
Drug Information
General Information of This Drug
| Drug ID | DRG00031 | |||||
|---|---|---|---|---|---|---|
| Drug Name | Mertansine DM1 | |||||
| Synonyms |
Mertansine; 139504-50-0; Maytansinoid DM 1; maytansinoid DM1; Mertansine (DM1); N2'-deacetyl-N2'-(3-mercapto-1-oxopropyl)-maytansine; DDZ29HGH0E; DM 1; [(1S,2R,3S,5S,6S,16E,18E,20R,21S)-11-chloro-21-hydroxy-12,20-dimethoxy-2,5,9,16-tetramethyl-8,23-dioxo-4,24-dioxa-9,22-diazatetracyclo[19.3.1.110,14.03,5]hexacosa-10,12,14(26),16,18-pentaen-6-yl] (2S)-2-[methyl(3-sulfanylpropanoyl)amino]propanoate; DM1; N2'-Deacetyl-N2'-(3-mercapto-1-oxopropyl)-maytansine, L-; Maytansine, N2'-deacetyl-N2'-(3-mercapto-1-oxopropyl)-; DM1 [Maytansinoid]; DM 1 [Maytansinoid]; UNII-DDZ29HGH0E; Mertasine; MFCD28398157; DM1;Maytansinoid; Maytansinoid DM1; Dm-1; MAYTANSINOID DM1 [MI]; CHEMBL4802230; SCHEMBL13558634; CHEBI:82755; ANZJBCHSOXCCRQ-FKUXLPTCSA-N; DTXSID801026968; DM1 , Maytansinoid DM1; s6773; CS-5804; DA-48536; HY-19792; Q4515649; (1S,2R,3S,5S,6S,16E,18E,20R,21S)-11-chloro-21-hydroxy-12,20-dimethoxy-2,5,9,16-tetramethyl-8,23-dioxo-4,24-dioxa-9,22-diazatetracyclo[19.3.1.1(10,14).0(3,5)]hexacosa-10(26),11,13,16,18-pentaen-6-yl (2S)-2-[methyl(3-sulfanylpropanoyl)amino]propanoate
Click to Show/Hide
|
|||||
| Target(s) | Microtubule (MT) | Target Info | ||||
| Structure |
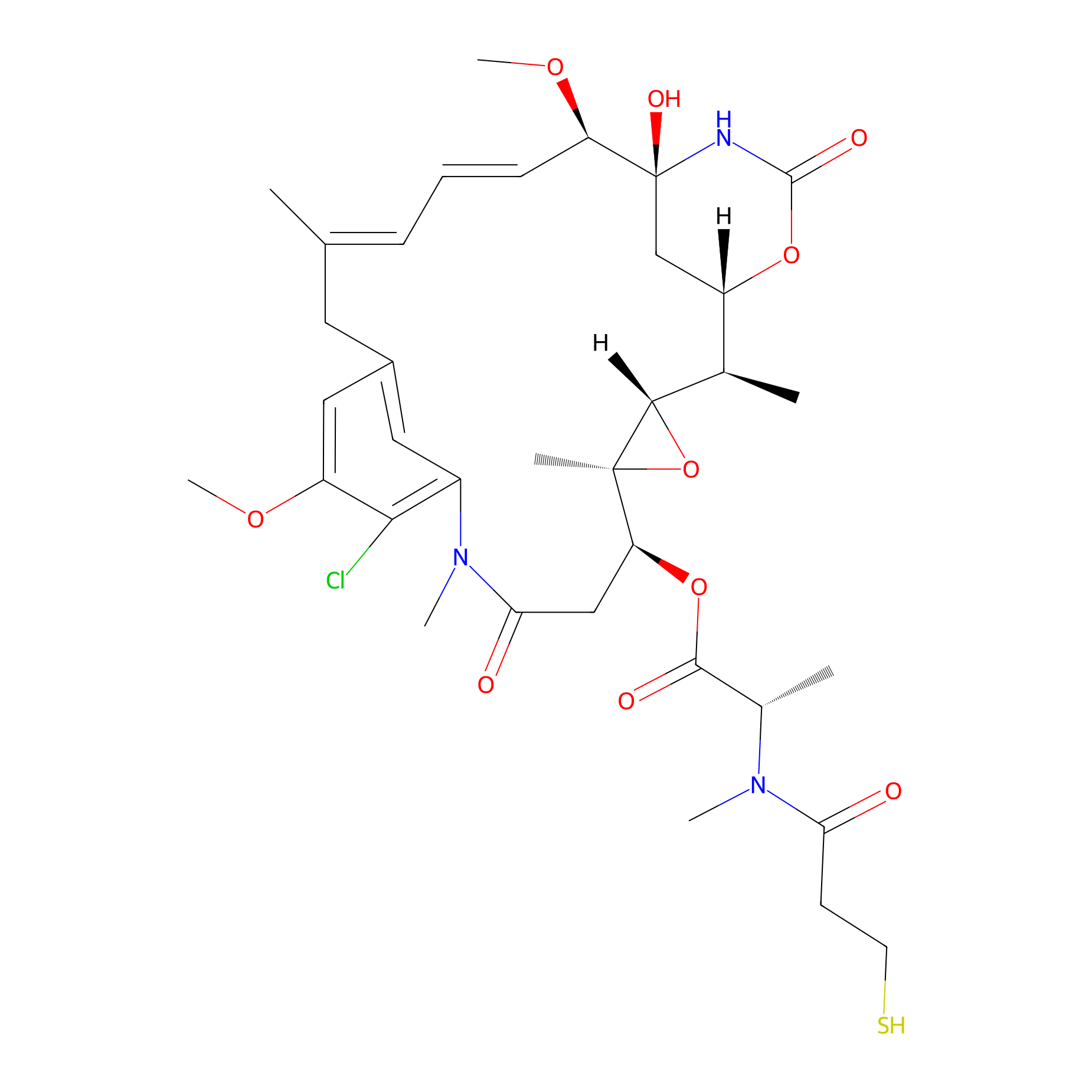
|
|||||
| Formula |
C35H48ClN3O10S
|
|||||
| #Ro5 Violations (Lipinski): 2 | Molecular Weight (mw) | 738.3 | ||||
| Lipid-water partition coefficient (xlogp) | 2.2 | |||||
| Hydrogen Bond Donor Count (hbonddonor) | 3 | |||||
| Hydrogen Bond Acceptor Count (hbondacc) | 11 | |||||
| Rotatable Bond Count (rotbonds) | 8 | |||||
| PubChem CID | ||||||
| Canonical smiles |
CC1C2CC(C(C=CC=C(CC3=CC(=C(C(=C3)OC)Cl)N(C(=O)CC(C4(C1O4)C)OC(=O)C(C)N(C)C(=O)CCS)C)C)OC)(NC(=O)O2)O
|
|||||
| InChI |
InChI=1S/C35H48ClN3O10S/c1-19-10-9-11-26(46-8)35(44)18-25(47-33(43)37-35)20(2)31-34(4,49-31)27(48-32(42)21(3)38(5)28(40)12-13-50)17-29(41)39(6)23-15-22(14-19)16-24(45-7)30(23)36/h9-11,15-16,20-21,25-27,31,44,50H,12-14,17-18H2,1-8H3,(H,37,43)/b11-9+,19-10+/t20-,21+,25+,26-,27+,31+,34+,35+/m1/s1
|
|||||
| InChIKey |
ANZJBCHSOXCCRQ-FKUXLPTCSA-N
|
|||||
| IUPAC Name |
[(1S,2R,3S,5S,6S,16E,18E,20R,21S)-11-chloro-21-hydroxy-12,20-dimethoxy-2,5,9,16-tetramethyl-8,23-dioxo-4,24-dioxa-9,22-diazatetracyclo[19.3.1.110,14.03,5]hexacosa-10,12,14(26),16,18-pentaen-6-yl] (2S)-2-[methyl(3-sulfanylpropanoyl)amino]propanoate
|
|||||
The activity data of This Drug
| Standard Type | Value | Vivo Model | Cell line | Cell line ID | Ref. | |
|---|---|---|---|---|---|---|
| Tumor Growth Inhibition value (TGI) | 27.10% | B16-F10 tumor-bearing mouse model. | B16-F10 cell | CVCL_0159 | [1] | |
Each Peptide-drug Conjugate Related to This Drug
Full Information of The Activity Data of The PDC(s) Related to This Drug
PEN-221 [Phase 2]
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Small cell lung cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.06 µmol/L | |||
| Administration Time | 6 h | ||||
| Description |
PEN-221 receptor dependent activity in vitro varied from strong (NCI-H524) to modest (HCC-33) but was consistent (N = 3) and correlated with in vivo activity (described below). Octreotide alone did not have an effect on cellular growth. These cell lines also demonstrated sensitivity to the cytotoxic payload, DM1, with sub-micromolar IC50's after a 6-hour exposure and 70-hour additional incubation period (Supplementary Fig. S2B).
Click to Show/Hide
|
||||
| In Vitro Model | Lung small cell carcinoma | NCI-H524 cell | CVCL_1568 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Small cell lung cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.108 µmol/L | |||
| Administration Time | 6 h | ||||
| Description |
PEN-221 receptor dependent activity in vitro varied from strong (NCI-H524) to modest (HCC-33) but was consistent (N = 3) and correlated with in vivo activity (described below). Octreotide alone did not have an effect on cellular growth. These cell lines also demonstrated sensitivity to the cytotoxic payload, DM1, with sub-micromolar IC50's after a 6-hour exposure and 70-hour additional incubation period (Supplementary Fig. S2B).
Click to Show/Hide
|
||||
| In Vitro Model | Small cell lung carcinoma | NCI-H69 cell | CVCL_1579 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Small cell lung cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.258 µmol/L | |||
| Administration Time | 6 h | ||||
| Description |
PEN-221 receptor dependent activity in vitro varied from strong (NCI-H524) to modest (HCC-33) but was consistent (N = 3) and correlated with in vivo activity (described below). Octreotide alone did not have an effect on cellular growth. These cell lines also demonstrated sensitivity to the cytotoxic payload, DM1, with sub-micromolar IC50's after a 6-hour exposure and 70-hour additional incubation period (Supplementary Fig. S2B).
Click to Show/Hide
|
||||
| In Vitro Model | Small cell lung carcinoma | NCI-H69 cell | CVCL_1579 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Small cell lung cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.333 µmol/L | |||
| Administration Time | 6 h | ||||
| Description |
PEN-221 receptor dependent activity in vitro varied from strong (NCI-H524) to modest (HCC-33) but was consistent (N = 3) and correlated with in vivo activity (described below). Octreotide alone did not have an effect on cellular growth. These cell lines also demonstrated sensitivity to the cytotoxic payload, DM1, with sub-micromolar IC50's after a 6-hour exposure and 70-hour additional incubation period (Supplementary Fig. S2B).
Click to Show/Hide
|
||||
| In Vitro Model | Lung small cell carcinoma | NCI-H524 cell | CVCL_1568 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Small cell lung cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.658 µmol/L | |||
| Administration Time | 6 h | ||||
| Description |
PEN-221 receptor dependent activity in vitro varied from strong (NCI-H524) to modest (HCC-33) but was consistent (N = 3) and correlated with in vivo activity (described below). Octreotide alone did not have an effect on cellular growth. These cell lines also demonstrated sensitivity to the cytotoxic payload, DM1, with sub-micromolar IC50's after a 6-hour exposure and 70-hour additional incubation period (Supplementary Fig. S2B).
Click to Show/Hide
|
||||
| In Vitro Model | Lung small cell carcinoma | HCC33 cell | CVCL_2058 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [2] | ||||
| Indication | Small cell lung cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 0.83 µmol/L | |||
| Administration Time | 6 h | ||||
| Description |
PEN-221 receptor dependent activity in vitro varied from strong (NCI-H524) to modest (HCC-33) but was consistent (N = 3) and correlated with in vivo activity (described below). Octreotide alone did not have an effect on cellular growth. These cell lines also demonstrated sensitivity to the cytotoxic payload, DM1, with sub-micromolar IC50's after a 6-hour exposure and 70-hour additional incubation period (Supplementary Fig. S2B).
Click to Show/Hide
|
||||
| In Vitro Model | Lung small cell carcinoma | HCC33 cell | CVCL_2058 | ||
LLC2B-Mal-DM1 [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 28.66% | |||
| Administration Time | 6 d | ||||
| Administration Dosage | 4.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 34.15% | |||
| Administration Time | 6 d | ||||
| Administration Dosage | 2.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 36.81% | |||
| Administration Time | 12 d | ||||
| Administration Dosage | 4.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 37.21% | |||
| Administration Time | 2 d | ||||
| Administration Dosage | 4.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 38.55% | |||
| Administration Time | 10 d | ||||
| Administration Dosage | 4.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 38.73% | |||
| Administration Time | 8 d | ||||
| Administration Dosage | 4.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 39.16% | |||
| Administration Time | Once a week for 3 weeks | ||||
| Administration Dosage | 4.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 44.93% | |||
| Administration Time | 8 d | ||||
| Administration Dosage | 2.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 46.77% | |||
| Administration Time | 18 d | ||||
| Administration Dosage | 1.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 47.60% | |||
| Administration Time | 16 d | ||||
| Administration Dosage | 4.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 47.63% | |||
| Administration Time | Once a week for 3 weeks | ||||
| Administration Dosage | 2.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 48.73% | |||
| Administration Time | 16 d | ||||
| Administration Dosage | 2.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 49.13% | |||
| Administration Time | 14 d | ||||
| Administration Dosage | 4.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 49.33% | |||
| Administration Time | 2 d | ||||
| Administration Dosage | 2.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 50.36% | |||
| Administration Time | 18 d | ||||
| Administration Dosage | 4.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 50.81% | |||
| Administration Time | 12 d | ||||
| Administration Dosage | 2.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 17 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 51.89% | |||
| Administration Time | 18 d | ||||
| Administration Dosage | 2.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 18 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 53.10% | |||
| Administration Time | 14 d | ||||
| Administration Dosage | 2.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 19 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 54.75% | |||
| Administration Time | 12 d | ||||
| Administration Dosage | 1.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 20 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 54.77% | |||
| Administration Time | 16 d | ||||
| Administration Dosage | 1.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 21 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 57.18% | |||
| Administration Time | 10 d | ||||
| Administration Dosage | 1.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 22 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 57.94% | |||
| Administration Time | 10 d | ||||
| Administration Dosage | 2.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 23 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 59.26% | |||
| Administration Time | 14 d | ||||
| Administration Dosage | 1.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 24 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 60.37% | |||
| Administration Time | 8 d | ||||
| Administration Dosage | 1.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 25 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 63.86% | |||
| Administration Time | 6 d | ||||
| Administration Dosage | 1.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 26 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 65.78% | |||
| Administration Time | Once a week for 3 weeks | ||||
| Administration Dosage | 1.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 27 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 71.61% | |||
| Administration Time | 2 d | ||||
| Administration Dosage | 1.0 mg DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Completely suppress concentration | 1 μg DM1 equiv./mL | |||
| Evaluation Method | Clone formation experiment | ||||
| Administration Time | 12 h | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
The results show that DM1 could completely suppress the clone formation of MCF-7 cells at 0.001 μg/mL, while LLC2B-SS-DM1 could completely suppress the clone formation of MCF-7 cells at 0.1 μg DM1 equiv./mL. A concentration of 1 μg DM1 equiv./mL was required for LLC2B-Mal-DM1 to achieve the same suppression effect.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
LLC2B-SS-DM1 [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 58.76% | |||
| Administration Time | 14 d | ||||
| Administration Dosage | 0.5 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 2 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 58.77% | |||
| Administration Time | 6 d | ||||
| Administration Dosage | 0.5 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 60.43% | |||
| Administration Time | 16 d | ||||
| Administration Dosage | 0.5 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 61.33% | |||
| Administration Time | 10 d | ||||
| Administration Dosage | 0.5 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 5 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 63.36% | |||
| Administration Time | Once a week for 3 weeks | ||||
| Administration Dosage | 2.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 6 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 64.57% | |||
| Administration Time | 8 d | ||||
| Administration Dosage | 0.5 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 7 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 65.31% | |||
| Administration Time | Once a week for 3 weeks | ||||
| Administration Dosage | 0.5 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 8 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 66.73% | |||
| Administration Time | 2 d | ||||
| Administration Dosage | 0.5 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 9 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 67.10% | |||
| Administration Time | 2 d | ||||
| Administration Dosage | 2.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 10 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 67.68% | |||
| Administration Time | Once a week for 3 weeks | ||||
| Administration Dosage | 1.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 11 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 68.27% | |||
| Administration Time | 12 d | ||||
| Administration Dosage | 0.5 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 12 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 69.20% | |||
| Administration Time | 20 d | ||||
| Administration Dosage | 0.5 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 13 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 69.39% | |||
| Administration Time | 10 d | ||||
| Administration Dosage | 1.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 14 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 70.04% | |||
| Administration Time | 8 d | ||||
| Administration Dosage | 2.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 15 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 70.26% | |||
| Administration Time | 6 d | ||||
| Administration Dosage | 2.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 16 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 71.16% | |||
| Administration Time | 6 d | ||||
| Administration Dosage | 1.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 17 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 72.14% | |||
| Administration Time | 16 d | ||||
| Administration Dosage | 1.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 18 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 72.20% | |||
| Administration Time | 8 d | ||||
| Administration Dosage | 1.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 19 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 72.45% | |||
| Administration Time | 10 d | ||||
| Administration Dosage | 2.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 20 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 72.90% | |||
| Administration Time | 2 d | ||||
| Administration Dosage | 1.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 21 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 72.91% | |||
| Administration Time | 16 d | ||||
| Administration Dosage | 2.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 22 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 72.98% | |||
| Administration Time | 12 d | ||||
| Administration Dosage | 2.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 23 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 73.90% | |||
| Administration Time | 20 d | ||||
| Administration Dosage | 2.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 24 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 75.40% | |||
| Administration Time | 14 d | ||||
| Administration Dosage | 1.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 25 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 75.49% | |||
| Administration Time | 20 d | ||||
| Administration Dosage | 1.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 26 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 77.25% | |||
| Administration Time | 12 d | ||||
| Administration Dosage | 1.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 27 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumor growth inhibition value (TGI) | 78.30% | |||
| Administration Time | 14 d | ||||
| Administration Dosage | 2.0 DM1 equiv/kg | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
We investigated the antitumor efficacy of those two PDCs on xenografted mice in vivo. LLC2B-Mal-DM1 at 1.0, 2.0 and 4.0 mg DM1 equiv./kg per week was injected into individual mice according to the tolerated concentrations of DM1 in mice: 1.0-1.5 mg/kg once per week. However, LLC2B-SS-DM1 at 0.5, 1.0, 2.0 and 4.0 mg DM1 equiv./kg was injected in the test due to its higher suppressive effect compared to LLC2B-Mal-DM in the clone formation experiment (the drug concentration used was based on the amount of the substance DM1). As shown in Figure 6A,C, LLC2B-Mal-DM1 at 2.0 and 4.0 mg DM1 equiv./kg and LLC2B-SS-DM1 with 0.5 mg DM1 equiv./kg had clear antitumor effects compared to the DM1 control groups. According to the mouse body weights, there were probably no acute life-threatening conditions in any of the groups. Moreover, the comparison of both PDCs indicated that LLC2B-SS-DM1 had a better effect than LLC2B-Mal-DM1 against the tumors.
Click to Show/Hide
|
||||
| In Vivo Model | BALB/c nude mice MCF-7 cells xenograft model. | ||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [3] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Completely suppress concentration | 0.1 μg DM1 equiv./mL | |||
| Evaluation Method | Clone formation experiment | ||||
| Administration Time | 12 h | ||||
| MOA of PDC |
A maytansin derivative, DM1, is a promising therapeutic compound for treating tumors, but is also a highly poisonous substance with various side effects. For clinical expansion, we tried to develop novel peptide-drug conjugates (PDCs) with DM1. In the study, a one-bead one-compound (OBOC) platform was used to screen and identify a novel, highly stable, non-natural amino acid peptide targeting the tyrosine receptor FGFR2. Then, the identified peptide, named LLC2B, was conjugated with the cytotoxin DM1. Our results show that LLC2B has high affinity for the FGFR2 protein according to an isothermal titration calorimetry (ITC) test. LLC2B-Cy5.5 binding to FGFR2-positive cancer cells was confirmed by fluorescent microscopic imaging and flow cytometry in vitro. Using xenografted nude mouse models established with breast cancer MCF-7 cells and esophageal squamous cell carcinoma KYSE180 cells, respectively, LLC2B-Cy5.5 was observed to specifically target tumor tissues 24 h after tail vein injection. Incubation assays, both in aqueous solution at room temperature and in human plasma at 37 °C, suggested that LLC2B has high stability and strong anti-proteolytic ability. Then, we used two different linkers, one of molecular disulfide bonds and another of a maleimide group, to couple LLC2B to the toxin DM1. The novel peptide-drug conjugates (PDCs) inhibited tumor growth and significantly increased the maximum tolerated dose of DM1 in xenografted mice. In brief, our results suggest that LLC2B-DM1 can be developed into a potential PDC for tumor treatment in the future.
Click to Show/Hide
|
||||
| Description |
The results show that DM1 could completely suppress the clone formation of MCF-7 cells at 0.001 μg/mL, while LLC2B-SS-DM1 could completely suppress the clone formation of MCF-7 cells at 0.1 μg DM1 equiv./mL. A concentration of 1 μg DM1 equiv./mL was required for LLC2B-Mal-DM1 to achieve the same suppression effect.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
cRGD-SS-DM1 [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Tumer volume | 1800 mm3 | |||
| Administration Time | Given every other day for a total of five times | ||||
| Administration Dosage | 400 µg/kg (calculated by free DM1) | ||||
| Description |
It was demonstrated here, RCCD@NPs and RSSD@NPs exhibited significantly better tumor-growth inhibition compared with that of the free DM1, QCCD@NPs or QSSD@NPs (Figure (Figure4A).4A). RSSD@NPs showed the most suppression on B16 tumor up to 25 days, and resulted in a tumor volume 4 times smaller than the saline group at the end of the experiment. The final tumor volumes in various nano-DDS groups ranked from the greatest to the least: QCCD@NPs, QSSD@NPs>DM1>RCCD@NPs>RSSD@NPs. Almost the same trends were found in terms of tumor weight and tumor size (Figures (Figures4B,4B, C).
Click to Show/Hide
|
||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 20.42 ± 4.70 nM | |||
| Administration Time | 48 h | ||||
| Description |
The IC50 of QCCD@NPs and QSSD@NPs (389.05±75.25 nM and 245.47±37.54 nM) on B16 cells seemed much higher than the values of RCCD@NPs and RSSD@NPs (102.33±38.92 nM and 21.38±4.32 nM). The cytotoxicity of APDC@NPs on HUVEC showed a similar pattern.
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cell | Homo sapiens | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 21.38 ± 4.32 nM | |||
| Administration Time | 48 h | ||||
| Description |
The IC50 of QCCD@NPs and QSSD@NPs (389.05±75.25 nM and 245.47±37.54 nM) on B16 cells seemed much higher than the values of RCCD@NPs and RSSD@NPs (102.33±38.92 nM and 21.38±4.32 nM). The cytotoxicity of APDC@NPs on HUVEC showed a similar pattern.
|
||||
| In Vitro Model | Melanoma | B16 cell | CVCL_F936 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 302.00 ± 72.91 nM | |||
| Administration Time | 48 h | ||||
| Description |
The IC50 of QCCD@NPs and QSSD@NPs (389.05±75.25 nM and 245.47±37.54 nM) on B16 cells seemed much higher than the values of RCCD@NPs and RSSD@NPs (102.33±38.92 nM and 21.38±4.32 nM). The cytotoxicity of APDC@NPs on HUVEC showed a similar pattern.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
cRPQ-SMCC-DM1 [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Tumer volume | 2400 mm3 | |||
| Administration Time | Given every other day for a total of five times | ||||
| Administration Dosage | 400 µg/kg (calculated by free DM1) | ||||
| Description |
It was demonstrated here, RCCD@NPs and RSSD@NPs exhibited significantly better tumor-growth inhibition compared with that of the free DM1, QCCD@NPs or QSSD@NPs (Figure (Figure4A).4A). RSSD@NPs showed the most suppression on B16 tumor up to 25 days, and resulted in a tumor volume 4 times smaller than the saline group at the end of the experiment. The final tumor volumes in various nano-DDS groups ranked from the greatest to the least: QCCD@NPs, QSSD@NPs>DM1>RCCD@NPs>RSSD@NPs. Almost the same trends were found in terms of tumor weight and tumor size (Figures (Figures4B,4B, C).
Click to Show/Hide
|
||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 67.61 ± 13.67 nM | |||
| Administration Time | 48 h | ||||
| Description |
The IC50 of QCCD@NPs and QSSD@NPs (389.05±75.25 nM and 245.47±37.54 nM) on B16 cells seemed much higher than the values of RCCD@NPs and RSSD@NPs (102.33±38.92 nM and 21.38±4.32 nM). The cytotoxicity of APDC@NPs on HUVEC showed a similar pattern.
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cell | Homo sapiens | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 331.13 ± 85.56 nM | |||
| Administration Time | 48 h | ||||
| Description |
The IC50 of QCCD@NPs and QSSD@NPs (389.05±75.25 nM and 245.47±37.54 nM) on B16 cells seemed much higher than the values of RCCD@NPs and RSSD@NPs (102.33±38.92 nM and 21.38±4.32 nM). The cytotoxicity of APDC@NPs on HUVEC showed a similar pattern.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 389.05 ± 75.25 nM | |||
| Administration Time | 48 h | ||||
| Description |
The IC50 of QCCD@NPs and QSSD@NPs (389.05±75.25 nM and 245.47±37.54 nM) on B16 cells seemed much higher than the values of RCCD@NPs and RSSD@NPs (102.33±38.92 nM and 21.38±4.32 nM). The cytotoxicity of APDC@NPs on HUVEC showed a similar pattern.
|
||||
| In Vitro Model | Melanoma | B16 cell | CVCL_F936 | ||
cRPQ-SS-DM1 [Investigative]
Discovered Using Cell Line-derived Xenograft Model
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Tumer volume | 5200 mm3 | |||
| Administration Time | Given every other day for a total of five times | ||||
| Administration Dosage | 400 µg/kg (calculated by free DM1) | ||||
| Description |
It was demonstrated here, RCCD@NPs and RSSD@NPs exhibited significantly better tumor-growth inhibition compared with that of the free DM1, QCCD@NPs or QSSD@NPs (Figure (Figure4A).4A). RSSD@NPs showed the most suppression on B16 tumor up to 25 days, and resulted in a tumor volume 4 times smaller than the saline group at the end of the experiment. The final tumor volumes in various nano-DDS groups ranked from the greatest to the least: QCCD@NPs, QSSD@NPs>DM1>RCCD@NPs>RSSD@NPs. Almost the same trends were found in terms of tumor weight and tumor size (Figures (Figures4B,4B, C).
Click to Show/Hide
|
||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 154.88 ± 31.85 nM | |||
| Administration Time | 48 h | ||||
| Description |
The IC50 of QCCD@NPs and QSSD@NPs (389.05±75.25 nM and 245.47±37.54 nM) on B16 cells seemed much higher than the values of RCCD@NPs and RSSD@NPs (102.33±38.92 nM and 21.38±4.32 nM). The cytotoxicity of APDC@NPs on HUVEC showed a similar pattern.
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cell | Homo sapiens | ||
| Experiment 2 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 245.47 ± 37.54 nM | |||
| Administration Time | 48 h | ||||
| Description |
The IC50 of QCCD@NPs and QSSD@NPs (389.05±75.25 nM and 245.47±37.54 nM) on B16 cells seemed much higher than the values of RCCD@NPs and RSSD@NPs (102.33±38.92 nM and 21.38±4.32 nM). The cytotoxicity of APDC@NPs on HUVEC showed a similar pattern.
|
||||
| In Vitro Model | Melanoma | B16 cell | CVCL_F936 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 371.53 ± 94.49 nM | |||
| Administration Time | 48 h | ||||
| Description |
The IC50 of QCCD@NPs and QSSD@NPs (389.05±75.25 nM and 245.47±37.54 nM) on B16 cells seemed much higher than the values of RCCD@NPs and RSSD@NPs (102.33±38.92 nM and 21.38±4.32 nM). The cytotoxicity of APDC@NPs on HUVEC showed a similar pattern.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
cRGD-SMCC-DM1 [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Uric acid levels | 185 | |||
| Administration Time | 21d | ||||
| Description |
Remarkably, leucopenia and myelosuppression did not occur during the use of PDC. In addition, the values of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and alkaline phosphatase (ALP), as the biomarkers of hepatic function, were all in the normal range in comparison to the saline group (Figure 5F-H). Meanwhile, normal renal function was also identified after treatment by monitoring CREA, UA, and BUN (Figure 5I-K).
Click to Show/Hide
|
||||
| In Vivo Model | Tumor-bearing nude mice. | ||||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Tumer volume | 1000 mm3 | |||
| Administration Time | 21d | ||||
| In Vivo Model | Tumor-bearing nude mice. | ||||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Tumer volume | 2100 mm3 | |||
| Administration Time | Given every other day for a total of five times | ||||
| Administration Dosage | 400 µg/kg (calculated by free DM1) | ||||
| Description |
It was demonstrated here, RCCD@NPs and RSSD@NPs exhibited significantly better tumor-growth inhibition compared with that of the free DM1, QCCD@NPs or QSSD@NPs (Figure (Figure4A).4A). RSSD@NPs showed the most suppression on B16 tumor up to 25 days, and resulted in a tumor volume 4 times smaller than the saline group at the end of the experiment. The final tumor volumes in various nano-DDS groups ranked from the greatest to the least: QCCD@NPs, QSSD@NPs>DM1>RCCD@NPs>RSSD@NPs. Almost the same trends were found in terms of tumor weight and tumor size (Figures (Figures4B,4B, C).
Click to Show/Hide
|
||||
| In Vivo Model | Tumor-bearing C57BL/6 mice model. | ||||
| Experiment 4 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | BUN level | 7 | |||
| Administration Time | 21d | ||||
| Description |
Remarkably, leucopenia and myelosuppression did not occur during the use of PDC. In addition, the values of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and alkaline phosphatase (ALP), as the biomarkers of hepatic function, were all in the normal range in comparison to the saline group (Figure 5F-H). Meanwhile, normal renal function was also identified after treatment by monitoring CREA, UA, and BUN (Figure 5I-K).
Click to Show/Hide
|
||||
| In Vivo Model | Tumor-bearing nude mice. | ||||
| Experiment 5 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | AST level | 175 | |||
| Administration Time | 21d | ||||
| Description |
Remarkably, leucopenia and myelosuppression did not occur during the use of PDC. In addition, the values of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and alkaline phosphatase (ALP), as the biomarkers of hepatic function, were all in the normal range in comparison to the saline group (Figure 5F-H). Meanwhile, normal renal function was also identified after treatment by monitoring CREA, UA, and BUN (Figure 5I-K).
Click to Show/Hide
|
||||
| In Vivo Model | Tumor-bearing nude mice. | ||||
| Experiment 6 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | ALT level | 60 | |||
| Administration Time | 21d | ||||
| Description |
Remarkably, leucopenia and myelosuppression did not occur during the use of PDC. In addition, the values of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and alkaline phosphatase (ALP), as the biomarkers of hepatic function, were all in the normal range in comparison to the saline group (Figure 5F-H). Meanwhile, normal renal function was also identified after treatment by monitoring CREA, UA, and BUN (Figure 5I-K).
Click to Show/Hide
|
||||
| In Vivo Model | Tumor-bearing nude mice. | ||||
| Experiment 7 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | ALP level | 80 | |||
| Administration Time | 21d | ||||
| Description |
Remarkably, leucopenia and myelosuppression did not occur during the use of PDC. In addition, the values of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and alkaline phosphatase (ALP), as the biomarkers of hepatic function, were all in the normal range in comparison to the saline group (Figure 5F-H). Meanwhile, normal renal function was also identified after treatment by monitoring CREA, UA, and BUN (Figure 5I-K).
Click to Show/Hide
|
||||
| In Vivo Model | Tumor-bearing nude mice. | ||||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 33.84 ± 6.30 nM | |||
| Administration Time | 48 h | ||||
| Description |
The IC50 of QCCD@NPs and QSSD@NPs (389.05±75.25 nM and 245.47±37.54 nM) on B16 cells seemed much higher than the values of RCCD@NPs and RSSD@NPs (102.33±38.92 nM and 21.38±4.32 nM). The cytotoxicity of APDC@NPs on HUVEC showed a similar pattern.
|
||||
| In Vitro Model | Normal | Human umbilical vein endothelial cell | Homo sapiens | ||
| Experiment 2 Reporting the Activity Data of This PDC | [5] | ||||
| Indication | Breast cancer | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 68.5 nM | |||
| Description |
After the MDA-MB-231 cell line was confirmed to be integrin v3-positive by immunofluorescence assay, it was demonstrated that cRGD-SMCC-DM1 had an IC50 value of 68.5 nM to MDA-MB-231 cells.
|
||||
| In Vitro Model | Breast adenocarcinoma | MDA-MB-231 cell | CVCL_0062 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 102.33 ± 38.92 nM | |||
| Administration Time | 48 h | ||||
| Description |
The IC50 of QCCD@NPs and QSSD@NPs (389.05±75.25 nM and 245.47±37.54 nM) on B16 cells seemed much higher than the values of RCCD@NPs and RSSD@NPs (102.33±38.92 nM and 21.38±4.32 nM). The cytotoxicity of APDC@NPs on HUVEC showed a similar pattern.
|
||||
| In Vitro Model | Melanoma | B16 cell | CVCL_F936 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [4] | ||||
| Indication | Tumor | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 407.38 ± 54.05 nM | |||
| Administration Time | 48 h | ||||
| Description |
The IC50 of QCCD@NPs and QSSD@NPs (389.05±75.25 nM and 245.47±37.54 nM) on B16 cells seemed much higher than the values of RCCD@NPs and RSSD@NPs (102.33±38.92 nM and 21.38±4.32 nM). The cytotoxicity of APDC@NPs on HUVEC showed a similar pattern.
|
||||
| In Vitro Model | Invasive breast carcinoma | MCF-7 cell | CVCL_0031 | ||
M-DM1 [Investigative]
Obtained from the Model Organism Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Tumor Growth Inhibition value (TGI) | 58.30% | |||
| MOA of PDC |
Recently, peptides that offer versatility in drug discovery for the successful treatment of cancers have emerged. Peptide-drug conjugates (PDCs) represent an important therapeutic strategy for increasing tumor penetration and selectivity. We previously reported that M, which is extracted from bee venom, targets M2 macrophages and improves tumor treatment in lung cancer. M is an amphipathic peptide with 26 amino acid residues (AIGAVLKVLTTGLPALISWIKRKRQQ) that specifically binds to M2-like TAMs. Mertansine (DM1) is a strong cytotoxic agent that interacts with tubulin and inhibits the assembly of tubulin into microtubules. Because it targets microtubules and inhibits cell cycle, its clinical efficacy as a potential anti-cancer agent has been studied. However, meaningful results have yet to be obtained in clinical trials, with patients also suffering several side effects, such as myelosuppression. Antibody drug conjugates (ADCs) are currently the most successful type of drug conjugates, consisting of antibody (targeting), linker (linking the antibody to the payload), and Payload (killing target cells). Payload is a cytotoxic compound and is divided into microtubule inhibitors, such as DM1, and DNA-damaging agents, such as anthracyclines. DM1 alone has not been developed as a drug but is currently used as an antibody-drug conjugate. It has been reported that the peptide LLC2B combined with DM1 exerts an anticancer effect in breast and esophageal squamous cell carcinoma, suggesting it could be developed as a potential PDC for tumor treatment. In the present study, we investigated the anti-cancer effects of M-DM1, which was synthesized using M as a carrier for targeting TAMs and DM1 as a payload. We found that M-DM1 exerts its therapeutic effects by specifically depleting M2-like TAMs in a melanoma mouse model. Through the regulation of M2-like TAMs, M-DM1 induced a significant increase in the infiltration of effector cells, such as CD8 T cells and natural killer (NK) cells, into the TME. Collectively, our results suggest that depleting M2-like TAMs in the TME by treatment with M-DM1 has immunotherapeutic effects in malignant melanoma.
Click to Show/Hide
|
||||
| Description |
To examine the therapeutic effects of M-DM1 in mice, we used the B16F10 tumor-bearing mouse model. We subcutaneously injected cancer cells into C57BL/6 mice and treated them with 20 nmol/kg M, DM1, or M-DM1 via intraperitoneal injections every 3 days. While slight growth inhibition was observed with M and DM1, M-DM1 was found to significantly suppress tumor growth. None of the mice showed any weight loss until the end of the study period, suggesting that there were no significant toxicities due to the drug treatments. At 21 days after tumor cell inoculation, the mice were sacrificed, and tumor volumes and weights were measured. A marked decrease in the tumor volumes and weights was observed in the M-DM1-treated mice group compared to that in the other groups. In addition, we determined the survival rate of B16-bearing mice following treatment with each peptide. The median survival of the M-DM1 group was significantly higher than that of the other groups. These results demonstrate that M-DM1 is highly effective in inhibiting tumor growth and prolonging survival in melanoma.
Click to Show/Hide
|
||||
| In Vivo Model | B16-F10 tumor-bearing mouse model. | ||||
| In Vitro Model | Mouse melanoma | B16-F10 cell | CVCL_0159 | ||
Revealed Based on the Cell Line Data
| Experiment 1 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.042 µM | |||
| Evaluation Method | MTS assay | ||||
| MOA of PDC |
Recently, peptides that offer versatility in drug discovery for the successful treatment of cancers have emerged. Peptide-drug conjugates (PDCs) represent an important therapeutic strategy for increasing tumor penetration and selectivity. We previously reported that M, which is extracted from bee venom, targets M2 macrophages and improves tumor treatment in lung cancer. M is an amphipathic peptide with 26 amino acid residues (AIGAVLKVLTTGLPALISWIKRKRQQ) that specifically binds to M2-like TAMs. Mertansine (DM1) is a strong cytotoxic agent that interacts with tubulin and inhibits the assembly of tubulin into microtubules. Because it targets microtubules and inhibits cell cycle, its clinical efficacy as a potential anti-cancer agent has been studied. However, meaningful results have yet to be obtained in clinical trials, with patients also suffering several side effects, such as myelosuppression. Antibody drug conjugates (ADCs) are currently the most successful type of drug conjugates, consisting of antibody (targeting), linker (linking the antibody to the payload), and Payload (killing target cells). Payload is a cytotoxic compound and is divided into microtubule inhibitors, such as DM1, and DNA-damaging agents, such as anthracyclines. DM1 alone has not been developed as a drug but is currently used as an antibody-drug conjugate. It has been reported that the peptide LLC2B combined with DM1 exerts an anticancer effect in breast and esophageal squamous cell carcinoma, suggesting it could be developed as a potential PDC for tumor treatment. In the present study, we investigated the anti-cancer effects of M-DM1, which was synthesized using M as a carrier for targeting TAMs and DM1 as a payload. We found that M-DM1 exerts its therapeutic effects by specifically depleting M2-like TAMs in a melanoma mouse model. Through the regulation of M2-like TAMs, M-DM1 induced a significant increase in the infiltration of effector cells, such as CD8 T cells and natural killer (NK) cells, into the TME. Collectively, our results suggest that depleting M2-like TAMs in the TME by treatment with M-DM1 has immunotherapeutic effects in malignant melanoma.
Click to Show/Hide
|
||||
| Description |
To evaluate the selective cytotoxicity of M-DM1 against M0, M1, and M2 macrophages and TAMs, the THP-1-derived macrophages were treated with different concentrations of M, DM1, and M-DM1 (0.05-10 uM) after differentiation into M0, M1, M2, and TAMs. A cytotoxicity assay was performed using the MTS assay. The half-maximal inhibitory concentrations (IC50) of M-DM1 were 2.104, 2.457, 1.488, and 1.042 uM for M0, M1, M2 macrophages, and TAMs, respectively. M-DM1 induced the apoptosis of M2-like TAMs at low concentrations compared to that observed with M2 macrophages, whereas DM1 alone did not have a cytotoxic effect on any of the macrophages.
Click to Show/Hide
|
||||
| In Vitro Model | Melanoma | Tumor-associated macrophages | Homo sapiens | ||
| Experiment 2 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 1.488 µM | |||
| Evaluation Method | MTS assay | ||||
| MOA of PDC |
Recently, peptides that offer versatility in drug discovery for the successful treatment of cancers have emerged. Peptide-drug conjugates (PDCs) represent an important therapeutic strategy for increasing tumor penetration and selectivity. We previously reported that M, which is extracted from bee venom, targets M2 macrophages and improves tumor treatment in lung cancer. M is an amphipathic peptide with 26 amino acid residues (AIGAVLKVLTTGLPALISWIKRKRQQ) that specifically binds to M2-like TAMs. Mertansine (DM1) is a strong cytotoxic agent that interacts with tubulin and inhibits the assembly of tubulin into microtubules. Because it targets microtubules and inhibits cell cycle, its clinical efficacy as a potential anti-cancer agent has been studied. However, meaningful results have yet to be obtained in clinical trials, with patients also suffering several side effects, such as myelosuppression. Antibody drug conjugates (ADCs) are currently the most successful type of drug conjugates, consisting of antibody (targeting), linker (linking the antibody to the payload), and Payload (killing target cells). Payload is a cytotoxic compound and is divided into microtubule inhibitors, such as DM1, and DNA-damaging agents, such as anthracyclines. DM1 alone has not been developed as a drug but is currently used as an antibody-drug conjugate. It has been reported that the peptide LLC2B combined with DM1 exerts an anticancer effect in breast and esophageal squamous cell carcinoma, suggesting it could be developed as a potential PDC for tumor treatment. In the present study, we investigated the anti-cancer effects of M-DM1, which was synthesized using M as a carrier for targeting TAMs and DM1 as a payload. We found that M-DM1 exerts its therapeutic effects by specifically depleting M2-like TAMs in a melanoma mouse model. Through the regulation of M2-like TAMs, M-DM1 induced a significant increase in the infiltration of effector cells, such as CD8 T cells and natural killer (NK) cells, into the TME. Collectively, our results suggest that depleting M2-like TAMs in the TME by treatment with M-DM1 has immunotherapeutic effects in malignant melanoma.
Click to Show/Hide
|
||||
| Description |
To evaluate the selective cytotoxicity of M-DM1 against M0, M1, and M2 macrophages and TAMs, the THP-1-derived macrophages were treated with different concentrations of M, DM1, and M-DM1 (0.05-10 uM) after differentiation into M0, M1, M2, and TAMs. A cytotoxicity assay was performed using the MTS assay. The half-maximal inhibitory concentrations (IC50) of M-DM1 were 2.104, 2.457, 1.488, and 1.042 uM for M0, M1, M2 macrophages, and TAMs, respectively. M-DM1 induced the apoptosis of M2-like TAMs at low concentrations compared to that observed with M2 macrophages, whereas DM1 alone did not have a cytotoxic effect on any of the macrophages.
Click to Show/Hide
|
||||
| In Vitro Model | Human monocytic leukemia | THP-1 M2 type macrophages | CVCL_0006 | ||
| Experiment 3 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 2.104 µM | |||
| Evaluation Method | MTS assay | ||||
| MOA of PDC |
Recently, peptides that offer versatility in drug discovery for the successful treatment of cancers have emerged. Peptide-drug conjugates (PDCs) represent an important therapeutic strategy for increasing tumor penetration and selectivity. We previously reported that M, which is extracted from bee venom, targets M2 macrophages and improves tumor treatment in lung cancer. M is an amphipathic peptide with 26 amino acid residues (AIGAVLKVLTTGLPALISWIKRKRQQ) that specifically binds to M2-like TAMs. Mertansine (DM1) is a strong cytotoxic agent that interacts with tubulin and inhibits the assembly of tubulin into microtubules. Because it targets microtubules and inhibits cell cycle, its clinical efficacy as a potential anti-cancer agent has been studied. However, meaningful results have yet to be obtained in clinical trials, with patients also suffering several side effects, such as myelosuppression. Antibody drug conjugates (ADCs) are currently the most successful type of drug conjugates, consisting of antibody (targeting), linker (linking the antibody to the payload), and Payload (killing target cells). Payload is a cytotoxic compound and is divided into microtubule inhibitors, such as DM1, and DNA-damaging agents, such as anthracyclines. DM1 alone has not been developed as a drug but is currently used as an antibody-drug conjugate. It has been reported that the peptide LLC2B combined with DM1 exerts an anticancer effect in breast and esophageal squamous cell carcinoma, suggesting it could be developed as a potential PDC for tumor treatment. In the present study, we investigated the anti-cancer effects of M-DM1, which was synthesized using M as a carrier for targeting TAMs and DM1 as a payload. We found that M-DM1 exerts its therapeutic effects by specifically depleting M2-like TAMs in a melanoma mouse model. Through the regulation of M2-like TAMs, M-DM1 induced a significant increase in the infiltration of effector cells, such as CD8 T cells and natural killer (NK) cells, into the TME. Collectively, our results suggest that depleting M2-like TAMs in the TME by treatment with M-DM1 has immunotherapeutic effects in malignant melanoma.
Click to Show/Hide
|
||||
| Description |
To evaluate the selective cytotoxicity of M-DM1 against M0, M1, and M2 macrophages and TAMs, the THP-1-derived macrophages were treated with different concentrations of M, DM1, and M-DM1 (0.05-10 uM) after differentiation into M0, M1, M2, and TAMs. A cytotoxicity assay was performed using the MTS assay. The half-maximal inhibitory concentrations (IC50) of M-DM1 were 2.104, 2.457, 1.488, and 1.042 uM for M0, M1, M2 macrophages, and TAMs, respectively. M-DM1 induced the apoptosis of M2-like TAMs at low concentrations compared to that observed with M2 macrophages, whereas DM1 alone did not have a cytotoxic effect on any of the macrophages.
Click to Show/Hide
|
||||
| In Vitro Model | Human monocytic leukemia | THP-1 M0 type macrophages | CVCL_0006 | ||
| Experiment 4 Reporting the Activity Data of This PDC | [1] | ||||
| Indication | Melanoma | ||||
| Efficacy Data | Half Maximal Inhibitory Concentration (IC50) | 2.457 µM | |||
| Evaluation Method | MTS assay | ||||
| MOA of PDC |
Recently, peptides that offer versatility in drug discovery for the successful treatment of cancers have emerged. Peptide-drug conjugates (PDCs) represent an important therapeutic strategy for increasing tumor penetration and selectivity. We previously reported that M, which is extracted from bee venom, targets M2 macrophages and improves tumor treatment in lung cancer. M is an amphipathic peptide with 26 amino acid residues (AIGAVLKVLTTGLPALISWIKRKRQQ) that specifically binds to M2-like TAMs. Mertansine (DM1) is a strong cytotoxic agent that interacts with tubulin and inhibits the assembly of tubulin into microtubules. Because it targets microtubules and inhibits cell cycle, its clinical efficacy as a potential anti-cancer agent has been studied. However, meaningful results have yet to be obtained in clinical trials, with patients also suffering several side effects, such as myelosuppression. Antibody drug conjugates (ADCs) are currently the most successful type of drug conjugates, consisting of antibody (targeting), linker (linking the antibody to the payload), and Payload (killing target cells). Payload is a cytotoxic compound and is divided into microtubule inhibitors, such as DM1, and DNA-damaging agents, such as anthracyclines. DM1 alone has not been developed as a drug but is currently used as an antibody-drug conjugate. It has been reported that the peptide LLC2B combined with DM1 exerts an anticancer effect in breast and esophageal squamous cell carcinoma, suggesting it could be developed as a potential PDC for tumor treatment. In the present study, we investigated the anti-cancer effects of M-DM1, which was synthesized using M as a carrier for targeting TAMs and DM1 as a payload. We found that M-DM1 exerts its therapeutic effects by specifically depleting M2-like TAMs in a melanoma mouse model. Through the regulation of M2-like TAMs, M-DM1 induced a significant increase in the infiltration of effector cells, such as CD8 T cells and natural killer (NK) cells, into the TME. Collectively, our results suggest that depleting M2-like TAMs in the TME by treatment with M-DM1 has immunotherapeutic effects in malignant melanoma.
Click to Show/Hide
|
||||
| Description |
To evaluate the selective cytotoxicity of M-DM1 against M0, M1, and M2 macrophages and TAMs, the THP-1-derived macrophages were treated with different concentrations of M, DM1, and M-DM1 (0.05-10 uM) after differentiation into M0, M1, M2, and TAMs. A cytotoxicity assay was performed using the MTS assay. The half-maximal inhibitory concentrations (IC50) of M-DM1 were 2.104, 2.457, 1.488, and 1.042 uM for M0, M1, M2 macrophages, and TAMs, respectively. M-DM1 induced the apoptosis of M2-like TAMs at low concentrations compared to that observed with M2 macrophages, whereas DM1 alone did not have a cytotoxic effect on any of the macrophages.
Click to Show/Hide
|
||||
| In Vitro Model | Human monocytic leukemia | THP-1 M1 type macrophages | CVCL_0006 | ||
References