
Linker Information
General Information of This Linker
| Linker ID |
LIN00091
|
|||||
|---|---|---|---|---|---|---|
| Linker Name |
Disulfide bond
|
|||||
| Linker Type |
GSH concentration-sensitive linkers
|
|||||
| Structure |
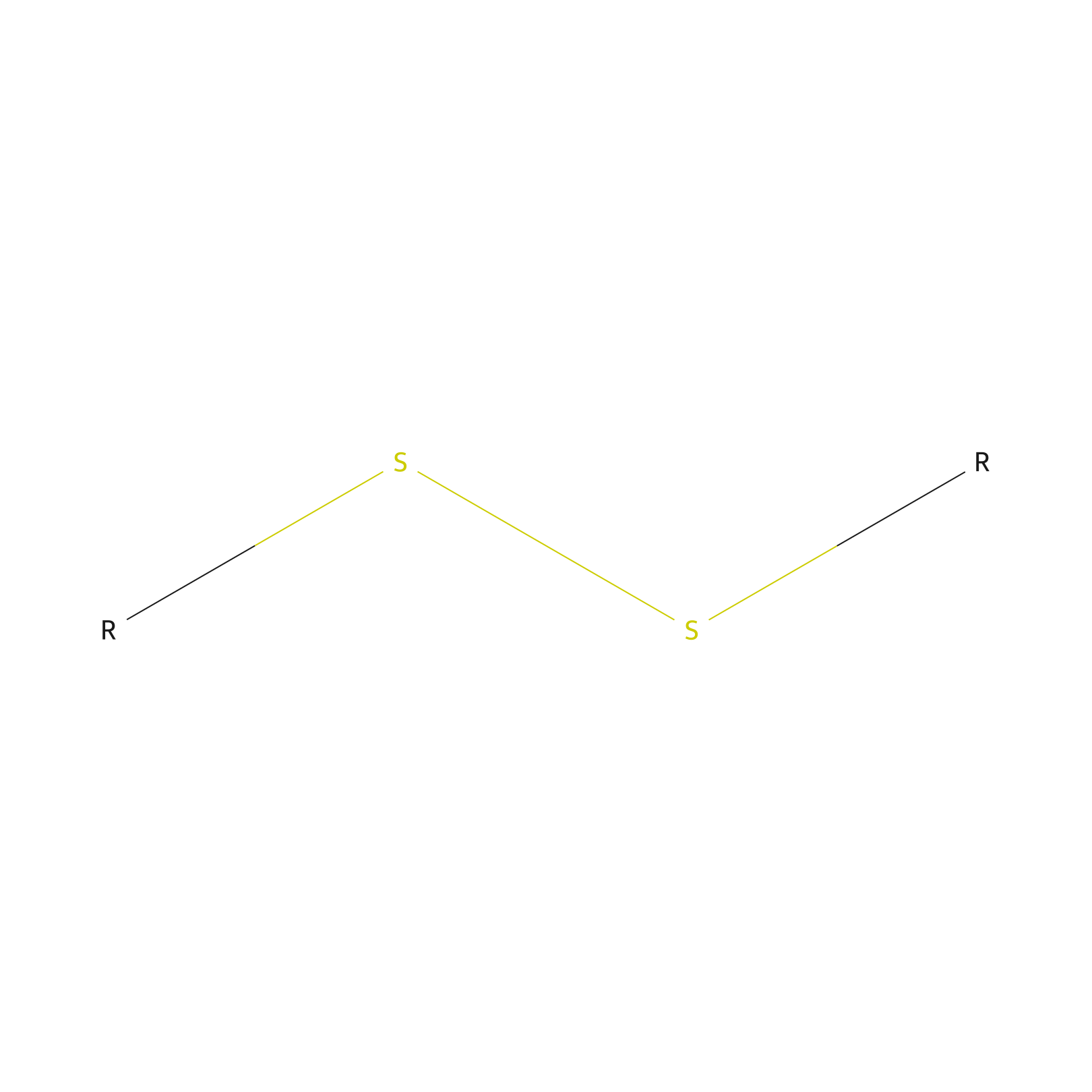
|
|||||
| Formula |
*2S2
|
|||||
| #Ro5 Violations (Lipinski): 0 | Molecular Weight (mw) | 64.134 | ||||
| Lipid-water partition coefficient (xlogp) | 1.2964 | |||||
| Hydrogen Bond Donor Count (hbonddonor) | 0 | |||||
| Hydrogen Bond Acceptor Count (hbondacc) | 2 | |||||
| Rotatable Bond Count (rotbonds) | 1 | |||||
| Canonical smiles |
*SS*
|
|||||
Each Peptide-drug Conjugate Related to This Linker
Full Information of The Activity Data of The PDC(s) Related to This Linker
BT1718 [Phase 2]
Identified from the Human Clinical Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Grade 3 increased GGT |
8.30%
|
|||
| Patients Enrolled |
24 patients with various types of solid tumors.
|
||||
| Administration Dosage | 9.6 mg/m2 BIW | ||||
| MOA of PDC |
BT1718 is a novel first in class bicyclic targeting peptide that selectively binds MT1 MMP (MMP-14) and is linked to the maytansinoid tubulin inhibitor DM1 by a cleavable disulfide linker. Bicycle Toxin Conjugates have a low molecular weight compared to other conjugated toxin approaches, enabling rapid tumour penetration and a short systemic half-life (up to 40 minutes in non-human primates) potentially reducing toxicity. The target MT1-MMP (MT1) is a surface metalloproteinase involved in tissue remodelling through proteolysis of extracellular matrix components: Highly expressed in tumours with unmet medical need, such as triple negative breast cancer, non small cell lung cancer and ovarian cancer. Strong link with cell invasion and metastasis. High tumour MT1 expression is correlated with poor outcomes in multiple tumour types. High adjacent stromal expression and low expression in adult normal tissue.
Click to Show/Hide
|
||||
| Description |
24 patients were enrolled with various types of solid tumors across both dose escalation cohorts (see table). BIW RP2D was determined as 7.2 mg/m2. The 2 patients with DLTs at 9.6 mg/m2 BIW experienced grade 3 increased GGT or fatigue. QW dose escalation continues at 20 mg/m2. Mean number of cycles received = 2.3 months (N = 24), with no objective responses observed to date in this unselected population.
Click to Show/Hide
|
||||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Fatigue |
8.30%
|
|||
| Patients Enrolled |
24 patients with various types of solid tumors.
|
||||
| Administration Dosage | 9.6 mg/m2 BIW | ||||
| MOA of PDC |
BT1718 is a novel first in class bicyclic targeting peptide that selectively binds MT1 MMP (MMP-14) and is linked to the maytansinoid tubulin inhibitor DM1 by a cleavable disulfide linker. Bicycle Toxin Conjugates have a low molecular weight compared to other conjugated toxin approaches, enabling rapid tumour penetration and a short systemic half-life (up to 40 minutes in non-human primates) potentially reducing toxicity. The target MT1-MMP (MT1) is a surface metalloproteinase involved in tissue remodelling through proteolysis of extracellular matrix components: Highly expressed in tumours with unmet medical need, such as triple negative breast cancer, non small cell lung cancer and ovarian cancer. Strong link with cell invasion and metastasis. High tumour MT1 expression is correlated with poor outcomes in multiple tumour types. High adjacent stromal expression and low expression in adult normal tissue.
Click to Show/Hide
|
||||
| Description |
24 patients were enrolled with various types of solid tumors across both dose escalation cohorts (see table). BIW RP2D was determined as 7.2 mg/m2. The 2 patients with DLTs at 9.6 mg/m2 BIW experienced grade 3 increased GGT or fatigue. QW dose escalation continues at 20 mg/m2. Mean number of cycles received = 2.3 months (N = 24), with no objective responses observed to date in this unselected population.
Click to Show/Hide
|
||||
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
12.50%
|
|||
| Administration Time | Twice a week for 12 days | ||||
| Administration Dosage | 1 mg/kg | ||||
| Evaluation Method | Tumor volume detection | ||||
| MOA of PDC |
BT1718 is a novel first in class bicyclic targeting peptide that selectively binds MT1 MMP (MMP-14) and is linked to the maytansinoid tubulin inhibitor DM1 by a cleavable disulfide linker. Bicycle Toxin Conjugates have a low molecular weight compared to other conjugated toxin approaches, enabling rapid tumour penetration and a short systemic half-life (up to 40 minutes in non-human primates) potentially reducing toxicity. The target MT1-MMP (MT1) is a surface metalloproteinase involved in tissue remodelling through proteolysis of extracellular matrix components: Highly expressed in tumours with unmet medical need, such as triple negative breast cancer, non small cell lung cancer and ovarian cancer. Strong link with cell invasion and metastasis. High tumour MT1 expression is correlated with poor outcomes in multiple tumour types. High adjacent stromal expression and low expression in adult normal tissue.
Click to Show/Hide
|
||||
| Description |
Testing of BT1718 in different dosing regimes in an additional model (HT-1080 cells) also demonstrated excellent tumour regression, with 10mg/kg biw leading to complete tumour clearance in all 3 animals within 23 days and no re-growth out to 72 days.
|
||||
| In Vivo Model | Mice implanted with MT1-positive HT-1080 cells | ||||
| In Vitro Model | Fibrosarcoma | HT-1080 cell | CVCL_0317 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
12.70%
|
|||
| Administration Time | Three times a week for 12 days | ||||
| Administration Dosage | 1 mg/kg | ||||
| Evaluation Method | Tumor volume detection | ||||
| MOA of PDC |
BT1718 is a novel first in class bicyclic targeting peptide that selectively binds MT1 MMP (MMP-14) and is linked to the maytansinoid tubulin inhibitor DM1 by a cleavable disulfide linker. Bicycle Toxin Conjugates have a low molecular weight compared to other conjugated toxin approaches, enabling rapid tumour penetration and a short systemic half-life (up to 40 minutes in non-human primates) potentially reducing toxicity. The target MT1-MMP (MT1) is a surface metalloproteinase involved in tissue remodelling through proteolysis of extracellular matrix components: Highly expressed in tumours with unmet medical need, such as triple negative breast cancer, non small cell lung cancer and ovarian cancer. Strong link with cell invasion and metastasis. High tumour MT1 expression is correlated with poor outcomes in multiple tumour types. High adjacent stromal expression and low expression in adult normal tissue.
Click to Show/Hide
|
||||
| Description |
Testing of BT1718 in different dosing regimes in an additional model (HT-1080 cells) also demonstrated excellent tumour regression, with 10mg/kg biw leading to complete tumour clearance in all 3 animals within 23 days and no re-growth out to 75 days.
|
||||
| In Vivo Model | Mice implanted with MT1-positive HT-1080 cells | ||||
| In Vitro Model | Fibrosarcoma | HT-1080 cell | CVCL_0317 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Fibrosarcoma | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
18%
|
|||
| Administration Time | 14 days | ||||
| Administration Dosage | 1 mg/kg qw | ||||
| MOA of PDC |
BT1718 is a novel first in class bicyclic targeting peptide that selectively binds MT1 MMP (MMP-14) and is linked to the maytansinoid tubulin inhibitor DM1 by a cleavable disulfide linker. Bicycle Toxin Conjugates have a low molecular weight compared to other conjugated toxin approaches, enabling rapid tumour penetration and a short systemic half-life (up to 40 minutes in non-human primates) potentially reducing toxicity. The target MT1-MMP (MT1) is a surface metalloproteinase involved in tissue remodelling through proteolysis of extracellular matrix components: Highly expressed in tumours with unmet medical need, such as triple negative breast cancer, non small cell lung cancer and ovarian cancer. Strong link with cell invasion and metastasis. High tumour MT1 expression is correlated with poor outcomes in multiple tumour types. High adjacent stromal expression and low expression in adult normal tissue.
Click to Show/Hide
|
||||
| Description |
Treatment with BDCs containing the most labile linkers (BT17BDC17 or BT1718) showed rapid and complete tumour clearance (EBC-1 cells), while BDCs containing more stabilised linkers showed comparatively reduced efficacy (fig. 5) suggesting that target internalisation is not the sole mechanism of action for BDC efficacy and that extracellular cleavage and release of toxin within the local tumour environment likely also contribute. Only the most labile BDC (BT17BDC17) caused any significant toxicity (17% 9.7 body weight loss); all others were well tolerated (<10% body weight loss at 10 mg/kg tiw). Optimal therapeutic index was achieved with BT1718. Testing of BT1718 in different dosing regimes in an additional model (HT-1080 cells) also demonstrated excellent tumour regression, with 10 mg/kg biw leading to complete tumour clearance in all 3 animals within 23 days and no re-growth out to 70 days.
Click to Show/Hide
|
||||
| In Vivo Model | Cell-derived xenograftmodel inmiceimplanted with MT1-positive HT-1080 cells. | ||||
| In Vitro Model | Fibrosarcoma | HT-1080 cell | CVCL_0317 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Fibrosarcoma | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
27%
|
|||
| Administration Time | 14 days | ||||
| Administration Dosage | 1 mg/kg biw | ||||
| MOA of PDC |
BT1718 is a novel first in class bicyclic targeting peptide that selectively binds MT1 MMP (MMP-14) and is linked to the maytansinoid tubulin inhibitor DM1 by a cleavable disulfide linker. Bicycle Toxin Conjugates have a low molecular weight compared to other conjugated toxin approaches, enabling rapid tumour penetration and a short systemic half-life (up to 40 minutes in non-human primates) potentially reducing toxicity. The target MT1-MMP (MT1) is a surface metalloproteinase involved in tissue remodelling through proteolysis of extracellular matrix components: Highly expressed in tumours with unmet medical need, such as triple negative breast cancer, non small cell lung cancer and ovarian cancer. Strong link with cell invasion and metastasis. High tumour MT1 expression is correlated with poor outcomes in multiple tumour types. High adjacent stromal expression and low expression in adult normal tissue.
Click to Show/Hide
|
||||
| Description |
Treatment with BDCs containing the most labile linkers (BT17BDC17 or BT1718) showed rapid and complete tumour clearance (EBC-1 cells), while BDCs containing more stabilised linkers showed comparatively reduced efficacy (fig. 5) suggesting that target internalisation is not the sole mechanism of action for BDC efficacy and that extracellular cleavage and release of toxin within the local tumour environment likely also contribute. Only the most labile BDC (BT17BDC17) caused any significant toxicity (17% 9.7 body weight loss); all others were well tolerated (<10% body weight loss at 10 mg/kg tiw). Optimal therapeutic index was achieved with BT1718. Testing of BT1718 in different dosing regimes in an additional model (HT-1080 cells) also demonstrated excellent tumour regression, with 10 mg/kg biw leading to complete tumour clearance in all 3 animals within 23 days and no re-growth out to 70 days.
Click to Show/Hide
|
||||
| In Vivo Model | Cell-derived xenograftmodel inmiceimplanted with MT1-positive HT-1080 cells. | ||||
| In Vitro Model | Fibrosarcoma | HT-1080 cell | CVCL_0317 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
85.40%
|
|||
| Administration Time | Three times a week for 12 days | ||||
| Administration Dosage | 3 mg/kg | ||||
| Evaluation Method | Tumor volume detection | ||||
| MOA of PDC |
BT1718 is a novel first in class bicyclic targeting peptide that selectively binds MT1 MMP (MMP-14) and is linked to the maytansinoid tubulin inhibitor DM1 by a cleavable disulfide linker. Bicycle Toxin Conjugates have a low molecular weight compared to other conjugated toxin approaches, enabling rapid tumour penetration and a short systemic half-life (up to 40 minutes in non-human primates) potentially reducing toxicity. The target MT1-MMP (MT1) is a surface metalloproteinase involved in tissue remodelling through proteolysis of extracellular matrix components: Highly expressed in tumours with unmet medical need, such as triple negative breast cancer, non small cell lung cancer and ovarian cancer. Strong link with cell invasion and metastasis. High tumour MT1 expression is correlated with poor outcomes in multiple tumour types. High adjacent stromal expression and low expression in adult normal tissue.
Click to Show/Hide
|
||||
| Description |
Testing of BT1718 in different dosing regimes in an additional model (HT-1080 cells) also demonstrated excellent tumour regression, with 10mg/kg biw leading to complete tumour clearance in all 3 animals within 23 days and no re-growth out to 74 days.
|
||||
| In Vivo Model | Mice implanted with MT1-positive HT-1080 cells | ||||
| In Vitro Model | Fibrosarcoma | HT-1080 cell | CVCL_0317 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Fibrosarcoma | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
88%
|
|||
| Administration Time | 14 days | ||||
| Administration Dosage | 3 mg/kg qw | ||||
| MOA of PDC |
BT1718 is a novel first in class bicyclic targeting peptide that selectively binds MT1 MMP (MMP-14) and is linked to the maytansinoid tubulin inhibitor DM1 by a cleavable disulfide linker. Bicycle Toxin Conjugates have a low molecular weight compared to other conjugated toxin approaches, enabling rapid tumour penetration and a short systemic half-life (up to 40 minutes in non-human primates) potentially reducing toxicity. The target MT1-MMP (MT1) is a surface metalloproteinase involved in tissue remodelling through proteolysis of extracellular matrix components: Highly expressed in tumours with unmet medical need, such as triple negative breast cancer, non small cell lung cancer and ovarian cancer. Strong link with cell invasion and metastasis. High tumour MT1 expression is correlated with poor outcomes in multiple tumour types. High adjacent stromal expression and low expression in adult normal tissue.
Click to Show/Hide
|
||||
| Description |
Treatment with BDCs containing the most labile linkers (BT17BDC17 or BT1718) showed rapid and complete tumour clearance (EBC-1 cells), while BDCs containing more stabilised linkers showed comparatively reduced efficacy (fig. 5) suggesting that target internalisation is not the sole mechanism of action for BDC efficacy and that extracellular cleavage and release of toxin within the local tumour environment likely also contribute. Only the most labile BDC (BT17BDC17) caused any significant toxicity (17% 9.7 body weight loss); all others were well tolerated (<10% body weight loss at 10 mg/kg tiw). Optimal therapeutic index was achieved with BT1718. Testing of BT1718 in different dosing regimes in an additional model (HT-1080 cells) also demonstrated excellent tumour regression, with 10 mg/kg biw leading to complete tumour clearance in all 3 animals within 23 days and no re-growth out to 70 days.
Click to Show/Hide
|
||||
| In Vivo Model | Cell-derived xenograftmodel inmiceimplanted with MT1-positive HT-1080 cells. | ||||
| In Vitro Model | Fibrosarcoma | HT-1080 cell | CVCL_0317 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Lung squamous cell carcinoma | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
100%
|
|||
| Administration Time | 14 days | ||||
| Administration Dosage | 10mg/kg | ||||
| MOA of PDC |
BT1718 is a novel first in class bicyclic targeting peptide that selectively binds MT1 MMP (MMP-14) and is linked to the maytansinoid tubulin inhibitor DM1 by a cleavable disulfide linker. Bicycle Toxin Conjugates have a low molecular weight compared to other conjugated toxin approaches, enabling rapid tumour penetration and a short systemic half-life (up to 40 minutes in non-human primates) potentially reducing toxicity. The target MT1-MMP (MT1) is a surface metalloproteinase involved in tissue remodelling through proteolysis of extracellular matrix components: Highly expressed in tumours with unmet medical need, such as triple negative breast cancer, non small cell lung cancer and ovarian cancer. Strong link with cell invasion and metastasis. High tumour MT1 expression is correlated with poor outcomes in multiple tumour types. High adjacent stromal expression and low expression in adult normal tissue.
Click to Show/Hide
|
||||
| Description |
Treatment with BDCs containing the most labile linkers (BT17BDC17 or BT1718) showed rapid and complete tumour clearance (EBC-1 cells), while BDCs containing more stabilised linkers showed comparatively reduced efficacy (fig. 5) suggesting that target internalisation is not the sole mechanism of action for BDC efficacy and that extracellular cleavage and release of toxin within the local tumour environment likely also contribute. Only the most labile BDC (BT17BDC17) caused any significant toxicity (17% 9.7 body weight loss); all others were well tolerated (<10% body weight loss at 10 mg/kg tiw). Optimal therapeutic index was achieved with BT1718. Testing of BT1718 in different dosing regimes in an additional model (HT-1080 cells) also demonstrated excellent tumour regression, with 10 mg/kg biw leading to complete tumour clearance in all 3 animals within 23 days and no re-growth out to 70 days.
Click to Show/Hide
|
||||
| In Vivo Model | Cell-derived xenograftmodel inmiceimplanted with MT1-positive EBC-1 cells. | ||||
| In Vitro Model | Lung squamous cell carcinoma | EBC-1 cell | CVCL_2891 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Fibrosarcoma | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
100%
|
|||
| Administration Time | 14 days | ||||
| Administration Dosage | 10 mg/kg biw | ||||
| MOA of PDC |
BT1718 is a novel first in class bicyclic targeting peptide that selectively binds MT1 MMP (MMP-14) and is linked to the maytansinoid tubulin inhibitor DM1 by a cleavable disulfide linker. Bicycle Toxin Conjugates have a low molecular weight compared to other conjugated toxin approaches, enabling rapid tumour penetration and a short systemic half-life (up to 40 minutes in non-human primates) potentially reducing toxicity. The target MT1-MMP (MT1) is a surface metalloproteinase involved in tissue remodelling through proteolysis of extracellular matrix components: Highly expressed in tumours with unmet medical need, such as triple negative breast cancer, non small cell lung cancer and ovarian cancer. Strong link with cell invasion and metastasis. High tumour MT1 expression is correlated with poor outcomes in multiple tumour types. High adjacent stromal expression and low expression in adult normal tissue.
Click to Show/Hide
|
||||
| Description |
Treatment with BDCs containing the most labile linkers (BT17BDC17 or BT1718) showed rapid and complete tumour clearance (EBC-1 cells), while BDCs containing more stabilised linkers showed comparatively reduced efficacy (fig. 5) suggesting that target internalisation is not the sole mechanism of action for BDC efficacy and that extracellular cleavage and release of toxin within the local tumour environment likely also contribute. Only the most labile BDC (BT17BDC17) caused any significant toxicity (17% 9.7 body weight loss); all others were well tolerated (<10% body weight loss at 10 mg/kg tiw). Optimal therapeutic index was achieved with BT1718. Testing of BT1718 in different dosing regimes in an additional model (HT-1080 cells) also demonstrated excellent tumour regression, with 10 mg/kg biw leading to complete tumour clearance in all 3 animals within 23 days and no re-growth out to 70 days.
Click to Show/Hide
|
||||
| In Vivo Model | Cell-derived xenograftmodel inmiceimplanted with MT1-positive HT-1080 cells. | ||||
| In Vitro Model | Fibrosarcoma | HT-1080 cell | CVCL_0317 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Fibrosarcoma | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
100%
|
|||
| Administration Time | 14 days | ||||
| Administration Dosage | 3 mg/kg biw | ||||
| MOA of PDC |
BT1718 is a novel first in class bicyclic targeting peptide that selectively binds MT1 MMP (MMP-14) and is linked to the maytansinoid tubulin inhibitor DM1 by a cleavable disulfide linker. Bicycle Toxin Conjugates have a low molecular weight compared to other conjugated toxin approaches, enabling rapid tumour penetration and a short systemic half-life (up to 40 minutes in non-human primates) potentially reducing toxicity. The target MT1-MMP (MT1) is a surface metalloproteinase involved in tissue remodelling through proteolysis of extracellular matrix components: Highly expressed in tumours with unmet medical need, such as triple negative breast cancer, non small cell lung cancer and ovarian cancer. Strong link with cell invasion and metastasis. High tumour MT1 expression is correlated with poor outcomes in multiple tumour types. High adjacent stromal expression and low expression in adult normal tissue.
Click to Show/Hide
|
||||
| Description |
Treatment with BDCs containing the most labile linkers (BT17BDC17 or BT1718) showed rapid and complete tumour clearance (EBC-1 cells), while BDCs containing more stabilised linkers showed comparatively reduced efficacy (fig. 5) suggesting that target internalisation is not the sole mechanism of action for BDC efficacy and that extracellular cleavage and release of toxin within the local tumour environment likely also contribute. Only the most labile BDC (BT17BDC17) caused any significant toxicity (17% 9.7 body weight loss); all others were well tolerated (<10% body weight loss at 10 mg/kg tiw). Optimal therapeutic index was achieved with BT1718. Testing of BT1718 in different dosing regimes in an additional model (HT-1080 cells) also demonstrated excellent tumour regression, with 10 mg/kg biw leading to complete tumour clearance in all 3 animals within 23 days and no re-growth out to 70 days.
Click to Show/Hide
|
||||
| In Vivo Model | Cell-derived xenograftmodel inmiceimplanted with MT1-positive HT-1080 cells. | ||||
| In Vitro Model | Fibrosarcoma | HT-1080 cell | CVCL_0317 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Fibrosarcoma | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
100%
|
|||
| Administration Time | 14 days | ||||
| Administration Dosage | 10 mg/kg qw | ||||
| MOA of PDC |
BT1718 is a novel first in class bicyclic targeting peptide that selectively binds MT1 MMP (MMP-14) and is linked to the maytansinoid tubulin inhibitor DM1 by a cleavable disulfide linker. Bicycle Toxin Conjugates have a low molecular weight compared to other conjugated toxin approaches, enabling rapid tumour penetration and a short systemic half-life (up to 40 minutes in non-human primates) potentially reducing toxicity. The target MT1-MMP (MT1) is a surface metalloproteinase involved in tissue remodelling through proteolysis of extracellular matrix components: Highly expressed in tumours with unmet medical need, such as triple negative breast cancer, non small cell lung cancer and ovarian cancer. Strong link with cell invasion and metastasis. High tumour MT1 expression is correlated with poor outcomes in multiple tumour types. High adjacent stromal expression and low expression in adult normal tissue.
Click to Show/Hide
|
||||
| Description |
Treatment with BDCs containing the most labile linkers (BT17BDC17 or BT1718) showed rapid and complete tumour clearance (EBC-1 cells), while BDCs containing more stabilised linkers showed comparatively reduced efficacy (fig. 5) suggesting that target internalisation is not the sole mechanism of action for BDC efficacy and that extracellular cleavage and release of toxin within the local tumour environment likely also contribute. Only the most labile BDC (BT17BDC17) caused any significant toxicity (17% 9.7 body weight loss); all others were well tolerated (<10% body weight loss at 10 mg/kg tiw). Optimal therapeutic index was achieved with BT1718. Testing of BT1718 in different dosing regimes in an additional model (HT-1080 cells) also demonstrated excellent tumour regression, with 10 mg/kg biw leading to complete tumour clearance in all 3 animals within 23 days and no re-growth out to 70 days.
Click to Show/Hide
|
||||
| In Vivo Model | Cell-derived xenograftmodel inmiceimplanted with MT1-positive HT-1080 cells. | ||||
| In Vitro Model | Fibrosarcoma | HT-1080 cell | CVCL_0317 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
100%
|
|||
| Administration Time | 12 days | ||||
| Administration Dosage | 10 mg/kg | ||||
| Evaluation Method | Tumor volume detection | ||||
| MOA of PDC |
BT1718 is a novel first in class bicyclic targeting peptide that selectively binds MT1 MMP (MMP-14) and is linked to the maytansinoid tubulin inhibitor DM1 by a cleavable disulfide linker. Bicycle Toxin Conjugates have a low molecular weight compared to other conjugated toxin approaches, enabling rapid tumour penetration and a short systemic half-life (up to 40 minutes in non-human primates) potentially reducing toxicity. The target MT1-MMP (MT1) is a surface metalloproteinase involved in tissue remodelling through proteolysis of extracellular matrix components: Highly expressed in tumours with unmet medical need, such as triple negative breast cancer, non small cell lung cancer and ovarian cancer. Strong link with cell invasion and metastasis. High tumour MT1 expression is correlated with poor outcomes in multiple tumour types. High adjacent stromal expression and low expression in adult normal tissue.
Click to Show/Hide
|
||||
| Description |
Treatment with BDCs containing the most labile linkers (BT17BDC17 orBT1718) showed rapid and complete tumour clearance (EBC-1 cells).
|
||||
| In Vivo Model | Mice implanted with MT1-positive EBC-1 cells | ||||
| In Vitro Model | Lung squamous cell carcinoma | EBC-1 cell | CVCL_2891 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
100%
|
|||
| Administration Time | Twice a week for 12 days | ||||
| Administration Dosage | 10 mg/kg | ||||
| Evaluation Method | Tumor volume detection | ||||
| MOA of PDC |
BT1718 is a novel first in class bicyclic targeting peptide that selectively binds MT1 MMP (MMP-14) and is linked to the maytansinoid tubulin inhibitor DM1 by a cleavable disulfide linker. Bicycle Toxin Conjugates have a low molecular weight compared to other conjugated toxin approaches, enabling rapid tumour penetration and a short systemic half-life (up to 40 minutes in non-human primates) potentially reducing toxicity. The target MT1-MMP (MT1) is a surface metalloproteinase involved in tissue remodelling through proteolysis of extracellular matrix components: Highly expressed in tumours with unmet medical need, such as triple negative breast cancer, non small cell lung cancer and ovarian cancer. Strong link with cell invasion and metastasis. High tumour MT1 expression is correlated with poor outcomes in multiple tumour types. High adjacent stromal expression and low expression in adult normal tissue.
Click to Show/Hide
|
||||
| Description |
Testing of BT1718 in different dosing regimes in an additional model (HT-1080 cells) also demonstrated excellent tumour regression, with 10mg/kg biw leading to complete tumour clearance in all 3 animals within 23 days and no re-growth out to 70 days.
|
||||
| In Vivo Model | Mice implanted with MT1-positive HT-1080 cells | ||||
| In Vitro Model | Fibrosarcoma | HT-1080 cell | CVCL_0317 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
100%
|
|||
| Administration Time | Twice a week for 12 days | ||||
| Administration Dosage | 3 mg/kg | ||||
| Evaluation Method | Tumor volume detection | ||||
| MOA of PDC |
BT1718 is a novel first in class bicyclic targeting peptide that selectively binds MT1 MMP (MMP-14) and is linked to the maytansinoid tubulin inhibitor DM1 by a cleavable disulfide linker. Bicycle Toxin Conjugates have a low molecular weight compared to other conjugated toxin approaches, enabling rapid tumour penetration and a short systemic half-life (up to 40 minutes in non-human primates) potentially reducing toxicity. The target MT1-MMP (MT1) is a surface metalloproteinase involved in tissue remodelling through proteolysis of extracellular matrix components: Highly expressed in tumours with unmet medical need, such as triple negative breast cancer, non small cell lung cancer and ovarian cancer. Strong link with cell invasion and metastasis. High tumour MT1 expression is correlated with poor outcomes in multiple tumour types. High adjacent stromal expression and low expression in adult normal tissue.
Click to Show/Hide
|
||||
| Description |
Testing of BT1718 in different dosing regimes in an additional model (HT-1080 cells) also demonstrated excellent tumour regression, with 10mg/kg biw leading to complete tumour clearance in all 3 animals within 23 days and no re-growth out to 71 days.
|
||||
| In Vivo Model | Mice implanted with MT1-positive HT-1080 cells | ||||
| In Vitro Model | Fibrosarcoma | HT-1080 cell | CVCL_0317 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
100%
|
|||
| Administration Time | Three times a week for 12 days | ||||
| Administration Dosage | 10 mg/kg | ||||
| Evaluation Method | Tumor volume detection | ||||
| MOA of PDC |
BT1718 is a novel first in class bicyclic targeting peptide that selectively binds MT1 MMP (MMP-14) and is linked to the maytansinoid tubulin inhibitor DM1 by a cleavable disulfide linker. Bicycle Toxin Conjugates have a low molecular weight compared to other conjugated toxin approaches, enabling rapid tumour penetration and a short systemic half-life (up to 40 minutes in non-human primates) potentially reducing toxicity. The target MT1-MMP (MT1) is a surface metalloproteinase involved in tissue remodelling through proteolysis of extracellular matrix components: Highly expressed in tumours with unmet medical need, such as triple negative breast cancer, non small cell lung cancer and ovarian cancer. Strong link with cell invasion and metastasis. High tumour MT1 expression is correlated with poor outcomes in multiple tumour types. High adjacent stromal expression and low expression in adult normal tissue.
Click to Show/Hide
|
||||
| Description |
Testing of BT1718 in different dosing regimes in an additional model (HT-1080 cells) also demonstrated excellent tumour regression, with 10mg/kg biw leading to complete tumour clearance in all 3 animals within 23 days and no re-growth out to 73 days.
|
||||
| In Vivo Model | Mice implanted with MT1-positive HT-1080 cells | ||||
| In Vitro Model | Fibrosarcoma | HT-1080 cell | CVCL_0317 | ||
CBX-12 [Phase 2]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
58.10%
|
|||
| Administration Time | 23 days | ||||
| Administration Dosage | 5 mg/kg | ||||
| Evaluation Method | Tumor volume detection | ||||
| Description |
Combination treatment significantly inhibited tumor growth without significant toxicity in both mouse xenografts compared with CBX-12 and ceralasertib monotherapy without significant toxicity.
|
||||
| In Vivo Model | HCT-116 xenografts | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
76.50%
|
|||
| Administration Time | 35 days | ||||
| Administration Dosage | 10 mg/kg | ||||
| Evaluation Method | Tumor volume detection | ||||
| Description |
Combination treatment significantly inhibited tumor growth without significant toxicity in both mouse xenografts compared with CBX-12 and ceralasertib monotherapy without significant toxicity.
|
||||
| In Vivo Model | MDA-MB-231 xenografts | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
81.40%
|
|||
| Administration Time | 23 days | ||||
| Administration Dosage | 5 mg/kg with ceralasertib 25 mg/kg | ||||
| Evaluation Method | Tumor volume detection | ||||
| Description |
Combination treatment significantly inhibited tumor growth without significant toxicity in both mouse xenografts compared with CBX-12 and ceralasertib monotherapy without significant toxicity.
|
||||
| In Vivo Model | HCT-116 xenografts | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
95%
|
|||
| Administration Time | 35 days | ||||
| Administration Dosage | 10 mg/kg with ceralasertib 25 mg/kg | ||||
| Evaluation Method | Tumor volume detection | ||||
| Description |
Combination treatment significantly inhibited tumor growth without significant toxicity in both mouse xenografts compared with CBX-12 and ceralasertib monotherapy without significant toxicity.
|
||||
| In Vivo Model | MDA-MB-231 xenografts | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Survival rate |
60 mm3
|
|||
| Administration Time | 45 days | ||||
| Administration Dosage | 5 mg/kg | ||||
| Description |
Combination treatment significantly inhibited tumor growth without significant toxicity in both mouse xenografts compared with CBX-12 and ceralasertib monotherapy without significant toxicity.
|
||||
| In Vivo Model | HCT-116 xenografts | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Survival rate |
75 mm3
|
|||
| Administration Time | 42 days | ||||
| Administration Dosage | 10 mg/kg | ||||
| Description |
Combination treatment significantly inhibited tumor growth without significant toxicity in both mouse xenografts compared with CBX-12 and ceralasertib monotherapy without significant toxicity.
|
||||
| In Vivo Model | MDA-MB-231 xenografts | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Survival rate |
100 mm3
|
|||
| Administration Time | 42 days | ||||
| Administration Dosage | 10 mg/kg with ceralasertib 25 mg/kg | ||||
| Description |
Combination treatment significantly inhibited tumor growth without significant toxicity in both mouse xenografts compared with CBX-12 and ceralasertib monotherapy without significant toxicity.
|
||||
| In Vivo Model | MDA-MB-231 xenografts | ||||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Survival rate |
100 mm3
|
|||
| Administration Time | 45 days | ||||
| Administration Dosage | 5 mg/kg with ceralasertib 25 mg/kg | ||||
| Description |
Combination treatment significantly inhibited tumor growth without significant toxicity in both mouse xenografts compared with CBX-12 and ceralasertib monotherapy without significant toxicity.
|
||||
| In Vivo Model | HCT-116 xenografts | ||||
PEN-221 [Phase 2]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Small cell lung cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
0.06 µmol/L
|
|||
| Administration Time | 6 h | ||||
| Description |
PEN-221 receptor dependent activity in vitro varied from strong (NCI-H524) to modest (HCC-33) but was consistent (N = 3) and correlated with in vivo activity (described below). Octreotide alone did not have an effect on cellular growth. These cell lines also demonstrated sensitivity to the cytotoxic payload, DM1, with sub-micromolar IC50's after a 6-hour exposure and 70-hour additional incubation period (Supplementary Fig. S2B).
Click to Show/Hide
|
||||
| In Vitro Model | Lung small cell carcinoma | NCI-H524 cell | CVCL_1568 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Small cell lung cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
0.108 µmol/L
|
|||
| Administration Time | 6 h | ||||
| Description |
PEN-221 receptor dependent activity in vitro varied from strong (NCI-H524) to modest (HCC-33) but was consistent (N = 3) and correlated with in vivo activity (described below). Octreotide alone did not have an effect on cellular growth. These cell lines also demonstrated sensitivity to the cytotoxic payload, DM1, with sub-micromolar IC50's after a 6-hour exposure and 70-hour additional incubation period (Supplementary Fig. S2B).
Click to Show/Hide
|
||||
| In Vitro Model | Small cell lung carcinoma | NCI-H69 cell | CVCL_1579 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Small cell lung cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
0.258 µmol/L
|
|||
| Administration Time | 6 h | ||||
| Description |
PEN-221 receptor dependent activity in vitro varied from strong (NCI-H524) to modest (HCC-33) but was consistent (N = 3) and correlated with in vivo activity (described below). Octreotide alone did not have an effect on cellular growth. These cell lines also demonstrated sensitivity to the cytotoxic payload, DM1, with sub-micromolar IC50's after a 6-hour exposure and 70-hour additional incubation period (Supplementary Fig. S2B).
Click to Show/Hide
|
||||
| In Vitro Model | Small cell lung carcinoma | NCI-H69 cell | CVCL_1579 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Small cell lung cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
0.333 µmol/L
|
|||
| Administration Time | 6 h | ||||
| Description |
PEN-221 receptor dependent activity in vitro varied from strong (NCI-H524) to modest (HCC-33) but was consistent (N = 3) and correlated with in vivo activity (described below). Octreotide alone did not have an effect on cellular growth. These cell lines also demonstrated sensitivity to the cytotoxic payload, DM1, with sub-micromolar IC50's after a 6-hour exposure and 70-hour additional incubation period (Supplementary Fig. S2B).
Click to Show/Hide
|
||||
| In Vitro Model | Lung small cell carcinoma | NCI-H524 cell | CVCL_1568 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Small cell lung cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
0.658 µmol/L
|
|||
| Administration Time | 6 h | ||||
| Description |
PEN-221 receptor dependent activity in vitro varied from strong (NCI-H524) to modest (HCC-33) but was consistent (N = 3) and correlated with in vivo activity (described below). Octreotide alone did not have an effect on cellular growth. These cell lines also demonstrated sensitivity to the cytotoxic payload, DM1, with sub-micromolar IC50's after a 6-hour exposure and 70-hour additional incubation period (Supplementary Fig. S2B).
Click to Show/Hide
|
||||
| In Vitro Model | Lung small cell carcinoma | HCC33 cell | CVCL_2058 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Small cell lung cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
0.83 µmol/L
|
|||
| Administration Time | 6 h | ||||
| Description |
PEN-221 receptor dependent activity in vitro varied from strong (NCI-H524) to modest (HCC-33) but was consistent (N = 3) and correlated with in vivo activity (described below). Octreotide alone did not have an effect on cellular growth. These cell lines also demonstrated sensitivity to the cytotoxic payload, DM1, with sub-micromolar IC50's after a 6-hour exposure and 70-hour additional incubation period (Supplementary Fig. S2B).
Click to Show/Hide
|
||||
| In Vitro Model | Lung small cell carcinoma | HCC33 cell | CVCL_2058 | ||
GLP-1-MK-801 [Preclinical]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Obesity | ||||
| Efficacy Data | Vehicle-corrected weight loss rate |
9.50%
|
|||
| Administration Time | 1 dose | ||||
| Administration Dosage | 0.22 nmol | ||||
| Evaluation Method | This study revealed a superior vehicle-corrected weight loss of 9.5% in response to the GLP-1-MK-801 infusion relative to a weight loss of 4.5% after semaglutide infusion (Fig. 4b). | ||||
| MOA of PDC |
Treatment with GLP-1-inactive MK-801 produced no additional weight loss efficacy relative to GLP-1 monotherapy (Fig. 2g-i), indicating that the pronounced weight loss induced by GLP-1-MK-801 is driven by concerted and site-directed pharmacological GLP-1 receptor agonism and NMDA receptor antagonism.
|
||||
| In Vivo Model | High-fat high-sucrose-fed mice | ||||
| Half life period | 1.9 h | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Obesity | ||||
| Efficacy Data | Vehicle-corrected weight loss rate |
15.20%
|
|||
| Administration Time | Once a day for 9 days | ||||
| Administration Dosage | 100 nmol kg-1 | ||||
| Evaluation Method | We observed a pronounced vehicle-corrected weight loss of 15.2% in the group treated with GLP-1-MK-801 compared with 3.5% in the group treated with the parent GLP-1 analogue after 9days of treatment, underscoring that the weight-lowering efficacy of the conjugate is intact in the absence of functional MC4R signalling (Fig. 3l,m). | ||||
| MOA of PDC |
Treatment with GLP-1-inactive MK-801 produced no additional weight loss efficacy relative to GLP-1 monotherapy (Fig. 2g-i), indicating that the pronounced weight loss induced by GLP-1-MK-801 is driven by concerted and site-directed pharmacological GLP-1 receptor agonism and NMDA receptor antagonism.
|
||||
| In Vivo Model | Mc4r-KO mice | ||||
| Half life period | 1.9 h | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Obesity | ||||
| Efficacy Data | Vehicle-corrected weight loss rate |
23.20%
|
|||
| Administration Time | Once a day for 14 days | ||||
| Administration Dosage | 100 nmol kg-1 | ||||
| Evaluation Method | Over a 14-day treatment period, GLP-1-MK-801 synergistically lowered body weight compared with the dose-matched monotherapies and produced a vehicle-corrected weight loss of 23.2%. | ||||
| MOA of PDC |
Treatment with GLP-1-inactive MK-801 produced no additional weight loss efficacy relative to GLP-1 monotherapy (Fig. 2g-i), indicating that the pronounced weight loss induced by GLP-1-MK-801 is driven by concerted and site-directed pharmacological GLP-1 receptor agonism and NMDA receptor antagonism.
|
||||
| In Vivo Model | DIO mice | ||||
| Half life period | 1.9 h | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Obesity | ||||
| Efficacy Data | Lean mass rate |
8%
|
|||
| Administration Time | Once a day for 14 days | ||||
| Administration Dosage | 100 nmol kg-1 | ||||
| Evaluation Method | GLP-1-MK-801 produced a vehicle-corrected reduction in body fat mass of 45%, accompanied by an 8% loss in lean mass. | ||||
| MOA of PDC |
Treatment with GLP-1-inactive MK-801 produced no additional weight loss efficacy relative to GLP-1 monotherapy (Fig. 2g-i), indicating that the pronounced weight loss induced by GLP-1-MK-801 is driven by concerted and site-directed pharmacological GLP-1 receptor agonism and NMDA receptor antagonism.
|
||||
| In Vivo Model | DIO mice | ||||
| Half life period | 1.9 h | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Obesity | ||||
| Efficacy Data | Fat mass reduction rate |
45%
|
|||
| Administration Time | Once a day for 14 days | ||||
| Administration Dosage | 100 nmol kg-1 | ||||
| Evaluation Method | GLP-1-MK-801 produced a vehicle-corrected reduction in body fat mass of 45%, accompanied by an 8% loss in lean mass. | ||||
| MOA of PDC |
Treatment with GLP-1-inactive MK-801 produced no additional weight loss efficacy relative to GLP-1 monotherapy (Fig. 2g-i), indicating that the pronounced weight loss induced by GLP-1-MK-801 is driven by concerted and site-directed pharmacological GLP-1 receptor agonism and NMDA receptor antagonism.
|
||||
| In Vivo Model | DIO mice | ||||
| Half life period | 1.9 h | ||||
PDC-Z8 [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) |
60% (Day 14)
|
|||
| Administration Time | Injected via tail vein every t hree days | ||||
| Administration Dosage | 8 µmol/kg | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To study the in vivo anti-tumor activity of Z8, SK-BR-3 tumor bearing nude mice were administered once every three days. As shown in Fig. 11A-11D, CPT and Z8 demonstrated significant inhibitory effects on SK-BR-3 tumor growth, with the Z8 group exhibiting more pronounced inhibition compared to CPT. Notably, the average tumor volume and weight in the Z8 group were the smallest among all groups, including the control and CPT groups. Moreover, to further study the toxicity of CPT and Z8, serums of mice were collected for blood biochemical analysis. As shown in Fig. 11E-H, treatment with both CPT and Z8 resulted in increased levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST), but the effect of Z8 group was relatively low, indicating that Z8 was safer than CPT in vivo. Furthermore, in CPT group, urea nitrogen levels increased significantly, whereas no significant change was observed in the Z8 group, indicating potential renal function impairment following CPT treatment, whereas Z8 showed better safety in this regard. Additionally, the levels of alkaline phosphatase (ALP), total protein (TP), albumin (ALB), globulin (GLOB) and creatinine (CREA) did not significantly differ between the control group and the treatment groups. Finally, compared to the control group, histological examination using H&E staining (Hematoxylin and Eosin staining) of the major organs in the treatment group did not reveal obvious cell necrosis and inflammatory cell infiltration, indicating that Z8 would not bring additional toxicity.
Click to Show/Hide
|
||||
| In Vivo Model | SK-BR-3 tumor-bearing nude mice xenograft model. | ||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.04 ± 0.24 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.91 ± 0.71 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
13.49 ± 3.59 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
26.34 ± 1.08 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Apoptosis rate |
27.10%
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 1 µM | ||||
| Evaluation Method | Flow cytometry assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
The proapoptotic activity of Z8 was studied by flow cytometry combined with Annexin V-FITC/PI double staining. As shown in Fig. 4, after treatment with 1 uM Z8, the apoptosis rate of SK-BR-3 cells was approximately 27.1 % which increased to 41.1 % following treatment with 2 uM Z8 (4.4 % in the blank control group and 55.8 % in CPT group. These findings suggest that Z8 can significantly induce apoptosis in a dose-dependent manner.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Apoptosis rate |
41.10%
|
|||
| Administration Time | 24 h | ||||
| Administration Dosage | 2 µM | ||||
| Evaluation Method | Flow cytometry assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
The proapoptotic activity of Z8 was studied by flow cytometry combined with Annexin V-FITC/PI double staining. As shown in Fig. 4, after treatment with 1 uM Z8, the apoptosis rate of SK-BR-3 cells was approximately 27.1 % which increased to 41.1 % following treatment with 2 uM Z8 (4.4 % in the blank control group and 55.8 % in CPT group. These findings suggest that Z8 can significantly induce apoptosis in a dose-dependent manner.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
LTP-1 [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
67.10%
|
|||
| Administration Time | Every two days for 2 weeks | ||||
| Administration Dosage | 8 μmol/kg | ||||
| MOA of PDC |
LHRH, also named gonadotropin-releasing hormone (GnRH), is an endogenous peptide agonist (primary sequence: pGlu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2) released from hypothalamus. LHRH-R (LHRH receptor), a member of the G protein-coupled receptor family, is overexpressed in various tumor types, while their expression in the corresponding normal tissues, apart from pituitary cells, is comparatively low. Given this, we chose LHRH as the TTP component of MSCPTP (TTP-CPP peptide). In this investigation, we combined LHRH (as the TTP part), peptide PLGLAG, T2 (as the CPP part) and cysteine (as linker binding site) into an MSCPTP named LT-1. Then PTX was conjugated with LT-1 via a GSH-cleavable module to produce the smart PDC, namely LTP-1 (TTP-CPP-PTX conjugate). In vitro, LTP-1 exhibited selective and stronger cytotoxicity than PTX against LHRH-R-positive tumor cells with little effect on normal cells. In vivo, LTP-1 was highly effective in suppressing tumor growth in an MCF-7 xenograft mouse model. Additional experiments on both cellular and molecular levels were carried out to unravel the possible antitumor mechanism of action of LTP-1.
Click to Show/Hide
|
||||
| Description |
At the dose of 8 μmol/kg, LTP-1 decreased the tumor volume and tumor weight by 77.2% and 67.1%.
|
||||
| In Vivo Model | MCF-7 xenograft mice. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
77.20%
|
|||
| Administration Time | Every two days for 2 weeks | ||||
| Administration Dosage | 8 μmol/kg | ||||
| Evaluation Method | Tumor volume detection | ||||
| MOA of PDC |
LHRH, also named gonadotropin-releasing hormone (GnRH), is an endogenous peptide agonist (primary sequence: pGlu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2) released from hypothalamus. LHRH-R (LHRH receptor), a member of the G protein-coupled receptor family, is overexpressed in various tumor types, while their expression in the corresponding normal tissues, apart from pituitary cells, is comparatively low. Given this, we chose LHRH as the TTP component of MSCPTP (TTP-CPP peptide). In this investigation, we combined LHRH (as the TTP part), peptide PLGLAG, T2 (as the CPP part) and cysteine (as linker binding site) into an MSCPTP named LT-1. Then PTX was conjugated with LT-1 via a GSH-cleavable module to produce the smart PDC, namely LTP-1 (TTP-CPP-PTX conjugate). In vitro, LTP-1 exhibited selective and stronger cytotoxicity than PTX against LHRH-R-positive tumor cells with little effect on normal cells. In vivo, LTP-1 was highly effective in suppressing tumor growth in an MCF-7 xenograft mouse model. Additional experiments on both cellular and molecular levels were carried out to unravel the possible antitumor mechanism of action of LTP-1.
Click to Show/Hide
|
||||
| Description |
At the dose of 8 μmol/kg, LTP-1 decreased the tumor volume and tumor weight by 77.2% and 67.1%.
|
||||
| In Vivo Model | MCF-7 xenograft mice. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
83.40%
|
|||
| Administration Time | Every two days for 2 weeks | ||||
| Administration Dosage | 12 μmol/kg | ||||
| MOA of PDC |
LHRH, also named gonadotropin-releasing hormone (GnRH), is an endogenous peptide agonist (primary sequence: pGlu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2) released from hypothalamus. LHRH-R (LHRH receptor), a member of the G protein-coupled receptor family, is overexpressed in various tumor types, while their expression in the corresponding normal tissues, apart from pituitary cells, is comparatively low. Given this, we chose LHRH as the TTP component of MSCPTP (TTP-CPP peptide). In this investigation, we combined LHRH (as the TTP part), peptide PLGLAG, T2 (as the CPP part) and cysteine (as linker binding site) into an MSCPTP named LT-1. Then PTX was conjugated with LT-1 via a GSH-cleavable module to produce the smart PDC, namely LTP-1 (TTP-CPP-PTX conjugate). In vitro, LTP-1 exhibited selective and stronger cytotoxicity than PTX against LHRH-R-positive tumor cells with little effect on normal cells. In vivo, LTP-1 was highly effective in suppressing tumor growth in an MCF-7 xenograft mouse model. Additional experiments on both cellular and molecular levels were carried out to unravel the possible antitumor mechanism of action of LTP-1.
Click to Show/Hide
|
||||
| Description |
At the dose of 12 μmol/kg, LTP-1 decreased the tumor volume and tumor weight by 90.1% and 83.4%, respectively.
|
||||
| In Vivo Model | MCF-7 xenograft mice. | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) |
90.10%
|
|||
| Administration Time | Every two days for 2 weeks | ||||
| Administration Dosage | 12 μmol/kg | ||||
| Evaluation Method | Tumor volume detection | ||||
| MOA of PDC |
LHRH, also named gonadotropin-releasing hormone (GnRH), is an endogenous peptide agonist (primary sequence: pGlu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2) released from hypothalamus. LHRH-R (LHRH receptor), a member of the G protein-coupled receptor family, is overexpressed in various tumor types, while their expression in the corresponding normal tissues, apart from pituitary cells, is comparatively low. Given this, we chose LHRH as the TTP component of MSCPTP (TTP-CPP peptide). In this investigation, we combined LHRH (as the TTP part), peptide PLGLAG, T2 (as the CPP part) and cysteine (as linker binding site) into an MSCPTP named LT-1. Then PTX was conjugated with LT-1 via a GSH-cleavable module to produce the smart PDC, namely LTP-1 (TTP-CPP-PTX conjugate). In vitro, LTP-1 exhibited selective and stronger cytotoxicity than PTX against LHRH-R-positive tumor cells with little effect on normal cells. In vivo, LTP-1 was highly effective in suppressing tumor growth in an MCF-7 xenograft mouse model. Additional experiments on both cellular and molecular levels were carried out to unravel the possible antitumor mechanism of action of LTP-1.
Click to Show/Hide
|
||||
| Description |
At the dose of 12 μmol/kg, LTP-1 decreased the tumor volume and tumor weight by 90.1% and 83.4%, respectively.
|
||||
| In Vivo Model | MCF-7 xenograft mice. | ||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
3.8 ± 0.3 nM
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
LHRH, also named gonadotropin-releasing hormone (GnRH), is an endogenous peptide agonist (primary sequence: pGlu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2) released from hypothalamus. LHRH-R (LHRH receptor), a member of the G protein-coupled receptor family, is overexpressed in various tumor types, while their expression in the corresponding normal tissues, apart from pituitary cells, is comparatively low. Given this, we chose LHRH as the TTP component of MSCPTP (TTP-CPP peptide). In this investigation, we combined LHRH (as the TTP part), peptide PLGLAG, T2 (as the CPP part) and cysteine (as linker binding site) into an MSCPTP named LT-1. Then PTX was conjugated with LT-1 via a GSH-cleavable module to produce the smart PDC, namely LTP-1 (TTP-CPP-PTX conjugate). In vitro, LTP-1 exhibited selective and stronger cytotoxicity than PTX against LHRH-R-positive tumor cells with little effect on normal cells. In vivo, LTP-1 was highly effective in suppressing tumor growth in an MCF-7 xenograft mouse model. Additional experiments on both cellular and molecular levels were carried out to unravel the possible antitumor mechanism of action of LTP-1.
Click to Show/Hide
|
||||
| Description |
LTP-1 exhibited greater anti-proliferative effects (IC50s of 3.8-20.3 nM) than PTX (IC50s of 6.6-28.6 nM) against most cancer cells except Hela, and less cytotoxicity to normal cells (IC50s of >80 nM and 66.0 nM for NCM460 and HEK-293, respectively). Thus, LTP-1 displayed not only enhanced anti-proliferative activity, but also higher selectivity for cancer cells over normal cells. It is also worthy of note that LTP-1 showed much higher activity against the paclitaxel-resistant A2780/PTX cells with an IC50 of 0.8 μM, as compared to PTX which is essentially inactive (IC50 = 23.9 μM). Hemolysis assay further testified that LTP-1 presented weak hemolytic activity even at a concentration up to 80 μM (as illustrated in Fig. 4a).
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
5.6 ± 0.2 nM
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
LHRH, also named gonadotropin-releasing hormone (GnRH), is an endogenous peptide agonist (primary sequence: pGlu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2) released from hypothalamus. LHRH-R (LHRH receptor), a member of the G protein-coupled receptor family, is overexpressed in various tumor types, while their expression in the corresponding normal tissues, apart from pituitary cells, is comparatively low. Given this, we chose LHRH as the TTP component of MSCPTP (TTP-CPP peptide). In this investigation, we combined LHRH (as the TTP part), peptide PLGLAG, T2 (as the CPP part) and cysteine (as linker binding site) into an MSCPTP named LT-1. Then PTX was conjugated with LT-1 via a GSH-cleavable module to produce the smart PDC, namely LTP-1 (TTP-CPP-PTX conjugate). In vitro, LTP-1 exhibited selective and stronger cytotoxicity than PTX against LHRH-R-positive tumor cells with little effect on normal cells. In vivo, LTP-1 was highly effective in suppressing tumor growth in an MCF-7 xenograft mouse model. Additional experiments on both cellular and molecular levels were carried out to unravel the possible antitumor mechanism of action of LTP-1.
Click to Show/Hide
|
||||
| Description |
LTP-1 exhibited greater anti-proliferative effects (IC50s of 3.8-20.3 nM) than PTX (IC50s of 6.6-28.6 nM) against most cancer cells except Hela, and less cytotoxicity to normal cells (IC50s of >80 nM and 66.0 nM for NCM460 and HEK-293, respectively). Thus, LTP-1 displayed not only enhanced anti-proliferative activity, but also higher selectivity for cancer cells over normal cells. It is also worthy of note that LTP-1 showed much higher activity against the paclitaxel-resistant A2780/PTX cells with an IC50 of 0.8 μM, as compared to PTX which is essentially inactive (IC50 = 23.9 μM). Hemolysis assay further testified that LTP-1 presented weak hemolytic activity even at a concentration up to 80 μM (as illustrated in Fig. 4a).
Click to Show/Hide
|
||||
| In Vitro Model | Colon adenocarcinoma | HT-29 cell | CVCL_0320 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
8.3 ± 0.5 nM
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
LHRH, also named gonadotropin-releasing hormone (GnRH), is an endogenous peptide agonist (primary sequence: pGlu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2) released from hypothalamus. LHRH-R (LHRH receptor), a member of the G protein-coupled receptor family, is overexpressed in various tumor types, while their expression in the corresponding normal tissues, apart from pituitary cells, is comparatively low. Given this, we chose LHRH as the TTP component of MSCPTP (TTP-CPP peptide). In this investigation, we combined LHRH (as the TTP part), peptide PLGLAG, T2 (as the CPP part) and cysteine (as linker binding site) into an MSCPTP named LT-1. Then PTX was conjugated with LT-1 via a GSH-cleavable module to produce the smart PDC, namely LTP-1 (TTP-CPP-PTX conjugate). In vitro, LTP-1 exhibited selective and stronger cytotoxicity than PTX against LHRH-R-positive tumor cells with little effect on normal cells. In vivo, LTP-1 was highly effective in suppressing tumor growth in an MCF-7 xenograft mouse model. Additional experiments on both cellular and molecular levels were carried out to unravel the possible antitumor mechanism of action of LTP-1.
Click to Show/Hide
|
||||
| Description |
LTP-1 exhibited greater anti-proliferative effects (IC50s of 3.8-20.3 nM) than PTX (IC50s of 6.6-28.6 nM) against most cancer cells except Hela, and less cytotoxicity to normal cells (IC50s of >80 nM and 66.0 nM for NCM460 and HEK-293, respectively). Thus, LTP-1 displayed not only enhanced anti-proliferative activity, but also higher selectivity for cancer cells over normal cells. It is also worthy of note that LTP-1 showed much higher activity against the paclitaxel-resistant A2780/PTX cells with an IC50 of 0.8 μM, as compared to PTX which is essentially inactive (IC50 = 23.9 μM). Hemolysis assay further testified that LTP-1 presented weak hemolytic activity even at a concentration up to 80 μM (as illustrated in Fig. 4a).
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
20.3 ± 3.3 nM
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
LHRH, also named gonadotropin-releasing hormone (GnRH), is an endogenous peptide agonist (primary sequence: pGlu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2) released from hypothalamus. LHRH-R (LHRH receptor), a member of the G protein-coupled receptor family, is overexpressed in various tumor types, while their expression in the corresponding normal tissues, apart from pituitary cells, is comparatively low. Given this, we chose LHRH as the TTP component of MSCPTP (TTP-CPP peptide). In this investigation, we combined LHRH (as the TTP part), peptide PLGLAG, T2 (as the CPP part) and cysteine (as linker binding site) into an MSCPTP named LT-1. Then PTX was conjugated with LT-1 via a GSH-cleavable module to produce the smart PDC, namely LTP-1 (TTP-CPP-PTX conjugate). In vitro, LTP-1 exhibited selective and stronger cytotoxicity than PTX against LHRH-R-positive tumor cells with little effect on normal cells. In vivo, LTP-1 was highly effective in suppressing tumor growth in an MCF-7 xenograft mouse model. Additional experiments on both cellular and molecular levels were carried out to unravel the possible antitumor mechanism of action of LTP-1.
Click to Show/Hide
|
||||
| Description |
LTP-1 exhibited greater anti-proliferative effects (IC50s of 3.8-20.3 nM) than PTX (IC50s of 6.6-28.6 nM) against most cancer cells except Hela, and less cytotoxicity to normal cells (IC50s of >80 nM and 66.0 nM for NCM460 and HEK-293, respectively). Thus, LTP-1 displayed not only enhanced anti-proliferative activity, but also higher selectivity for cancer cells over normal cells. It is also worthy of note that LTP-1 showed much higher activity against the paclitaxel-resistant A2780/PTX cells with an IC50 of 0.8 μM, as compared to PTX which is essentially inactive (IC50 = 23.9 μM). Hemolysis assay further testified that LTP-1 presented weak hemolytic activity even at a concentration up to 80 μM (as illustrated in Fig. 4a).
Click to Show/Hide
|
||||
| In Vitro Model | Human papillomavirus-related cervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
66.0 ± 8.3 nM
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
LHRH, also named gonadotropin-releasing hormone (GnRH), is an endogenous peptide agonist (primary sequence: pGlu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2) released from hypothalamus. LHRH-R (LHRH receptor), a member of the G protein-coupled receptor family, is overexpressed in various tumor types, while their expression in the corresponding normal tissues, apart from pituitary cells, is comparatively low. Given this, we chose LHRH as the TTP component of MSCPTP (TTP-CPP peptide). In this investigation, we combined LHRH (as the TTP part), peptide PLGLAG, T2 (as the CPP part) and cysteine (as linker binding site) into an MSCPTP named LT-1. Then PTX was conjugated with LT-1 via a GSH-cleavable module to produce the smart PDC, namely LTP-1 (TTP-CPP-PTX conjugate). In vitro, LTP-1 exhibited selective and stronger cytotoxicity than PTX against LHRH-R-positive tumor cells with little effect on normal cells. In vivo, LTP-1 was highly effective in suppressing tumor growth in an MCF-7 xenograft mouse model. Additional experiments on both cellular and molecular levels were carried out to unravel the possible antitumor mechanism of action of LTP-1.
Click to Show/Hide
|
||||
| Description |
LTP-1 exhibited greater anti-proliferative effects (IC50s of 3.8-20.3 nM) than PTX (IC50s of 6.6-28.6 nM) against most cancer cells except Hela, and less cytotoxicity to normal cells (IC50s of >80 nM and 66.0 nM for NCM460 and HEK-293, respectively). Thus, LTP-1 displayed not only enhanced anti-proliferative activity, but also higher selectivity for cancer cells over normal cells. It is also worthy of note that LTP-1 showed much higher activity against the paclitaxel-resistant A2780/PTX cells with an IC50 of 0.8 μM, as compared to PTX which is essentially inactive (IC50 = 23.9 μM). Hemolysis assay further testified that LTP-1 presented weak hemolytic activity even at a concentration up to 80 μM (as illustrated in Fig. 4a).
Click to Show/Hide
|
||||
| In Vitro Model | Normal | HEK293 cell | CVCL_0045 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | > 80.0 nM | |||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
LHRH, also named gonadotropin-releasing hormone (GnRH), is an endogenous peptide agonist (primary sequence: pGlu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2) released from hypothalamus. LHRH-R (LHRH receptor), a member of the G protein-coupled receptor family, is overexpressed in various tumor types, while their expression in the corresponding normal tissues, apart from pituitary cells, is comparatively low. Given this, we chose LHRH as the TTP component of MSCPTP (TTP-CPP peptide). In this investigation, we combined LHRH (as the TTP part), peptide PLGLAG, T2 (as the CPP part) and cysteine (as linker binding site) into an MSCPTP named LT-1. Then PTX was conjugated with LT-1 via a GSH-cleavable module to produce the smart PDC, namely LTP-1 (TTP-CPP-PTX conjugate). In vitro, LTP-1 exhibited selective and stronger cytotoxicity than PTX against LHRH-R-positive tumor cells with little effect on normal cells. In vivo, LTP-1 was highly effective in suppressing tumor growth in an MCF-7 xenograft mouse model. Additional experiments on both cellular and molecular levels were carried out to unravel the possible antitumor mechanism of action of LTP-1.
Click to Show/Hide
|
||||
| Description |
LTP-1 exhibited greater anti-proliferative effects (IC50s of 3.8-20.3 nM) than PTX (IC50s of 6.6-28.6 nM) against most cancer cells except Hela, and less cytotoxicity to normal cells (IC50s of >80 nM and 66.0 nM for NCM460 and HEK-293, respectively). Thus, LTP-1 displayed not only enhanced anti-proliferative activity, but also higher selectivity for cancer cells over normal cells. It is also worthy of note that LTP-1 showed much higher activity against the paclitaxel-resistant A2780/PTX cells with an IC50 of 0.8 μM, as compared to PTX which is essentially inactive (IC50 = 23.9 μM). Hemolysis assay further testified that LTP-1 presented weak hemolytic activity even at a concentration up to 80 μM (as illustrated in Fig. 4a).
Click to Show/Hide
|
||||
| In Vitro Model | Normal | NCM460 cell | CVCL_0460 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [7] | ||||
| Indication | Solid tumor | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
800 ± 100 nM
|
|||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
LHRH, also named gonadotropin-releasing hormone (GnRH), is an endogenous peptide agonist (primary sequence: pGlu-His-Trp-Ser-Tyr-Gly-Leu-Arg-Pro-Gly-NH2) released from hypothalamus. LHRH-R (LHRH receptor), a member of the G protein-coupled receptor family, is overexpressed in various tumor types, while their expression in the corresponding normal tissues, apart from pituitary cells, is comparatively low. Given this, we chose LHRH as the TTP component of MSCPTP (TTP-CPP peptide). In this investigation, we combined LHRH (as the TTP part), peptide PLGLAG, T2 (as the CPP part) and cysteine (as linker binding site) into an MSCPTP named LT-1. Then PTX was conjugated with LT-1 via a GSH-cleavable module to produce the smart PDC, namely LTP-1 (TTP-CPP-PTX conjugate). In vitro, LTP-1 exhibited selective and stronger cytotoxicity than PTX against LHRH-R-positive tumor cells with little effect on normal cells. In vivo, LTP-1 was highly effective in suppressing tumor growth in an MCF-7 xenograft mouse model. Additional experiments on both cellular and molecular levels were carried out to unravel the possible antitumor mechanism of action of LTP-1.
Click to Show/Hide
|
||||
| Description |
LTP-1 exhibited greater anti-proliferative effects (IC50s of 3.8-20.3 nM) than PTX (IC50s of 6.6-28.6 nM) against most cancer cells except Hela, and less cytotoxicity to normal cells (IC50s of >80 nM and 66.0 nM for NCM460 and HEK-293, respectively). Thus, LTP-1 displayed not only enhanced anti-proliferative activity, but also higher selectivity for cancer cells over normal cells. It is also worthy of note that LTP-1 showed much higher activity against the paclitaxel-resistant A2780/PTX cells with an IC50 of 0.8 μM, as compared to PTX which is essentially inactive (IC50 = 23.9 μM). Hemolysis assay further testified that LTP-1 presented weak hemolytic activity even at a concentration up to 80 μM (as illustrated in Fig. 4a).
Click to Show/Hide
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780/PTX cell | CVCL_C0D6 | ||
cRGD-SS-DM1 [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Tumer volume |
1800 mm3
|
|||
| Administration Time | Given every other day for a total of five times | ||||
| Administration Dosage | 400 µg/kg (calculated by free DM1) | ||||
| Description |
It was demonstrated here, RCCD@NPs and RSSD@NPs exhibited significantly better tumor-growth inhibition compared with that of the free DM1, QCCD@NPs or QSSD@NPs (Figure (Figure4A).4A). RSSD@NPs showed the most suppression on B16 tumor up to 25 days, and resulted in a tumor volume 4 times smaller than the saline group at the end of the experiment. The final tumor volumes in various nano-DDS groups ranked from the greatest to the least: QCCD@NPs, QSSD@NPs>DM1>RCCD@NPs>RSSD@NPs. Almost the same trends were found in terms of tumor weight and tumor size (Figures (Figures4B,4B, C).
Click to Show/Hide
|
||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
20.42 ± 4.70 nM
|
|||
| Administration Time | 48 h | ||||
| Description |
The IC50 of QCCD@NPs and QSSD@NPs (389.05±75.25 nM and 245.47±37.54 nM) on B16 cells seemed much higher than the values of RCCD@NPs and RSSD@NPs (102.33±38.92 nM and 21.38±4.32 nM). The cytotoxicity of APDC@NPs on HUVEC showed a similar pattern.
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cell | Homo sapiens | ||
| Experiment 2 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
21.38 ± 4.32 nM
|
|||
| Administration Time | 48 h | ||||
| Description |
The IC50 of QCCD@NPs and QSSD@NPs (389.05±75.25 nM and 245.47±37.54 nM) on B16 cells seemed much higher than the values of RCCD@NPs and RSSD@NPs (102.33±38.92 nM and 21.38±4.32 nM). The cytotoxicity of APDC@NPs on HUVEC showed a similar pattern.
|
||||
| In Vitro Model | Melanoma | B16 cell | CVCL_F936 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
302.00 ± 72.91 nM
|
|||
| Administration Time | 48 h | ||||
| Description |
The IC50 of QCCD@NPs and QSSD@NPs (389.05±75.25 nM and 245.47±37.54 nM) on B16 cells seemed much higher than the values of RCCD@NPs and RSSD@NPs (102.33±38.92 nM and 21.38±4.32 nM). The cytotoxicity of APDC@NPs on HUVEC showed a similar pattern.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
cRPQ-SS-DM1 [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Tumer volume |
5200 mm3
|
|||
| Administration Time | Given every other day for a total of five times | ||||
| Administration Dosage | 400 µg/kg (calculated by free DM1) | ||||
| Description |
It was demonstrated here, RCCD@NPs and RSSD@NPs exhibited significantly better tumor-growth inhibition compared with that of the free DM1, QCCD@NPs or QSSD@NPs (Figure (Figure4A).4A). RSSD@NPs showed the most suppression on B16 tumor up to 25 days, and resulted in a tumor volume 4 times smaller than the saline group at the end of the experiment. The final tumor volumes in various nano-DDS groups ranked from the greatest to the least: QCCD@NPs, QSSD@NPs>DM1>RCCD@NPs>RSSD@NPs. Almost the same trends were found in terms of tumor weight and tumor size (Figures (Figures4B,4B, C).
Click to Show/Hide
|
||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
154.88 ± 31.85 nM
|
|||
| Administration Time | 48 h | ||||
| Description |
The IC50 of QCCD@NPs and QSSD@NPs (389.05±75.25 nM and 245.47±37.54 nM) on B16 cells seemed much higher than the values of RCCD@NPs and RSSD@NPs (102.33±38.92 nM and 21.38±4.32 nM). The cytotoxicity of APDC@NPs on HUVEC showed a similar pattern.
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cell | Homo sapiens | ||
| Experiment 2 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
245.47 ± 37.54 nM
|
|||
| Administration Time | 48 h | ||||
| Description |
The IC50 of QCCD@NPs and QSSD@NPs (389.05±75.25 nM and 245.47±37.54 nM) on B16 cells seemed much higher than the values of RCCD@NPs and RSSD@NPs (102.33±38.92 nM and 21.38±4.32 nM). The cytotoxicity of APDC@NPs on HUVEC showed a similar pattern.
|
||||
| In Vitro Model | Melanoma | B16 cell | CVCL_F936 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [8] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
371.53 ± 94.49 nM
|
|||
| Administration Time | 48 h | ||||
| Description |
The IC50 of QCCD@NPs and QSSD@NPs (389.05±75.25 nM and 245.47±37.54 nM) on B16 cells seemed much higher than the values of RCCD@NPs and RSSD@NPs (102.33±38.92 nM and 21.38±4.32 nM). The cytotoxicity of APDC@NPs on HUVEC showed a similar pattern.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
Tubugi-1-NPY [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
205 ± 49 nM
|
|||
| Description |
As shown in Table 1, the cytotoxic potency of the tubugi-1-SH was - in case of HT-29 and PC-3 - by factors 5 to 8 higher compared to the entire peptide-toxin conjugate 8. The only slight increase of cytotoxic activity of compound 9 compared to the complete conjugate 8 in Colo320 cells is most likely caused by a generally weak responsiveness of Colo320 cells towards tubugi-1-SH and the entire conjugate tubugi-1-SS-NPY. When compared with HT-29 and PC-3 cells, the IC50 value of tubugi-1-SH is by factor 10 higher in Colo320. Since the membrane passage of tubugi-1-SH is not depending on a NPY receptor, there have to be other explanations for the reduced cytotoxic impact of tubugi-1 and corresponding derivatives in Colo320, rather than the NPY Y1 receptor expression level.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
452 ± 60 nM
|
|||
| Description |
As shown in Table 1, the cytotoxic potency of the tubugi-1-SH was - in case of HT-29 and PC-3 - by factors 5 to 8 higher compared to the entire peptide-toxin conjugate 8. The only slight increase of cytotoxic activity of compound 9 compared to the complete conjugate 8 in Colo320 cells is most likely caused by a generally weak responsiveness of Colo320 cells towards tubugi-1-SH and the entire conjugate tubugi-1-SS-NPY. When compared with HT-29 and PC-3 cells, the IC50 value of tubugi-1-SH is by factor 10 higher in Colo320. Since the membrane passage of tubugi-1-SH is not depending on a NPY receptor, there have to be other explanations for the reduced cytotoxic impact of tubugi-1 and corresponding derivatives in Colo320, rather than the NPY Y1 receptor expression level.
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
| Experiment 3 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
706 ± 185 nM
|
|||
| Description |
As shown in Table 1, the cytotoxic potency of the tubugi-1-SH was - in case of HT-29 and PC-3 - by factors 5 to 8 higher compared to the entire peptide-toxin conjugate 8. The only slight increase of cytotoxic activity of compound 9 compared to the complete conjugate 8 in Colo320 cells is most likely caused by a generally weak responsiveness of Colo320 cells towards tubugi-1-SH and the entire conjugate tubugi-1-SS-NPY. When compared with HT-29 and PC-3 cells, the IC50 value of tubugi-1-SH is by factor 10 higher in Colo320. Since the membrane passage of tubugi-1-SH is not depending on a NPY receptor, there have to be other explanations for the reduced cytotoxic impact of tubugi-1 and corresponding derivatives in Colo320, rather than the NPY Y1 receptor expression level.
Click to Show/Hide
|
||||
| In Vitro Model | Colon adenocarcinoma | COLO 320 cell | CVCL_1989 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
0%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 10 µM | ||||
| Description |
The 72 h treatment is more effective than the 6 h pulse treatment. Notably, in vitro antitumor activities of 8 were found to correlate very good with the hY1R expression levels, as detected by gene expression analyses using RT-qPCR. Both the cytotoxic activity and the hY1R expression level rank in the order SK-N-MC > MDA-MB-468 > MDA-MB-231 > 184B5, what proofs the hY1R-specific and -selective nature of the mode of antitumor action of the designed PDC 8. Importantly, the activity of 8 against the selected normal breast cell line 184B5 is in the same order of magnitude as for the hY1R-deficient tumor cell line (MDA-MB-231), both tested at even higher concentration of the PDC than for the Y1 cell lines.
Click to Show/Hide
|
||||
| In Vitro Model | Askin tumor | SK-N-MC cell | CVCL_0530 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
2%
|
|||
| Administration Time | 6 h | ||||
| Administration Dosage | 10 µM | ||||
| Description |
The 72 h treatment is more effective than the 6 h pulse treatment. Notably, in vitro antitumor activities of 8 were found to correlate very good with the hY1R expression levels, as detected by gene expression analyses using RT-qPCR. Both the cytotoxic activity and the hY1R expression level rank in the order SK-N-MC > MDA-MB-468 > MDA-MB-231 > 184B5, what proofs the hY1R-specific and -selective nature of the mode of antitumor action of the designed PDC 8. Importantly, the activity of 8 against the selected normal breast cell line 184B5 is in the same order of magnitude as for the hY1R-deficient tumor cell line (MDA-MB-231), both tested at even higher concentration of the PDC than for the Y1 cell lines.
Click to Show/Hide
|
||||
| In Vitro Model | Askin tumor | SK-N-MC cell | CVCL_0530 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
5%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 10 µM | ||||
| Description |
The 72 h treatment is more effective than the 6 h pulse treatment. Notably, in vitro antitumor activities of 8 were found to correlate very good with the hY1R expression levels, as detected by gene expression analyses using RT-qPCR. Both the cytotoxic activity and the hY1R expression level rank in the order SK-N-MC > MDA-MB-468 > MDA-MB-231 > 184B5, what proofs the hY1R-specific and -selective nature of the mode of antitumor action of the designed PDC 8. Importantly, the activity of 8 against the selected normal breast cell line 184B5 is in the same order of magnitude as for the hY1R-deficient tumor cell line (MDA-MB-231), both tested at even higher concentration of the PDC than for the Y1 cell lines.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-468 cell | CVCL_0419 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
10%
|
|||
| Administration Time | 6 h | ||||
| Administration Dosage | 10 µM | ||||
| Description |
The 72 h treatment is more effective than the 6 h pulse treatment. Notably, in vitro antitumor activities of 8 were found to correlate very good with the hY1R expression levels, as detected by gene expression analyses using RT-qPCR. Both the cytotoxic activity and the hY1R expression level rank in the order SK-N-MC > MDA-MB-468 > MDA-MB-231 > 184B5, what proofs the hY1R-specific and -selective nature of the mode of antitumor action of the designed PDC 8. Importantly, the activity of 8 against the selected normal breast cell line 184B5 is in the same order of magnitude as for the hY1R-deficient tumor cell line (MDA-MB-231), both tested at even higher concentration of the PDC than for the Y1 cell lines.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-468 cell | CVCL_0419 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
20%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 10 µM | ||||
| Description |
The 72 h treatment is more effective than the 6 h pulse treatment. Notably, in vitro antitumor activities of 8 were found to correlate very good with the hY1R expression levels, as detected by gene expression analyses using RT-qPCR. Both the cytotoxic activity and the hY1R expression level rank in the order SK-N-MC > MDA-MB-468 > MDA-MB-231 > 184B5, what proofs the hY1R-specific and -selective nature of the mode of antitumor action of the designed PDC 8. Importantly, the activity of 8 against the selected normal breast cell line 184B5 is in the same order of magnitude as for the hY1R-deficient tumor cell line (MDA-MB-231), both tested at even higher concentration of the PDC than for the Y1 cell lines.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
20%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 10 µM | ||||
| Description |
The 72 h treatment is more effective than the 6 h pulse treatment. Notably, in vitro antitumor activities of 8 were found to correlate very good with the hY1R expression levels, as detected by gene expression analyses using RT-qPCR. Both the cytotoxic activity and the hY1R expression level rank in the order SK-N-MC > MDA-MB-468 > MDA-MB-231 > 184B5, what proofs the hY1R-specific and -selective nature of the mode of antitumor action of the designed PDC 8. Importantly, the activity of 8 against the selected normal breast cell line 184B5 is in the same order of magnitude as for the hY1R-deficient tumor cell line (MDA-MB-231), both tested at even higher concentration of the PDC than for the Y1 cell lines.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Normal mammary gland epithelium | Homo sapiens | ||
| Experiment 10 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
35%
|
|||
| Administration Time | 6 h | ||||
| Administration Dosage | 10 µM | ||||
| Description |
The 72 h treatment is more effective than the 6 h pulse treatment. Notably, in vitro antitumor activities of 8 were found to correlate very good with the hY1R expression levels, as detected by gene expression analyses using RT-qPCR. Both the cytotoxic activity and the hY1R expression level rank in the order SK-N-MC > MDA-MB-468 > MDA-MB-231 > 184B5, what proofs the hY1R-specific and -selective nature of the mode of antitumor action of the designed PDC 8. Importantly, the activity of 8 against the selected normal breast cell line 184B5 is in the same order of magnitude as for the hY1R-deficient tumor cell line (MDA-MB-231), both tested at even higher concentration of the PDC than for the Y1 cell lines.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [9] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
50%
|
|||
| Administration Time | 6 h | ||||
| Administration Dosage | 10 µM | ||||
| Description |
The 72 h treatment is more effective than the 6 h pulse treatment. Notably, in vitro antitumor activities of 8 were found to correlate very good with the hY1R expression levels, as detected by gene expression analyses using RT-qPCR. Both the cytotoxic activity and the hY1R expression level rank in the order SK-N-MC > MDA-MB-468 > MDA-MB-231 > 184B5, what proofs the hY1R-specific and -selective nature of the mode of antitumor action of the designed PDC 8. Importantly, the activity of 8 against the selected normal breast cell line 184B5 is in the same order of magnitude as for the hY1R-deficient tumor cell line (MDA-MB-231), both tested at even higher concentration of the PDC than for the Y1 cell lines.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | Normal mammary gland epithelium | Homo sapiens | ||
Cytolysin[F7,P34]-NPY bioconjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [10] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
236.1 nM
|
|||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-468 cell | CVCL_0419 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [10] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
405.3 nM
|
|||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
LWJ-M30 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [11] | ||||
| Indication | Colorectal cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
0.047 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
Given these findings, we employed tissue-specific drug delivery approaches PDC to overcome those adverse effects. Several TfR-targeted peptides sequence with different affinity have been reported, so we synthesized a series of TfR-targeted peptide-DM1 conjugates for screening the effects of TfR-targeted drug candidates. It is speculated that the conjugates can specifically target tumor with high expression of TfR and can be uptake more than that of the normal cells. We succeeded found LWJ-M30, a conjugate of DM1 and B6, had a significant therapeutic effect after preliminary screening. In order to further reveal the improved therapeutic effects and mechanism of the LWJ-M30 on cancer with high TfR expression, in this study, we investigated the role of the LWJ-M30 in the targeted therapy for colorectal cancer in vitro and in vivo and explored the therapeutic mechanism.
Click to Show/Hide
|
||||
| Description |
To determine the activity of the compounds, we analyzed the effects of them on cell growth and migration in HCT116 cells. We found that LWJ-M30 displayed significant inhibition of cell proliferation and clone formation ability. The IC50 of LWJ-M30 was 0.047 uM which was close to DM1 (0.026 uM). According to the results of wound-healing assay and transwell migration assay, LWJ-M30 also dramatically showed a inhibitory effect on cells migration. These results suggested that LWJ-M30 could inhibit the cells proliferation and migration.
Click to Show/Hide
|
||||
| In Vitro Model | Colon carcinoma | HCT 116 cell | CVCL_0291 | ||
PDC-Z11 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
0.41 ± 0.13 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.93 ± 1.41 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
10.25 ± 1.34 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
12.93 ± 1.15 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
GE11-DOX conjugate [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | Hepatocellular carcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
0.87 µM
|
|||
| In Vitro Model | Hepatocellular carcinoma | SMMC-7721 cell | CVCL_0534 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.66 µM
|
|||
| In Vitro Model | Invasive breast carcinoma of no special type | EGFR-overexpressing MCF-7 cell | CVCL_0031 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [12] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
32.2µM
|
|||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
PDC-Z4 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.37 ± 0.42 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.45 ± 1.09 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
13.91 ± 2.61 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
28.25 ± 3.12 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
PDC-Z5 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.44 ± 0.63 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
7.83 ± 1.77 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
17.84 ± 3.14 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
20.36 ± 1.82 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
PDC-Z12 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
1.76 ± 0.33 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
6.12 ± 0.45 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
13.21 ± 1.15 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
15.25 ± 2.08 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
PDC-Z2 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.37 ± 0.32 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
7.25 ± 1.75 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
19.10 ± 2.94 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
24.45 ± 2.04 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
Peptide-drug conjugate 10 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Neuroblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.47 ± 0.91 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | CellTiter-Glo luminescent cell viability assay | ||||
| Description |
The cytotoxic effect of the peptide-drug conjugate on Kelly-WT cells was comparable to that of 9, but not as strong as the one of the native drug. Contrary to the other utilized substances, the antiproliferative action of the bioconjugate in wild-type and drug-resistant cells was nearly the same. In comparison with doxorubicin the cytotoxicity of 10 against Kelly-ADR cells was increased by a factor of 3 according to the obtained IC50 values. Even the unmodified dimer had roughly the same cytotoxic effect on Kelly-ADR cells as doxorubicin.
Click to Show/Hide
|
||||
| In Vitro Model | Neuroblastoma | Kelly-WT cell | CVCL_2092 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Neuroblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
2.76 ± 0.33 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | CellTiter-Glo luminescent cell viability assay | ||||
| Description |
The cytotoxic effect of the peptide-drug conjugate on Kelly-WT cells was comparable to that of 9, but not as strong as the one of the native drug. Contrary to the other utilized substances, the antiproliferative action of the bioconjugate in wild-type and drug-resistant cells was nearly the same. In comparison with doxorubicin the cytotoxicity of 10 against Kelly-ADR cells was increased by a factor of 3 according to the obtained IC50 values. Even the unmodified dimer had roughly the same cytotoxic effect on Kelly-ADR cells as doxorubicin.
Click to Show/Hide
|
||||
| In Vitro Model | Neuroblastoma | Kelly-ADR cell | CVCL_2092 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [13] | ||||
| Indication | Neuroblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
3.66 ± 0.77 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | CellTiter-Glo luminescent cell viability assay | ||||
| Description |
The obtained IC50 values for 9 (6.73 ± 2.44 uM) and 10 (3.66 ± 0.77 uM) were higher than for doxorubicin (0.90 ± 0.12 uM).
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
TATE-SS-Ama [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [14] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) |
5.0 ±0.6 μM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTS assay | ||||
| MOA of PDC |
Towards these ends, we report on the design of a modular and user-friendly approach to enable bio-orthogonal conjugation of synthetic amanitins to three different linkers that are elaborated to three potent peptide-amanitin conjugates that differ by their mode of toxin release, as defined by the linker. Octreotate (TATE-N3, 2) was chosen as it is a circulation-stable octapeptide somatostatin analog that agonizes somatostatin receptors (Kd &tide; 0.4 nM for sstr2). Targeting sstr2 has been successfully exploited for drug delivery, cancer imaging, and radiotherapy owing to the high levels of sstr2 expression on neuroendocrine tumors (e.g. carcinoids, pancreatic islet cell tumors, thyroid carcinomas, and small lung cancer to name a few). While this is the first report of an octreotate-amanitin conjugate, herein, octreotate serves as an exemplar to highlight the potential for developing peptide-amanitin conjugates in targeted applications.
Click to Show/Hide
|
||||
| Description |
With bioconjugates 5-7 in hand, we evaluated their cytotoxicity by assaying cell viability on an sstr2-positive rat pancreatic cancer Ar42J cells, using free amatoxins as controls. The MTS cell viability assay showed that all three PDCs were effective at reducing the viability of Ar42J cells in the low nM range, providing calculated EC50 values of 4.2, and 2.3 nM for conjugates 6 and 7, respectively; these displayed up to 1000-fold enhancement in apparent cytotoxicity compared to free amatoxins 3 and 4, both of which gave calculated EC50 values of &tide;2 μM. This points to successful targeting and intracellular accumulation of toxin inside cells through an sstr2-mediated uptake inside the cells. Further evidence supporting target-mediated uptake is the micromolar toxicity of bioconjugates to the sstr2-negative CHO cell line that is otherwise sensitive to non-targeted amanitin. In comparison to -amanitin and synthetic amatoxins 3 and 4, bioconjugates 5-7 are 7- to 14-fold less toxic to CHO cells (control cell line), likely owing to impaired uptake of octreotate-amanitin conjugates that must enter via diffusion mediated processes or other non-specific import mechanisms that are currently unknown. In addition, a blocking study was run in the presence of free TATE-N32, which led to the expected reduction in apparent toxicity.
Click to Show/Hide
|
||||
| In Vitro Model | Digestive system neoplasms | AR42J (SSTR2+) cell | CVCL_0143 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [14] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half maximal effective concentration (EC50) |
4.2 ±0.6 nM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTS assay | ||||
| MOA of PDC |
Towards these ends, we report on the design of a modular and user-friendly approach to enable bio-orthogonal conjugation of synthetic amanitins to three different linkers that are elaborated to three potent peptide-amanitin conjugates that differ by their mode of toxin release, as defined by the linker. Octreotate (TATE-N3, 2) was chosen as it is a circulation-stable octapeptide somatostatin analog that agonizes somatostatin receptors (Kd &tide; 0.4 nM for sstr2). Targeting sstr2 has been successfully exploited for drug delivery, cancer imaging, and radiotherapy owing to the high levels of sstr2 expression on neuroendocrine tumors (e.g. carcinoids, pancreatic islet cell tumors, thyroid carcinomas, and small lung cancer to name a few). While this is the first report of an octreotate-amanitin conjugate, herein, octreotate serves as an exemplar to highlight the potential for developing peptide-amanitin conjugates in targeted applications.
Click to Show/Hide
|
||||
| Description |
With bioconjugates 5-7 in hand, we evaluated their cytotoxicity by assaying cell viability on an sstr2-positive rat pancreatic cancer Ar42J cells, using free amatoxins as controls. The MTS cell viability assay showed that all three PDCs were effective at reducing the viability of Ar42J cells in the low nM range, providing calculated EC50 values of 4.2, and 2.3 nM for conjugates 6 and 7, respectively; these displayed up to 1000-fold enhancement in apparent cytotoxicity compared to free amatoxins 3 and 4, both of which gave calculated EC50 values of &tide;2 μM. This points to successful targeting and intracellular accumulation of toxin inside cells through an sstr2-mediated uptake inside the cells. Further evidence supporting target-mediated uptake is the micromolar toxicity of bioconjugates to the sstr2-negative CHO cell line that is otherwise sensitive to non-targeted amanitin. In comparison to -amanitin and synthetic amatoxins 3 and 4, bioconjugates 5-7 are 7- to 14-fold less toxic to CHO cells (control cell line), likely owing to impaired uptake of octreotate-amanitin conjugates that must enter via diffusion mediated processes or other non-specific import mechanisms that are currently unknown. In addition, a blocking study was run in the presence of free TATE-N32, which led to the expected reduction in apparent toxicity.
Click to Show/Hide
|
||||
| In Vitro Model | Digestive system neoplasms | AR42J (SSTR2+) cell | CVCL_0143 | ||
PDC-Z7 [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
5.96 ± 2.41 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | SK-BR-3 cell | CVCL_0033 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Gastric cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
10.23 ± 1.65 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Gastric tubular adenocarcinoma | NCI-N87 cell | CVCL_1603 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
15.44 ± 2.41 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [6] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
33.12 ± 3.84 µM
|
|||
| Administration Time | 48 h | ||||
| Evaluation Method | CCK-8 assay | ||||
| MOA of PDC |
In the previous study, we focused on the discovery of novel cell targeting peptides, leading to the identification of the preferred HER2-targeting peptide P1 (NPNWGRSWYNQRFK) derived from pertuzumab, with the Kd of 5.70 10-6 M. Here, we designed and synthesized a series of PDCs choosing Camptothecin (CPT) as the payload and P1 as the peptide carrier. We aimed to discover novel PDC molecules with potent inhibitory activity and selectivity targeting HER2 for effective cancer treatment. Conjugate Z8, which balanced the activity and the biosafety, showed good antiproliferative activity (IC50 = 1.91 ± 0.71 M), comparable to that of CPT (IC50 = 2.10 ± 1.34 M), against NCI-N87 cells. Moreover, subsequent verification tests indicated that Z8 operated through a mechanism similar to CPT, but the results from in vivo anti-tumor activity assay suggested that Z8 exhibited greater safety compared to CPT to some extent. These findings highlight Z8 as a promising candidate for further investigation in the treatment of HER2-positive tumors.
Click to Show/Hide
|
||||
| Description |
To evaluated the in vitro anti-tumor activity of the conjugates, HER2-positive SK-BR-3, NCI-N87, HER2-negative MDA-MB-231 and normal MCF-10A cells were selected. As shown in Table 2, nearly all conjugates exhibited obvious anti-proliferation activity against SK-BR-3 cells, while most of the conjugates, except for Z6 and Z9, demonstrated different degrees of inhibitory activity on NCI-N87 cells. Taken together, Z8 and Z11 exhibited superior anti-tumor activity. The antiproliferative activity of Z8 against NCI-N87 cells (IC50 = 1.91 ± 0.71 uM) was comparable to that of CPT (IC50 = 2.10 ± 1.34 uM), while the antiproliferative activity of Z11 against SK-BR-3 cells (IC50 = 0.41 ± 0.13 uM) was comparable to that of CPT (IC50 = 0.26 ± 0.06 uM). Additionally, the IC50 values of these two conjugates against MDA-MB-231 and MCF-10A cells were significantly increased, showing their ability to selectively inhibit tumor cells. Based on the above results, Z8 and Z11, which exhibited better activity and apparent selectivity, were selected for subsequent studies.
Click to Show/Hide
|
||||
| In Vitro Model | Normal | MCF-10A cell | CVCL_0598 | ||
BP9a-SS-DOX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [15] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
6.21 ± 1.12 µM
|
|||
| Evaluation Method | CCK-8 assay | ||||
| Description |
The BP9a-SS-DOX conjugate exhibited impressed antiproliferative activity against HepG2 cells with an IC50 value of 6.21 ± 1.12 uM which was lower than that of free DOX.
|
||||
| In Vitro Model | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | ||
DOXoxmCPP [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [16] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
11.4 μM
|
|||
| Description |
Doxorubicin binds with high affinity to DNA by intercalation, which leads to DNA damage and subsequent growth inhibition44,45. We performed MTT assays in order to determine the viability of the tumor cells and additionally to assess the proliferative potential of the cells after drug treatment. MCF-7 and HT-29 cells were incubated with doxorubicin and the peptide conjugates at various concentrations in the range of 0.1 and 50 μmfor 72 h (in a serum-containing medium), and the results were expressed as IC50-values (Table 1).
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [16] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
27 μM
|
|||
| Description |
Doxorubicin binds with high affinity to DNA by intercalation, which leads to DNA damage and subsequent growth inhibition44,45. We performed MTT assays in order to determine the viability of the tumor cells and additionally to assess the proliferative potential of the cells after drug treatment. MCF-7 and HT-29 cells were incubated with doxorubicin and the peptide conjugates at various concentrations in the range of 0.1 and 50 μmfor 72 h (in a serum-containing medium), and the results were expressed as IC50-values (Table 1).
Click to Show/Hide
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
DOXoxmCPPamph [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [16] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
19 μM
|
|||
| Description |
Doxorubicin binds with high affinity to DNA by intercalation, which leads to DNA damage and subsequent growth inhibition44,45. We performed MTT assays in order to determine the viability of the tumor cells and additionally to assess the proliferative potential of the cells after drug treatment. MCF-7 and HT-29 cells were incubated with doxorubicin and the peptide conjugates at various concentrations in the range of 0.1 and 50 μmfor 72 h (in a serum-containing medium), and the results were expressed as IC50-values (Table 1).
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
| Experiment 2 Reporting the Activity Data of This PDC | [16] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
24.7 μM
|
|||
| Description |
Doxorubicin binds with high affinity to DNA by intercalation, which leads to DNA damage and subsequent growth inhibition44,45. We performed MTT assays in order to determine the viability of the tumor cells and additionally to assess the proliferative potential of the cells after drug treatment. MCF-7 and HT-29 cells were incubated with doxorubicin and the peptide conjugates at various concentrations in the range of 0.1 and 50 μmfor 72 h (in a serum-containing medium), and the results were expressed as IC50-values (Table 1).
Click to Show/Hide
|
||||
| In Vitro Model | Colon cancer | HT29 cell | CVCL_A8EZ | ||
pHLIP-SS-DOX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [17] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 400 µM | |||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
pHLIP-S-S-DOX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [18] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
18%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 10 µM | ||||
| Evaluation Method | MTS assay | ||||
| Description |
HeLa cells treated with pHLIP-S-S-DOX at low pH showed significant toxicity (˜50% viability at the lowest concentration tested (0.16 μM pHLIP)) which, in this construct, corresponds to the DOX concentration. Cellular viability tracked inversely with increasing pHLIP conjugate concentration, peaking at ˜18% viability at 16 μM.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [18] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
20%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 1.3 µM | ||||
| Evaluation Method | MTS assay | ||||
| Description |
HeLa cells treated with pHLIP-S-S-DOX at low pH showed significant toxicity (˜50% viability at the lowest concentration tested (0.16 μM pHLIP)) which, in this construct, corresponds to the DOX concentration. Cellular viability tracked inversely with increasing pHLIP conjugate concentration, peaking at ˜18% viability at 13 μM.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [18] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
22%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 2.5 µM | ||||
| Evaluation Method | MTS assay | ||||
| Description |
HeLa cells treated with pHLIP-S-S-DOX at low pH showed significant toxicity (˜50% viability at the lowest concentration tested (0.16 μM pHLIP)) which, in this construct, corresponds to the DOX concentration. Cellular viability tracked inversely with increasing pHLIP conjugate concentration, peaking at ˜18% viability at 14 μM.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [18] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
22%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 5.0 µM | ||||
| Evaluation Method | MTS assay | ||||
| Description |
HeLa cells treated with pHLIP-S-S-DOX at low pH showed significant toxicity (˜50% viability at the lowest concentration tested (0.16 μM pHLIP)) which, in this construct, corresponds to the DOX concentration. Cellular viability tracked inversely with increasing pHLIP conjugate concentration, peaking at ˜18% viability at 15 μM.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [18] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
30%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 0.31 µM | ||||
| Evaluation Method | MTS assay | ||||
| Description |
The cytotoxicity data for the pHLIP-PAMAM-DOX conjugate mirrored quite closely that of the pHLIP-S-S-DOX conjugate, particularly at the higher concentrations (>1.25 μM). However, at lower pHLIP concentrations (0.16 μM-0.63 μM), the PAMAM conjugate exhibited higher cytotoxicity than the single DOX conjugate (˜up to 17% higher toxicity).
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [18] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
30%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 0.63 µM | ||||
| Evaluation Method | MTS assay | ||||
| Description |
The cytotoxicity data for the pHLIP-PAMAM-DOX conjugate mirrored quite closely that of the pHLIP-S-S-DOX conjugate, particularly at the higher concentrations (>1.25 μM). However, at lower pHLIP concentrations (0.16 μM-0.63 μM), the PAMAM conjugate exhibited higher cytotoxicity than the single DOX conjugate (˜up to 17% higher toxicity).
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [18] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
30%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 1.3 µM | ||||
| Evaluation Method | MTS assay | ||||
| Description |
The cytotoxicity data for the pHLIP-PAMAM-DOX conjugate mirrored quite closely that of the pHLIP-S-S-DOX conjugate, particularly at the higher concentrations (>1.25 μM). However, at lower pHLIP concentrations (0.16 μM-0.63 μM), the PAMAM conjugate exhibited higher cytotoxicity than the single DOX conjugate (˜up to 17% higher toxicity).
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [18] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
38%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 0.16 µM | ||||
| Evaluation Method | MTS assay | ||||
| Description |
The cytotoxicity data for the pHLIP-PAMAM-DOX conjugate mirrored quite closely that of the pHLIP-S-S-DOX conjugate, particularly at the higher concentrations (>1.25 μM). However, at lower pHLIP concentrations (0.16 μM-0.63 μM), the PAMAM conjugate exhibited higher cytotoxicity than the single DOX conjugate (˜up to 17% higher toxicity).
Click to Show/Hide
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [18] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
42%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 0.63 µM | ||||
| Evaluation Method | MTS assay | ||||
| Description |
HeLa cells treated with pHLIP-S-S-DOX at low pH showed significant toxicity (˜50% viability at the lowest concentration tested (0.16 μM pHLIP)) which, in this construct, corresponds to the DOX concentration. Cellular viability tracked inversely with increasing pHLIP conjugate concentration, peaking at ˜18% viability at 12 μM.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [18] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
45%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 0.31 µM | ||||
| Evaluation Method | MTS assay | ||||
| Description |
HeLa cells treated with pHLIP-S-S-DOX at low pH showed significant toxicity (˜50% viability at the lowest concentration tested (0.16 μM pHLIP)) which, in this construct, corresponds to the DOX concentration. Cellular viability tracked inversely with increasing pHLIP conjugate concentration, peaking at ˜18% viability at 11 μM.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [18] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
50%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 0.16 µM | ||||
| Evaluation Method | MTS assay | ||||
| Description |
HeLa cells treated with pHLIP-S-S-DOX at low pH showed significant toxicity (˜50% viability at the lowest concentration tested (0.16 μM pHLIP)) which, in this construct, corresponds to the DOX concentration. Cellular viability tracked inversely with increasing pHLIP conjugate concentration, peaking at ˜18% viability at 10 μM.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [18] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
90%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 0.63 µM | ||||
| Evaluation Method | MTS assay | ||||
| Description |
HeLa cells treated with pHLIP-S-S-DOX at low pH showed significant toxicity (˜50% viability at the lowest concentration tested (0.16 μM pHLIP)) which, in this construct, corresponds to the DOX concentration. Cellular viability tracked inversely with increasing pHLIP conjugate concentration, peaking at ˜18% viability at 19 μM.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [18] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
95%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 0.16 µM | ||||
| Evaluation Method | MTS assay | ||||
| Description |
HeLa cells treated with pHLIP-S-S-DOX at low pH showed significant toxicity (˜50% viability at the lowest concentration tested (0.16 μM pHLIP)) which, in this construct, corresponds to the DOX concentration. Cellular viability tracked inversely with increasing pHLIP conjugate concentration, peaking at ˜18% viability at 17 μM.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [18] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
98%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 0.31 µM | ||||
| Evaluation Method | MTS assay | ||||
| Description |
HeLa cells treated with pHLIP-S-S-DOX at low pH showed significant toxicity (˜50% viability at the lowest concentration tested (0.16 μM pHLIP)) which, in this construct, corresponds to the DOX concentration. Cellular viability tracked inversely with increasing pHLIP conjugate concentration, peaking at ˜18% viability at 18 μM.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [18] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
99%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 10 µM | ||||
| Evaluation Method | MTS assay | ||||
| Description |
HeLa cells treated with pHLIP-S-S-DOX at low pH showed significant toxicity (˜50% viability at the lowest concentration tested (0.16 μM pHLIP)) which, in this construct, corresponds to the DOX concentration. Cellular viability tracked inversely with increasing pHLIP conjugate concentration, peaking at ˜18% viability at 23 μM.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [18] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
100%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 1.3 µM | ||||
| Evaluation Method | MTS assay | ||||
| Description |
HeLa cells treated with pHLIP-S-S-DOX at low pH showed significant toxicity (˜50% viability at the lowest concentration tested (0.16 μM pHLIP)) which, in this construct, corresponds to the DOX concentration. Cellular viability tracked inversely with increasing pHLIP conjugate concentration, peaking at ˜18% viability at 20 μM.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 17 Reporting the Activity Data of This PDC | [18] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
100%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 2.5 µM | ||||
| Evaluation Method | MTS assay | ||||
| Description |
HeLa cells treated with pHLIP-S-S-DOX at low pH showed significant toxicity (˜50% viability at the lowest concentration tested (0.16 μM pHLIP)) which, in this construct, corresponds to the DOX concentration. Cellular viability tracked inversely with increasing pHLIP conjugate concentration, peaking at ˜18% viability at 21 μM.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
| Experiment 18 Reporting the Activity Data of This PDC | [18] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Cell viability |
100%
|
|||
| Administration Time | 72 h | ||||
| Administration Dosage | 5.0 µM | ||||
| Evaluation Method | MTS assay | ||||
| Description |
HeLa cells treated with pHLIP-S-S-DOX at low pH showed significant toxicity (˜50% viability at the lowest concentration tested (0.16 μM pHLIP)) which, in this construct, corresponds to the DOX concentration. Cellular viability tracked inversely with increasing pHLIP conjugate concentration, peaking at ˜18% viability at 22 μM.
|
||||
| In Vitro Model | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | ||
References