
Peptide Information
General Information of This Peptide
| Peptide ID |
PEP00162
|
|||||
|---|---|---|---|---|---|---|
| Peptide Name |
Angiopep-2
|
|||||
| Structure |
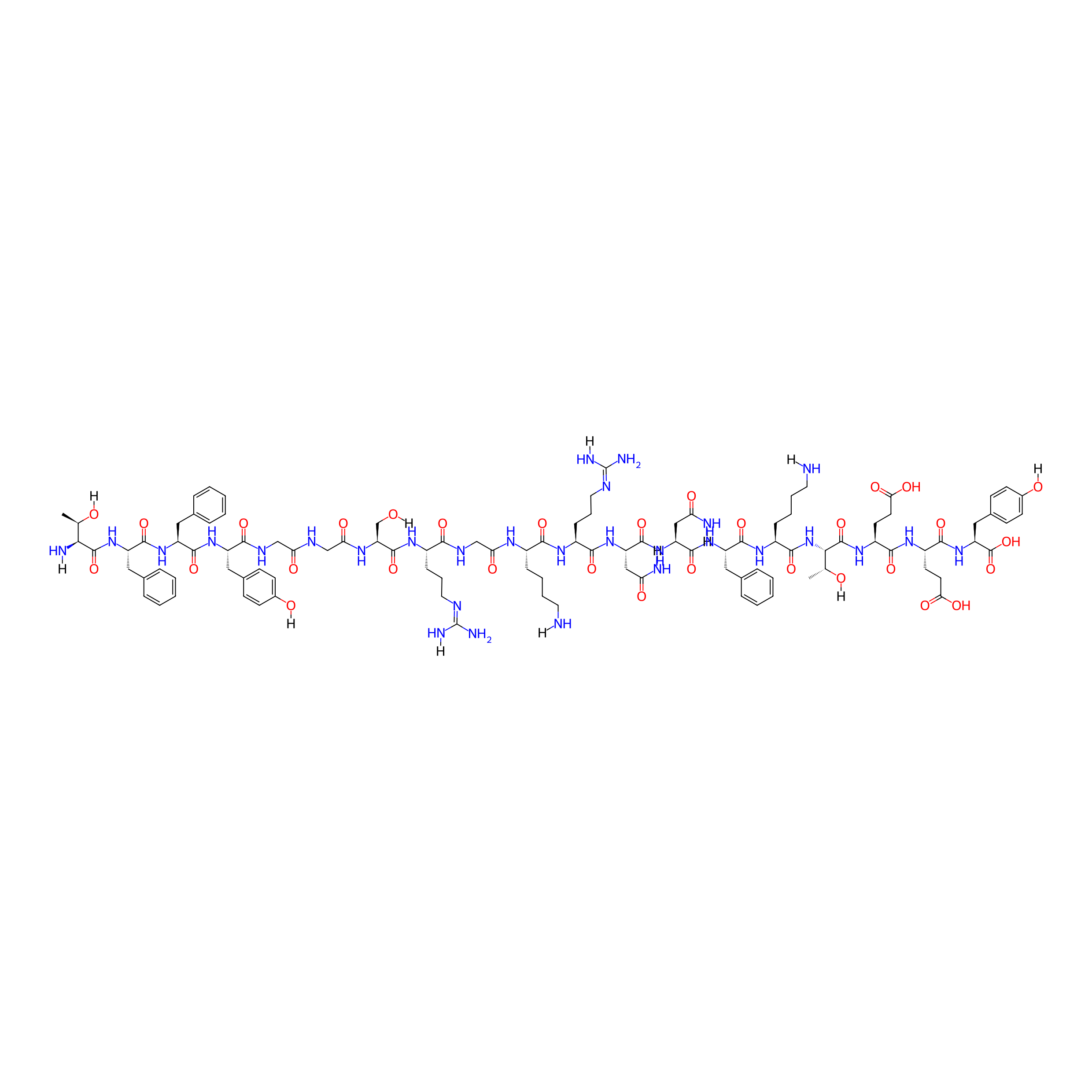
|
|||||
| Sequence |
TFFYGGSRGKRNNFKTEEY
|
|||||
| Peptide Type |
Linear
|
|||||
| Receptor Name |
Prolow-density lipoprotein receptor-related protein 1; Low-density lipoprotein receptor-related protein 2 (LRP1; LRP2)
|
Receptor Info | ||||
| PDC Transmembrane Types | Cell-penetrating peptides (CPPs) | |||||
| Formula |
C104H149N29O31
|
|||||
| Isosmiles |
[H]NCCCC[C@H](NC(=O)CNC(=O)[C@H](CCC/N=C(/N)N[H])NC(=O)[C@H](CO[H])NC(=O)CNC(=O)CNC(=O)[C@H](Cc1ccc(O[H])cc1)NC(=O)[C@H](Cc1ccccc1)NC(=O)[C@H](Cc1ccccc1)NC(=O)[C@@]([H])(N[H])[C@@H](C)O[H])C(=O)N[C@@H](CCC/N=C(/N)N[H])C(=O)N[C@@H](CC(=O)N[H])C(=O)N[C@@H](CC(=O)N[H])C(=O)N[C@@H](Cc1ccccc1)C(=O)N[C@@H](CCCCN[H])C(=O)N[C@]([H])(C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](CCC(=O)O)C(=O)N[C@@H](Cc1ccc(O[H])cc1)C(=O)O)[C@@H](C)O[H]
|
|||||
| InChI |
InChI=1S/C104H149N29O31/c1-55(135)85(109)100(161)131-73(46-59-22-10-5-11-23-59)96(157)127-72(45-58-20-8-4-9-21-58)95(156)126-70(47-60-28-32-62(137)33-29-60)88(149)117-51-80(141)116-52-81(142)120-77(54-134)99(160)121-64(26-16-42-114-103(110)111)87(148)118-53-82(143)119-65(24-12-14-40-105)89(150)122-67(27-17-43-115-104(112)113)90(151)129-74(49-78(107)139)98(159)130-75(50-79(108)140)97(158)128-71(44-57-18-6-3-7-19-57)94(155)123-66(25-13-15-41-106)93(154)133-86(56(2)136)101(162)125-69(37-39-84(146)147)91(152)124-68(36-38-83(144)145)92(153)132-76(102(163)164)48-61-30-34-63(138)35-31-61/h3-11,18-23,28-35,55-56,64-77,85-86,134-138H,12-17,24-27,36-54,105-106,109H2,1-2H3,(H2,107,139)(H2,108,140)(H,116,141)(H,117,149)(H,118,148)(H,119,143)(H,120,142)(H,121,160)(H,122,150)(H,123,155)(H,124,152)(H,125,162)(H,126,156)(H,127,157)(H,128,158)(H,129,151)(H,130,159)(H,131,161)(H,132,153)(H,133,154)(H,144,145)(H,146,147)(H,163,164)(H4,110,111,114)(H4,112,113,115)/t55-,56-,64+,65+,66+,67+,68+,69+,70+,71+,72+,73+,74+,75+,76+,77+,85+,86+/m1/s1
|
|||||
| InChIKey |
RCVVJXYDPKEONQ-JVFWYHQTSA-N
|
|||||
| Pharmaceutical Properties |
Molecule Weight
|
2301.508
|
Polar area
|
1029.89
|
||
|
Complexity
|
2300.097429
|
xlogp Value
|
-11.7569
|
|||
|
Heavy Count
|
164
|
Rot Bonds
|
84
|
|||
|
Hbond acc
|
33
|
Hbond Donor
|
35
|
|||
The Activity Data of This Peptide
| Peptide Activity Information 1 | [1] | |||||
| KD | 36.8 nM | |||||
|---|---|---|---|---|---|---|
| Binding Affinity Assay |
We therefore measured the binding kinetics of the peptide to LRP1 in the format of protein fusion.
|
|||||
| Experimental Condition | Low-density LRP1 | |||||
Each Peptide-drug Conjugate Related to This Peptide
Full Information of The Activity Data of The PDC(s) Related to This Peptide
ANG1005 [Phase 3]
Identified from the Human Clinical Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Objective response rate (ORR) |
15%
|
|||
| Patients Enrolled |
Adult patients with measurable recurrent brain metastases from breast cancer.
|
||||
| Administration Time | Every 3 weeks | ||||
| Administration Dosage | 600 mg/m2 | ||||
| MOA of PDC |
Because LRP1 is also expressed on tumor cells in both CNS and systemic metastases, ANG1005 gains entry via LRP1 mediated endocytosis, where paclitaxel is cleaved from the peptide backbone by lysosomal esterases.
|
||||
| Description |
On the basis of the CNS tumor response assessment, performed by local investigators, there were nine (15%) evaluable patients with PR including five (8%) confirmed PR (to confirm PR, it was required that the response was sustained for ≥4 weeks), and 32 (53%) evaluable patients with SD, resulting in an overall iORR of 15% and iCBR of 68%.
|
||||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Objective response rate (ORR) |
29%
|
|||
| Patients Enrolled |
Patients with leptomeningeal carcinomatosis.
|
||||
| Administration Time | Every 3 weeks | ||||
| Administration Dosage | 600 mg/m2 | ||||
| MOA of PDC |
Because LRP1 is also expressed on tumor cells in both CNS and systemic metastases, ANG1005 gains entry via LRP1 mediated endocytosis, where paclitaxel is cleaved from the peptide backbone by lysosomal esterases.
|
||||
| Description |
Investigator determined ORR was 29% and the iCBR was 67%.
|
||||
| Experiment 3 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
2.8 months
|
|||
| Patients Enrolled |
Patients with leptomeningeal carcinomatosis.
|
||||
| Administration Time | Every 3 weeks | ||||
| Administration Dosage | 600 mg/m2 | ||||
| MOA of PDC |
Because LRP1 is also expressed on tumor cells in both CNS and systemic metastases, ANG1005 gains entry via LRP1 mediated endocytosis, where paclitaxel is cleaved from the peptide backbone by lysosomal esterases.
|
||||
| Description |
The investigator determined intracranial median PFS was 2.8 months and the 3-month PFS rate was 54% (Table 3). Median duration of response was 18 weeks (7.3-26.3).
|
||||
| Experiment 4 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Median progression-free survival (mPFS) |
12.1 weeks
|
|||
| Patients Enrolled |
Adult patients with measurable recurrent brain metastases from breast cancer.
|
||||
| Administration Time | Every 3 weeks | ||||
| Administration Dosage | 600 mg/m2 | ||||
| MOA of PDC |
Because LRP1 is also expressed on tumor cells in both CNS and systemic metastases, ANG1005 gains entry via LRP1 mediated endocytosis, where paclitaxel is cleaved from the peptide backbone by lysosomal esterases.
|
||||
| Description |
Investigator assessments resulted in median intracranial PFS of 2.8 months and the 3-month intracranial PFS rate was 52%.
|
||||
| Experiment 5 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Median duration of response |
18 weeks
|
|||
| Patients Enrolled |
Patients with leptomeningeal carcinomatosis.
|
||||
| Administration Time | Every 3 weeks | ||||
| Administration Dosage | 600 mg/m2 | ||||
| MOA of PDC |
Because LRP1 is also expressed on tumor cells in both CNS and systemic metastases, ANG1005 gains entry via LRP1 mediated endocytosis, where paclitaxel is cleaved from the peptide backbone by lysosomal esterases.
|
||||
| Description |
The investigator determined intracranial median PFS was 2.8 months and the 3-month PFS rate was 54% (Table 3). Median duration of response was 18 weeks (7.3-26.3).
|
||||
| Experiment 6 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Clinical benefit rate (CBR) |
67%
|
|||
| Patients Enrolled |
Patients with leptomeningeal carcinomatosis.
|
||||
| Administration Time | Every 3 weeks | ||||
| Administration Dosage | 600 mg/m2 | ||||
| MOA of PDC |
Because LRP1 is also expressed on tumor cells in both CNS and systemic metastases, ANG1005 gains entry via LRP1 mediated endocytosis, where paclitaxel is cleaved from the peptide backbone by lysosomal esterases.
|
||||
| Description |
Investigator determined ORR was 29% and the iCBR was 67%.
|
||||
| Experiment 7 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Clinical benefit rate (CBR) |
68%
|
|||
| Patients Enrolled |
Adult patients with measurable recurrent brain metastases from breast cancer.
|
||||
| Administration Time | Every 3 weeks | ||||
| Administration Dosage | 600 mg/m2 | ||||
| MOA of PDC |
Because LRP1 is also expressed on tumor cells in both CNS and systemic metastases, ANG1005 gains entry via LRP1 mediated endocytosis, where paclitaxel is cleaved from the peptide backbone by lysosomal esterases.
|
||||
| Description |
On the basis of the CNS tumor response assessment, performed by local investigators, there were nine (15%) evaluable patients with PR including five (8%) confirmed PR (to confirm PR, it was required that the response was sustained for ≥4 weeks), and 32 (53%) evaluable patients with SD, resulting in an overall iORR of 15% and iCBR of 68%.
|
||||
| Experiment 8 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | 6-month progression-free survival rate |
18.70%
|
|||
| Patients Enrolled |
Adult patients with measurable recurrent brain metastases from breast cancer.
|
||||
| Administration Time | Every 3 weeks | ||||
| Administration Dosage | 600 mg/m2 | ||||
| MOA of PDC |
Because LRP1 is also expressed on tumor cells in both CNS and systemic metastases, ANG1005 gains entry via LRP1 mediated endocytosis, where paclitaxel is cleaved from the peptide backbone by lysosomal esterases.
|
||||
| Description |
Investigator assessments resulted in median intracranial PFS of 2.8 months and the 3-month intracranial PFS rate was 52%.
|
||||
| Experiment 9 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | 3-month progression-free survival rate |
52.00%
|
|||
| Patients Enrolled |
Adult patients with measurable recurrent brain metastases from breast cancer.
|
||||
| Administration Time | Every 3 weeks | ||||
| Administration Dosage | 600 mg/m2 | ||||
| MOA of PDC |
Because LRP1 is also expressed on tumor cells in both CNS and systemic metastases, ANG1005 gains entry via LRP1 mediated endocytosis, where paclitaxel is cleaved from the peptide backbone by lysosomal esterases.
|
||||
| Description |
Investigator assessments resulted in median intracranial PFS of 2.8 months and the 3-month intracranial PFS rate was 52%.
|
||||
| Experiment 10 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | 3-month progression-free survival rate |
54.00%
|
|||
| Patients Enrolled |
Patients with leptomeningeal carcinomatosis.
|
||||
| Administration Time | Every 3 weeks | ||||
| Administration Dosage | 600 mg/m2 | ||||
| MOA of PDC |
Because LRP1 is also expressed on tumor cells in both CNS and systemic metastases, ANG1005 gains entry via LRP1 mediated endocytosis, where paclitaxel is cleaved from the peptide backbone by lysosomal esterases.
|
||||
| Description |
The investigator determined intracranial median PFS was 2.8 months and the 3-month PFS rate was 54% (Table 3). Median duration of response was 18 weeks (7.3-26.3).
|
||||
An2-M6G [Preclinical]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Severe pain | ||||
| Efficacy Data | Maximal antinociceptive effect |
34.90%
|
|||
| Administration Time | Intravenous administration 60 min | ||||
| Administration Dosage | 4 mg/kg | ||||
| Evaluation Method | Rat tail-flick test assay | ||||
| MOA of PDC |
Given the high analgesic potency of M6G, without induction of the M3G metabolite that antagonizes the analgesic effect of morphine, M6G could be a promising drug to treat moderate to severe pain. The major issue of systemic use of M6G is its poor BBB permeability. In this study, we proposed to increase the BBB penetration of M6G and morphine by conjugation to the shuttle angiopep-2 peptide (An2). Morphine and M6G were first conjugated to An2, a 19-mer peptide that crosses the BBB by low-density lipoprotein receptor-related protein 1 (LRP1) receptor-mediated transcytosis.
Click to Show/Hide
|
||||
| Description |
Indeed, at an equimolar dose of 3 mg/kg of morphine (i.e., 12 mg/kg), An2-M6G produced a latency to tail withdrawal reaching the cutoff (i.e., 10 seconds) after 30 minutes, an effect lasting at least 3 hours. The %MPE calculated at 60 minutes after the intravenous injection of An2-M6G at 4, 8, and 12 mg/kg (equivalent to 1, 2, and 3 mg/kg of morphine and to 1.5, 3, and 4.5 mg/kg of M6G) was 34.9%, 66.2%, and 100%, respectively.
Click to Show/Hide
|
||||
| In Vivo Model | Rat model. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Severe pain | ||||
| Efficacy Data | Maximal antinociceptive effect |
66.20%
|
|||
| Administration Time | Intravenous administration 60 min | ||||
| Administration Dosage | 8 mg/kg | ||||
| Evaluation Method | Rat tail-flick test assay | ||||
| MOA of PDC |
Given the high analgesic potency of M6G, without induction of the M3G metabolite that antagonizes the analgesic effect of morphine, M6G could be a promising drug to treat moderate to severe pain. The major issue of systemic use of M6G is its poor BBB permeability. In this study, we proposed to increase the BBB penetration of M6G and morphine by conjugation to the shuttle angiopep-2 peptide (An2). Morphine and M6G were first conjugated to An2, a 19-mer peptide that crosses the BBB by low-density lipoprotein receptor-related protein 1 (LRP1) receptor-mediated transcytosis.
Click to Show/Hide
|
||||
| Description |
Indeed, at an equimolar dose of 3 mg/kg of morphine (i.e., 12 mg/kg), An2-M6G produced a latency to tail withdrawal reaching the cutoff (i.e., 10 seconds) after 30 minutes, an effect lasting at least 3 hours. The %MPE calculated at 60 minutes after the intravenous injection of An2-M6G at 4, 8, and 12 mg/kg (equivalent to 1, 2, and 3 mg/kg of morphine and to 1.5, 3, and 4.5 mg/kg of M6G) was 34.9%, 66.2%, and 100%, respectively.
Click to Show/Hide
|
||||
| In Vivo Model | Rat model. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Severe pain | ||||
| Efficacy Data | Maximal antinociceptive effect |
74%
|
|||
| Administration Time | Subcutaneous administration 200 min | ||||
| Administration Dosage | 12 mg/kg | ||||
| Evaluation Method | Rat tail-flick test assay | ||||
| MOA of PDC |
Given the high analgesic potency of M6G, without induction of the M3G metabolite that antagonizes the analgesic effect of morphine, M6G could be a promising drug to treat moderate to severe pain. The major issue of systemic use of M6G is its poor BBB permeability. In this study, we proposed to increase the BBB penetration of M6G and morphine by conjugation to the shuttle angiopep-2 peptide (An2). Morphine and M6G were first conjugated to An2, a 19-mer peptide that crosses the BBB by low-density lipoprotein receptor-related protein 1 (LRP1) receptor-mediated transcytosis.
Click to Show/Hide
|
||||
| Description |
We also measured the analgesic effect of An2-morphine and An2-M6G after subcutaneous injections. Despite similar MPE at the peak effect, subcutaneous injection of 20 mg/kg An2-morphine (equivalent to 5.5 mg/kg of morphine) produced an analgesic effect that was more prolonged over the time than what was observed with an equimolar dose of morphine.
|
||||
| In Vivo Model | Rat model. | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Severe pain | ||||
| Efficacy Data | Maximal antinociceptive effect |
100%
|
|||
| Administration Time | Intravenous administration 60 min | ||||
| Administration Dosage | 12 mg/kg | ||||
| Evaluation Method | Rat tail-flick test assay | ||||
| MOA of PDC |
Given the high analgesic potency of M6G, without induction of the M3G metabolite that antagonizes the analgesic effect of morphine, M6G could be a promising drug to treat moderate to severe pain. The major issue of systemic use of M6G is its poor BBB permeability. In this study, we proposed to increase the BBB penetration of M6G and morphine by conjugation to the shuttle angiopep-2 peptide (An2). Morphine and M6G were first conjugated to An2, a 19-mer peptide that crosses the BBB by low-density lipoprotein receptor-related protein 1 (LRP1) receptor-mediated transcytosis.
Click to Show/Hide
|
||||
| Description |
Indeed, at an equimolar dose of 3 mg/kg of morphine (i.e., 12 mg/kg), An2-M6G produced a latency to tail withdrawal reaching the cutoff (i.e., 10 seconds) after 30 minutes, an effect lasting at least 3 hours. The %MPE calculated at 60 minutes after the intravenous injection of An2-M6G at 4, 8, and 12 mg/kg (equivalent to 1, 2, and 3 mg/kg of morphine and to 1.5, 3, and 4.5 mg/kg of M6G) was 34.9%, 66.2%, and 100%, respectively.
Click to Show/Hide
|
||||
| In Vivo Model | Rat model. | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Severe pain | ||||
| Efficacy Data | Maximal antinociceptive effect |
100%
|
|||
| Administration Time | Intravenous administration 30 min | ||||
| Administration Dosage | 6 mg/kg | ||||
| Evaluation Method | Hot-plate test assay | ||||
| MOA of PDC |
Given the high analgesic potency of M6G, without induction of the M3G metabolite that antagonizes the analgesic effect of morphine, M6G could be a promising drug to treat moderate to severe pain. The major issue of systemic use of M6G is its poor BBB permeability. In this study, we proposed to increase the BBB penetration of M6G and morphine by conjugation to the shuttle angiopep-2 peptide (An2). Morphine and M6G were first conjugated to An2, a 19-mer peptide that crosses the BBB by low-density lipoprotein receptor-related protein 1 (LRP1) receptor-mediated transcytosis.
Click to Show/Hide
|
||||
| Description |
Similar results were also obtained in the hot-plate test using male CD1 mice. Over a 2-hour period, both morphine and An2-morphine caused similar increases in hot-plate latencies. Likewise, mice receiving An2-M6G (6 mg/kg i.v.) also exhibited a sustained and superior analgesic effect compared with equimolar doses of either morphine or M6G.
|
||||
| In Vivo Model | Male CD1 mice. | ||||
An2-morphine [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Severe pain | ||||
| Efficacy Data | Maximal antinociceptive effect |
46%
|
|||
| Administration Time | Intravenous administration 60 min | ||||
| Administration Dosage | 3 mg/kg | ||||
| Evaluation Method | Rat tail-flick test assay | ||||
| MOA of PDC |
Given the high analgesic potency of M6G, without induction of the M3G metabolite that antagonizes the analgesic effect of morphine, M6G could be a promising drug to treat moderate to severe pain. The major issue of systemic use of M6G is its poor BBB permeability. In this study, we proposed to increase the BBB penetration of M6G and morphine by conjugation to the shuttle angiopep-2 peptide (An2). Morphine and M6G were first conjugated to An2, a 19-mer peptide that crosses the BBB by low-density lipoprotein receptor-related protein 1 (LRP1) receptor-mediated transcytosis.
Click to Show/Hide
|
||||
| Description |
When equimolar doses of morphine conjugated to An2 (An2-morphine; 3 mg/kg) were compared with unconjugated morphine (1 mg/kg) at the peak effect, similar levels of antinociception were observed, reaching 46% and 49.7% of MPE, respectively.
|
||||
| In Vivo Model | Rat model. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Severe pain | ||||
| Efficacy Data | Maximal antinociceptive effect |
79%
|
|||
| Administration Time | Intravenous administration 30 min | ||||
| Administration Dosage | 30 mg/kg | ||||
| Evaluation Method | Hot-plate test assay | ||||
| MOA of PDC |
Given the high analgesic potency of M6G, without induction of the M3G metabolite that antagonizes the analgesic effect of morphine, M6G could be a promising drug to treat moderate to severe pain. The major issue of systemic use of M6G is its poor BBB permeability. In this study, we proposed to increase the BBB penetration of M6G and morphine by conjugation to the shuttle angiopep-2 peptide (An2). Morphine and M6G were first conjugated to An2, a 19-mer peptide that crosses the BBB by low-density lipoprotein receptor-related protein 1 (LRP1) receptor-mediated transcytosis.
Click to Show/Hide
|
||||
| Description |
Similar results were also obtained in the hot-plate test using male CD1 mice. Over a 2-hour period, both morphine and An2-morphine caused similar increases in hot-plate latencies. Likewise, mice receiving An2-M6G (6 mg/kg i.v.) also exhibited a sustained and superior analgesic effect compared with equimolar doses of either morphine or M6G.
|
||||
| In Vivo Model | Male CD1 mice. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Severe pain | ||||
| Efficacy Data | Maximal antinociceptive effect |
79%
|
|||
| Administration Time | Subcutaneous administration 95 min | ||||
| Administration Dosage | 20 mg/kg | ||||
| Evaluation Method | Rat tail-flick test assay | ||||
| MOA of PDC |
Given the high analgesic potency of M6G, without induction of the M3G metabolite that antagonizes the analgesic effect of morphine, M6G could be a promising drug to treat moderate to severe pain. The major issue of systemic use of M6G is its poor BBB permeability. In this study, we proposed to increase the BBB penetration of M6G and morphine by conjugation to the shuttle angiopep-2 peptide (An2). Morphine and M6G were first conjugated to An2, a 19-mer peptide that crosses the BBB by low-density lipoprotein receptor-related protein 1 (LRP1) receptor-mediated transcytosis.
Click to Show/Hide
|
||||
| Description |
We also measured the analgesic effect of An2-morphine and An2-M6G after subcutaneous injections. Despite similar MPE at the peak effect, subcutaneous injection of 20 mg/kg An2-morphine (equivalent to 5.5 mg/kg of morphine) produced an analgesic effect that was more prolonged over the time than what was observed with an equimolar dose of morphine.
|
||||
| In Vivo Model | Rat model. | ||||
ANG-PTX [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Central nervous system disease | ||||
| Efficacy Data | Inhinition rate |
33.21 ± 3.32%
|
|||
| Administration Time | 24 h | ||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Central nervous system disease | ||||
| Efficacy Data | Inhinition rate |
50.24 ± 4.75%
|
|||
| Administration Time | 48 h | ||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
Dau=Aoa-TFFYGGSRGK(Dau=Aoa)RNNFKTEEY-OH [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
7.8 ± 6.3 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The blood-brain barrier (BBB) is a semipermeable system, and, therefore, most of the active substances are poorly transported through this barrier, resulting in decreased therapeutic effects. Angiopep-2 (TFFYGGSRGKRNNFKTEEY) is a peptide ligand of low-density lipoprotein receptor-related protein-1 (LRP1), which can cross the BBB via receptor-mediated transcytosis and simultaneously target glioblastomas. Angiopep-2 contains three amino groups that have previously been used to produce drug-peptide conjugates, although the role and importance of each position have not yet been investigated. Thus, we studied the number and position of drug molecules in Angiopep-2 based conjugates. Conjugates containing one, two, and three daunomycin molecules conjugated via oxime linkage in all possible variations were prepared. The in vitro cytostatic effect and cellular uptake of the conjugates were investigated on U87 human glioblastoma cells. Degradation studies in the presence of rat liver lysosomal homogenates were also performed in order for us to better understand the structure-activity relationship and to determine the smallest metabolites. Conjugates with the best cytostatic effects had a drug molecule at the N-terminus. We demonstrated that the increasing number of drug molecules does not necessarily increase the efficacy of the conjugates, and proved that modification of the different conjugation sites results in differing biological effectiveness.
Click to Show/Hide
|
||||
| Description |
The in vitro cytostatic effect of the synthesized peptide (1) and daunomycin-peptide conjugates (2-8) was investigated on U87 human glioblastoma cells. The cells were treated with the peptide and conjugates at different concentrations (0.05-50 uM) for 24 h, and after a washing step, the cells were incubated for another 48 h at 37 C. The cytostatic effect of the compounds was determined using the MTT test. The measured IC50values are shown in. The free Angiopep-2 did not show any effect on tumor cells up to a 50 μM concentration. However, no clear correlation could be obtained between the number of the daunomycin and the cytostatic efficacy. Among the conjugates with only one Dau, compound3,in which Dau was attached to theN-terminus, showed the highest in vitro cytostatic effect on glioblastoma cells. When the Dau was conjugated to the Lys side chain in position 15 (compound2), the effect decreased significantly, and conjugate4was also not significantly better. In case of conjugates with two drug molecules, a similar tendency was observed. Compound7,with Dau at theN-terminus and on the Lys side chain in position 10, showed the highest activity. When the Lys side chains were used as conjugation sites, the formed compound (6) had moderate activity on U87 cells. Surprisingly, conjugate5,with Dau at theN-terminus and Lys side chain in position 15, showed the lowest efficacy on the U87 cells. Compound8,with three Dau, showed moderate activity. Thus, the effectiveness of Angiopep-2-daunomycin conjugates does not primarily depend on the number of drug molecules, but rather on their position within the molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
Dau=Aoa-TFFYGGSRGKRNNFKTEEY-OH [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
10.9 ± 2.8 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The blood-brain barrier (BBB) is a semipermeable system, and, therefore, most of the active substances are poorly transported through this barrier, resulting in decreased therapeutic effects. Angiopep-2 (TFFYGGSRGKRNNFKTEEY) is a peptide ligand of low-density lipoprotein receptor-related protein-1 (LRP1), which can cross the BBB via receptor-mediated transcytosis and simultaneously target glioblastomas. Angiopep-2 contains three amino groups that have previously been used to produce drug-peptide conjugates, although the role and importance of each position have not yet been investigated. Thus, we studied the number and position of drug molecules in Angiopep-2 based conjugates. Conjugates containing one, two, and three daunomycin molecules conjugated via oxime linkage in all possible variations were prepared. The in vitro cytostatic effect and cellular uptake of the conjugates were investigated on U87 human glioblastoma cells. Degradation studies in the presence of rat liver lysosomal homogenates were also performed in order for us to better understand the structure-activity relationship and to determine the smallest metabolites. Conjugates with the best cytostatic effects had a drug molecule at the N-terminus. We demonstrated that the increasing number of drug molecules does not necessarily increase the efficacy of the conjugates, and proved that modification of the different conjugation sites results in differing biological effectiveness.
Click to Show/Hide
|
||||
| Description |
The in vitro cytostatic effect of the synthesized peptide (1) and daunomycin-peptide conjugates (2-8) was investigated on U87 human glioblastoma cells. The cells were treated with the peptide and conjugates at different concentrations (0.05-50 uM) for 24 h, and after a washing step, the cells were incubated for another 48 h at 37 C. The cytostatic effect of the compounds was determined using the MTT test. The measured IC50values are shown in. The free Angiopep-2 did not show any effect on tumor cells up to a 50 μM concentration. However, no clear correlation could be obtained between the number of the daunomycin and the cytostatic efficacy. Among the conjugates with only one Dau, compound3,in which Dau was attached to theN-terminus, showed the highest in vitro cytostatic effect on glioblastoma cells. When the Dau was conjugated to the Lys side chain in position 15 (compound2), the effect decreased significantly, and conjugate4was also not significantly better. In case of conjugates with two drug molecules, a similar tendency was observed. Compound7,with Dau at theN-terminus and on the Lys side chain in position 10, showed the highest activity. When the Lys side chains were used as conjugation sites, the formed compound (6) had moderate activity on U87 cells. Surprisingly, conjugate5,with Dau at theN-terminus and Lys side chain in position 15, showed the lowest efficacy on the U87 cells. Compound8,with three Dau, showed moderate activity. Thus, the effectiveness of Angiopep-2-daunomycin conjugates does not primarily depend on the number of drug molecules, but rather on their position within the molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
H-TFFYGGSRGK(Dau=Aoa-GFLG)RNNFKTEEY-OH [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
16.9 ± 5.6 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The blood-brain barrier (BBB) is a semipermeable system, and, therefore, most of the active substances are poorly transported through this barrier, resulting in decreased therapeutic effects. Angiopep-2 (TFFYGGSRGKRNNFKTEEY) is a peptide ligand of low-density lipoprotein receptor-related protein-1 (LRP1), which can cross the BBB via receptor-mediated transcytosis and simultaneously target glioblastomas. Angiopep-2 contains three amino groups that have previously been used to produce drug-peptide conjugates, although the role and importance of each position have not yet been investigated. Thus, we studied the number and position of drug molecules in Angiopep-2 based conjugates. Conjugates containing one, two, and three daunomycin molecules conjugated via oxime linkage in all possible variations were prepared. The in vitro cytostatic effect and cellular uptake of the conjugates were investigated on U87 human glioblastoma cells. Degradation studies in the presence of rat liver lysosomal homogenates were also performed in order for us to better understand the structure-activity relationship and to determine the smallest metabolites. Conjugates with the best cytostatic effects had a drug molecule at the N-terminus. We demonstrated that the increasing number of drug molecules does not necessarily increase the efficacy of the conjugates, and proved that modification of the different conjugation sites results in differing biological effectiveness.
Click to Show/Hide
|
||||
| Description |
The in vitro cytostatic effect and the in vitro cellular uptake of the spacer-containing daunomycin-peptide conjugates (9-11) were studied on U87 human glioblastoma cells as previously described. The measured IC50 values are shown in Table 2, while the measured cellular uptake at 10 μM and 50 μM concentration are given in Figure 4. The fluorescence intensity values showed good correlation with the percentage of daunomycin-positive cells in the case of these conjugates as well. Conjugate 9 showed slight toxicity during the measurements at a concentration of 50 μM.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
H-TFFYGGSRGK(Dau=Aoa-VAGG)RNNFKTEEY-OH [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
20.2 ± 3.0 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The blood-brain barrier (BBB) is a semipermeable system, and, therefore, most of the active substances are poorly transported through this barrier, resulting in decreased therapeutic effects. Angiopep-2 (TFFYGGSRGKRNNFKTEEY) is a peptide ligand of low-density lipoprotein receptor-related protein-1 (LRP1), which can cross the BBB via receptor-mediated transcytosis and simultaneously target glioblastomas. Angiopep-2 contains three amino groups that have previously been used to produce drug-peptide conjugates, although the role and importance of each position have not yet been investigated. Thus, we studied the number and position of drug molecules in Angiopep-2 based conjugates. Conjugates containing one, two, and three daunomycin molecules conjugated via oxime linkage in all possible variations were prepared. The in vitro cytostatic effect and cellular uptake of the conjugates were investigated on U87 human glioblastoma cells. Degradation studies in the presence of rat liver lysosomal homogenates were also performed in order for us to better understand the structure-activity relationship and to determine the smallest metabolites. Conjugates with the best cytostatic effects had a drug molecule at the N-terminus. We demonstrated that the increasing number of drug molecules does not necessarily increase the efficacy of the conjugates, and proved that modification of the different conjugation sites results in differing biological effectiveness.
Click to Show/Hide
|
||||
| Description |
The in vitro cytostatic effect and the in vitro cellular uptake of the spacer-containing daunomycin-peptide conjugates (9-11) were studied on U87 human glioblastoma cells as previously described. The measured IC50 values are shown in Table 2, while the measured cellular uptake at 10 μM and 50 μM concentration are given in Figure 4. The fluorescence intensity values showed good correlation with the percentage of daunomycin-positive cells in the case of these conjugates as well. Conjugate 9 showed slight toxicity during the measurements at a concentration of 50 μM.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
H-TFFYGGSRGK(Dau=Aoa)RNNFK(Dau=Aoa)TEEY-OH [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
21.6 ± 5.4 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The blood-brain barrier (BBB) is a semipermeable system, and, therefore, most of the active substances are poorly transported through this barrier, resulting in decreased therapeutic effects. Angiopep-2 (TFFYGGSRGKRNNFKTEEY) is a peptide ligand of low-density lipoprotein receptor-related protein-1 (LRP1), which can cross the BBB via receptor-mediated transcytosis and simultaneously target glioblastomas. Angiopep-2 contains three amino groups that have previously been used to produce drug-peptide conjugates, although the role and importance of each position have not yet been investigated. Thus, we studied the number and position of drug molecules in Angiopep-2 based conjugates. Conjugates containing one, two, and three daunomycin molecules conjugated via oxime linkage in all possible variations were prepared. The in vitro cytostatic effect and cellular uptake of the conjugates were investigated on U87 human glioblastoma cells. Degradation studies in the presence of rat liver lysosomal homogenates were also performed in order for us to better understand the structure-activity relationship and to determine the smallest metabolites. Conjugates with the best cytostatic effects had a drug molecule at the N-terminus. We demonstrated that the increasing number of drug molecules does not necessarily increase the efficacy of the conjugates, and proved that modification of the different conjugation sites results in differing biological effectiveness.
Click to Show/Hide
|
||||
| Description |
The in vitro cytostatic effect of the synthesized peptide (1) and daunomycin-peptide conjugates (2-8) was investigated on U87 human glioblastoma cells. The cells were treated with the peptide and conjugates at different concentrations (0.05-50 uM) for 24 h, and after a washing step, the cells were incubated for another 48 h at 37 C. The cytostatic effect of the compounds was determined using the MTT test. The measured IC50values are shown in. The free Angiopep-2 did not show any effect on tumor cells up to a 50 μM concentration. However, no clear correlation could be obtained between the number of the daunomycin and the cytostatic efficacy. Among the conjugates with only one Dau, compound3,in which Dau was attached to theN-terminus, showed the highest in vitro cytostatic effect on glioblastoma cells. When the Dau was conjugated to the Lys side chain in position 15 (compound2), the effect decreased significantly, and conjugate4was also not significantly better. In case of conjugates with two drug molecules, a similar tendency was observed. Compound7,with Dau at theN-terminus and on the Lys side chain in position 10, showed the highest activity. When the Lys side chains were used as conjugation sites, the formed compound (6) had moderate activity on U87 cells. Surprisingly, conjugate5,with Dau at theN-terminus and Lys side chain in position 15, showed the lowest efficacy on the U87 cells. Compound8,with three Dau, showed moderate activity. Thus, the effectiveness of Angiopep-2-daunomycin conjugates does not primarily depend on the number of drug molecules, but rather on their position within the molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
Dau=Aoa-TFFYGGSRGK(Dau=Aoa)RNNFK(Dau=Aoa)TEEY-OH [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
23.6 ± 6.3 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The blood-brain barrier (BBB) is a semipermeable system, and, therefore, most of the active substances are poorly transported through this barrier, resulting in decreased therapeutic effects. Angiopep-2 (TFFYGGSRGKRNNFKTEEY) is a peptide ligand of low-density lipoprotein receptor-related protein-1 (LRP1), which can cross the BBB via receptor-mediated transcytosis and simultaneously target glioblastomas. Angiopep-2 contains three amino groups that have previously been used to produce drug-peptide conjugates, although the role and importance of each position have not yet been investigated. Thus, we studied the number and position of drug molecules in Angiopep-2 based conjugates. Conjugates containing one, two, and three daunomycin molecules conjugated via oxime linkage in all possible variations were prepared. The in vitro cytostatic effect and cellular uptake of the conjugates were investigated on U87 human glioblastoma cells. Degradation studies in the presence of rat liver lysosomal homogenates were also performed in order for us to better understand the structure-activity relationship and to determine the smallest metabolites. Conjugates with the best cytostatic effects had a drug molecule at the N-terminus. We demonstrated that the increasing number of drug molecules does not necessarily increase the efficacy of the conjugates, and proved that modification of the different conjugation sites results in differing biological effectiveness.
Click to Show/Hide
|
||||
| Description |
The in vitro cytostatic effect of the synthesized peptide (1) and daunomycin-peptide conjugates (2-8) was investigated on U87 human glioblastoma cells. The cells were treated with the peptide and conjugates at different concentrations (0.05-50 uM) for 24 h, and after a washing step, the cells were incubated for another 48 h at 37 C. The cytostatic effect of the compounds was determined using the MTT test. The measured IC50values are shown in. The free Angiopep-2 did not show any effect on tumor cells up to a 50 μM concentration. However, no clear correlation could be obtained between the number of the daunomycin and the cytostatic efficacy. Among the conjugates with only one Dau, compound3,in which Dau was attached to theN-terminus, showed the highest in vitro cytostatic effect on glioblastoma cells. When the Dau was conjugated to the Lys side chain in position 15 (compound2), the effect decreased significantly, and conjugate4was also not significantly better. In case of conjugates with two drug molecules, a similar tendency was observed. Compound7,with Dau at theN-terminus and on the Lys side chain in position 10, showed the highest activity. When the Lys side chains were used as conjugation sites, the formed compound (6) had moderate activity on U87 cells. Surprisingly, conjugate5,with Dau at theN-terminus and Lys side chain in position 15, showed the lowest efficacy on the U87 cells. Compound8,with three Dau, showed moderate activity. Thus, the effectiveness of Angiopep-2-daunomycin conjugates does not primarily depend on the number of drug molecules, but rather on their position within the molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
H-TFFYGGSRGK(Dau=Aoa)RNNFKTEEY-OH [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
24.0 ± 5.9 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The blood-brain barrier (BBB) is a semipermeable system, and, therefore, most of the active substances are poorly transported through this barrier, resulting in decreased therapeutic effects. Angiopep-2 (TFFYGGSRGKRNNFKTEEY) is a peptide ligand of low-density lipoprotein receptor-related protein-1 (LRP1), which can cross the BBB via receptor-mediated transcytosis and simultaneously target glioblastomas. Angiopep-2 contains three amino groups that have previously been used to produce drug-peptide conjugates, although the role and importance of each position have not yet been investigated. Thus, we studied the number and position of drug molecules in Angiopep-2 based conjugates. Conjugates containing one, two, and three daunomycin molecules conjugated via oxime linkage in all possible variations were prepared. The in vitro cytostatic effect and cellular uptake of the conjugates were investigated on U87 human glioblastoma cells. Degradation studies in the presence of rat liver lysosomal homogenates were also performed in order for us to better understand the structure-activity relationship and to determine the smallest metabolites. Conjugates with the best cytostatic effects had a drug molecule at the N-terminus. We demonstrated that the increasing number of drug molecules does not necessarily increase the efficacy of the conjugates, and proved that modification of the different conjugation sites results in differing biological effectiveness.
Click to Show/Hide
|
||||
| Description |
The in vitro cytostatic effect of the synthesized peptide (1) and daunomycin-peptide conjugates (2-8) was investigated on U87 human glioblastoma cells. The cells were treated with the peptide and conjugates at different concentrations (0.05-50 uM) for 24 h, and after a washing step, the cells were incubated for another 48 h at 37 C. The cytostatic effect of the compounds was determined using the MTT test. The measured IC50values are shown in. The free Angiopep-2 did not show any effect on tumor cells up to a 50 μM concentration. However, no clear correlation could be obtained between the number of the daunomycin and the cytostatic efficacy. Among the conjugates with only one Dau, compound3,in which Dau was attached to theN-terminus, showed the highest in vitro cytostatic effect on glioblastoma cells. When the Dau was conjugated to the Lys side chain in position 15 (compound2), the effect decreased significantly, and conjugate4was also not significantly better. In case of conjugates with two drug molecules, a similar tendency was observed. Compound7,with Dau at theN-terminus and on the Lys side chain in position 10, showed the highest activity. When the Lys side chains were used as conjugation sites, the formed compound (6) had moderate activity on U87 cells. Surprisingly, conjugate5,with Dau at theN-terminus and Lys side chain in position 15, showed the lowest efficacy on the U87 cells. Compound8,with three Dau, showed moderate activity. Thus, the effectiveness of Angiopep-2-daunomycin conjugates does not primarily depend on the number of drug molecules, but rather on their position within the molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
H-TFFYGGSRGKRNNFK(Dau=Aoa)TEEY-OH [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
30.2 ± 6.4 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The blood-brain barrier (BBB) is a semipermeable system, and, therefore, most of the active substances are poorly transported through this barrier, resulting in decreased therapeutic effects. Angiopep-2 (TFFYGGSRGKRNNFKTEEY) is a peptide ligand of low-density lipoprotein receptor-related protein-1 (LRP1), which can cross the BBB via receptor-mediated transcytosis and simultaneously target glioblastomas. Angiopep-2 contains three amino groups that have previously been used to produce drug-peptide conjugates, although the role and importance of each position have not yet been investigated. Thus, we studied the number and position of drug molecules in Angiopep-2 based conjugates. Conjugates containing one, two, and three daunomycin molecules conjugated via oxime linkage in all possible variations were prepared. The in vitro cytostatic effect and cellular uptake of the conjugates were investigated on U87 human glioblastoma cells. Degradation studies in the presence of rat liver lysosomal homogenates were also performed in order for us to better understand the structure-activity relationship and to determine the smallest metabolites. Conjugates with the best cytostatic effects had a drug molecule at the N-terminus. We demonstrated that the increasing number of drug molecules does not necessarily increase the efficacy of the conjugates, and proved that modification of the different conjugation sites results in differing biological effectiveness.
Click to Show/Hide
|
||||
| Description |
The in vitro cytostatic effect of the synthesized peptide (1) and daunomycin-peptide conjugates (2-8) was investigated on U87 human glioblastoma cells. The cells were treated with the peptide and conjugates at different concentrations (0.05-50 uM) for 24 h, and after a washing step, the cells were incubated for another 48 h at 37 C. The cytostatic effect of the compounds was determined using the MTT test. The measured IC50values are shown in. The free Angiopep-2 did not show any effect on tumor cells up to a 50 μM concentration. However, no clear correlation could be obtained between the number of the daunomycin and the cytostatic efficacy. Among the conjugates with only one Dau, compound3,in which Dau was attached to theN-terminus, showed the highest in vitro cytostatic effect on glioblastoma cells. When the Dau was conjugated to the Lys side chain in position 15 (compound2), the effect decreased significantly, and conjugate4was also not significantly better. In case of conjugates with two drug molecules, a similar tendency was observed. Compound7,with Dau at theN-terminus and on the Lys side chain in position 10, showed the highest activity. When the Lys side chains were used as conjugation sites, the formed compound (6) had moderate activity on U87 cells. Surprisingly, conjugate5,with Dau at theN-terminus and Lys side chain in position 15, showed the lowest efficacy on the U87 cells. Compound8,with three Dau, showed moderate activity. Thus, the effectiveness of Angiopep-2-daunomycin conjugates does not primarily depend on the number of drug molecules, but rather on their position within the molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
Dau=Aoa-TFFYGGSRGKRNNFK(Dau=Aoa)TEEY-OH [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) |
32.3 ± 8.1 µM
|
|||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The blood-brain barrier (BBB) is a semipermeable system, and, therefore, most of the active substances are poorly transported through this barrier, resulting in decreased therapeutic effects. Angiopep-2 (TFFYGGSRGKRNNFKTEEY) is a peptide ligand of low-density lipoprotein receptor-related protein-1 (LRP1), which can cross the BBB via receptor-mediated transcytosis and simultaneously target glioblastomas. Angiopep-2 contains three amino groups that have previously been used to produce drug-peptide conjugates, although the role and importance of each position have not yet been investigated. Thus, we studied the number and position of drug molecules in Angiopep-2 based conjugates. Conjugates containing one, two, and three daunomycin molecules conjugated via oxime linkage in all possible variations were prepared. The in vitro cytostatic effect and cellular uptake of the conjugates were investigated on U87 human glioblastoma cells. Degradation studies in the presence of rat liver lysosomal homogenates were also performed in order for us to better understand the structure-activity relationship and to determine the smallest metabolites. Conjugates with the best cytostatic effects had a drug molecule at the N-terminus. We demonstrated that the increasing number of drug molecules does not necessarily increase the efficacy of the conjugates, and proved that modification of the different conjugation sites results in differing biological effectiveness.
Click to Show/Hide
|
||||
| Description |
The in vitro cytostatic effect of the synthesized peptide (1) and daunomycin-peptide conjugates (2-8) was investigated on U87 human glioblastoma cells. The cells were treated with the peptide and conjugates at different concentrations (0.05-50 uM) for 24 h, and after a washing step, the cells were incubated for another 48 h at 37 C. The cytostatic effect of the compounds was determined using the MTT test. The measured IC50values are shown in. The free Angiopep-2 did not show any effect on tumor cells up to a 50 μM concentration. However, no clear correlation could be obtained between the number of the daunomycin and the cytostatic efficacy. Among the conjugates with only one Dau, compound3,in which Dau was attached to theN-terminus, showed the highest in vitro cytostatic effect on glioblastoma cells. When the Dau was conjugated to the Lys side chain in position 15 (compound2), the effect decreased significantly, and conjugate4was also not significantly better. In case of conjugates with two drug molecules, a similar tendency was observed. Compound7,with Dau at theN-terminus and on the Lys side chain in position 10, showed the highest activity. When the Lys side chains were used as conjugation sites, the formed compound (6) had moderate activity on U87 cells. Surprisingly, conjugate5,with Dau at theN-terminus and Lys side chain in position 15, showed the lowest efficacy on the U87 cells. Compound8,with three Dau, showed moderate activity. Thus, the effectiveness of Angiopep-2-daunomycin conjugates does not primarily depend on the number of drug molecules, but rather on their position within the molecule.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
H-TFFYGGSRGK(Dau=Aoa-VA)RNNFKTEEY-OH [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 50 µM | |||
| Administration Time | 72 h | ||||
| Evaluation Method | MTT assay | ||||
| MOA of PDC |
The blood-brain barrier (BBB) is a semipermeable system, and, therefore, most of the active substances are poorly transported through this barrier, resulting in decreased therapeutic effects. Angiopep-2 (TFFYGGSRGKRNNFKTEEY) is a peptide ligand of low-density lipoprotein receptor-related protein-1 (LRP1), which can cross the BBB via receptor-mediated transcytosis and simultaneously target glioblastomas. Angiopep-2 contains three amino groups that have previously been used to produce drug-peptide conjugates, although the role and importance of each position have not yet been investigated. Thus, we studied the number and position of drug molecules in Angiopep-2 based conjugates. Conjugates containing one, two, and three daunomycin molecules conjugated via oxime linkage in all possible variations were prepared. The in vitro cytostatic effect and cellular uptake of the conjugates were investigated on U87 human glioblastoma cells. Degradation studies in the presence of rat liver lysosomal homogenates were also performed in order for us to better understand the structure-activity relationship and to determine the smallest metabolites. Conjugates with the best cytostatic effects had a drug molecule at the N-terminus. We demonstrated that the increasing number of drug molecules does not necessarily increase the efficacy of the conjugates, and proved that modification of the different conjugation sites results in differing biological effectiveness.
Click to Show/Hide
|
||||
| Description |
The in vitro cytostatic effect and the in vitro cellular uptake of the spacer-containing daunomycin-peptide conjugates (9-11) were studied on U87 human glioblastoma cells as previously described. The measured IC50 values are shown in Table 2, while the measured cellular uptake at 10 μM and 50 μM concentration are given in Figure 4. The fluorescence intensity values showed good correlation with the percentage of daunomycin-positive cells in the case of these conjugates as well. Conjugate 9 showed slight toxicity during the measurements at a concentration of 50 μM.
Click to Show/Hide
|
||||
| In Vitro Model | Glioblastoma | U87 cell | CVCL_3429 | ||
References