
Drug Information
General Information of This Drug
| Drug ID | DRG00013 | |||||
|---|---|---|---|---|---|---|
| Drug Name | Gemcitabine | |||||
| Synonyms |
gemcitabine; 95058-81-4; 2'-Deoxy-2',2'-difluorocytidine; 2',2'-Difluorodeoxycytidine; dFdC; Cytidine, 2'-deoxy-2',2'-difluoro-; Gemcitabine free base; 2',2'-difluoro-2'-deoxycytidine; 103882-84-4; 4-amino-1-[(2R,4R,5R)-3,3-difluoro-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-1,2-dihydropyrimidin-2-one; LY188011; 4-amino-1-((2R,4R,5R)-3,3-difluoro-4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)pyrimidin-2(1H)-one; 95058-81-4 (free base); B76N6SBZ8R; LY 188011; 4-Amino-1-[(2R,4R,5R)-3,3-difluoro-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidin-2-one; Gamcitabine; Gemcitabina; CHEBI:175901; DFdCyd; NSC-613327; Gemcitabinum; Folfugem; Gemcel; Zefei; GemLip; 4-amino-1-((2R,4R,5R)-3,3-difluoro-4-hydroxy-5-(hydroxymethyl)-tetrahydrofuran-2-yl)pyrimidin-2(1H)-one; Gemcitabinum [INN-Latin]; Gemcitabina [INN-Spanish]; C9H11F2N3O4; Gemzar (hydrochloride); SR-05000001491; Gemcitabine (USAN/INN); UNII-B76N6SBZ8R; 2',2'-DiF-dC; Gemcitabine [USAN:INN:BAN]; CCRIS 8984; MFCD00869720; HSDB 7567; 2vpp; gemcitabine (Gemzar); NSC 613327; 4pd5; GEMCITABINE [MI]; GEMCITABINE [INN]; 2'-Deoxy-.beta.-D-2',2'-difluorocytidine; GEMCITABINE [HSDB]; GEMCITABINE [USAN]; CHEMBL888; GEMCITABINE [VANDF]; SCHEMBL4295; GEMCITABINE [WHO-DD]; GTPL4793; DTXSID3040487; 2'deoxy-2',2'-difluorocytidine; BCPP000219; BDBM429521; GLXC-04598; HMS2089P10; HMS3715N07; 2`-Deoxy-2`,2`-difluorocytidine; med.21724, Compound Gemcitabine; DL-215; s1714; AKOS015920303; Cytidine, 2'-deoxy-2',2'-difluoro-2'-Deoxy-.beta.-D-2',2'-difluorocytidine; BCP9000721; CCG-221183; CS-0643; DB00441; GS-3582; 4-Amino-1-[3,3-difluoro-4-hydroxy-5-(hydroxymethyl) tetrahydrofuran-2-yl]-1H-pyrimidin-2-one; NCGC00168784-02; NCGC00168784-08; NCGC00168784-12; BP-58640; HY-17026; G0544; NS00000342; SW199649-2; C07650; D02368; EN300-267822; AB01274777-01; AB01274777-02; AB01274777_05; AB01274777_06; Q414143; J-001056; SR-05000001491-1; SR-05000001491-2; BRD-K15108141-001-04-1; Z1741982024; 4-AMINO-1-[(2R,4R,5R)-3,3-DIFLUORO-4-HYDROXY-5-(HYDROXYMETHYL)TETRAHYDRO-2-FURANYL]-2(1H)-PYRIMIDINONE; 4-amino-1-[(2R,4R,5R)-3,3-difluoro-4-hydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl]pyrimidin-2-one
Click to Show/Hide
|
|||||
| Target(s) | Ribonucleoside-diphosphate reductase subunit M2 (RRM2) | Target Info | ||||
| Structure |
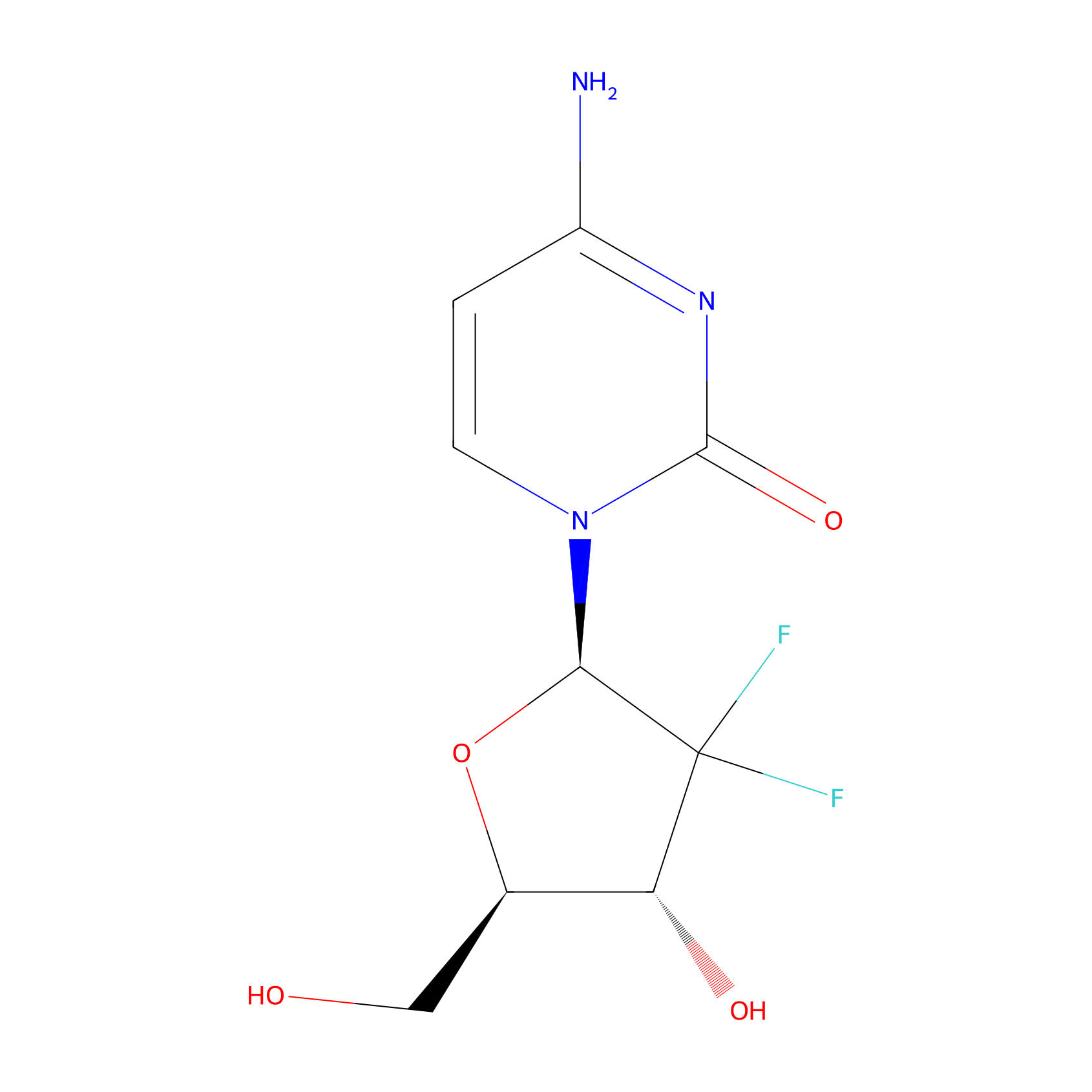
|
|||||
| Formula |
C9H11F2N3O4
|
|||||
| #Ro5 Violations (Lipinski): 0 | Molecular Weight (mw) | 263.2 | ||||
| Lipid-water partition coefficient (xlogp) | -1.5 | |||||
| Hydrogen Bond Donor Count (hbonddonor) | 3 | |||||
| Hydrogen Bond Acceptor Count (hbondacc) | 6 | |||||
| Rotatable Bond Count (rotbonds) | 2 | |||||
| PubChem CID | ||||||
| Canonical smiles |
C1=CN(C(=O)N=C1N)C2C(C(C(O2)CO)O)(F)F
|
|||||
| InChI |
InChI=1S/C9H11F2N3O4/c10-9(11)6(16)4(3-15)18-7(9)14-2-1-5(12)13-8(14)17/h1-2,4,6-7,15-16H,3H2,(H2,12,13,17)/t4-,6-,7-/m1/s1
|
|||||
| InChIKey |
SDUQYLNIPVEERB-QPPQHZFASA-N
|
|||||
| IUPAC Name |
4-amino-1-[(2R,4R,5R)-3,3-difluoro-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidin-2-one
|
|||||
The activity data of This Drug
| Standard Type | Value | Disease Model | Cell line | Cell line ID | Ref. | |
|---|---|---|---|---|---|---|
| Half Maximal Growth Inhibition (GI50) | 2 nM | Lung adenocarcinoma | NCI-H23 cell | CVCL_1547 | [1] | |
| Half Maximal Growth Inhibition (GI50) | 2.085 nM | Lung large cell carcinoma | NCI-H460 cell | CVCL_0459 | [2] | |
| Half Maximal Growth Inhibition (GI50) | 3 nM | Colon adenocarcinoma | HCT 15 cell | CVCL_0292 | [1] | |
| Half Maximal Growth Inhibition (GI50) | 4 nM | Breast ductal carcinoma | BT-549 cell | CVCL_1092 | [1] | |
| Half Maximal Growth Inhibition (GI50) | 6 nM | Prostate carcinoma | PC-3 cell | CVCL_0035 | [1] | |
| Half Maximal Growth Inhibition (GI50) | 10 nM | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | [3] | |
| Half Maximal Inhibitory Concentration (IC50) | 1.5 nM | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | [4] | |
| Half Maximal Inhibitory Concentration (IC50) | 2.6 nM | High grade ovarian serous adenocarcinoma | OVCAR-8 cell | CVCL_1629 | [5] | |
| Half Maximal Inhibitory Concentration (IC50) | 6 nM | Pancreatic ductal adenocarcinoma | BxPC-3 cell | CVCL_0186 | [6] | |
| Half Maximal Inhibitory Concentration (IC50) | 6 nM | Colon carcinoma | CT26 cell | CVCL_7254 | [7] | |
| Half Maximal Inhibitory Concentration (IC50) | 6 nM | Colon carcinoma | HCT 116 cell | CVCL_0291 | [8] | |
| Half Maximal Inhibitory Concentration (IC50) | 7 nM | Thymoma | EL4 cell | CVCL_0255 | [7] | |
| Half Maximal Inhibitory Concentration (IC50) | 7 nM | Lymphoblastic leukemia | L1210 cell | CVCL_0382 | [7] | |
| Half Maximal Inhibitory Concentration (IC50) | 8 nM | Breast ductal carcinoma | BT-549 cell | CVCL_1092 | [7] | |
| Half Maximal Inhibitory Concentration (IC50) | 9 nM | Lung small cell carcinoma | DMS-53 cell | CVCL_1177 | [9] | |
| Half Maximal Inhibitory Concentration (IC50) | 9.2 nM | Uterine sarcoma | MES-SA/Dx5 cell | CVCL_2598 | [10] | |
| Half Maximal Inhibitory Concentration (IC50) | 9.7 nM | Colon carcinoma | HCT 116 cell | CVCL_0291 | [5] | |
| Half Maximal Inhibitory Concentration (IC50) | 9.9 nM | Colon adenocarcinoma | HCT 15 cell | CVCL_0292 | [5] | |
| Half Maximal Inhibitory Concentration (IC50) | 10.3 nM | Astrocytoma | SF268 cell | CVCL_1689 | [5] | |
| Half Maximal Inhibitory Concentration (IC50) | 11.4 nM | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | [11] | |
| Half Maximal Inhibitory Concentration (IC50) | 13.6 nM | Colon adenocarcinoma | SW480 cell | CVCL_0546 | [5] | |
| Half Maximal Inhibitory Concentration (IC50) | 16 nM | Pancreatic ductal adenocarcinoma | MIA PaCa-2 cell | CVCL_0428 | [12] | |
| Half Maximal Inhibitory Concentration (IC50) | 16 nM | Lung carcinoma | SW1573 cell | CVCL_1720 | [4] | |
| Half Maximal Inhibitory Concentration (IC50) | 19 nM | Pancreatic ductal adenocarcinoma | Capan-1 cell | CVCL_0237 | [6] | |
| Half Maximal Inhibitory Concentration (IC50) | 19.8 nM | Gliosarcoma | SF539 cell | CVCL_1691 | [5] | |
| Half Maximal Inhibitory Concentration (IC50) | 20 nM | Lung adenocarcinoma | A-549 cell | CVCL_0023 | [9] | |
| Half Maximal Inhibitory Concentration (IC50) | 25 nM | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | [13] | |
| Half Maximal Inhibitory Concentration (IC50) | 30 nM | Colon carcinoma | HCT 116 cell | CVCL_0291 | [14] | |
| Half Maximal Inhibitory Concentration (IC50) | 35 nM | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | [8] | |
| Half Maximal Inhibitory Concentration (IC50) | 40 nM | Prostate carcinoma | PC-3 cell | CVCL_0035 | [15] | |
| Half Maximal Inhibitory Concentration (IC50) | 47 nM | Squamous carcinoma | SCC-25 cell | CVCL_1682 | [16] | |
| Half Maximal Inhibitory Concentration (IC50) | 50 nM | Lung adenocarcinoma | A-549 cell | CVCL_0023 | [17] | |
| Half Maximal Inhibitory Concentration (IC50) | 50 nM | Chronic myeloid leukemia | K562 cell | CVCL_0004 | [18] | |
| Half Maximal Inhibitory Concentration (IC50) | 51.4 nM | Normal | COLO205 cell | CVCL_F402 | [5] | |
| Half Maximal Inhibitory Concentration (IC50) | 70 nM | Acute monocytic leukemia | U-937 cell | CVCL_0007 | [19] | |
| Half Maximal Inhibitory Concentration (IC50) | 90.7 nM | Osteosarcoma | U2OS cell | CVCL_0042 | [5] | |
| Half Maximal Inhibitory Concentration (IC50) | 100 nM | Chronic myeloid leukemia | K562 cell | CVCL_0004 | [20] | |
| Half Maximal Inhibitory Concentration (IC50) | 120 nM | Pancreatic ductal adenocarcinoma | MIA PaCa-2 cell | CVCL_0428 | [21] | |
| Half Maximal Inhibitory Concentration (IC50) | 150 nM | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | [22] | |
| Half Maximal Inhibitory Concentration (IC50) | 180 nM | Osteosarcoma | U2OS cell | CVCL_0042 | [20] | |
| Half Maximal Inhibitory Concentration (IC50) | 275 nM | Lung carcinoma | SW1573 cell | CVCL_1720 | [4] | |
| Half Maximal Inhibitory Concentration (IC50) | 310 nM | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | [23] | |
| Half Maximal Inhibitory Concentration (IC50) | 410 nM | Colon carcinoma | HCT 116 cell | CVCL_0291 | [20] | |
| Half Maximal Inhibitory Concentration (IC50) | 512 nM | Prostate carcinoma | LNCaP cell | CVCL_0395 | [7] | |
| Half Maximal Inhibitory Concentration (IC50) | 570 nM | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | [24] | |
| Half Maximal Inhibitory Concentration (IC50) | 718 nM | Chronic myeloid leukemia | K562 cell | CVCL_0004 | [7] | |
| Half Maximal Inhibitory Concentration (IC50) | 840 nM | Hepatoma | Bel-7402 cell | CVCL_5492 | [23] | |
| Half Maximal Inhibitory Concentration (IC50) | 1000 nM | T acute lymphoblastic leukemia | CCRF-CEM cell | CVCL_0207 | [4] | |
| Half Maximal Inhibitory Concentration (IC50) | 1.1 uM | Neuroblastoma | SK-N-AS cell | CVCL_1700 | [7] | |
| Half Maximal Inhibitory Concentration (IC50) | 1.4 uM | Lung adenocarcinoma | A-549 cell | CVCL_0023 | [23] | |
| Half Maximal Inhibitory Concentration (IC50) | 2.1 uM | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | [25] | |
| Half Maximal Inhibitory Concentration (IC50) | 3 uM | Normal | COLO205 cell | CVCL_F402 | [8] | |
| Half Maximal Inhibitory Concentration (IC50) | 3.28 uM | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | [23] | |
| Half Maximal Inhibitory Concentration (IC50) | 4.12 uM | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | [7] | |
| Half Maximal Inhibitory Concentration (IC50) | 4.9 uM | Hepatoblastoma | Hep-G2 cell | CVCL_0027 | [26] | |
| Half Maximal Inhibitory Concentration (IC50) | 5.05 uM | Lung adenocarcinoma | A-549 cell | CVCL_0023 | [26] | |
| Half Maximal Inhibitory Concentration (IC50) | >10 uM | Endocervical adenocarcinoma | HeLa cell | CVCL_0030 | [8] | |
| Half Maximal Inhibitory Concentration (IC50) | 40.791 uM | Pancreatic ductal adenocarcinoma | Capan-2 cell | CVCL_0026 | [12] | |
| Half Maximal Inhibitory Concentration (IC50) | 50 uM | T acute lymphoblastic leukemia | CCRF-CEM cell | CVCL_0207 | [4] | |
| Half Maximal Inhibitory Concentration (IC50) | >50 uM | Normal | BJ cell | CVCL_E483 | [27] | |
| Tumor Growth Inhibition value (TGI) | 100 uM | Pancreatic ductal adenocarcinoma | MIA PaCa-2 cell | CVCL_0428 | [28] | |
Each Peptide-drug Conjugate Related to This Drug
Full Information of The Activity Data of The PDC(s) Related to This Drug
GOXG2 [Investigative]
Identified from the Human Clinical Data
| Experiment 1 Reporting the Activity Data of This PDC | [29] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Relative intensity | 5% | |||
| Administration Time | 60 min | ||||
| Administration Dosage | 1 μg/ml | ||||
| MOA of PDC |
We aimed to construct PDCs with linker controllable drug release rates simply by manipulating the linker unit. For a more rapid drug-release rate we developed GOXG1 and GOXG2. These conjugates bear a carboxylate ester linker directly attached to the primary and the secondary alcohol group of the drug respectively, followed by oxime and amide bond. The primary alcohol of gemcitabine has been used since it is involved in the phosphorylation process, through which gemcitabine exerts its cytotoxic effect. Therefore, we aimed to block the primary alcohol and examine its effect (GOXG1) and also take advantage of the secondary alcohol which could lead to a PDC with a completely different profile (GOXG2), although they share structural similarities. For a slower drug release rate, we designed and developed the PDC GN4OXG that contains an amide bond on the 4-N position of the parent drug followed by click oxime ligation and another amide bond. The stability of this molecule should be enhanced since it is devoid of rapidly hydrolyzable ester bonds. Furthermore, in this PDC since the 4-NH2 moiety of gemcitabine is capped it could further surmount the rapid gemcitabine metabolism that leads to the formation of dFdU, after the enzymatic 4-N deamination of gemcitabine by cytidine deaminase (CDA).
Click to Show/Hide
|
||||
| Description |
GOXG2 was the least stable, followed by GOXG1, while GN4OXG was the most stable.
|
||||
| In Vivo Model | Human plasma. | ||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [29] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Relative intensity | 0% | |||
| Administration Time | 10 h | ||||
| Administration Dosage | 1 μM | ||||
| MOA of PDC |
We aimed to construct PDCs with linker controllable drug release rates simply by manipulating the linker unit. For a more rapid drug-release rate we developed GOXG1 and GOXG2. These conjugates bear a carboxylate ester linker directly attached to the primary and the secondary alcohol group of the drug respectively, followed by oxime and amide bond. The primary alcohol of gemcitabine has been used since it is involved in the phosphorylation process, through which gemcitabine exerts its cytotoxic effect. Therefore, we aimed to block the primary alcohol and examine its effect (GOXG1) and also take advantage of the secondary alcohol which could lead to a PDC with a completely different profile (GOXG2), although they share structural similarities. For a slower drug release rate, we designed and developed the PDC GN4OXG that contains an amide bond on the 4-N position of the parent drug followed by click oxime ligation and another amide bond. The stability of this molecule should be enhanced since it is devoid of rapidly hydrolyzable ester bonds. Furthermore, in this PDC since the 4-NH2 moiety of gemcitabine is capped it could further surmount the rapid gemcitabine metabolism that leads to the formation of dFdU, after the enzymatic 4-N deamination of gemcitabine by cytidine deaminase (CDA).
Click to Show/Hide
|
||||
| Description |
This experiment showed that the more labile pro-drug was GOXG2, which was almost fully degraded in less than 10 hours while GN4OXG and GOXG1 showed similar, enhanced stability compared to GOXG2.
|
||||
| In Vitro Model | Prostate carcinoma | DU145 cell | CVCL_0105 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [29] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 494 ± 93 nM | |||
| MOA of PDC |
We aimed to construct PDCs with linker controllable drug release rates simply by manipulating the linker unit. For a more rapid drug-release rate we developed GOXG1 and GOXG2. These conjugates bear a carboxylate ester linker directly attached to the primary and the secondary alcohol group of the drug respectively, followed by oxime and amide bond. The primary alcohol of gemcitabine has been used since it is involved in the phosphorylation process, through which gemcitabine exerts its cytotoxic effect. Therefore, we aimed to block the primary alcohol and examine its effect (GOXG1) and also take advantage of the secondary alcohol which could lead to a PDC with a completely different profile (GOXG2), although they share structural similarities. For a slower drug release rate, we designed and developed the PDC GN4OXG that contains an amide bond on the 4-N position of the parent drug followed by click oxime ligation and another amide bond. The stability of this molecule should be enhanced since it is devoid of rapidly hydrolyzable ester bonds. Furthermore, in this PDC since the 4-NH2 moiety of gemcitabine is capped it could further surmount the rapid gemcitabine metabolism that leads to the formation of dFdU, after the enzymatic 4-N deamination of gemcitabine by cytidine deaminase (CDA).
Click to Show/Hide
|
||||
| Description |
The most potent among them was found to be pro-drug GOXG2 with IC50 494 ± 93 nM and 675 ± 82 nM against DU145 and PC3, respectively. Regarding the other two conjugates, GN4OXG was found to be the next most cytotoxic compound against DU145 with IC50 590 ± 62 nM, followed by the least toxic GOXG1 with IC50 611 ± 80 nM. In contrast, GOXG1 was the second more toxic against PC3 with IC50 754 ± 142 nM, followed by the least toxic GN4OXG which showed IC50 833 ± 27 nM. The results are summarized in Table 2.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | DU145 cell | CVCL_0105 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [29] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 675 ± 82 nM | |||
| MOA of PDC |
We aimed to construct PDCs with linker controllable drug release rates simply by manipulating the linker unit. For a more rapid drug-release rate we developed GOXG1 and GOXG2. These conjugates bear a carboxylate ester linker directly attached to the primary and the secondary alcohol group of the drug respectively, followed by oxime and amide bond. The primary alcohol of gemcitabine has been used since it is involved in the phosphorylation process, through which gemcitabine exerts its cytotoxic effect. Therefore, we aimed to block the primary alcohol and examine its effect (GOXG1) and also take advantage of the secondary alcohol which could lead to a PDC with a completely different profile (GOXG2), although they share structural similarities. For a slower drug release rate, we designed and developed the PDC GN4OXG that contains an amide bond on the 4-N position of the parent drug followed by click oxime ligation and another amide bond. The stability of this molecule should be enhanced since it is devoid of rapidly hydrolyzable ester bonds. Furthermore, in this PDC since the 4-NH2 moiety of gemcitabine is capped it could further surmount the rapid gemcitabine metabolism that leads to the formation of dFdU, after the enzymatic 4-N deamination of gemcitabine by cytidine deaminase (CDA).
Click to Show/Hide
|
||||
| Description |
The most potent among them was found to be pro-drug GOXG2 with IC50 494 ± 93 nM and 675 ± 82 nM against DU145 and PC3, respectively. Regarding the other two conjugates, GN4OXG was found to be the next most cytotoxic compound against DU145 with IC50 590 ± 62 nM, followed by the least toxic GOXG1 with IC50 611 ± 80 nM. In contrast, GOXG1 was the second more toxic against PC3 with IC50 754 ± 142 nM, followed by the least toxic GN4OXG which showed IC50 833 ± 27 nM. The results are summarized in Table 2.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
GOXG1 [Investigative]
Identified from the Human Clinical Data
| Experiment 1 Reporting the Activity Data of This PDC | [29] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Relative intensity | 30% | |||
| Administration Time | 60 min | ||||
| Administration Dosage | 1 μg/ml | ||||
| MOA of PDC |
We aimed to construct PDCs with linker controllable drug release rates simply by manipulating the linker unit. For a more rapid drug-release rate we developed GOXG1 and GOXG2. These conjugates bear a carboxylate ester linker directly attached to the primary and the secondary alcohol group of the drug respectively, followed by oxime and amide bond. The primary alcohol of gemcitabine has been used since it is involved in the phosphorylation process, through which gemcitabine exerts its cytotoxic effect. Therefore, we aimed to block the primary alcohol and examine its effect (GOXG1) and also take advantage of the secondary alcohol which could lead to a PDC with a completely different profile (GOXG2), although they share structural similarities. For a slower drug release rate, we designed and developed the PDC GN4OXG that contains an amide bond on the 4-N position of the parent drug followed by click oxime ligation and another amide bond. The stability of this molecule should be enhanced since it is devoid of rapidly hydrolyzable ester bonds. Furthermore, in this PDC since the 4-NH2 moiety of gemcitabine is capped it could further surmount the rapid gemcitabine metabolism that leads to the formation of dFdU, after the enzymatic 4-N deamination of gemcitabine by cytidine deaminase (CDA).
Click to Show/Hide
|
||||
| Description |
GOXG2 was the least stable, followed by GOXG1, while GN4OXG was the most stable.
|
||||
| In Vivo Model | Human plasma. | ||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [29] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Relative intensity | 10% | |||
| Administration Time | 10 h | ||||
| Administration Dosage | 1 μM | ||||
| MOA of PDC |
We aimed to construct PDCs with linker controllable drug release rates simply by manipulating the linker unit. For a more rapid drug-release rate we developed GOXG1 and GOXG2. These conjugates bear a carboxylate ester linker directly attached to the primary and the secondary alcohol group of the drug respectively, followed by oxime and amide bond. The primary alcohol of gemcitabine has been used since it is involved in the phosphorylation process, through which gemcitabine exerts its cytotoxic effect. Therefore, we aimed to block the primary alcohol and examine its effect (GOXG1) and also take advantage of the secondary alcohol which could lead to a PDC with a completely different profile (GOXG2), although they share structural similarities. For a slower drug release rate, we designed and developed the PDC GN4OXG that contains an amide bond on the 4-N position of the parent drug followed by click oxime ligation and another amide bond. The stability of this molecule should be enhanced since it is devoid of rapidly hydrolyzable ester bonds. Furthermore, in this PDC since the 4-NH2 moiety of gemcitabine is capped it could further surmount the rapid gemcitabine metabolism that leads to the formation of dFdU, after the enzymatic 4-N deamination of gemcitabine by cytidine deaminase (CDA).
Click to Show/Hide
|
||||
| Description |
This experiment showed that the more labile pro-drug was GOXG2, which was almost fully degraded in less than 10 hours while GN4OXG and GOXG1 showed similar, enhanced stability compared to GOXG2.
|
||||
| In Vitro Model | Prostate carcinoma | DU145 cell | CVCL_0105 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [29] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 611 ± 80 nM | |||
| MOA of PDC |
We aimed to construct PDCs with linker controllable drug release rates simply by manipulating the linker unit. For a more rapid drug-release rate we developed GOXG1 and GOXG2. These conjugates bear a carboxylate ester linker directly attached to the primary and the secondary alcohol group of the drug respectively, followed by oxime and amide bond. The primary alcohol of gemcitabine has been used since it is involved in the phosphorylation process, through which gemcitabine exerts its cytotoxic effect. Therefore, we aimed to block the primary alcohol and examine its effect (GOXG1) and also take advantage of the secondary alcohol which could lead to a PDC with a completely different profile (GOXG2), although they share structural similarities. For a slower drug release rate, we designed and developed the PDC GN4OXG that contains an amide bond on the 4-N position of the parent drug followed by click oxime ligation and another amide bond. The stability of this molecule should be enhanced since it is devoid of rapidly hydrolyzable ester bonds. Furthermore, in this PDC since the 4-NH2 moiety of gemcitabine is capped it could further surmount the rapid gemcitabine metabolism that leads to the formation of dFdU, after the enzymatic 4-N deamination of gemcitabine by cytidine deaminase (CDA).
Click to Show/Hide
|
||||
| Description |
The most potent among them was found to be pro-drug GOXG2 with IC50 494 ± 93 nM and 675 ± 82 nM against DU145 and PC3, respectively. Regarding the other two conjugates, GN4OXG was found to be the next most cytotoxic compound against DU145 with IC50 590 ± 62 nM, followed by the least toxic GOXG1 with IC50 611 ± 80 nM. In contrast, GOXG1 was the second more toxic against PC3 with IC50 754 ± 142 nM, followed by the least toxic GN4OXG which showed IC50 833 ± 27 nM. The results are summarized in Table 2.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | DU145 cell | CVCL_0105 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [29] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 754 ± 142 nM | |||
| MOA of PDC |
We aimed to construct PDCs with linker controllable drug release rates simply by manipulating the linker unit. For a more rapid drug-release rate we developed GOXG1 and GOXG2. These conjugates bear a carboxylate ester linker directly attached to the primary and the secondary alcohol group of the drug respectively, followed by oxime and amide bond. The primary alcohol of gemcitabine has been used since it is involved in the phosphorylation process, through which gemcitabine exerts its cytotoxic effect. Therefore, we aimed to block the primary alcohol and examine its effect (GOXG1) and also take advantage of the secondary alcohol which could lead to a PDC with a completely different profile (GOXG2), although they share structural similarities. For a slower drug release rate, we designed and developed the PDC GN4OXG that contains an amide bond on the 4-N position of the parent drug followed by click oxime ligation and another amide bond. The stability of this molecule should be enhanced since it is devoid of rapidly hydrolyzable ester bonds. Furthermore, in this PDC since the 4-NH2 moiety of gemcitabine is capped it could further surmount the rapid gemcitabine metabolism that leads to the formation of dFdU, after the enzymatic 4-N deamination of gemcitabine by cytidine deaminase (CDA).
Click to Show/Hide
|
||||
| Description |
The most potent among them was found to be pro-drug GOXG2 with IC50 494 ± 93 nM and 675 ± 82 nM against DU145 and PC3, respectively. Regarding the other two conjugates, GN4OXG was found to be the next most cytotoxic compound against DU145 with IC50 590 ± 62 nM, followed by the least toxic GOXG1 with IC50 611 ± 80 nM. In contrast, GOXG1 was the second more toxic against PC3 with IC50 754 ± 142 nM, followed by the least toxic GN4OXG which showed IC50 833 ± 27 nM. The results are summarized in Table 2.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
GN4OXG [Investigative]
Identified from the Human Clinical Data
| Experiment 1 Reporting the Activity Data of This PDC | [29] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Relative intensity | 75% | |||
| Administration Time | 60 min | ||||
| Administration Dosage | 1 μg/ml | ||||
| MOA of PDC |
We aimed to construct PDCs with linker controllable drug release rates simply by manipulating the linker unit. For a more rapid drug-release rate we developed GOXG1 and GOXG2. These conjugates bear a carboxylate ester linker directly attached to the primary and the secondary alcohol group of the drug respectively, followed by oxime and amide bond. The primary alcohol of gemcitabine has been used since it is involved in the phosphorylation process, through which gemcitabine exerts its cytotoxic effect. Therefore, we aimed to block the primary alcohol and examine its effect (GOXG1) and also take advantage of the secondary alcohol which could lead to a PDC with a completely different profile (GOXG2), although they share structural similarities. For a slower drug release rate, we designed and developed the PDC GN4OXG that contains an amide bond on the 4-N position of the parent drug followed by click oxime ligation and another amide bond. The stability of this molecule should be enhanced since it is devoid of rapidly hydrolyzable ester bonds. Furthermore, in this PDC since the 4-NH2 moiety of gemcitabine is capped it could further surmount the rapid gemcitabine metabolism that leads to the formation of dFdU, after the enzymatic 4-N deamination of gemcitabine by cytidine deaminase (CDA).
Click to Show/Hide
|
||||
| Description |
GOXG2 was the least stable, followed by GOXG1, while GN4OXG was the most stable.
|
||||
| In Vivo Model | Human plasma. | ||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [29] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Relative intensity | 12% | |||
| Administration Time | 10 h | ||||
| Administration Dosage | 1 μM | ||||
| MOA of PDC |
We aimed to construct PDCs with linker controllable drug release rates simply by manipulating the linker unit. For a more rapid drug-release rate we developed GOXG1 and GOXG2. These conjugates bear a carboxylate ester linker directly attached to the primary and the secondary alcohol group of the drug respectively, followed by oxime and amide bond. The primary alcohol of gemcitabine has been used since it is involved in the phosphorylation process, through which gemcitabine exerts its cytotoxic effect. Therefore, we aimed to block the primary alcohol and examine its effect (GOXG1) and also take advantage of the secondary alcohol which could lead to a PDC with a completely different profile (GOXG2), although they share structural similarities. For a slower drug release rate, we designed and developed the PDC GN4OXG that contains an amide bond on the 4-N position of the parent drug followed by click oxime ligation and another amide bond. The stability of this molecule should be enhanced since it is devoid of rapidly hydrolyzable ester bonds. Furthermore, in this PDC since the 4-NH2 moiety of gemcitabine is capped it could further surmount the rapid gemcitabine metabolism that leads to the formation of dFdU, after the enzymatic 4-N deamination of gemcitabine by cytidine deaminase (CDA).
Click to Show/Hide
|
||||
| Description |
This experiment showed that the more labile pro-drug was GOXG2, which was almost fully degraded in less than 10 hours while GN4OXG and GOXG1 showed similar, enhanced stability compared to GOXG2.
|
||||
| In Vitro Model | Prostate carcinoma | DU145 cell | CVCL_0105 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [29] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 590 ± 62 nM | |||
| MOA of PDC |
We aimed to construct PDCs with linker controllable drug release rates simply by manipulating the linker unit. For a more rapid drug-release rate we developed GOXG1 and GOXG2. These conjugates bear a carboxylate ester linker directly attached to the primary and the secondary alcohol group of the drug respectively, followed by oxime and amide bond. The primary alcohol of gemcitabine has been used since it is involved in the phosphorylation process, through which gemcitabine exerts its cytotoxic effect. Therefore, we aimed to block the primary alcohol and examine its effect (GOXG1) and also take advantage of the secondary alcohol which could lead to a PDC with a completely different profile (GOXG2), although they share structural similarities. For a slower drug release rate, we designed and developed the PDC GN4OXG that contains an amide bond on the 4-N position of the parent drug followed by click oxime ligation and another amide bond. The stability of this molecule should be enhanced since it is devoid of rapidly hydrolyzable ester bonds. Furthermore, in this PDC since the 4-NH2 moiety of gemcitabine is capped it could further surmount the rapid gemcitabine metabolism that leads to the formation of dFdU, after the enzymatic 4-N deamination of gemcitabine by cytidine deaminase (CDA).
Click to Show/Hide
|
||||
| Description |
The most potent among them was found to be pro-drug GOXG2 with IC50 494 ± 93 nM and 675 ± 82 nM against DU145 and PC3, respectively. Regarding the other two conjugates, GN4OXG was found to be the next most cytotoxic compound against DU145 with IC50 590 ± 62 nM, followed by the least toxic GOXG1 with IC50 611 ± 80 nM. In contrast, GOXG1 was the second more toxic against PC3 with IC50 754 ± 142 nM, followed by the least toxic GN4OXG which showed IC50 833 ± 27 nM. The results are summarized in Table 2.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | DU145 cell | CVCL_0105 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [29] | ||||
| Indication | Prostate cancer | ||||
| Efficacy Data | Half maximal inhibitory concentration (IC50) | 833 ± 27 nM | |||
| MOA of PDC |
We aimed to construct PDCs with linker controllable drug release rates simply by manipulating the linker unit. For a more rapid drug-release rate we developed GOXG1 and GOXG2. These conjugates bear a carboxylate ester linker directly attached to the primary and the secondary alcohol group of the drug respectively, followed by oxime and amide bond. The primary alcohol of gemcitabine has been used since it is involved in the phosphorylation process, through which gemcitabine exerts its cytotoxic effect. Therefore, we aimed to block the primary alcohol and examine its effect (GOXG1) and also take advantage of the secondary alcohol which could lead to a PDC with a completely different profile (GOXG2), although they share structural similarities. For a slower drug release rate, we designed and developed the PDC GN4OXG that contains an amide bond on the 4-N position of the parent drug followed by click oxime ligation and another amide bond. The stability of this molecule should be enhanced since it is devoid of rapidly hydrolyzable ester bonds. Furthermore, in this PDC since the 4-NH2 moiety of gemcitabine is capped it could further surmount the rapid gemcitabine metabolism that leads to the formation of dFdU, after the enzymatic 4-N deamination of gemcitabine by cytidine deaminase (CDA).
Click to Show/Hide
|
||||
| Description |
The most potent among them was found to be pro-drug GOXG2 with IC50 494 ± 93 nM and 675 ± 82 nM against DU145 and PC3, respectively. Regarding the other two conjugates, GN4OXG was found to be the next most cytotoxic compound against DU145 with IC50 590 ± 62 nM, followed by the least toxic GOXG1 with IC50 611 ± 80 nM. In contrast, GOXG1 was the second more toxic against PC3 with IC50 754 ± 142 nM, followed by the least toxic GN4OXG which showed IC50 833 ± 27 nM. The results are summarized in Table 2.
Click to Show/Hide
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
D-Lys6-GnRH-gemcitabine(2G2) [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [30] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.11 nM | |||
| Description |
The presented data show that 2G2, 2G1 and GSHG bind to GnRH-R with 95.5-, 15.2-, and 4.4-fold higher affinity, respectively, than that of the native peptide D-Lys6-GnRH (10.5 ± 0.2 nM, according to our former study [3]).
|
||||
| In Vitro Model | Normal | HEK293 cell | CVCL_0045 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [30] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 449.1 nM | |||
| Description |
GSHGpossesses the highest cytotoxic effect among the three conjugates, which is comparable with that of gemcitabine in the examined cell lines and especially regarding MCF-7 cells.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [30] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 9761 nM | |||
| Description |
GSHGpossesses the highest cytotoxic effect among the three conjugates, which is comparable with that of gemcitabine in the examined cell lines and especially regarding MCF-7 cells.
|
||||
| In Vitro Model | Prostate carcinoma | DU145 cell | CVCL_0105 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [30] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 40000 nM | |||
| Description |
GSHGpossesses the highest cytotoxic effect among the three conjugates, which is comparable with that of gemcitabine in the examined cell lines and especially regarding MCF-7 cells.
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [30] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 40000 nM | |||
| Description |
GSHGpossesses the highest cytotoxic effect among the three conjugates, which is comparable with that of gemcitabine in the examined cell lines and especially regarding MCF-7 cells.
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
D-Lys6-GnRH-gemcitabine(2G1) [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [30] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.69 nM | |||
| Description |
The presented data show that 2G2, 2G1 and GSHG bind to GnRH-R with 95.5-, 15.2-, and 4.4-fold higher affinity, respectively, than that of the native peptide D-Lys6-GnRH (10.5 ± 0.2 nM, according to our former study [3]).
|
||||
| In Vitro Model | Normal | HEK293 cell | CVCL_0045 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [30] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 621.3 nM | |||
| Description |
GSHGpossesses the highest cytotoxic effect among the three conjugates, which is comparable with that of gemcitabine in the examined cell lines and especially regarding MCF-7 cells.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [30] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 40000 nM | |||
| Description |
GSHGpossesses the highest cytotoxic effect among the three conjugates, which is comparable with that of gemcitabine in the examined cell lines and especially regarding MCF-7 cells.
|
||||
| In Vitro Model | Prostate carcinoma | DU145 cell | CVCL_0105 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [30] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 40000 nM | |||
| Description |
GSHGpossesses the highest cytotoxic effect among the three conjugates, which is comparable with that of gemcitabine in the examined cell lines and especially regarding MCF-7 cells.
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [30] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | > 40000 nM | |||
| Description |
GSHGpossesses the highest cytotoxic effect among the three conjugates, which is comparable with that of gemcitabine in the examined cell lines and especially regarding MCF-7 cells.
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
EETI-2.5Z-Val-Ala-PAB-gemcitabine [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [31] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.5 ± 0.2 nM | |||
| Evaluation Method | CCK-8 assay | ||||
| Description |
Cell proliferation was quantified 4 d after treatment with each compoundusing CCK-8 colorimetric assays and compared to the untreated control.
|
||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [31] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) | 0.6 ± 0.1 nM | |||
| Evaluation Method | CCK-8 assay | ||||
| Description |
Cell proliferation was quantified 4 d after treatment with 5 d or 3 using CCK-8 colorimetric assays and compared to the untreated control. Metabolic activity measured by CCK-8 was validated by quantifying celldeath using Trypan Blue.
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-468 cell | CVCL_0419 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [31] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) | 1.8 ± 0.8 nM | |||
| Evaluation Method | CCK-8 assay | ||||
| Description |
Cell proliferation was quantified 4 d after treatment with 5 d or 3 using CCK-8 colorimetric assays and compared to the untreated control. Metabolic activity measured by CCK-8 was validated by quantifying celldeath using Trypan Blue.
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | BxPC-3 cell | CVCL_0186 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [31] | ||||
| Indication | Pancreatic cancer | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) | 2.1 ± 0.2 nM | |||
| Evaluation Method | CCK-8 assay | ||||
| Description |
Cell proliferation was quantified 4 d after treatment with 5 d or 3 using CCK-8 colorimetric assays and compared to the untreated control. Metabolic activity measured by CCK-8 was validated by quantifying celldeath using Trypan Blue.
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | PANC-1 cell | CVCL_0480 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [31] | ||||
| Indication | Ovarian cancer | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) | 2.3 ± 0.5 nM | |||
| Evaluation Method | CCK-8 assay | ||||
| Description |
Cell proliferation was quantified 4 d after treatment with 5 d or 3 using CCK-8 colorimetric assays and compared to the untreated control. Metabolic activity measured by CCK-8 was validated by quantifying celldeath using Trypan Blue.
|
||||
| In Vitro Model | Ovarian endometrioid adenocarcinoma | A2780 cell | CVCL_0134 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [31] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) | 7.9 ± 0.8 nM | |||
| Evaluation Method | CCK-8 assay | ||||
| Description |
Cell proliferation was quantified 4 d after treatment with 5 d or 3 using CCK-8 colorimetric assays and compared to the untreated control. Metabolic activity measured by CCK-8 was validated by quantifying celldeath using Trypan Blue.
|
||||
| In Vitro Model | Glioblastoma | D-270MG cell | CVCL_S751 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [31] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) | 9.0 ± 1.8 nM | |||
| Evaluation Method | CCK-8 assay | ||||
| Description |
Cell proliferation was quantified 4 d after treatment with 5 d or 3 using CCK-8 colorimetric assays and compared to the untreated control. Metabolic activity measured by CCK-8 was validated by quantifying celldeath using Trypan Blue.
|
||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
D-Lys6-GnRH-gemcitabine(GSHG) [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [30] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 2.40 nM | |||
| Description |
The presented data show that 2G2, 2G1 and GSHG bind to GnRH-R with 95.5-, 15.2-, and 4.4-fold higher affinity, respectively, than that of the native peptide D-Lys6-GnRH (10.5 ± 0.2 nM, according to our former study [3]).
|
||||
| In Vitro Model | Normal | HEK293 cell | CVCL_0045 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [30] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 55.5 nM | |||
| Description |
GSHGpossesses the highest cytotoxic effect among the three conjugates, which is comparable with that of gemcitabine in the examined cell lines and especially regarding MCF-7 cells.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [30] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 684 nM | |||
| Description |
GSHGpossesses the highest cytotoxic effect among the three conjugates, which is comparable with that of gemcitabine in the examined cell lines and especially regarding MCF-7 cells.
|
||||
| In Vitro Model | Prostate carcinoma | DU145 cell | CVCL_0105 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [30] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 937 nM | |||
| Description |
GSHGpossesses the highest cytotoxic effect among the three conjugates, which is comparable with that of gemcitabine in the examined cell lines and especially regarding MCF-7 cells.
|
||||
| In Vitro Model | Prostate carcinoma | PC-3 cell | CVCL_0035 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [30] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 2387 nM | |||
| Description |
GSHGpossesses the highest cytotoxic effect among the three conjugates, which is comparable with that of gemcitabine in the examined cell lines and especially regarding MCF-7 cells.
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
EETI-2.5Z-amide -gemcitabine [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [31] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 2.8 ± 0.2 nM | |||
| Evaluation Method | CCK-8 assay | ||||
| Description |
Cell proliferation was quantified 4 d after treatment with each compoundusing CCK-8 colorimetric assays and compared to the untreated control.
|
||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [31] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) | 8.9 ± 1.2 nM | |||
| Evaluation Method | CCK-8 assay | ||||
| Description |
Cell proliferation was quantified 4 d after treatment with each compoundusing CCK-8 colorimetric assays and compared to the untreated control.
|
||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
EETI-2.5Z-carbamate -gemcitabine [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [31] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 3.3 ± 0.2 nM | |||
| Evaluation Method | CCK-8 assay | ||||
| Description |
Cell proliferation was quantified 4 d after treatment with each compoundusing CCK-8 colorimetric assays and compared to the untreated control.
|
||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [31] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) | > 1000 nM | |||
| Evaluation Method | CCK-8 assay | ||||
| Description |
Cell proliferation was quantified 4 d after treatment with each compoundusing CCK-8 colorimetric assays and compared to the untreated control.
|
||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
EETI-2.5Z-ester-gemcitabine [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [31] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 5.2 ± 3.6 nM | |||
| Evaluation Method | CCK-8 assay | ||||
| Description |
Cell proliferation was quantified 4 d after treatment with each compoundusing CCK-8 colorimetric assays and compared to the untreated control.
|
||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [31] | ||||
| Indication | Glioblastoma | ||||
| Efficacy Data | Half Maximal Effective Concentration (EC50) | 8.5 ± 3.3 nM | |||
| Evaluation Method | CCK-8 assay | ||||
| Description |
Cell proliferation was quantified 4 d after treatment with each compoundusing CCK-8 colorimetric assays and compared to the untreated control.
|
||||
| In Vitro Model | Glioblastoma | U-87MG cell | CVCL_0022 | ||
OGF-Gem [Investigative]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [32] | ||||
| Indication | Pancreatic ductal adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 17.63 ± 2.334 nM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
Therefore, we designed, synthesized, and characterized an OGF-Gem conjugate, where OGF and Gem are tethered by an organic linker (Figure 1). Gem was subjected to selective protection using the tert-butoxycarbonyl (Boc) group and prepared as gemcitabine hemisuccinate. 5-O-diBoc-gemcitabine hemisuccinate was conjugated with the OGF peptide in solution. We demonstrated the cytotoxic activity of the OGF-Gem conjugate against pancreatic cancer cell lines, including the metastatic line (MIA PaCa-2 and AsPC-1). Furthermore, we confirmed that OGF-Gem is either not cytotoxic or significantly less cytotoxic to two non-tumor-transformed human cellskidney (HEK-293) and skin fibroblast cells (HDFa). We also determined the effect of OGF-Gem on cell cycle inhibition, and the inhibition of cell proliferation, senescent cells, and apoptosis. We have demonstrated that OGF-Gem has antimetastatic potential due to inhibited pancreatic tumor cell (AsPC-1)-induced platelet aggregation. This can significantly impact the inhibition of disease progression (metastasis) of pancreatic cancer.
Click to Show/Hide
|
||||
| Description |
The tested compounds cytotoxic activity was determined using the MTT test, which is based on the ability to convert tetrazole salts to water insoluble formazan through mitochondrial dehydrogenases. Our results show a high cytotoxic effect on all pancreatic cancer cell lines. Exposing pancreatic cell lines MIA PaCa-2 and AsPC-1 to OGF-Gem decreased viability. Importantly, the OGF-Gem conjugate demonstrated a more pronounced cytotoxic effect against the metastatic pancreatic cancer cell line AsPC-1 compared to the commonly used chemotherapeutic agent. The results obtained for non-tumor-transformed cellsa human embryonic kidney line HEK-293 and human primary dermal fibroblast line HDFa presented a slight cytotoxicity effect from the OGF-Gem derivative within 3 days of incubation for all tested concentrations. Interestingly, an 80% reduction in HEK-293 cell viability was observed for the 100 nM Gem compared to the 100 nM OGF-Gem derivative. In HDFa cells, 100 nM Gem reduced viability to 35%, while the OGF-Gem conjugate slightly decreased the viability (to 75% viability) after 72 h of incubation. Based on the analysis of the results, concentrations of 3.125, 12.5, 50, and 100 nM, as well as an incubation time of 72 h, were selected for further experiments on the three pancreatic cancer cell lines.
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | MIA PaCa-2 cell | CVCL_0428 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [32] | ||||
| Indication | Pancreatic ductal adenocarcinoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 27.44 ± 9.161 nM | |||
| Evaluation Method | MTT assay | ||||
| Administration Time | 72 h | ||||
| MOA of PDC |
Therefore, we designed, synthesized, and characterized an OGF-Gem conjugate, where OGF and Gem are tethered by an organic linker (Figure 1). Gem was subjected to selective protection using the tert-butoxycarbonyl (Boc) group and prepared as gemcitabine hemisuccinate. 5-O-diBoc-gemcitabine hemisuccinate was conjugated with the OGF peptide in solution. We demonstrated the cytotoxic activity of the OGF-Gem conjugate against pancreatic cancer cell lines, including the metastatic line (MIA PaCa-2 and AsPC-1). Furthermore, we confirmed that OGF-Gem is either not cytotoxic or significantly less cytotoxic to two non-tumor-transformed human cellskidney (HEK-293) and skin fibroblast cells (HDFa). We also determined the effect of OGF-Gem on cell cycle inhibition, and the inhibition of cell proliferation, senescent cells, and apoptosis. We have demonstrated that OGF-Gem has antimetastatic potential due to inhibited pancreatic tumor cell (AsPC-1)-induced platelet aggregation. This can significantly impact the inhibition of disease progression (metastasis) of pancreatic cancer.
Click to Show/Hide
|
||||
| Description |
The tested compounds cytotoxic activity was determined using the MTT test, which is based on the ability to convert tetrazole salts to water insoluble formazan through mitochondrial dehydrogenases. Our results show a high cytotoxic effect on all pancreatic cancer cell lines. Exposing pancreatic cell lines MIA PaCa-2 and AsPC-1 to OGF-Gem decreased viability. Importantly, the OGF-Gem conjugate demonstrated a more pronounced cytotoxic effect against the metastatic pancreatic cancer cell line AsPC-1 compared to the commonly used chemotherapeutic agent. The results obtained for non-tumor-transformed cellsa human embryonic kidney line HEK-293 and human primary dermal fibroblast line HDFa presented a slight cytotoxicity effect from the OGF-Gem derivative within 3 days of incubation for all tested concentrations. Interestingly, an 80% reduction in HEK-293 cell viability was observed for the 100 nM Gem compared to the 100 nM OGF-Gem derivative. In HDFa cells, 100 nM Gem reduced viability to 35%, while the OGF-Gem conjugate slightly decreased the viability (to 75% viability) after 72 h of incubation. Based on the analysis of the results, concentrations of 3.125, 12.5, 50, and 100 nM, as well as an incubation time of 72 h, were selected for further experiments on the three pancreatic cancer cell lines.
Click to Show/Hide
|
||||
| In Vitro Model | Pancreatic ductal adenocarcinoma | AsPC-1 cell | CVCL_0152 | ||
References