentration. Cellular viability tracked inversely with increasing pHLIP conjugate concentration, peaking at ˜18% viability at 15 μM.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 30% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 0.31 µM | ||||
| Description |
The cytotoxicity data for the pHLIP-PAMAM-DOX conjugate mirrored quite closely that of the pHLIP-S-S-DOX conjugate, particularly at the higher concentrations (>1.25 μM). However, at lower pHLIP concentrations (0.16 μM-0.63 μM), the PAMAM conjugate exhibited higher cytotoxicity than the single DOX conjugate (˜up to 17% higher toxicity).
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 30% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 0.63 µM | ||||
| Description |
The cytotoxicity data for the pHLIP-PAMAM-DOX conjugate mirrored quite closely that of the pHLIP-S-S-DOX conjugate, particularly at the higher concentrations (>1.25 μM). However, at lower pHLIP concentrations (0.16 μM-0.63 μM), the PAMAM conjugate exhibited higher cytotoxicity than the single DOX conjugate (˜up to 17% higher toxicity).
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 30% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 1.3 µM | ||||
| Description |
The cytotoxicity data for the pHLIP-PAMAM-DOX conjugate mirrored quite closely that of the pHLIP-S-S-DOX conjugate, particularly at the higher concentrations (>1.25 μM). However, at lower pHLIP concentrations (0.16 μM-0.63 μM), the PAMAM conjugate exhibited higher cytotoxicity than the single DOX conjugate (˜up to 17% higher toxicity).
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 38% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 0.16 µM | ||||
| Description |
The cytotoxicity data for the pHLIP-PAMAM-DOX conjugate mirrored quite closely that of the pHLIP-S-S-DOX conjugate, particularly at the higher concentrations (>1.25 μM). However, at lower pHLIP concentrations (0.16 μM-0.63 μM), the PAMAM conjugate exhibited higher cytotoxicity than the single DOX conjugate (˜up to 17% higher toxicity).
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 42% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 0.63 µM | ||||
| Description |
HeLa cells treated with pHLIP-S-S-DOX at low pH showed significant toxicity (˜50% viability at the lowest concentration tested (0.16 μM pHLIP)) which, in this construct, corresponds to the DOX concentration. Cellular viability tracked inversely with increasing pHLIP conjugate concentration, peaking at ˜18% viability at 12 μM.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 45% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 0.31 µM | ||||
| Description |
HeLa cells treated with pHLIP-S-S-DOX at low pH showed significant toxicity (˜50% viability at the lowest concentration tested (0.16 μM pHLIP)) which, in this construct, corresponds to the DOX concentration. Cellular viability tracked inversely with increasing pHLIP conjugate concentration, peaking at ˜18% viability at 11 μM.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 50% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 0.16 µM | ||||
| Description |
HeLa cells treated with pHLIP-S-S-DOX at low pH showed significant toxicity (˜50% viability at the lowest concentration tested (0.16 μM pHLIP)) which, in this construct, corresponds to the DOX concentration. Cellular viability tracked inversely with increasing pHLIP conjugate concentration, peaking at ˜18% viability at 10 μM.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 90% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 0.63 µM | ||||
| Description |
HeLa cells treated with pHLIP-S-S-DOX at low pH showed significant toxicity (˜50% viability at the lowest concentration tested (0.16 μM pHLIP)) which, in this construct, corresponds to the DOX concentration. Cellular viability tracked inversely with increasing pHLIP conjugate concentration, peaking at ˜18% viability at 19 μM.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 95% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 0.16 µM | ||||
| Description |
HeLa cells treated with pHLIP-S-S-DOX at low pH showed significant toxicity (˜50% viability at the lowest concentration tested (0.16 μM pHLIP)) which, in this construct, corresponds to the DOX concentration. Cellular viability tracked inversely with increasing pHLIP conjugate concentration, peaking at ˜18% viability at 17 μM.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 98% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 0.31 µM | ||||
| Description |
HeLa cells treated with pHLIP-S-S-DOX at low pH showed significant toxicity (˜50% viability at the lowest concentration tested (0.16 μM pHLIP)) which, in this construct, corresponds to the DOX concentration. Cellular viability tracked inversely with increasing pHLIP conjugate concentration, peaking at ˜18% viability at 18 μM.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 99% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 10 µM | ||||
| Description |
HeLa cells treated with pHLIP-S-S-DOX at low pH showed significant toxicity (˜50% viability at the lowest concentration tested (0.16 μM pHLIP)) which, in this construct, corresponds to the DOX concentration. Cellular viability tracked inversely with increasing pHLIP conjugate concentration, peaking at ˜18% viability at 23 μM.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 100% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 1.3 µM | ||||
| Description |
HeLa cells treated with pHLIP-S-S-DOX at low pH showed significant toxicity (˜50% viability at the lowest concentration tested (0.16 μM pHLIP)) which, in this construct, corresponds to the DOX concentration. Cellular viability tracked inversely with increasing pHLIP conjugate concentration, peaking at ˜18% viability at 20 μM.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 17 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 100% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 2.5 µM | ||||
| Description |
HeLa cells treated with pHLIP-S-S-DOX at low pH showed significant toxicity (˜50% viability at the lowest concentration tested (0.16 μM pHLIP)) which, in this construct, corresponds to the DOX concentration. Cellular viability tracked inversely with increasing pHLIP conjugate concentration, peaking at ˜18% viability at 21 μM.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 18 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 100% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 5.0 µM | ||||
| Description |
HeLa cells treated with pHLIP-S-S-DOX at low pH showed significant toxicity (˜50% viability at the lowest concentration tested (0.16 μM pHLIP)) which, in this construct, corresponds to the DOX concentration. Cellular viability tracked inversely with increasing pHLIP conjugate concentration, peaking at ˜18% viability at 22 μM.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
Dox-S-S-GFLG-C6-[KTVRTSADE] [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [999] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Cell viability | 22% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 5 µM | ||||
| Description |
Dox conjugate 13 was moderately toxic with a reduced cell proliferation to a range of 25-35% as compared to Dox which reduced cell proliferation in the range of 20-34% for all selected four cell lines. However, it was interesting to observe that Doce conjugate 14 was almost nontoxic (cell proliferation within the range of 89-96%) in all the cell lines as compared to Doce alone which reduced the cell proliferation in the range of 54-61% (Figure 9).
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | LNCaP cell | CVCL_0395 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [999] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Cell viability | 25% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 5 µM | ||||
| Description |
Dox conjugate 13 was moderately toxic with a reduced cell proliferation to a range of 25-35% as compared to Dox which reduced cell proliferation in the range of 20-34% for all selected four cell lines. However, it was interesting to observe that Doce conjugate 14 was almost nontoxic (cell proliferation within the range of 89-96%) in all the cell lines as compared to Doce alone which reduced the cell proliferation in the range of 54-61% (Figure 9).
Click to Show/Hide
|
||||
| In Vitro Model | Normal | RWPE-1 cell | CVCL_3791 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [999] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Cell viability | 28% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 5 µM | ||||
| Description |
Dox conjugate 13 was moderately toxic with a reduced cell proliferation to a range of 25-35% as compared to Dox which reduced cell proliferation in the range of 20-34% for all selected four cell lines. However, it was interesting to observe that Doce conjugate 14 was almost nontoxic (cell proliferation within the range of 89-96%) in all the cell lines as compared to Doce alone which reduced the cell proliferation in the range of 54-61% (Figure 9).
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | DU145 cell | CVCL_0105 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [999] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Cell viability | 38% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 5 µM | ||||
| Description |
Dox conjugate 13 was moderately toxic with a reduced cell proliferation to a range of 25-35% as compared to Dox which reduced cell proliferation in the range of 20-34% for all selected four cell lines. However, it was interesting to observe that Doce conjugate 14 was almost nontoxic (cell proliferation within the range of 89-96%) in all the cell lines as compared to Doce alone which reduced the cell proliferation in the range of 54-61% (Figure 9).
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [999] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Cell viability | 48% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Description |
An increase in the cytotoxicity of Dox and Dox/peptide 9 physical mixture as compared to the conjugate 13 over an incubation period of 24 h to 72 h. Conjugates 13 and 14 were found to be less cytotoxic as compared to drug alone in 24-72 h. These cells were not treated with TGF-, so very minimal or no overexpression of EDB-FN. Figure 10b showed the effect of overexpression of EDB-FN in the cell viability. There was no observed effect of TGF- treatment for the cytotoxicity of Dox and physical mixture of Dox/peptide 9 on the cell viability as compared to the TGF- untreated cell lines. However, conjugate 13 showed a decrease in cell viability by 17% after 72 h as compared to untreated cell lines. Similarly, Doce and Doce conjugate 14 showed decrease in cell viability by 16 and 10%, respectively, after 72 h. The physical mixtures of Doce/peptide 9 showed a decrease in cell viability by 16% as compared to untreated cells.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | LNCaP C4-2 cell | CVCL_4782 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [999] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Cell viability | 70% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Description |
An increase in the cytotoxicity of Dox and Dox/peptide 9 physical mixture as compared to the conjugate 13 over an incubation period of 24 h to 72 h. Conjugates 13 and 14 were found to be less cytotoxic as compared to drug alone in 24-72 h. These cells were not treated with TGF-, so very minimal or no overexpression of EDB-FN. Figure 10b showed the effect of overexpression of EDB-FN in the cell viability. There was no observed effect of TGF- treatment for the cytotoxicity of Dox and physical mixture of Dox/peptide 9 on the cell viability as compared to the TGF- untreated cell lines. However, conjugate 13 showed a decrease in cell viability by 17% after 72 h as compared to untreated cell lines. Similarly, Doce and Doce conjugate 14 showed decrease in cell viability by 16 and 10%, respectively, after 72 h. The physical mixtures of Doce/peptide 9 showed a decrease in cell viability by 16% as compared to untreated cells.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | LNCaP C4-2 cell | CVCL_4782 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [999] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Cell viability | 74% | |||
| Administration Time | 2 h | ||||
| Administration Dosage | 100 nM | ||||
| Description |
The conjugate 13, peptide 9, and Dox showed no significant toxicity at or below 10 μM, possibly due to the shorter incubation time of 2 h.
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [999] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Cell viability | 75% | |||
| Administration Time | 2 h | ||||
| Administration Dosage | 1 µM | ||||
| Description |
The conjugate 13, peptide 9, and Dox showed no significant toxicity at or below 10 μM, possibly due to the shorter incubation time of 2 h.
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [999] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Cell viability | 77% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 48 h | ||||
| Description |
An increase in the cytotoxicity of Dox and Dox/peptide 9 physical mixture as compared to the conjugate 13 over an incubation period of 24 h to 72 h. Conjugates 13 and 14 were found to be less cytotoxic as compared to drug alone in 24-72 h. These cells were not treated with TGF-, so very minimal or no overexpression of EDB-FN. Figure 10b showed the effect of overexpression of EDB-FN in the cell viability. There was no observed effect of TGF- treatment for the cytotoxicity of Dox and physical mixture of Dox/peptide 9 on the cell viability as compared to the TGF- untreated cell lines. However, conjugate 13 showed a decrease in cell viability by 17% after 72 h as compared to untreated cell lines. Similarly, Doce and Doce conjugate 14 showed decrease in cell viability by 16 and 10%, respectively, after 72 h. The physical mixtures of Doce/peptide 9 showed a decrease in cell viability by 16% as compared to untreated cells.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | LNCaP C4-2 cell | CVCL_4782 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [999] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Cell viability | 78% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 48 h | ||||
| Description |
An increase in the cytotoxicity of Dox and Dox/peptide 9 physical mixture as compared to the conjugate 13 over an incubation period of 24 h to 72 h. Conjugates 13 and 14 were found to be less cytotoxic as compared to drug alone in 24-72 h. These cells were not treated with TGF-, so very minimal or no overexpression of EDB-FN. Figure 10b showed the effect of overexpression of EDB-FN in the cell viability. There was no observed effect of TGF- treatment for the cytotoxicity of Dox and physical mixture of Dox/peptide 9 on the cell viability as compared to the TGF- untreated cell lines. However, conjugate 13 showed a decrease in cell viability by 17% after 72 h as compared to untreated cell lines. Similarly, Doce and Doce conjugate 14 showed decrease in cell viability by 16 and 10%, respectively, after 72 h. The physical mixtures of Doce/peptide 9 showed a decrease in cell viability by 16% as compared to untreated cells.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | LNCaP C4-2 cell | CVCL_4782 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [999] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Cell viability | 98% | |||
| Administration Time | 2 h | ||||
| Administration Dosage | 100 µM | ||||
| Description |
The conjugate 13, peptide 9, and Dox showed no significant toxicity at or below 10 μM, possibly due to the shorter incubation time of 2 h.
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [999] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Cell viability | 102% | |||
| Administration Time | 2 h | ||||
| Administration Dosage | 10 nM | ||||
| Description |
The conjugate 13, peptide 9, and Dox showed no significant toxicity at or below 10 μM, possibly due to the shorter incubation time of 2 h.
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [999] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Cell viability | 105% | |||
| Administration Time | 2 h | ||||
| Administration Dosage | 10 µM | ||||
| Description |
The conjugate 13, peptide 9, and Dox showed no significant toxicity at or below 10 μM, possibly due to the shorter incubation time of 2 h.
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [999] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Cell viability | 108% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 24 h | ||||
| Description |
An increase in the cytotoxicity of Dox and Dox/peptide 9 physical mixture as compared to the conjugate 13 over an incubation period of 24 h to 72 h. Conjugates 13 and 14 were found to be less cytotoxic as compared to drug alone in 24-72 h. These cells were not treated with TGF-, so very minimal or no overexpression of EDB-FN. Figure 10b showed the effect of overexpression of EDB-FN in the cell viability. There was no observed effect of TGF- treatment for the cytotoxicity of Dox and physical mixture of Dox/peptide 9 on the cell viability as compared to the TGF- untreated cell lines. However, conjugate 13 showed a decrease in cell viability by 17% after 72 h as compared to untreated cell lines. Similarly, Doce and Doce conjugate 14 showed decrease in cell viability by 16 and 10%, respectively, after 72 h. The physical mixtures of Doce/peptide 9 showed a decrease in cell viability by 16% as compared to untreated cells.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | LNCaP C4-2 cell | CVCL_4782 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [999] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Cell viability | 110% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 24 h | ||||
| Description |
An increase in the cytotoxicity of Dox and Dox/peptide 9 physical mixture as compared to the conjugate 13 over an incubation period of 24 h to 72 h. Conjugates 13 and 14 were found to be less cytotoxic as compared to drug alone in 24-72 h. These cells were not treated with TGF-, so very minimal or no overexpression of EDB-FN. Figure 10b showed the effect of overexpression of EDB-FN in the cell viability. There was no observed effect of TGF- treatment for the cytotoxicity of Dox and physical mixture of Dox/peptide 9 on the cell viability as compared to the TGF- untreated cell lines. However, conjugate 13 showed a decrease in cell viability by 17% after 72 h as compared to untreated cell lines. Similarly, Doce and Doce conjugate 14 showed decrease in cell viability by 16 and 10%, respectively, after 72 h. The physical mixtures of Doce/peptide 9 showed a decrease in cell viability by 16% as compared to untreated cells.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | LNCaP C4-2 cell | CVCL_4782 | ||
[d-Cys6-des-Gly10-Pro9-NH2]-GnRH-Dox [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [997] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 30% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 100 µM | ||||
| Description |
The results showed that all the conjugates had lower antiproliferative effects than Dox at the tested concentrations (Figure 2A); this may be related to inefficient release of Dox from the conjugate caused by the relative stable thioether bond linkage between Dox-SMP and GnRH analog. The antiproliferative effects of the three conjugates were close at 25, 50, and 75 uM. While at 100 uM, conjugate II exhibited higher inhibitory effect than that of I and III.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 4.67 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [997] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 38% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 75 µM | ||||
| MOA of PDC |
lthough RNT with 177Lu-DOTATATE/PSMA is known as a novel and effective therapy option for cancer that significantly improves the quality of life and survival of patients, it may have acute or chronic side effects. Therefore, any method that can ameliorate these side effects is useful in the RNT process. For this purpose, a few clinical studies have reported that antioxidants as free radical scavengers such as amifostine and vitamins C and E can reduce radioiodine-related side effects, particularly in salivary glands in thyroid cancer patients.
Click to Show/Hide
|
||||
| Description |
The results showed that all the conjugates had lower antiproliferative effects than Dox at the tested concentrations (Figure 2A); this may be related to inefficient release of Dox from the conjugate caused by the relative stable thioether bond linkage between Dox-SMP and GnRH analog. The antiproliferative effects of the three conjugates were close at 25, 50, and 75 uM. While at 100 uM, conjugate II exhibited higher inhibitory effect than that of I and III.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 4.67 h | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [997] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 50% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 50 µM | ||||
| Description |
The results showed that all the conjugates had lower antiproliferative effects than Dox at the tested concentrations (Figure 2A); this may be related to inefficient release of Dox from the conjugate caused by the relative stable thioether bond linkage between Dox-SMP and GnRH analog. The antiproliferative effects of the three conjugates were close at 25, 50, and 75 uM. While at 100 uM, conjugate II exhibited higher inhibitory effect than that of I and III.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 4.67 h | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [997] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 55% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 25 µM | ||||
| Description |
The results showed that all the conjugates had lower antiproliferative effects than Dox at the tested concentrations (Figure 2A); this may be related to inefficient release of Dox from the conjugate caused by the relative stable thioether bond linkage between Dox-SMP and GnRH analog. The antiproliferative effects of the three conjugates were close at 25, 50, and 75 uM. While at 100 uM, conjugate II exhibited higher inhibitory effect than that of I and III.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 4.67 h | ||||
[d-Cys6-des-Gly10-Pro9-α-azaGly-NH2]-GnRH-Dox [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [997] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 0.3 | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 100 µM | ||||
| Description |
The results showed that all the conjugates had lower antiproliferative effects than Dox at the tested concentrations (Figure 2A); this may be related to inefficient release of Dox from the conjugate caused by the relative stable thioether bond linkage between Dox-SMP and GnRH analog. The antiproliferative effects of the three conjugates were close at 25, 50, and 75 uM. While at 100 uM, conjugate II exhibited higher inhibitory effect than that of I and III.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 5.23 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [997] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 0.38 | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 75 µM | ||||
| MOA of PDC |
Vitamin C as a water-soluble vitamin is the reduced form of ascorbic acid. No significant adverse effect of taking high doses of vitamin C (over 2000 mg/day) has been reported due to the water-soluble feature of vitamin C. Vitamin C directly reacts with hydroxy, alkoxyl, and lipid peroxyl radicals and converts them to alcohol, water, and hydroperoxide lipid, respectively. It has been shown that taking vitamin C before radioiodine therapy can ameliorate the oxidative stress effect of radioiodine. The radioprotective effects of vitamin C are mainly due to its free radical scavenging activity.
Click to Show/Hide
|
||||
| Description |
The results showed that all the conjugates had lower antiproliferative effects than Dox at the tested concentrations (Figure 2A); this may be related to inefficient release of Dox from the conjugate caused by the relative stable thioether bond linkage between Dox-SMP and GnRH analog. The antiproliferative effects of the three conjugates were close at 25, 50, and 75 uM. While at 100 uM, conjugate II exhibited higher inhibitory effect than that of I and III.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 5.23 h | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [997] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 0.5 | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 50 µM | ||||
| Description |
The results showed that all the conjugates had lower antiproliferative effects than Dox at the tested concentrations (Figure 2A); this may be related to inefficient release of Dox from the conjugate caused by the relative stable thioether bond linkage between Dox-SMP and GnRH analog. The antiproliferative effects of the three conjugates were close at 25, 50, and 75 uM. While at 100 uM, conjugate II exhibited higher inhibitory effect than that of I and III.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 5.23 h | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [997] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 0.59 | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 25 µM | ||||
| Description |
The results showed that all the conjugates had lower antiproliferative effects than Dox at the tested concentrations (Figure 2A); this may be related to inefficient release of Dox from the conjugate caused by the relative stable thioether bond linkage between Dox-SMP and GnRH analog. The antiproliferative effects of the three conjugates were close at 25, 50, and 75 uM. While at 100 uM, conjugate II exhibited higher inhibitory effect than that of I and III.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Half life period | 5.23 h | ||||
[C15]-NPY-Doxo-MBS [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1000] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability | 90.00% | |||
| Evaluation Method | XTT assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 1 μM | ||||
| MOA of PDC |
In this study, we investigate the usefulness of peptides as carriers for which the receptors are overexpressed on tumor cells. Covalent linking of the drug to the peptide could be used for chemotherapy and would lead to selective addressing of the tumor cells. As a model peptide, we used neuropeptide Y (NPY) because its receptors are produced in a number of neuroblastoma and the thereof derived cell lines. NPY is a 36-amino acid peptide of the pancreatic polypeptide family. It is expressed in the peripheral and central nervous systems and is one of the most abundant neuropeptides in the brain. Five distinct NPY receptors have been cloned, which have been named Y1, Y2, Y4, Y5, and y6 receptor subtypes. Upon binding to the G-protein coupled receptors, the ligand-receptor complex is internalized, which provides a convenient way to enter the cell by receptor-mediated endocytosis. Because structure-activity relationships (SARs) of NPY are well-known, position 15 of NPY was used for attaching maleimide anthracycline derivatives which would presumably not lead to a significant loss of binding activity for the Y1 receptor. Because the NPY-receptor complex is internalized and undergoes a pH shift from 7.4 to approximately 5.0 during endosomal trafficking, two different anthracycline derivatives that differ in the acid sensitivity of the bond between the drug and the spacer were selected (see Figure 1). Doxo-MBS and Dauno-MBS are characterized by a stable amide bond at the 3-amino position of the anthracycline; Dauno-HYD is a daunorubicin derivative incorporating an acid-sensitive hydrazone linker at the 13-keto position. The maleimide moiety was introduced into daunorubicin and doxorubicin in order to selectively label the sulfhydryl group of [C15]-NPY.
Click to Show/Hide
|
||||
| Description |
[C15]-NPY, [C15]-NPY-Dauno-MBS, and [C15]-NPY-Doxo-MBS showed no or only marginal effects. In contrast, [C15]-NPY-Dauno-HYD was as effective as free daunorubicin with respect to cytotoxicity, and the compounds were able to reduce cell growth by 66.9 ± 2.5% and 68.6 ± 0.4%, respectively.
|
||||
| In Vitro Model | Askin tumor | SK-N-MC cell | CVCL_0530 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1000] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Cell viability | 98.00% | |||
| Administration Time | 36 h | ||||
| Administration Dosage | 1 μM with BIBP 3226 (100 μM) | ||||
| MOA of PDC |
In this study, we investigate the usefulness of peptides as carriers for which the receptors are overexpressed on tumor cells. Covalent linking of the drug to the peptide could be used for chemotherapy and would lead to selective addressing of the tumor cells. As a model peptide, we used neuropeptide Y (NPY) because its receptors are produced in a number of neuroblastoma and the thereof derived cell lines. NPY is a 36-amino acid peptide of the pancreatic polypeptide family. It is expressed in the peripheral and central nervous systems and is one of the most abundant neuropeptides in the brain. Five distinct NPY receptors have been cloned, which have been named Y1, Y2, Y4, Y5, and y6 receptor subtypes. Upon binding to the G-protein coupled receptors, the ligand-receptor complex is internalized, which provides a convenient way to enter the cell by receptor-mediated endocytosis. Because structure-activity relationships (SARs) of NPY are well-known, position 15 of NPY was used for attaching maleimide anthracycline derivatives which would presumably not lead to a significant loss of binding activity for the Y1 receptor. Because the NPY-receptor complex is internalized and undergoes a pH shift from 7.4 to approximately 5.0 during endosomal trafficking, two different anthracycline derivatives that differ in the acid sensitivity of the bond between the drug and the spacer were selected (see Figure 1). Doxo-MBS and Dauno-MBS are characterized by a stable amide bond at the 3-amino position of the anthracycline; Dauno-HYD is a daunorubicin derivative incorporating an acid-sensitive hydrazone linker at the 13-keto position. The maleimide moiety was introduced into daunorubicin and doxorubicin in order to selectively label the sulfhydryl group of [C15]-NPY.
Click to Show/Hide
|
||||
| Description |
Pretreatment with 100 μM of the Y1 receptor antagonist BIBP 322629,30 antagonized the cytotoxicity of [C15]-NPY-Dauno-HYD (cell viability remained at 89.1 ± 4.6%), but not that of daunorubicin. To investigate the minimal required concentration of the administered conjugates, dilutions were made and tested on SK-N-MC cells; the cytotoxic effects of [C15]-NPY-Dauno-HYD and daunorubicin were visible starting at 1 μM concentrations.
Click to Show/Hide
|
||||
| In Vitro Model | Askin tumor | SK-N-MC cell | CVCL_0530 | ||
pHLIP-M-DOX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 97% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 5 µM | ||||
| Description |
pHLIP-M-DOX had a negligible effect on HeLa cell proliferation when incubated on cells at either pH at pHLIP concentrations as high as 10 μM. This is consistent with our imaging data that showed no release of DOX into the cell interior at either pH.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 97% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 10 µM | ||||
| Description |
pHLIP-M-DOX had a negligible effect on HeLa cell proliferation when incubated on cells at either pH at pHLIP concentrations as high as 10 μM. This is consistent with our imaging data that showed no release of DOX into the cell interior at either pH.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 98% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 2.5 µM | ||||
| Description |
pHLIP-M-DOX had a negligible effect on HeLa cell proliferation when incubated on cells at either pH at pHLIP concentrations as high as 10 μM. This is consistent with our imaging data that showed no release of DOX into the cell interior at either pH.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 99% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 5 µM | ||||
| Description |
pHLIP-M-DOX had a negligible effect on HeLa cell proliferation when incubated on cells at either pH at pHLIP concentrations as high as 10 μM. This is consistent with our imaging data that showed no release of DOX into the cell interior at either pH.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [998] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability | 100% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 2.5 µM | ||||
| Description |
pHLIP-M-DOX had a negligible effect on HeLa cell proliferation when incubated on cells at either pH at pHLIP concentrations as high as 10 μM. This is consistent with our imaging data that showed no release of DOX into the cell interior at either pH.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
PNS-SS-DOX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell survival rate | 12% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 24 h | ||||
| Administration Dosage | 20 µg/mL | ||||
| MOA of PDC |
Therefore, in this study, we present an intelligent drug delivery system based on 2D PNSs, utilizing a thermosensitive chitosan (CS) hydrogel as a sustained release platform to achieve the goal of long-term and effective drug treatment in the body. Under specific conditions, the peptide denoted by the sequence of Fmoc-FKKGSHC undergoes self-assembly, forming 2D PNSs with uniform nanostructure. PNSs are then successfully covalently linked to the thiol-modified DOX via the disulfide bonds, resulting in the synthesis of a 2D PDCs (PNS-SS-DOX). Subsequently, the PNS-SS-DOX PDCs are encapsulated within the injectable CS-based thermosensitive hydrogels. To validate the feasibility of this novel intelligent responsive drug delivery system, we conduct invitro release testing using the CS hydrogels and tested the GSH-responsive release of the PNS-SS-DOX, simulating the tumor cell environment. The results of the study indicate that the hydrogels exhibited pH-responsive release and swelling, providing favorable conditions for the controlled release of the PNS-SS-DOX. Furthermore, the outstanding sustained release effect also facilitated the effective accumulation of the PNS-SS-DOX at the tumor site, reducing inflammation reactions caused by multiple dosages.
Click to Show/Hide
|
||||
| Description |
Subsequently, the cell toxicity of the DOX, DOX-SH, and PNS-SS-DOX was also investigated using the CCK-8 method. As shown in Figure8b, all three materials display significant concentration-dependent cytotoxicity. Additionally, it can be observed that at the same concentration, the cytotoxicity of DOX-SH to MCF-7 cells was nearly identical to that of DOX, indicating that thiolation did not affect the efficacy of DOX. Furthermore, the PNS-SS-DOX group exhibited stronger cytotoxicity to MCF-7 cells. Particularly, at a material concentration of 20ug mL-1, the cell survival rate for the PNS-SS-DOX group was only 12 %, while that of the DOX-SH group was 20 %. We suggest that this effect is possibly due to the modification of PNSs with DOX-SH. On one hand, the hydrophilic peptides effectively improved the solubility of the hydrophobic drug DOX-SH, and on the other hand, the positively charged PNSs enhanced the drug uptake by cancer cells, thus increasing the cancer cell killing efficacy.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell survival rate | 32% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 24 h | ||||
| Administration Dosage | 15 µg/mL | ||||
| MOA of PDC |
Therefore, in this study, we present an intelligent drug delivery system based on 2D PNSs, utilizing a thermosensitive chitosan (CS) hydrogel as a sustained release platform to achieve the goal of long-term and effective drug treatment in the body. Under specific conditions, the peptide denoted by the sequence of Fmoc-FKKGSHC undergoes self-assembly, forming 2D PNSs with uniform nanostructure. PNSs are then successfully covalently linked to the thiol-modified DOX via the disulfide bonds, resulting in the synthesis of a 2D PDCs (PNS-SS-DOX). Subsequently, the PNS-SS-DOX PDCs are encapsulated within the injectable CS-based thermosensitive hydrogels. To validate the feasibility of this novel intelligent responsive drug delivery system, we conduct invitro release testing using the CS hydrogels and tested the GSH-responsive release of the PNS-SS-DOX, simulating the tumor cell environment. The results of the study indicate that the hydrogels exhibited pH-responsive release and swelling, providing favorable conditions for the controlled release of the PNS-SS-DOX. Furthermore, the outstanding sustained release effect also facilitated the effective accumulation of the PNS-SS-DOX at the tumor site, reducing inflammation reactions caused by multiple dosages.
Click to Show/Hide
|
||||
| Description |
Subsequently, the cell toxicity of the DOX, DOX-SH, and PNS-SS-DOX was also investigated using the CCK-8 method. As shown in Figure8b, all three materials display significant concentration-dependent cytotoxicity. Additionally, it can be observed that at the same concentration, the cytotoxicity of DOX-SH to MCF-7 cells was nearly identical to that of DOX, indicating that thiolation did not affect the efficacy of DOX. Furthermore, the PNS-SS-DOX group exhibited stronger cytotoxicity to MCF-7 cells. Particularly, at a material concentration of 20ug mL-1, the cell survival rate for the PNS-SS-DOX group was only 12 %, while that of the DOX-SH group was 20 %. We suggest that this effect is possibly due to the modification of PNSs with DOX-SH. On one hand, the hydrophilic peptides effectively improved the solubility of the hydrophobic drug DOX-SH, and on the other hand, the positively charged PNSs enhanced the drug uptake by cancer cells, thus increasing the cancer cell killing efficacy.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell survival rate | 55% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 24 h | ||||
| Administration Dosage | 10 µg/mL | ||||
| MOA of PDC |
Therefore, in this study, we present an intelligent drug delivery system based on 2D PNSs, utilizing a thermosensitive chitosan (CS) hydrogel as a sustained release platform to achieve the goal of long-term and effective drug treatment in the body. Under specific conditions, the peptide denoted by the sequence of Fmoc-FKKGSHC undergoes self-assembly, forming 2D PNSs with uniform nanostructure. PNSs are then successfully covalently linked to the thiol-modified DOX via the disulfide bonds, resulting in the synthesis of a 2D PDCs (PNS-SS-DOX). Subsequently, the PNS-SS-DOX PDCs are encapsulated within the injectable CS-based thermosensitive hydrogels. To validate the feasibility of this novel intelligent responsive drug delivery system, we conduct invitro release testing using the CS hydrogels and tested the GSH-responsive release of the PNS-SS-DOX, simulating the tumor cell environment. The results of the study indicate that the hydrogels exhibited pH-responsive release and swelling, providing favorable conditions for the controlled release of the PNS-SS-DOX. Furthermore, the outstanding sustained release effect also facilitated the effective accumulation of the PNS-SS-DOX at the tumor site, reducing inflammation reactions caused by multiple dosages.
Click to Show/Hide
|
||||
| Description |
Subsequently, the cell toxicity of the DOX, DOX-SH, and PNS-SS-DOX was also investigated using the CCK-8 method. As shown in Figure8b, all three materials display significant concentration-dependent cytotoxicity. Additionally, it can be observed that at the same concentration, the cytotoxicity of DOX-SH to MCF-7 cells was nearly identical to that of DOX, indicating that thiolation did not affect the efficacy of DOX. Furthermore, the PNS-SS-DOX group exhibited stronger cytotoxicity to MCF-7 cells. Particularly, at a material concentration of 20ug mL-1, the cell survival rate for the PNS-SS-DOX group was only 12 %, while that of the DOX-SH group was 20 %. We suggest that this effect is possibly due to the modification of PNSs with DOX-SH. On one hand, the hydrophilic peptides effectively improved the solubility of the hydrophobic drug DOX-SH, and on the other hand, the positively charged PNSs enhanced the drug uptake by cancer cells, thus increasing the cancer cell killing efficacy.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Cell survival rate | 75% | |||
| Evaluation Method | CCK-8 assay | ||||
| Administration Time | 24 h | ||||
| Administration Dosage | 5 µg/mL | ||||
| MOA of PDC |
Therefore, in this study, we present an intelligent drug delivery system based on 2D PNSs, utilizing a thermosensitive chitosan (CS) hydrogel as a sustained release platform to achieve the goal of long-term and effective drug treatment in the body. Under specific conditions, the peptide denoted by the sequence of Fmoc-FKKGSHC undergoes self-assembly, forming 2D PNSs with uniform nanostructure. PNSs are then successfully covalently linked to the thiol-modified DOX via the disulfide bonds, resulting in the synthesis of a 2D PDCs (PNS-SS-DOX). Subsequently, the PNS-SS-DOX PDCs are encapsulated within the injectable CS-based thermosensitive hydrogels. To validate the feasibility of this novel intelligent responsive drug delivery system, we conduct invitro release testing using the CS hydrogels and tested the GSH-responsive release of the PNS-SS-DOX, simulating the tumor cell environment. The results of the study indicate that the hydrogels exhibited pH-responsive release and swelling, providing favorable conditions for the controlled release of the PNS-SS-DOX. Furthermore, the outstanding sustained release effect also facilitated the effective accumulation of the PNS-SS-DOX at the tumor site, reducing inflammation reactions caused by multiple dosages.
Click to Show/Hide
|
||||
| Description |
Subsequently, the cell toxicity of the DOX, DOX-SH, and PNS-SS-DOX was also investigated using the CCK-8 method. As shown in Figure8b, all three materials display significant concentration-dependent cytotoxicity. Additionally, it can be observed that at the same concentration, the cytotoxicity of DOX-SH to MCF-7 cells was nearly identical to that of DOX, indicating that thiolation did not affect the efficacy of DOX. Furthermore, the PNS-SS-DOX group exhibited stronger cytotoxicity to MCF-7 cells. Particularly, at a material concentration of 20ug mL-1, the cell survival rate for the PNS-SS-DOX group was only 12 %, while that of the DOX-SH group was 20 %. We suggest that this effect is possibly due to the modification of PNSs with DOX-SH. On one hand, the hydrophilic peptides effectively improved the solubility of the hydrophobic drug DOX-SH, and on the other hand, the positively charged PNSs enhanced the drug uptake by cancer cells, thus increasing the cancer cell killing efficacy.
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
AEZS-108 [Terminated in Phase 3]
Identified from the Human Clinical Data
| Experiment 1 Reporting the Activity Data of This PDC | [1001] | ||||
| Indication | Castration and taxane resistant prostate cancer | ||||
| Efficacy Data | Vomit | 32% | |||
| Description |
The most common all-grade adverse events were hematologic (22; 88%), fatigue (19; 76%), anorexia (13; 52%),alopecia(13; 52%), nausea (13; 52%), and vomiting (8;32%).
|
||||
| In Vivo Model | Men with histologically confirmed prostatic adenocarcinoma. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [1001] | ||||
| Indication | Castration and taxane resistant prostate cancer | ||||
| Efficacy Data | Neutropenia | 56% | |||
| Description |
The most common hematologic adverse event was neutropenia at 56% (all grades).
|
||||
| In Vivo Model | Men with histologically confirmed prostatic adenocarcinoma. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [1001] | ||||
| Indication | Castration and taxane resistant prostate cancer | ||||
| Efficacy Data | Nausea toxicity | 52% | |||
| Description |
The most common all-grade adverse events were hematologic (22; 88%), fatigue (19; 76%), anorexia (13; 52%),alopecia(13; 52%), nausea (13; 52%), and vomiting (8;32%).
|
||||
| In Vivo Model | Men with histologically confirmed prostatic adenocarcinoma. | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [1001] | ||||
| Indication | Castration and taxane resistant prostate cancer | ||||
| Efficacy Data | Median progression-free survival (mPFS) | 3.8 months | |||
| Description |
With a median follow-up of 16.1 months (range, 3.2-36.1), the median PFS was 3.8 months (95% confidence interval [CI], 2.1-4.4) and median OS was 6.0 months (95% CI, 4.2-10.1; Figure 3).
|
||||
| In Vivo Model | Men with histologically confirmed prostatic adenocarcinoma. | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [1001] | ||||
| Indication | Castration and taxane resistant prostate cancer | ||||
| Efficacy Data | Median overall survival (mOS) | 6.0 months | |||
| Description |
With a median follow-up of 16.1 months (range, 3.2-36.1), the median PFS was 3.8 months (95% confidence interval [CI], 2.1-4.4) and median OS was 6.0 months (95% CI, 4.2-10.1; Figure 3).
|
||||
| In Vivo Model | Men with histologically confirmed prostatic adenocarcinoma. | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [1001] | ||||
| Indication | Castration and taxane resistant prostate cancer | ||||
| Efficacy Data | Hematologic toxicity | 88% | |||
| Description |
The most common all-grade adverse events were hematologic (22; 88%), fatigue (19; 76%), anorexia (13; 52%),alopecia(13; 52%), nausea (13; 52%), and vomiting (8;32%).
|
||||
| In Vivo Model | Men with histologically confirmed prostatic adenocarcinoma. | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [1001] | ||||
| Indication | Castration and taxane resistant prostate cancer | ||||
| Efficacy Data | Grade 3/4 nonhematologic toxicities | 24% | |||
| Description |
Six of 25 patients (24%) experienced Grade 3 or 4 nonhematologic toxicities
|
||||
| In Vivo Model | Men with histologically confirmed prostatic adenocarcinoma. | ||||
| Experiment 8 Reporting the Activity Data of This PDC | [1001] | ||||
| Indication | Castration and taxane resistant prostate cancer | ||||
| Efficacy Data | Grade ≥ 3 hematologic toxicity | 56% | |||
| Description |
Fourteen of 25 patients (56%) experienced a Grade ≥ 3 hematologic toxicity
|
||||
| In Vivo Model | Men with histologically confirmed prostatic adenocarcinoma. | ||||
| Experiment 9 Reporting the Activity Data of This PDC | [1001] | ||||
| Indication | Castration and taxane resistant prostate cancer | ||||
| Efficacy Data | Fatigue | 76% | |||
| Description |
The most common all-grade adverse events were hematologic (22; 88%), fatigue (19; 76%), anorexia (13; 52%),alopecia(13; 52%), nausea (13; 52%), and vomiting (8;32%).
|
||||
| In Vivo Model | Men with histologically confirmed prostatic adenocarcinoma. | ||||
| Experiment 10 Reporting the Activity Data of This PDC | [1001] | ||||
| Indication | Castration and taxane resistant prostate cancer | ||||
| Efficacy Data | Alopecia toxicity | 52.00% | |||
| Description |
The most common all-grade adverse events were hematologic (22; 88%), fatigue (19; 76%), anorexia (13; 52%),alopecia(13; 52%), nausea (13; 52%), and vomiting (8;32%).
|
||||
| In Vivo Model | Men with histologically confirmed prostatic adenocarcinoma. | ||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1002] | ||||
| Indication | Uveal melanoma | ||||
| Efficacy Data | Cell viability | 0% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 5 µM | ||||
| Description |
OCM3DOX320cells did not show significant difference in cell viability in the presence of 1 uM DOX when compared to untreated control cells. 1 uM DOX induced significant cell death of OCM3 cells but did not cause significant cell death in OCM3DOX320cells, confirming DOX resistance. AN-152 at lower concentration (40 nM, 320 nM) increased cell proliferation significantly compared to equimolar dose of DOX which does not have any effect on cell viability at this concentration in OCM3 cells. However, higher concentrations of AN-152 can effectively inhibit cell proliferation in both cell lines. Higher concentrations (1-5 uM) of DOX and AN-152 showed no significantly different effect either on OCM3 or OCM3DOX320cells (Fig. 3).
Click to Show/Hide
|
||||
| In Vitro Model | Cutaneous melanoma | OCM-3 cell | CVCL_6937 | ||
| Half life period | 2 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [1002] | ||||
| Indication | Uveal melanoma | ||||
| Efficacy Data | Cell viability | 5% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 1 µM | ||||
| Description |
OCM3DOX320cells did not show significant difference in cell viability in the presence of 1 uM DOX when compared to untreated control cells. 1 uM DOX induced significant cell death of OCM3 cells but did not cause significant cell death in OCM3DOX320cells, confirming DOX resistance. AN-152 at lower concentration (40 nM, 320 nM) increased cell proliferation significantly compared to equimolar dose of DOX which does not have any effect on cell viability at this concentration in OCM3 cells. However, higher concentrations of AN-152 can effectively inhibit cell proliferation in both cell lines. Higher concentrations (1-5 uM) of DOX and AN-152 showed no significantly different effect either on OCM3 or OCM3DOX320cells (Fig. 3).
Click to Show/Hide
|
||||
| In Vitro Model | Cutaneous melanoma | OCM-3 cell | CVCL_6937 | ||
| Half life period | 2 h | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [1002] | ||||
| Indication | Uveal melanoma | ||||
| Efficacy Data | Cell viability | 6% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 2.5 µM | ||||
| Description |
OCM3DOX320cells did not show significant difference in cell viability in the presence of 1 uM DOX when compared to untreated control cells. 1 uM DOX induced significant cell death of OCM3 cells but did not cause significant cell death in OCM3DOX320cells, confirming DOX resistance. AN-152 at lower concentration (40 nM, 320 nM) increased cell proliferation significantly compared to equimolar dose of DOX which does not have any effect on cell viability at this concentration in OCM3 cells. However, higher concentrations of AN-152 can effectively inhibit cell proliferation in both cell lines. Higher concentrations (1-5 uM) of DOX and AN-152 showed no significantly different effect either on OCM3 or OCM3DOX320cells (Fig. 3).
Click to Show/Hide
|
||||
| In Vitro Model | Cutaneous melanoma | OCM-3 cell | CVCL_6937 | ||
| Half life period | 2 h | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [1002] | ||||
| Indication | Uveal melanoma | ||||
| Efficacy Data | Cell viability | 25% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 5 µM | ||||
| Description |
OCM3DOX320cells did not show significant difference in cell viability in the presence of 1 uM DOX when compared to untreated control cells. 1 uM DOX induced significant cell death of OCM3 cells but did not cause significant cell death in OCM3DOX320cells, confirming DOX resistance. AN-152 at lower concentration (40 nM, 320 nM) increased cell proliferation significantly compared to equimolar dose of DOX which does not have any effect on cell viability at this concentration in OCM3 cells. However, higher concentrations of AN-152 can effectively inhibit cell proliferation in both cell lines. Higher concentrations (1-5 uM) of DOX and AN-152 showed no significantly different effect either on OCM3 or OCM3DOX320cells (Fig. 3).
Click to Show/Hide
|
||||
| In Vitro Model | Cutaneous melanoma | OCM3DOX320 cell | CVCL_6937 | ||
| Half life period | 2 h | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Uveal melanoma | ||||
| Efficacy Data | Cell viability | 36.30% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 24 h | ||||
| Administration Dosage | 5 µM | ||||
| MOA of PDC |
Our results show that AEZS-108 upregulates the expression of MASPIN/SERPINB5 tumor suppressor gene, which is downregulated in normal uvea and UM specimens independently from the LHRH receptor-ligand interaction. AEZS-108 also substantially downregulates hypoxia-inducible factor 1 alpha (HIF1A) expression.
|
||||
| Description |
In order to investigate whether AEZS-108 inhibits cell proliferation and its extent, OCM3 cells were treated either with 5 M AEZS-108 or equal amount of doxorubicin. MTS assay was performed after 24 and 48 hours of treatment. AEZS-108 and doxorubicin have been shown to reduce cell proliferation by 36.3% (p< 0.001) and 62.9% (p< 0.001) respectively after 24 hours, and by 84.7% (p< 0.001) and 89.7% (p< 0.001) respectively after 48 hours, (Figure 2).
Click to Show/Hide
|
||||
| In Vitro Model | Cutaneous melanoma | OCM-3 cell | CVCL_6937 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [1002] | ||||
| Indication | Uveal melanoma | ||||
| Efficacy Data | Cell viability | 48% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 2.5 µM | ||||
| Description |
OCM3DOX320cells did not show significant difference in cell viability in the presence of 1 uM DOX when compared to untreated control cells. 1 uM DOX induced significant cell death of OCM3 cells but did not cause significant cell death in OCM3DOX320cells, confirming DOX resistance. AN-152 at lower concentration (40 nM, 320 nM) increased cell proliferation significantly compared to equimolar dose of DOX which does not have any effect on cell viability at this concentration in OCM3 cells. However, higher concentrations of AN-152 can effectively inhibit cell proliferation in both cell lines. Higher concentrations (1-5 uM) of DOX and AN-152 showed no significantly different effect either on OCM3 or OCM3DOX320cells (Fig. 3).
Click to Show/Hide
|
||||
| In Vitro Model | Cutaneous melanoma | OCM3DOX320 cell | CVCL_6937 | ||
| Half life period | 2 h | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [1002] | ||||
| Indication | Uveal melanoma | ||||
| Efficacy Data | Cell viability | 80% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 1 µM | ||||
| Description |
OCM3DOX320cells did not show significant difference in cell viability in the presence of 1 uM DOX when compared to untreated control cells. 1 uM DOX induced significant cell death of OCM3 cells but did not cause significant cell death in OCM3DOX320cells, confirming DOX resistance. AN-152 at lower concentration (40 nM, 320 nM) increased cell proliferation significantly compared to equimolar dose of DOX which does not have any effect on cell viability at this concentration in OCM3 cells. However, higher concentrations of AN-152 can effectively inhibit cell proliferation in both cell lines. Higher concentrations (1-5 uM) of DOX and AN-152 showed no significantly different effect either on OCM3 or OCM3DOX320cells (Fig. 3).
Click to Show/Hide
|
||||
| In Vitro Model | Cutaneous melanoma | OCM3DOX320 cell | CVCL_6937 | ||
| Half life period | 2 h | ||||
| Experiment 8 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Uveal melanoma | ||||
| Efficacy Data | Cell viability | 84.70% | |||
| Evaluation Method | MTS assay | ||||
| Administration Time | 48 h | ||||
| Administration Dosage | 5 µM | ||||
| MOA of PDC |
Our results show that AEZS-108 upregulates the expression of MASPIN/SERPINB5 tumor suppressor gene, which is downregulated in normal uvea and UM specimens independently from the LHRH receptor-ligand interaction. AEZS-108 also substantially downregulates hypoxia-inducible factor 1 alpha (HIF1A) expression.
|
||||
| Description |
In order to investigate whether AEZS-108 inhibits cell proliferation and its extent, OCM3 cells were treated either with 5 M AEZS-108 or equal amount of doxorubicin. MTS assay was performed after 24 and 48 hours of treatment. AEZS-108 and doxorubicin have been shown to reduce cell proliferation by 36.3% (p< 0.001) and 62.9% (p< 0.001) respectively after 24 hours, and by 84.7% (p< 0.001) and 89.7% (p< 0.001) respectively after 48 hours, (Figure 2).
Click to Show/Hide
|
||||
| In Vitro Model | Cutaneous melanoma | OCM-3 cell | CVCL_6937 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [1002] | ||||
| Indication | Uveal melanoma | ||||
| Efficacy Data | Cell viability | 105% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 320 nM | ||||
| Description |
OCM3DOX320cells did not show significant difference in cell viability in the presence of 1 uM DOX when compared to untreated control cells. 1 uM DOX induced significant cell death of OCM3 cells but did not cause significant cell death in OCM3DOX320cells, confirming DOX resistance. AN-152 at lower concentration (40 nM, 320 nM) increased cell proliferation significantly compared to equimolar dose of DOX which does not have any effect on cell viability at this concentration in OCM3 cells. However, higher concentrations of AN-152 can effectively inhibit cell proliferation in both cell lines. Higher concentrations (1-5 uM) of DOX and AN-152 showed no significantly different effect either on OCM3 or OCM3DOX320cells (Fig. 3).
Click to Show/Hide
|
||||
| In Vitro Model | Cutaneous melanoma | OCM3DOX320 cell | CVCL_6937 | ||
| Half life period | 2 h | ||||
| Experiment 10 Reporting the Activity Data of This PDC | [1002] | ||||
| Indication | Uveal melanoma | ||||
| Efficacy Data | Cell viability | 120% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 40 nM | ||||
| Description |
OCM3DOX320cells did not show significant difference in cell viability in the presence of 1 uM DOX when compared to untreated control cells. 1 uM DOX induced significant cell death of OCM3 cells but did not cause significant cell death in OCM3DOX320cells, confirming DOX resistance. AN-152 at lower concentration (40 nM, 320 nM) increased cell proliferation significantly compared to equimolar dose of DOX which does not have any effect on cell viability at this concentration in OCM3 cells. However, higher concentrations of AN-152 can effectively inhibit cell proliferation in both cell lines. Higher concentrations (1-5 uM) of DOX and AN-152 showed no significantly different effect either on OCM3 or OCM3DOX320cells (Fig. 3).
Click to Show/Hide
|
||||
| In Vitro Model | Cutaneous melanoma | OCM3DOX320 cell | CVCL_6937 | ||
| Half life period | 2 h | ||||
| Experiment 11 Reporting the Activity Data of This PDC | [1002] | ||||
| Indication | Uveal melanoma | ||||
| Efficacy Data | Cell viability | 130% | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| Administration Dosage | 40 nM | ||||
| Description |
OCM3DOX320cells did not show significant difference in cell viability in the presence of 1 uM DOX when compared to untreated control cells. 1 uM DOX induced significant cell death of OCM3 cells but did not cause significant cell death in OCM3DOX320cells, confirming DOX resistance. AN-152 at lower concentration (40 nM, 320 nM) increased cell proliferation significantly compared to equimolar dose of DOX which does not have any effect on cell viability at this concentration in OCM3 cells. However, higher concentrations of AN-152 can effectively inhibit cell proliferation in both cell lines. Higher concentrations (1-5 uM) of DOX and AN-152 showed no significantly different effect either on OCM3 or OCM3DOX320cells (Fig. 3).
Click to Show/Hide
|
||||
| In Vitro Model | Cutaneous melanoma | OCM-3 cell | CVCL_6937 | ||
| Half life period | 2 h | ||||
References